
Abstract
In this study, the in vitro and in vivo transfection capacity of novel pH-sensitive sugar-based gemini surfactants was investigated. In an aqueous environment at physiological pH, these compounds form bilayer vesicles, but they undergo a lamellar-to-micellar phase transition in the endosomal pH range as a consequence of an increased protonation state. In the same way, lipoplexes made with these amphiphiles exhibit a lamellar morphology at physiological pH and a non-lamellar phase at acidic pH. In this study, we confirm that the gemini surfactants are able to form complexes with plasmid DNA at physiological pH and are able to transfect efficiently CHO cells in vitro. Out of the five compounds tested here, two of these amphiphiles, GS1 and GS2, led to 70% of transfected cells with a good cell survival. These two compounds were tested further for in vivo applications. Because of their lamellar organisation, these lipoplexes exhibited a good colloidal stability in salt and in serum at physiological pH compatible with a prolonged stability in vivo. Indeed, when injected intravenously to mice, these stable lipoplexes apparently did not substantially accumulate, as inferred from the observation that transfection of the lungs was not detectable, as examined by in vivo bioluminescence. This potential of avoiding ‘preliminary capture’ in the lungs may, thus, be further exploited in developing devices for specific targeting of gemini lipoplexes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although viral vectors display a highly effective gene delivery and transfection efficacy in vivo, these systems, nevertheless, suffer from several drawbacks, particularly with regard to biohazard and safety [1, 2]. Consequently, further research in the development of improved non-viral vectors for therapeutic applications appears justified, as these nanocarriers often show negligible immunogenicity. In addition, the synthetic organic compounds, constituting the carriers’ core, are readily chemically modified for target purposes, while they can also accommodate a wide variety of cargo ranging from small interfering RNA, plasmid DNA, antisense oligodesoxynucleotides to proteins [3–10]. For decades, cationic lipids have been exploited as non-viral DNA vectors (‘lipoplexes’), and have been shown to give rise to good transfection efficiency in vitro; however, they mostly failed to sustain this efficiency in vivo [11, 12].
One of the major hurdles for cationic lipid-based gene delivery in vivo is the interaction of the lipoplexes with serum and their uncontrolled and avid interaction with cellular surfaces. Both electrostatic neutralization and the presence of negatively charged proteins can affect the colloidal stability of lipoplexes. It is commonly observed for cationic entities that aggregation and interaction with blood components result in a relatively enhanced gene expression in the lungs, compared to other organs [13–16]. This is due to the fact that after intravenous injection, lipoplexes first encounter the pulmonary capillary, where the aggregated complexes are effectively retained [17].
PEGylation of cationic lipoplexes, relying on mixing of lipids coupled with poly(ethylene glycol) (PEG-lipids) with cationic lipids, is often used to minimize the interaction with blood components, thereby reducing aggregation and accumulation in the lungs [18, 19] and concomitantly extending the circulation time. Indeed, we and others have previously shown that PEG-lipids act via stabilizing the lamellar phase of the lipoplex lipid phase while this relatively hydrophilic coating to the lipoplex shields it from interactions with protein or other blood components [20–23]. Nevertheless, the presence of a lamellar-phase stabilizing PEG coating can impair intracellular gene delivery by frustrating endosomal release of the cargo from cationic lipoplexes [21, 24]. Consequently, strategies have been developed to trigger the timely removal of this PEG coating to accomplish effective cellular transfection. These strategies involve release of either the PEG group via pH sensitive cleavage [25–27] or the PEG-lipids as such, relying on acyl chain length-dependent lipid exchange [28, 29].
However, the application of a novel class of pH-sensitive sugar-based gemini surfactants may provide a simpler and better programmable alternative for the use of a multicomponent PEGylated lipoplex for in vivo gene delivery. Such gemini surfactants [30] do not require helper lipids, as is the case for numerous cationic lipid-based systems, and, most importantly, liposomes prepared from such surfactants show a pH-dependent transition from the lamellar to a micellar phase [30–32]. In fact, helper lipids, like DOPE, frustrate this transition, thus causing a relative stabilization of the bilayer structure and emphasizing the membrane destabilizing micellar phase as a prerequisite for cytoplasmic DNA release [33]. The potential of the gemini surfactant systems for gene delivery in vitro has been demonstrated [34, 35].
The present work was undertaken to investigate the mechanism of gemini-mediated transfection, i.e., whether lipoplex destabilization follows a similar pH-sensitive pattern as observed for liposomal bilayers. If so, we would anticipate an enhanced stability of such complexes in vivo, endosomal membrane destabilization being particularly favored at mild acidic pH after cellular internalization. Our data show that distinct gemini surfactants can be prepared, which meet these criteria, showing no massive aggregation and, hence, accumulation in lung tissue. The observed transfection in vivo of the oral tract may potentially open new applications for sugar-based gemini surfactants.
Materials and methods
Cell culture
CHO-K1 cells were grown in Dulbecco’s Modified Eagle medium (Gibco) supplemented with 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells were maintained in a humidified incubator at 37°C and 5% CO2 atmosphere.
Preparation of lipid vesicles
The sugar-based gemini surfactants were synthesized as previously described [30, 31]. Liposomes were prepared 1 day before the experiment. Briefly, the appropriate amount of lipids in methanol for a final concentration of 1 mM was put in a glass tube. The methanol was evaporated under a stream of nitrogen and the lipid film was further dried under vacuum for 4 h. Subsequently, the film was resuspended in a buffer containing MES, HEPES, and sodium acetate at 5 mM each, pH 6.5. The obtained liposomal suspension was then freeze-thawed five times and sonicated for 5 min in a bath sonicator.
Transfection in vitro
The plasmid DNA used is pEGFP-N1 from Clontech laboratories, which contains a luciferase reporter gene inserted at the multiple cloning site. The plasmid, which codes for both the Green Fluorescent Protein (GFP) and the luciferase enzyme, was propagated in Escherichia coli strains, and plasmid DNA was extracted using a GenElute plasmid midi-prep kit from Sigma.
For transfection, cells were plated in six-well plate at a density of 3×105 cells per well 18 h before transfection. For one well, transfection medium was prepared as follows: 1 μg of plasmid was mixed with 25 nmol of gemini surfactant vesicles, prepared as described above, in an equal volume of serum-free medium. The medium was supplemented with 25 mM HEPES and filtered after adjustment at pH 7.0. The molar ratio of positive charges to negative charges was 8 to 1. After 15 min of incubation at room temperature, the lipoplexes were incubated with the cells for 4 h at 37°C. Subsequently, the transfection medium was removed and fresh medium containing 10% fetal calf serum was added to the cells. Twenty-four hours after initiating transfection, the cell medium was refreshed again and after 48 h, the expression of the reporter gene GFP was measured with a flow cytometer Epics Elite from Beckman Coulter. Transfection efficiency is expressed as the percentage of GFP-positive cells on the total of surviving cells. Potential toxicity and cