
Abstract
Neuroblastoma (NB) is the most common extracranial solid tumor in children. Combining passive immunotherapy with an antibody to the disialoganglioside GD2 (ch14.18/SP2/0) and cytokines with 13-cis-retinoic acid for post-myeloablative maintenance therapy increased survival in high-risk NB, but the overall prognosis for these children is still in need of improvement. Fenretinide (4-HPR) is a synthetic retinoid that has shown clinical activity in recurrent NB and is cytotoxic to a variety of cancer cells, in part via the accumulation of dihydroceramides, which are precursors of GD2. We investigated the effect of 4-HPR on CHO-derived, ch14.18-mediated anti-NB effector functions, complement-dependent cytotoxicity (CDC), and antibody-dependent and antibody-independent cellular cytotoxicity (ADCC and AICC, respectively). Here, we demonstrate for the first time that pretreatment of fenretinide-resistant NB cells with 4-HPR significantly enhanced ch14.18/CHO-mediated CDC and ADCC and AICC by both human natural killer cells and peripheral blood mononuclear cells. Treatment with 4-HPR increased GD2 and death receptor (DR) expression in resistant NB cells and induced an enhanced granzyme B and perforin production by effector cells. Blocking of ganglioside synthesis with a glucosylceramide synthase inhibitor abrogated the increased ADCC response but had no effect on the AICC, indicating that GD2 induced by 4-HPR mediates the sensitization of NB cells for ADCC. We also showed that 4-HPR induced increased GD2 and DR expression in a resistant NB xenograft model that was associated with an increased ADCC and AICC response using explanted tumor target cells from 4-HPR-treated mice. In summary, these findings provide an important baseline for the combination of 4-HPR and passive immunotherapy with ch14.18/CHO in future clinical trials for high-risk NB patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuroblastoma (NB) is one of the most aggressive cancers of childhood, accounting for 9 % of all childhood malignancies [1]. The tumor originates from neural crest cells and expresses the ganglioside GD2. In a recent phase III study in high-risk NB patients, immunotherapy with anti-GD2 antibody ch14.18 (SP2/0) and cytokines revealed a significant increase in the 2-year event-free survival (EFS), rising from 46 to 66 % [2]. Despite this success, there is a need to further improve survival rates since more than one third of high-risk NB patients will not survive the disease. One approach is to evaluate combinations of immunotherapeutic agents with established drugs or new agents that demonstrate additive or synergistic interactions with antibody and cytokines in preclinical models.
The ganglioside GD2 is a glycolipid antigen abundantly expressed by NB [3]. The fact that its natural expression is restricted to tissue of neuroectodermal origin such as melanocytes, peripheral nerve fibers, and neurons [4] makes GD2 an interesting target for passive immunotherapy, and several antibody strategies have been tested in clinical trials over the past decade [5, 6]. As described above, the chimeric antibody ch14.18/SP2/0 demonstrated efficacy against minimal residual disease in a recent phase III trial of high-risk NB patients post-myeloablative therapy [2].
One important mechanism of antibody-based immunotherapy is tumor-directed antibody-dependent cell-mediated cytotoxicity (ADCC), which is mediated by CD16 (Fcγ receptor III) expressed on effector cells of the innate immune system, primarily by natural killer (NK) cells. A second antibody-independent cellular cytotoxicity (AICC) mechanism also plays an important role in NK cell-mediated elimination of tumor cells and pathogens supporting ADCC. AICC by NK cells is initiated via the interaction of NKG2D, a C-type lectin-like receptor, activating killer cell immunoglobulin-like receptors (KIR), and/or natural cytotoxicity receptors with their respective ligands. It is also tightly regulated by the presence of inhibitory KIRs and their ligands on a target cell. An activated NK cell can induce target cell killing through the release of lytic granules (granzymes and perforin). While perforin is presumably responsible for pore formation in the target cell membrane, the main component of lytic granules, granzyme B (GrB), is the inductor of apoptosis. GrB induces two different apoptotic pathways in target cells: first, GrB mediates a direct cleavage and activation of caspases, and second, it triggers mitochondrial permeabilization followed by cytochrome c release [7].
Another mechanism of cell killing is mediated via the interaction of death receptors (DRs) with the Fas ligand or tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) expressed by NK cells. DRs belong to the TNF receptor superfamily. At least five different DRs are identified: Fas (or CD95), TRAIL receptors 1 and 2 (or DR4 and DR5, respectively), DR3, and TNF receptor [8]. Binding of TNF superfamily ligands to their receptors induces receptor trimerization and recruitment of the Fas-associated death domain, which subsequently recruits procaspase-8 to form the death-inducing signaling complex. This association of proteins activates caspase-8, which subsequently cleaves caspase-3 to induce a nonmitochondrial apoptotic pathway [9].
Fenretinide or N-(4-hydroxyphenyl)retinamide (4-HPR) is a synthetic derivative of vitamin A that has shown antitumor activity with minimal systemic toxicity in preclinical and clinical studies [10, 11]. One mechanism by which 4-HPR induces cytotoxicity in cancer cells is via intracellular accumulation of ceramides as a result of de novo synthesis, especially dihydroceramides [12]. However, a common evasion mechanism of multidrug-resistant cancer cells to prevent the accumulation of ceramides is the upregulation of enzymes involved in their cellular clearance, such as sphingosine kinase (converts sphingosine into sphingosine-1-phosphate) [13, 14], sphingomyelin synthase (converts ceramide into sphingomyelin), and glucosylceramide synthase (GCS) [15–18], which catalyzes the glycosylation of ceramide, leading to glucosylceramide, the precursor for glycosphingolipids, i.e., gangliosides. Interestingly, GD2 is a ganglioside directly generated from GD3, a metabolite of ceramides, and increased GD2 expression was demonstrated in NB cell lines after treatment with 4-HPR in vitro [18]. Furthermore, 4-HPR treatment led to an upregulation of DRs in Ewing sarcoma, which subsequently enhanced apoptosis induced by DR ligands [19]. These findings suggest a potential for 4-HPR to improve ADCC and AICC.
Here, we demonstrate for the first time that both ADCC mediated by CHO-derived ch14.18 and AICC mediated by NK cells could be increased significantly by 4-HPR treatment in vitro and in vivo, suggesting that a direct combination of ch14.18/CHO immunotherapy with 4-HPR could be beneficial for NB patients.
Material and methods
Cell culture
4-HPR-resistant (IC90 > 6.2 μM) GD2+ NB cell lines (CHLA-15, CHLA-20, CHLA-79, CHLA-136, and CHLA-172) [20] were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Hyclone Laboratories, Logan, UT, USA), completed with 20 % fetal bovine serum (FBS; Gibco, Invitrogen, Carlsbad, CA, USA), l-glutamine (4 mM; CellGro, Mediatech Inc., Manassas, VA, USA), insulin (5 mg/ml), transferrin (5 mg/ml), and selenious acid (5 ng/ml) (all from ITS Culture Supplement, BD Biosciences, San Jose, CA, USA). SK-N-BE(2) was cultured in RPMI 1640 with 10 % FBS. All cell lines were cultured at 37 °C in a humidified incubator (5 % O2 and 5 % CO2). Cell line identities were confirmed by short tandem repeat profiling by the Children’s Oncology Group Cell Culture and Xenograft Repository (http://www.COGcell.org) as previously described [21].
Peripheral blood mononuclear cells (PBMCs) were isolated from the blood of healthy volunteers, obtained with written informed consent, using Ficoll-Paque™ PLUS (GE Healthcare, Pittsburgh, PA, USA). PBMCs were washed and plated out into cell culture flasks and cultured for 3 days with 100 IU/ml interleukin-2 (IL-2) (Aldesleukin, Novartis, Emeryville, CA, USA) at 37 °C (5 % O2 and 5 % CO2) before use in cytotoxicity assays.
NK cells were expanded from the PBMCs isolated from a healthy donor as previously described [22]. Briefly, PBMCs were stimulated weekly with irradiated (100 Gy) K562 Cl9mIL21 at an effector-to-target (E/T) ratio of 1:2 in NK cell expansion media [comprised of RPMI, 10 % FBS, 100 IU/ml penicillin, 100 μg/ml streptomycin (all from Cellgro), 2 mM l-glutamine (Gibco), and 50 IU/ml of IL-2 (Proleukin, Novartis Vaccines and Diagnostics, Inc.)] at 37 °C in 5 % CO2 to selectively expand NK cells. At the end of two stimulations, NK cells were isolated using RosetteSep Human NK Cell Enrichment Cocktail (Stemcell Technologies) [23] and expanded further for 1 week following a third stimulation with irradiated K562 Cl9mIL21. The NK cells were phenotyped to assess purity by flow cytometry (BD FACSCalibur) using anti-CD3, anti-CD16, and anti-CD56 antibodies (BD Biosciences) and then cryopreserved. RosetteSep purification depletes CD3 cells and purified, expanded NK cells were <1 % CD3+. Expanded frozen NK cells were cultured in RPMI 1640 including 10 % FBS and 100 IU/ml IL-2 for 4–5 days prior to a cytotoxicity assay.
Experiments involving human specimens were approved by the Institutional Review Board of the Texas Tech University Health Sciences Center and/or the University of Texas MD Anderson Cancer Center.
Drug treatment
For all assays, cells were pretreated with either 4-HPR or D/L-Threo-1-phenyl-2-hexadecanoylamino-3-pyrrolidino-1-propanol (PPPP) (Matreya Inc., Pleasant Gap, PA, USA) or both. Drugs were diluted in 100 % ethanol. NB cells were plated out at a concentration of 5–10 × 105 into six-well plates in 3 ml culture medium and allowed to attach for 24 h. 4-HPR was tested in two different concentrations (5 or 10 μM) and cells were incubated at 37 °C in a humidified incubator (5 % O2; 5 % CO2; 48 h). 4-HPR concentrations of up to 10 μM are clinically achievable; however, since drug concentrations in human sera often vary depending on the patient, we additionally used a lower dose (5 μM) in order to provide for a broader range of effective concentrations [24]. In order to block GCS function, cells were treated with 1 μM PPPP for 4–5 days as previously described [25]. Dead cells in the supernatant were removed by pipetting and gentle washing with medium and adherent cells were counted and only cell populations with >95 % viable cells, as determined by trypan blue staining, were used for further analysis. 4-HPR for in vivo studies was employed in the Lym-X-Sorb oral powder formulation (4-HPR-LXS). 4-HPR-LXS provides an optimal method for achieving high plasma and tissue levels of 4-HPR in mice and has achieved clinical responses in recurrent high-risk NB [26]. ch14.18/CHO is a mouse/human chimeric anti-GD2 antibody. It was manufactured in CHO cells by Polymun (Vienna, Austria) and is used in clinical phase I to III trials in Europe. The antibody was kindly provided by the SIOPEN group (International Society of Pediatric Oncology European Neuroblastoma).
Flow cytometric analysis
GD2 expression on NB cells was analyzed by staining of the cells for 15 min with 1 μg ch14.18/CHO followed by incubation for 15 min with a phycoerythrin (PE)-labeled anti-human IgG secondary antibody at a dilution of 1:5 (cat. no. 555787, BD Biosciences, San Jose, CA, USA). As an isotype control, we used chimeric anti-CD20 antibody (rituximab; MabThera, Roche, Germany). From live and PE-labeled cells, we calculated the GD2 binding index (BI; which represents the ability of anti-GD2 antibody ch14.18/CHO to bind to its antigen) for basal cell surface expression and for expression after treatment with 4-HPR based on the percentage of GD2-positive cells and mean fluorescence (mean fluorescence × n% positive cells/100) [27].
DRs were stained with the following antibodies for 45 min: 0.5 μg anti-CD95 (Fas) (cat. no. MC-063, Kamiya Biomedical, Seattle, WA, USA), 0.5 μg anti-TRAIL-R1 (cat. no. ALX-804-297), and 1 μg anti-TRAIL-R2 (cat. no.ALX-804-298, both from Enzo Life Sciences, Plymouth Meeting, PA, USA). As a secondary antibody, we used a PE-labeled rat anti-mouse IgG1 antibody in a dilution of 1:200 (cat. no. 550083, BD Biosciences) for 20 min.
To exclude dead cells from the analysis, cells were stained with 4 μl 4′,6-diamidino-2-phenylindole (DAPI; 0.1 μg/ml), which was added shortly before analysis. For each cell sample, 20,000 live cells were analyzed by the LSRII flow cytometer using FACSDiva software (BD Biosciences). Final data analysis was done with FlowJo software (TreeStar, Ashland, OR, USA). From live and PE-labeled cells, we calculated the binding indices as described above.
Cytotoxicity assays
All assays were performed using the CytoTox 96 nonradioactive cytotoxicity assay from Promega (Madison, WI, USA), according to the manufacturer’s protocol. Each experiment including spontaneous and maximum release determination and controls was performed in eight replicates in a U-bottom microtiter plate (BD Biosciences). Absorbance was measured at 490 nm using a standard microplate reader (Bio-Rad Benchmark Microplate Reader). To induce complement-dependent cytotoxicity (CDC) and ADCC, ch14.18/CHO was used in a concentration of 1 or 10 μg/ml.
CDC
Nonheat-inactivated, frozen serum from one healthy donor was added to RPMI medium (10 % final serum concentration) and target cells were used in a concentration of 1 × 104 cells in 100 μl. Antibody ch14.18/CHO and anti-CD20 control (MabThera, Roche, Germany) were added in a final concentration of 10 μg/ml and plates were incubated at 37 °C (5 % O2 and 5 % CO2) for 6 h. Lysis solution was added after 5 h and 15 min.
ADCC
Briefly, effector cells (PBMCs or expanded NK cells) were harvested and plated at an E/T ratio of 25:1 (for PBMCs) and 5:1 or 2.5:1 (for NK cells) in 75 μl RPMI supplemented with 5 % heat-inactivated FBS. Target cells were added at a concentration of 1 × 104 cells in 100 μl RPMI (10 % FBS). Antibody ch14.18/CHO and anti-CD20 control were added in a final concentration of 1 μg/ml and plates were incubated at 37 °C (5 % O2 and 5 % CO2) for 4 h. Lysis solution was added after 3 h and 15 min.
AICC
The same procedure was used as described for ADCC, but we added medium or medium/rituximab instead of ch14.18/CHO antibody.
Blocking of death receptors in AICC
We used 1 × 106 target cells in 100 μl that were preincubated with 1 μg anti-TRAIL-R2 (cat. no. ALX-804-298, from Enzo Life Sciences, Plymouth Meeting, PA, USA) and anti-CD95 (cat. no. MC-117, Kamiya Biomedical) for 1 h on ice. The antibody concentrations were employed according to the manufacturer’s recommendations. To block Fc receptors on expanded NK cells, we used 10 μl human FcR Blocking Reagent (Miltenyi) per 2 × 106 cells in 100 μl FACS buffer. The mixture was incubated on ice for 10–15 min. Effector and target cells were washed twice to remove residual antibodies and then employed in an AICC assay, as described above, in an E/T ratio of 2.5:1.
Ex vivo ADCC/AICC
Tumor cells were isolated from tumors with a volume of 1,200–1,400 mm3 by using a 70-μm cell strainer (cat. no. 352350, BD Bioscience). Suspended tumor cells were washed twice in complete IMDM and red blood cells were removed by incubation with ACK lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA in distilled water; all Sigma-Aldrich) for 5 min at room temperature (RT). Cells were washed twice in complete IMDM and dead cells were removed by density gradient centrifugation using Ficoll (400×g without brake for 30 min at 4 °C). Target cells were then employed in an ADCC/AICC assay as described above. Cytotoxicity was calculated according to the following formulas:
Granzyme B and perforin ELISA
For measuring GrB and perforin release from effector (NK) cells upon coculture with fenretinide-treated NB cells, cells were incubated for 6 h in a 24-well plate at an E/T ratio of 2.5:1. We used 1 × 105 NB cells in 350 μl RPMI complete medium and accordingly 2.5 × 105 NK cells (in 350 μl) in each well. To measure spontaneous GrB and perforin release by effector cells, we incubated NK cells alone in the same volume that was used in the other wells (700 μl). As a positive control, NK cells were activated with 10 ng/ml PMA (Calbiochem, EMD Biosciences Inc., La Jolla, CA, USA) and 1 μg/ml ionomycin (Sigma Aldrich, St. Louis, MO, USA). The enzyme-linked immunosorbent assay (ELISA) was performed with standards and samples in duplicates according to the manufacturer’s protocol (Human Granzyme B and Human Perforin ELISA Kit, MabTech, Sweden). Briefly, on day 1, a high protein-binding ELISA plate was coated with anti-GrB or anti-perforin antibody (1.5 and 4 μg/ml, respectively) in phosphate-buffered saline (PBS) and incubated overnight at 4 °C. Then, the plate was washed twice with washing buffer (0.05 % Tween 20 in PBS) followed by blocking with assay diluent (PBS containing 0.1 % BSA) for 1 h at RT. The plate was washed five times with washing buffer. One hundred microliters of standards and samples diluted in assay diluent were added and incubated for 2 h at RT. After washing five times, biotinylated monoclonal antibody (0.5 μg/ml) was added to each well and incubated for 1 h at RT. After washing, the plate was incubated with streptavidin-HRP (1:1,000 in assay diluent) for 1 h at RT. The plate was washed again five times and 75 μl/well of substrate reagent (R&D Systems, Color Reagent A and Color Reagent B mixed in equal volumes) was added and incubated for 5–20 min in the dark. After a suitable developing time, 75 μl/well of stop solution (2 N H2SO4) was added and optical density was measured at 450 nm using a standard microplate reader. Antibody-dependent GrB and perforin release was calculated by subtracting the spontaneous release from the experimental release values.
Western blot analysis
NB cells (5 × 105 per well) were plated in complete medium (3 ml) into flat-bottom six-well plates. Cells were exposed to 4-HPR or PPPP alone or in combination as described above. Cells were harvested, washed twice (PBS), and centrifuged. Protein content was isolated as previously described [28]. Forty micrograms of cell protein was mixed with 4× SDS Sample Buffer (Novagen, San Diego, CA, USA) and heated for 3 min at 96 °C. Proteins were resolved by electrophoresis on 10–20 % triethanolamine sodium dodecyl sulfate precast gel (C.B.S. Scientific, Del Mar, CA, USA) and transferred onto polyvinylidene difluoride membrane (Protran, Keene, NH, USA). For the detection of GCS, we incubated the membrane overnight at 4 °C with a monoclonal mouse anti-human GCS antibody (1:200; Abnova, Taipei City, Taiwan) in blocking solution (5 % nonfat dry milk (Bio-Rad Laboratories), 0.1 % v/v Tween 20 in PBS). For FLICE-inhibitory protein (FLIP) detection, the membrane was incubated overnight at 4 °C with a monoclonal anti-human FLIP antibody (1:400, cat. no. ALX-804-428-C100 Enzo Life Science, Lörrach, Germany) in blocking solution (Roti®-Block (Roth, Karlsruhe, Germany), 0.1 % v/v Tween 20 in PBS). For the detection of GCS, a secondary peroxidase-conjugated goat anti-mouse IgG1 antibody (1:2,000, GE Healthcare, Amersham, Buckinghamshire, UK) was used. For the detection of FLIP, a secondary peroxidase-conjugated goat anti-mouse IgG1 antibody (1:3,000, cat. no. 166-2408EDU, Bio-Rad Laboratories, Munich, Germany) was used. Both reactions were visualized using the enhanced chemiluminescence detection system from Amersham (Buckinghamshire, UK) according to the manufacturer’s protocol. As a control, we employed a monoclonal mouse anti-β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and a peroxidase-conjugated goat anti-mouse IgG1 (Amersham, Buckinghamshire, UK).
Mouse xenograft experiments
Female Fox Chase SCID mice (CB17/Icr-Prkdc scid/IcrIcoCrl) were obtained at 4–6 weeks of age from Charles River (strain code 236) and maintained under specific pathogen-free conditions. Housing and treatment protocols were approved by the Institutional Animal Care and Use Committee of our institution. For tumor induction, 1 × 107cells CHLA-136 cells were injected subcutaneously in 200 μl. Shortly before injection, the cell suspension was diluted 1:2 with Matrigel™ Basement Membrane Matrix (BD Biosciences, Bedford, MA, USA). Tumors were measured every second day using a microcaliper and tumor volumes were calculated according to the formula:\( {{1} \left/ {2} \right.}{\mathrm{width}} \times {\mathrm{length}} \times {\mathrm{width}} \). When the tumors reached a volume of 150–200 mm3, mice were treated with a daily oral gavage of 4-HPR-LXS oral powder suspended in water (240 mg/kg/day) or sterile water (controls) for 5 days. After a 2-day break, treatment continued for 5 days. Animals were sacrificed by carbon dioxide necrosis when tumor volumes reached 1,300 mm3. Tumors were extracted and processed for the following ex vivo assays.
Analysis of tumor-infiltrating NK cells
To determine whether 4-HPR treatment has an effect on the amount of tumor-infiltrating NK cells, we isolated all live cells from tumors as described above in the ex vivo cytotoxicity assay section. Cells (1 × 106) were stained with 1 μl of an anti-mouse NK1.1 fluorescein isothiocyanate (FITC)-conjugated antibody (cat. no. 553164, BD Biosciences) for 45 min. As an isotype control, we used an FITC-labeled mouse IgG2A, K isotype antibody (cat. no. 553456, BD Biosciences). After incubation with the antibody, cells were washed two times, counterstained with DAPI as described above, and analyzed by flow cytometry.
Statistical analysis
All experiments were verified at least three times. Data are expressed as the means ± standard deviations (SD). Statistical analysis was performed using Student’s t test. Differences were considered significant at a p value of <0.05 (*) and highly significant at p values <0.01 (**) or <0.005 (***).
Results
GD2 expression of 4-HPR-resistant human NB cells increases upon treatment with 4-HPR
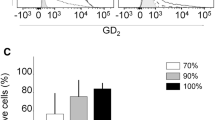
Since fenretinide induces (dihydro)ceramide accumulation [12], which is a ganglioside precursor, we analyzed the GD2 expression upon treatment with 4-HPR in 4-HPR-resistant NB cells. We found an increase of the GD2 BI in all six human 4-HPR-resistant cell lines (CHLA-136, CHLA-172, SK-N-BE(2), CHLA-79, CHLA-15, and CHLA-20). The 4-HPR-induced average increase of the GD2 BI, compared to vehicle-treated controls, was calculated from five independent experiments for each cell line (Fig. 1). Interestingly, in cell lines with a more heterogeneous basal expression of GD2 (CHLA-136 and SK-N-BE(2)), 4-HPR induced an increase in the percentage of GD2-positive cells (data not shown) and in the mean fluorescence intensity, resulting in a 50 to >80 % increase of the average GD2 BI, respectively. In case of cell lines with high and homogeneous expression of GD2 (e.g., CHLA-15 and CHLA-20, basal expression of GD2 was 92–95 %), treatment with 4-HPR induced a 10–20 % increase in the GD2 BI, which is mostly dependent on an increase in the mean fluorescence. Thus, NB cell lines having the lowest basal levels of GD2 expression demonstrated the greatest 4-HPR-mediated increase in GD2 expression.

GD2 expression of 4-HPR-resistant human NB cells increases upon treatment with 4-HPR. GD2 expression was analyzed in 4-HPR-resistant NB cells after exposure to 10 μM 4-HPR (5 μM for CHLA-15) for 48 h as described in the “Material and methods” section. Cells were stained with ch14.18/CHO (1 μg) and results were compared to rituximab (1 μg) as isotype control. GD2 BI was calculated according to the formula: \( {{{\left( {{\mathrm{mean}}\;{\mathrm{fluorescence}} \times \% \;{\mathrm{GD}2}\;{\mathrm{positive}}\;{\mathrm{cells}}} \right)}} \left/ {{{1}00}} \right.} \). Results are presented as percentage of increase of GD2 BI of 4-HPR-treated cells from vehicle control. Data represent the mean increase and SD of five independent experiments. *p < 0.05, calculated using the Student’s t test
4-HPR enhances ch14.18/CHO-mediated CDC, ADCC, and AICC of 4-HPR-resistant human NB cells
CDC was analyzed after 4-HPR treatment using five 4-HPR-resistant NB cell lines (CHLA-136, CHLA-172, SK-N-BE(2), CHLA-79, and CHLA-20). Cells were treated with 5 μM 4-HPR for 48 h, resulting in an increase of CDC with ch14.18/CHO of up to 45 % (CHLA-136, CHLA-172, and SK-N-BE(2)) and up to 25 % (CHLA-79 and CHLA-20) compared to vehicle controls (Fig. 2a and Supplementary Fig. S3), while isotype control rituximab did not elicit a CDC response at all. Interestingly, the level of ch14.18/CHO-mediated CDC correlated with the GD2 BI (Fig. 1): CHLA-136, CHLA-172, and SK-N-BE(2) showed the highest increase in CDC compared to the other cell lines tested, i.e., CHLA-20 and CHLA-79. We were able to confirm this result with serum from another donor as complement source and 1 μg/ml ch14.18/CHO (data not shown); both antibody concentrations are achievable in a clinical setting ([29] and H.N. Lode, personal communication).

Fenretinide treatment enhances ch14.18/CHO-mediated CDC and ADCC by PBMCs towards 4-HPR-resistant human NB cell lines. The effect of 4-HPR on ch14.18/CHO-mediated CDC by human complement and ADCC by PBMCs was analyzed using human NB target cells after exposure to 4-HPR. ADCC with healthy donor PBMCs was analyzed at an E/T ratio of 25:1 in the presence of ch14.18/CHO antibody (1 μg/ml). a CDC was performed with 10 % of human serum in the presence of ch14.18/CHO or isotype control rituximab (10 μg/ml each) and NB cells previously treated with 5 μM 4-HPR for 48 h. Each bar represents the mean value for eight replicates. The results from one representative experiment out of three independent experiments are presented as mean percent CDC ± SD. ***p < 0.001, **p < 0.01, statistically significant differences of ch14.18/CHO-mediated CDC and controls (Student’s t test). b Varying human NB cell lines were exposed to vehicle (black bar) or 10 μM 4-HPR (gray bar) for 48 h and then used in an ADCC assay with PBMCs from donor 1. c Varying donors were used as effector cells after treatment of CHLA-20 with vehicle (black bar) or 10 μM 4-HPR (gray bar). Each bar represents the mean value for eight replicates. Results from one representative experiment out of three independent experiments are presented as percent cell lysis by PBMCs ± SD. ***p < 0.001, **p < 0.01, *p < 0.05, statistically significant differential findings between 4-HPR-treated cells and controls (Student’s t test)
Next, we used whole PBMC populations of several healthy donors as effector cells. First, overall cytotoxicity with ch14.18/CHO was analyzed with PBMCs from one healthy donor and cell lines CHLA-20, CHLA-172, SK-N-BE(2), and CHLA-79 treated with 4-HPR (10 μM). All cell lines were lysed more effectively after 4-HPR treatment (Fig. 2b and Supplementary Fig. S2). Importantly, similar effects were observed when we determined the ADCC by PBMCs obtained from four healthy donors towards CHLA-20 cells that were treated with 4-HPR (10 μM) for 48 h. All four donor PBMCs showed an increase in lytic activity towards CHLA-20 after treatment with 4-HPR (Fig. 2c and Supplementary Fig. S2). In order to assess the overall ability of 4-HPR to increase the cytotoxicity of PBMCs from different donors, we pooled our data for four donors from three experiments, respectively, and calculated the percentage of increase of cytotoxicity towards 4-HPR-treated CHLA-20 cells compared to vehicle-treated CHLA-20, revealing an average increase in cytotoxicity of 102 % (±48 % SD, data not shown).
We wanted to confirm the results obtained with different PBMCs with a purified NK cell population from one healthy donor. For this purpose, we evaluated the cytotoxicity of expanded NK cells with ch14.18/CHO (ADCC) and without antibody (AICC) in six NB cell lines (CHLA-136, CHLA-172, SK-N-BE(2), CHLA-79, CHLA-15, and CHLA-20) after exposure to 10 μM 4-HPR for 48 h. Since rituximab did not show any specific activity in CDC assays and rituximab-mediated ADCC of NK cells and PBMCs was identical to controls without any antibody in preliminary experiments (data not shown), we only employed no-antibody controls in the following assays to determine the effect of 4-HPR treatment on ADCC/AICC.
The expanded NK effector cells showed a significantly higher overall lytic activity towards all fenretinide-treated NB cells compared to the vehicle control with an increase in AICC (black bars represent lysis mediated solely by effector cells without the addition of ch14.18 antibody) and an enhanced GD2-specific ADCC (gray bars represent the additional lysis effect occurring when ch14.18 is present) (Fig. 3). In all cell lines, the AICC response was markedly increased. The increase in GD2-specific ADCC was more pronounced in CHLA-136, CHLA-172, and SK-N-BE(2), while in CHLA-79, CHLA-15, and CHLA-20, ADCC mediated by ch14.18/CHO was only slightly higher upon treatment with 4-HPR compared to vehicle-treated cells. This correlates with the limited effect of 4-HPR on GD2 expression in these cells and is similar to observations in CDC experiments. Importantly, the absence of AICC by NK cells towards untreated SK-N-BE(2) was reversed by treatment with 4-HPR, again indicating that there is a second mechanism involved in the sensitization of NB cells to NK cell lysis by 4-HPR treatment. The results for the overall cytotoxicity (ADCC plus AICC) from all experiments with expanded NK cells are additionally shown in Supplementary Fig. S2.

Fenretinide treatment enhances AICC and ch14.18/CHO-mediated ADCC by purified NK cells towards 4-HPR-resistant human NB cell lines. Overall cytotoxicity (AICC + ADCC) was analyzed with expanded NK cells and human NB cells that were treated with 10 μM 4-HPR for 48 h at an E/T ratio of 5:1 in the presence (ADCC, gray part of stacked bar) or absence (AICC, black part of stacked bar) of ch14.18/CHO antibody (1 μg/ml). Each bar represents the mean value for eight replicates. Results are presented as percent cytotoxicity ± SD (from one representative experiment out of three independent experiments). ***p < 0.001, **p < 0.01, *p < 0.05, statistically significant differences in ADCC and AICC with and without 4-HPR treatment (Student’s t test)
Effect of 4-HPR treatment on death receptor expression on neuroblastoma cells
Since 4-HPR treatment increased the susceptibility of human NB cells to effector cell killing by AICC, we examined the expression of DR molecules on our human NB cell lines and found a 4-HPR-mediated increase of Fas and TRAIL-R2 expression compared to vehicle-treated cells. TRAIL-R1 expression was very low or absent in the cell lines tested in our panel (data not shown). Fas expression was also relatively low in all cell lines, but could be enhanced >3-fold in CHLA-15, SK-N-BE(2), and CHLA-136 cells after 4-HPR treatment (Fig. 4a, left panel; b, right panel). Overall, TRAIL-R2 expression was higher in all cell lines compared to Fas, with the highest percentage of positive cells in CHLA-136 and could be enhanced in all cell lines (Fig. 4a, right panel), even in cell lines that showed only very low expression levels (<1 %) when untreated, such as SK-N-BE(2) (Fig. 4b, right panel).

Effect of 4-HPR treatment on Fas and TRAIL-R2 expression on NB cells and on GrB and perforin production by NK effector cells. Fas and TRAIL-R2 expression on NB cells was analyzed by flow cytometry as described in the “Material and methods” section. a Fas and TRAIL-R2 binding indices were calculated and results are presented as fold increase of BI of 4-HPR-treated cells compared to vehicle control. Horizontal line indicates control expression. Data represent the mean increase and SD from five independent experiments. b Left panel inhibition of AICC after DR blocking. NB cells CHLA-136 and SK-N-BE(2) were exposed to 5 μM 4-HPR or vehicle for 48 h. After treatment, cells were harvested and incubated with non-apoptosis-inducing anti-TRAIL-R2 and anti-CD95 (Fas) antibodies for 1 h on ice (1 μg each). The lytic activity of NK cells was assessed at an E/T ratio of 2.5:1. Each bar represents the mean value for eight replicates. Results are presented as percent cell lysis by NK cells of 4-HPR-treated cells (dark gray column) compared to vehicle-treated cells (black column) from one representative experiment out of three independent experiments ± SD. *p < 0.05, statistically significant difference between TRAIL-R2 blocking and AICC in 4-HPR-treated CHLA-136 cells (Student’s t test). Right panel mean Fas and TRAIL-R2 expression levels of CHLA-136 and SK-N-BE(2) in vehicle controls and following 4-HPR treatment, in percent positive cells ± SD. c Left panel GrB production by expanded NK cells upon 6 h co-culture with vehicle-treated (black bars) or 4-HPR-treated (gray bars) NB cells (E/T ratio, 2.5:1) in picograms per milliliter (from one representative experiment out of three independent experiments). Right panel mean percent increase of perforin production by expanded NK cells upon co-incubation (6 h; E/T ratio, 2.5:1) with vehicle-treated or 4-HPR-treated NB cell lines (±SD)
In order to determine a functional role for DR expression after 4-HPR treatment in AICC, we blocked the respective DRs during an AICC assay by using non-apoptosis-inducing blocking antibodies for Fas and TRAIL-R2. AICC was analyzed with CHLA-136 and SK-N-BE(2) cells as a target and expanded human NK effector cells, which were pretreated with FcR block prior to the assay in order to prevent a Fas/TRAIL-R2 antibody-mediated ADCC response. Inhibition of TRAIL-R2 resulted in a significantly decreased AICC in CHLA-136 cells in contrast to Fas in the conditions used in this setting. This result suggests a partial role for a 4-HPR-stimulated TRAIL/TRAIL-R2 pathway as a mechanism for the increased AICC. In SK-N-BE(2) cells that reacted with a significant increase of TRAIL-R2 and Fas expression upon 4-HPR treatment, the effect of DR blocking was not significant, although blocking of TRAIL-R2 was more effective than of Fas (Fig. 4b), which, however, can be explained by the generally low expression of these DR molecules in this cell line (Fig. 4b, right panel). In another set of experiments, we tested whether 4-HPR might be responsible for a change in the expression of the FLICE inhibitory protein cFLIP that is known to be an inhibitor of DR-mediated apoptosis [30, 31] and, thus, might be involved in the sensitization of NB cells towards AICC. However, our experiments did not lead to unanimous results in all six NB cell lines tested (see Supplementary Fig. S5), suggesting that cFLIP does not play a major role in the discovered sensitization effect of 4-HPR.
NK effector cells show enhanced effector molecule production upon co-culture with 4-HPR-treated NB cells
In our attempt to further characterize the mechanisms that underlie the enhanced antibody-independent cell-mediated killing of fenretinide-treated NB cells, we determined the production of effector molecules GrB and perforin by effector cells, i.e., expanded NK cells, upon co-incubation with 4-HPR-pretreated NB cell lines. As exemplified in Fig. 4c, NK cells are stimulated to produce an increased amount of GrB and perforin as a reaction to co-culture with fenretinide-treated cells. Interestingly, this increase is dependent on the NB cell line: While CHLA-136 and CHLA-172 mediated an increase of GrB production by NK cells by >100 % (and >200 % for perforin; Fig. 4c, right panel), the increase induced by CHLA-15 and CHLA-20 is much lower, both for GrB and perforin. We also tested whole PBMCs from healthy donors in this respect and found comparable increases of GrB and perforin, although the strength of GrB and perforin production in general was highly variable depending on the donor (data not shown).
Downregulation of GD2 reverses the 4-HPR-mediated increase in ch14.18/CHO-dependent ADCC and CDC
GCS catalyzes the conversion of ceramides to glucosylceramides, which is the first-step enzyme in GD2 synthesis. Protein expression of GCS showed an apparent increase in 4-HPR-treated CHLA-136 cells relative to vehicle-treated controls (Fig. 5a), while exposure to PPPP during 4-HPR exposure decreased GCS protein expression (Fig. 5a) and resulted in lower GD2 expression levels compared to vehicle-treated cells (Fig. 5b). PPPP inhibited 4-HPR-mediated increases of ch14.18/CHO-mediated CDC (Fig. 5c) and ADCC (10 and 1 μg/ml, respectively) (Fig. 5d and Supplementary Fig. S4, gray part of stacked bars), but not AICC (Fig. 5d and Supplementary Fig. S4, black part of stacked bars), indicating a crucial role for GD2 expression as a mechanism for 4-HPR-mediated sensitization of NB for ch14.18/CHO-mediated immunotherapy.

Inhibition of GD2 expression reverses the 4-HPR-mediated increase in ch14.18/CHO-dependent ADCC and CDC. The role of GD2 in 4-HPR-mediated increase in ch14.18/CHO-dependent ADCC and AICC was analyzed by inhibition of GCS with PPPP. a Effect of 4-HPR and PPPP on GCS expression. CHLA-136 cells were treated with vehicle (ethanol; final concentration, 0.1 %) or 5 μM 4-HPR or 1 μM PPPP/5 μM 4-HPR for 48 h and then used to prepare lysates for Western blot analysis to detect GCS (52 kDa). β-Actin was used as a control for equal protein loading. b PPPP-mediated inhibition of GD2 expression is shown in representative histograms from three independent experiments with vehicle (solid black line), PPPP and 4-HPR (dotted black line, tinted gray curve), and an isotype control (solid black line, filled black curve). c CDC was analyzed with 10 % human serum and NB target cells treated with 4-HPR (5 μM) or 4-HPR/PPPP (5 μM/1 μM) in the presence of ch14.18/CHO or rituximab control (10 μg/ml). Each bar represents the mean value for eight replicates. Results from one representative experiment out of three independent experiments are presented as percentage of cell lysis of target cells ± SD. ***p < 0.001, **p < 0.01, statistically significant differences in CDC with and without 4-HPR treatment (Student’s t test). d Human NB cell line CHLA-136 was treated with 1 μM PPPP/5 μM 4-HPR (right column), vehicle (ethanol; final concentration, 0.1 %; left column) or 5 μM 4-HPR alone for 48 h (middle column). Expanded NK cells were employed as effector cells at an E/T ratio of 5:1 in the absence or presence of ch14.18/CHO antibody (1 μg/ml). Each bar represents the mean value for eight replicates. Results from one representative experiment out of three independent experiments are presented as percent AICC by NK cells (black part of stacked bars) and ch14.18/CHO-mediated lysis (gray part of stacked bars) ± SD. ***p < 0.001, **p < 0.01, statistically significant difference in overall cytotoxicity between 4-HPR/PPPP-treated and 4-HPR-treated cells; *p < 0.05, statistically significant difference in GD2-specific ADCC between groups (Student’s t test; not indicated in the figure)
Effect of 4-HPR treatment on NK cell-mediated killing in neuroblastoma xenografts
To assess whether the observed in vitro effects of fenretinide on NB cells also occurred in vivo, Fox Chase SCID mice with established, palpable CHLA-136 NB xenografts (200 mm3) were assigned to treatment with 240 mg/kg 4-HPR or sterile water (diluent). When tumors reached a volume of 1,300 mm3, mice were terminated and tumor cells were isolated and analyzed for GD2, Fas, and TRAIL-R2 expression by flow cytometry and the binding indices were calculated. Tumors of mice treated with 4-HPR showed a significantly higher GD2 BI compared to tumors of control mice, which increased on average by 65 % (Fig. 6a). Furthermore, both Fas and TRAIL-R2 binding indices were increased in 4-HPR-treated tumors compared to control tumors, by 60 % for Fas and by 25 % for TRAIL-R2 (Fig. 6a). 4-HPR-mediated increase of GD2 and DR expression in vivo also translated into effective cell killing by effector cells. Isolated tumor cells from 4-HPR-treated mice were tested ex vivo in a cytotoxicity assay with expanded human NK cells as effector cells. Both AICC and GD2-specific ADCC by NK cells were increased after treatment of mice with 4-HPR in vivo (Fig. 6b), resulting in a significantly enhanced overall cytotoxicity by NK cells. Most importantly, the GD2-specific ADCC of NB cells isolated from primary tumors (gray part of stacked bars) was much higher than the AICC response (black part of stacked bars), which was reversed in our in vitro experiments (see Fig. 3). Indeed, CHLA-136 cells isolated from primary tumors showed a more homogenous and higher GD2 expression compared to CHLA-136 cells grown in vitro (data not shown).

Effect of 4-HPR treatment on NK cell-mediated killing in NB xenografts. Female Fox Chase SCID mice were inoculated subcutaneously into the right flank with 1 × 107 CHLA-136. Mice with established tumors (200 mm3) were treated with 240 mg/kg 4-HPR as oral LYM-X-SORB formulation or water as described in the “Material and methods” section. GD2, Fas, and TRAIL-R2 expression (a), ex vivo ADCC (b), and NK cell infiltration (c, d) were analyzed when tumors reached a volume of 1,300 mm3. a GD2, Fas, and TRAIL-R2 BI were calculated and expressed as mean percentage increase of BI of tumors from six 4-HPR-treated animals compared to controls ± SD. b ADCC was analyzed with NK cells as effector cells and target cells isolated from explanted tumors at an E/T ratio of 5:1 in the presence (ADCC) or absence (AICC) of ch14.18/CHO (1 μg/ml). Results are presented as mean percentage AICC (black part of stacked bars) and ADCC (gray part of stacked bars) of NK cells for six control tumors and six tumors of 4-HPR-treated mice (eight replicates each). *p < 0.05, statistically significant difference in overall cytotoxicity (ADCC + AICC) between vehicle-treated and 4-HPR-treated tumors (Student’s t test). c Tumor-infiltrating NK cells after 4-HPR treatment were analyzed by flow cytometry. Representative dot blots are shown for diluent controls and 4-HPR-treated tumors (three mice, respectively); isotype controls using FITC-labeled mouse IgG2A were subtracted. d Fold increase of tumor-infiltrating NK cells in 4-HPR-treated tumors compared to controls was calculated from six mice of each group. Results are presented as fold increase of infiltrating NK cells in 4-HPR tumors compared to vehicle controls. Horizontal line indicates control. **p < 0.01, statistically significant difference from vehicle-treated mice between infiltrating NK cells in 4-HPR-treated mice (Student’s t test)
We also determined the amount of infiltrating NK cells by analyzing with flow cytometry the percentage of NK1.1+ cells of total live cells in xenograft suspensions. Interestingly, tumors from 4-HPR-treated mice showed a significantly higher percentage of tumor-infiltrating NK cells compared to control tumors (representative dot blots of three mice of each group are shown in Fig. 6c) resulting in a sixfold increase compared to control tumors (Fig. 6d, p = 0.002).
Discussion
Anti-GD2 antibody ch14.18/SP2/0 combined with IL-2 and GM-CSF achieved an increase of the 2-year EFS rate from 46 to 66 % [2], emphasizing the role of immunotherapy in high-risk NB. The primary mechanism of action of the anti-GD2 antibody is the induction of ADCC, which can be efficiently augmented by stimulating effector cells with cytokines [32]. A similar strategy to improve antibody immunotherapy in NB is to use other stimulators of the innate immune system, such as β-glucan, a glucose polymer that can induce TNF-α secretion and ADCC by NK cells and monocytes and neutrophils [33]. All co-stimulatory strategies in NB immunotherapy so far have focused on the “effector side” of the immune response.
Here, we demonstrate for the first time that sensitization of the “target cell” towards antibody-mediated effects like ADCC and CDC provides a new strategy to further improve anti-NB immunotherapy. It is important to mention that we were able to induce this effect with nontoxic concentrations of fenretinide in tumor cells that were highly multidrug-resistant or partially drug-resistant.
4-HPR treatment is known to induce an increase of gangliosides on 4-HPR-resistant ovarian cancer [34] and SH-SY5Y cells [18]. Consistent with those results, we show that 4-HPR induced GD2 target antigen expression by up to 100 %, which resulted in a significantly enhanced GD2-specific ADCC and complement-dependent killing. 4-HPR also increased the expression of DRs Fas and TRAIL-R2, the latter of which was partly involved in an improved antibody-independent killing of NB cells (AICC) (Figs. 2 and 3). Importantly, the enhancing effect of 4-HPR was visible at two concentrations (5 and 10 μM) that are clinically achievable in NB patient sera [24].
One mechanism of drug-resistant NB cells to evade a ceramide-mediated cell death is shunting the accumulating toxic molecules into several pathways [16, 18]. Overexpression of GCS in a breast cancer line has been shown to confer resistance to doxorubicin [17]. Interestingly, it was shown previously that a combination of fenretinide, as a ceramide inducer, and inhibitors of the ceramide glucosylation pathway, such as PPMP, resulted in a synergistic cytotoxicity in cancer cells [35]. However, when we inhibited GCS by employing the GCS inhibitor PPPP, we found a downregulation of the ch14.18/CHO target antigen GD2, resulting in a nearly complete abrogation of the 4-HPR-mediated increase of ch14.18/CHO-dependent cytotoxicity of effector cells and complement (Fig. 5). Our data indicate that GCS is at least partially responsible for the increase of GD2-specific ADCC and CDC towards NB cells after 4-HPR treatment by contributing to an increased GD2 expression. Thus, while combining 4-HPR with a GCS inhibitor may be advantageous in terms of cytotoxicity, our data would suggest that combinations of 4-HPR and anti-GD2 immunotherapy should not include a GCS inhibitor.
We observed that 4-HPR treatment of target cells resulted in an enhanced AICC of effector cells, a mechanism partly depending on NK cell-mediated DR-induced apoptosis. Similar to the previously reported effect of 4-HPR on increasing DR expression in colon cancer [36] and Ewing sarcoma cell lines [19], we observed a similar increase in DR binding indices in NB cells (Fig. 4a). Differential findings in this respect in ovarian cancer cells may relate to the low concentration of 4-HPR (1 μM) used for the experiments [37]. The mechanism of increased AICC by 4-HPR-treatment in our study was not dependent on Fas, which can be explained by the generally low expression of this DR in all cell lines tested. A partial involvement of TRAIL-R2 could be observed, which is consistent with our findings that, in all NB cell lines tested, TRAIL-R2 expression was much higher, ranging from 3 % (in 4-HPR-treated CHLA-172) to 56.8 % (in 4-HPR-treated CHLA-136) TRAIL-R2-positive cells. However, the merely partial inhibition of AICC after TRAIL-R2 blocking indicates that other mechanisms are involved, e.g. expression of activating ligands for NK cell function, such as MICA/B or ULBPs [38]. This hypothesis is supported by the fact that immune effector cells produce increasing amounts of the effector molecules GrB and perforin upon co-culture with 4-HPR-treated NB cells. Although in our set of NB cells we could not identify a major role for the inhibitor of DR-mediated apoptosis cFLIP after 4-HPR treatment, it might be possible that other pro-apoptotic or anti-apoptotic protein levels are affected by 4-HPR. The identification of the definitive mechanisms involved, however, is still under way in our laboratory.
Interestingly, the analysis of multidrug-resistant NB xenografts following treatment of mice with 4-HPR revealed that not only GD2 expression increases significantly in the tumor tissue (Fig. 6a), but also that the GD2-specific ADCC in our setting is a major effector mechanism in vivo compared to the AICC response (Fig. 6b), which is reversed in the data we obtained in vitro (Fig. 3). This result is very promising since the AICC activity of NB patient NK cells is very donor-dependent based on donor KIR, HLA, and licensing patterns and varies greatly among patients (N.H. and H.N.L., unpublished data). The fact that in vivo 4-HPR treatment is correlated with an increased NK cell infiltration into tumor tissue further indicates that the effects of fenretinide are much more diverse than we thought and that more research is needed to characterize these effects in detail.
In summary, we have shown that a direct combination of 4-HPR with ch14.18/CHO-based immunotherapy sensitizes NB cells for ch14.18-dependent NK cell and complement killing, two well-established effector mechanisms in anti-NB immunotherapy with ch14.18. Additionally, 4-HPR increases NB cell sensitivity for AICC, which is in part mediated by a DR upregulation (TRAIL-R2), but also through an increased GrB and perforin production by effector cells. Thus, our data suggest that a direct combination of ch14.18/CHO immunotherapy with 4-HPR could be beneficial for high-risk NB patients.
Abbreviations
- NB:
-
Neuroblastoma
- 4-HPR:
-
N-(4-hydroxyphenyl)retinamide
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- AICC:
-
Antibody-independent cell-mediated cytotoxicity
- CDC:
-
Complement-dependent cytotoxicity
- NK cell:
-
Natural killer cell
- PBMCs:
-
Peripheral blood mononuclear cells
- DR:
-
Death receptor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- CHO:
-
Chinese hamster ovary
- IL-2:
-
Interleukin-2
- PPPP:
-
D/L-Threo-1-phenyl-2-hexadecanoylamino-3-pyrrolidino-1-propanol
- GCS:
-
Glucosylceramide synthase
- GD2 BI:
-
GD2 binding index
- FLIP:
-
FLICE-inhibitory protein
References
Westermann F, Schwab M (2002) Genetic parameters of neuroblastomas. Cancer Lett 184:127–147
Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman S, Chen H, Smith M, Anderson B, Villablanca J, Matthay KK et al (2010) Chimeric anti-GD2 antibody with GM-CSF, IL2 and 13-cis retinoic acid for high-risk neuroblastoma: a Children’s Oncology Group (COG) phase 3 study. N Engl J Med 363(14):1324–1334
Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA (1984) Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res 44(12 Pt 1):5914–5920
Svennerholm L, Bostrom K, Fredman P, Jungbjer B, Lekman A, Mansson JE, Rynmark BM (1994) Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta 1214(2):115–123
Handgretinger R, Baader P, Dopfer R, Klingebiel T, Reuland P, Treuner J, Reisfeld RA, Niethammer D (1992) A phase I study of neuroblastoma with the anti-ganglioside GD2 antibody 14.G2a. Cancer Immunol Immunother 35(3):199–204
Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R (2006) FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol 24(18):2885–2890
Cullen SP, Martin SJ (2008) Mechanisms of granule-dependent killing. Cell Death Differ 15(2):251–262
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME (1998) Apoptosis signaling by death receptors. Eur J Biochem 254(3):439–459
Ashkenazi A (2008) Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 7(12):1001–1012
Veronesi U, Mariani L, Decensi A, Formelli F, Camerini T, Miceli R, Di Mauro MG, Costa A, Marubini E, Sporn MB et al (2006) Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Ann Oncol 17(7):1065–1071
Villablanca JG, Krailo MD, Ames MM, Reid JM, Reaman GH, Reynolds CP (2006) Phase I trial of oral fenretinide in children with high-risk solid tumors: a report from the Children’s Oncology Group (CCG 09709). J Clin Oncol 24(21):3423–3430
Maurer BJ, Metelitsa LS, Seeger RC, Cabot MC, Reynolds CP (1999) Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)-retinamide in neuroblastoma cell lines. J Natl Cancer Inst 91(13):1138–1146
Illuzzi G, Bernacchioni C, Aureli M, Prioni S, Frera G, Donati C, Valsecchi M, Chigorno V, Bruni P, Sonnino S et al (2010) Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J Biol Chem 285(24):18594–18602
Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, Mazerolles C, Rischmann P, Teissie J, Malavaud B et al (2005) Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res 65(24):11667–11675
Itoh M, Kitano T, Watanabe M, Kondo T, Yabu T, Taguchi Y, Iwai K, Tashima M, Uchiyama T, Okazaki T (2003) Possible role of ceramide as an indicator of chemoresistance: decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin Cancer Res 9(1):415–423
Gouaze-Andersson V, Cabot MC (2006) Glycosphingolipids and drug resistance. Biochim Biophys Acta 1758(12):2096–2103
Liu YY, Han TY, Giuliano AE, Hansen N, Cabot MC (2000) Uncoupling ceramide glycosylation by transfection of glucosylceramide synthase antisense reverses adriamycin resistance. J Biol Chem 275(10):7138–7143
Lovat PE, Di SF, Corazzari M, Fazi B, Donnorso RP, Pearson AD, Hall AG, Redfern CP, Piacentini M (2004) Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. J Natl Cancer Inst 96(17):1288–1299
White DE, Burchill SA (2010) Fenretinide-dependent upregulation of death receptors through ASK1 and p38alpha enhances death receptor ligand-induced cell death in Ewing’s sarcoma family of tumours. Br J Cancer 103:1380–1390
Reynolds CP, Wang Y, Melton LJ, Einhorn PA, Slamon DJ, Maurer BJ (2000) Retinoic-acid-resistant neuroblastoma cell lines show altered MYC regulation and high sensitivity to fenretinide. Med Pediatr Oncol 35:597–602
Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF et al (2001) Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci U S A 98(14):8012–8017
Somanchi SS, Senyukov VV, Denman CJ, Lee DA (2011) Expansion, purification, and functional assessment of human peripheral blood NK cells. J Vis Exp (48). doi:10.3791/2540
Warren HS, Rana PM (2003) An economical adaptation of the RosetteSep procedure for NK cell enrichment from whole blood, and its use with liquid nitrogen stored peripheral blood mononuclear cells. J Immunol Methods 280(1–2):135–138
Garaventa A, Luksch R, Lo Piccolo MS, Cavadini E, Montaldo PG, Pizzitola MR, Boni L, Ponzoni M, Decensi A, De BB et al (2003) Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin Cancer Res 9(6):2032–2039
Deng W, Li R, Ladisch S (2000) Influence of cellular ganglioside depletion on tumor formation. J Natl Cancer Inst 92(11):912–917
Marachelian A, Kang MH, Hwang K, Villablanca JG, Groshen S, Matthay KK, Maris JM, Desantis KB, Reynolds CP, Maurer BJ (2009) Phase I study of fenretinide (4-HPR) oral powder in patients with recurrent or resistant neuroblastoma: New Approaches to Neuroblastoma Therapy (NANT) Consortium trial. J Clin Oncol 27:15S, abstr 10009
Hoon DS, Kuo CT, Wen S, Wang H, Metelitsa L, Reynolds CP, Seeger RC (2001) Ganglioside GM2/GD2 synthetase mRNA is a marker for detection of infrequent neuroblastoma cells in bone marrow. Am J Pathol 159(2):493–500
Kang MH, Wan Z, Kang YH, Sposto R, Reynolds CP (2008) Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst 100:580–595
Gilman AL, Ozkaynak MF, Matthay KK, Krailo M, Yu AL, Gan J, Sternberg A, Hank JA, Seeger R, Reaman GH et al (2009) Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children’s Oncology Group. J Clin Oncol 27(1):85–91
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C et al (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388(6638):190–195
Kavuri SM, Geserick P, Berg D, Dimitrova DP, Feoktistova M, Siegmund D, Gollnick H, Neumann M, Wajant H, Leverkus M (2011) Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J Biol Chem 286(19):16631–16646
Zeng Y, Huebener N, Fest S, Weixler S, Schroeder U, Gaedicke G, Xiang R, Schramm A, Eggert A, Reisfeld RA et al (2007) Fractalkine (CX3CL1)- and interleukin-2-enriched neuroblastoma microenvironment induces eradication of metastases mediated by T cells and natural killer cells. Cancer Res 67(5):2331–2338
Cheung NK, Modak S (2002) Oral (1→3),(1→4)-beta-d-glucan synergizes with antiganglioside GD2 monoclonal antibody 3F8 in the therapy of neuroblastoma. Clin Cancer Res 8(5):1217–1223
Prinetti A, Basso L, Appierto V, Villani MG, Valsecchi M, Loberto N, Prioni S, Chigorno V, Cavadini E, Formelli F et al (2003) Altered sphingolipid metabolism in N-(4-hydroxyphenyl)-retinamide-resistant A2780 human ovarian carcinoma cells. J Biol Chem 278(8):5574–5583
Maurer BJ, Melton L, Billups C, Cabot MC, Reynolds CP (2000) Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J Natl Cancer Inst 92(23):1897–1909
Kouhara J, Yoshida T, Nakata S, Horinaka M, Wakada M, Ueda Y, Yamagishi H, Sakai T (2007) Fenretinide up-regulates DR5/TRAIL-R2 expression via the induction of the transcription factor CHOP and combined treatment with fenretinide and TRAIL induces synergistic apoptosis in colon cancer cell lines. Int J Oncol 30(3):679–687
Cuello M, Coats AO, Darko I, Ettenberg SA, Gardner GJ, Nau MM, Liu JR, Birrer MJ, Lipkowitz S (2004) N-(4-hydroxyphenyl) retinamide (4HPR) enhances TRAIL-mediated apoptosis through enhancement of a mitochondrial-dependent amplification loop in ovarian cancer cell lines. Cell Death Differ 11(5):527–541
Bahram S, Inoko H, Shiina T, Radosavljevic M (2005) MIC and other NKG2D ligands: from none to too many. Curr Opin Immunol 17(5):505–509
Acknowledgments
Further, we want to thank Tito Woodburn, Malkanthi Mudannayake, and Isabelle Lichy for the excellent technical assistance. We also want to thank the TTUHSC Cancer Center and the South Plains Foundation for the financial support. Support also came in part by CA82830 from the National Cancer Institute and by RP 100762 from the Cancer Prevention & Research Institute of Texas. We extend our special thanks to the SIOPEN group for providing ch14.18/CHO to conduct these experiments.
Conflict of interest
CPR and BJM are inventors on issued patents for both intravenous and LXS oral powder fenretinide formulations for clinical use.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 204 kb)
Rights and permissions
About this article
Cite this article
Shibina, A., Seidel, D., Somanchi, S.S. et al. Fenretinide sensitizes multidrug-resistant human neuroblastoma cells to antibody-independent and ch14.18-mediated NK cell cytotoxicity. J Mol Med 91, 459–472 (2013). https://doi.org/10.1007/s00109-012-0958-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-012-0958-0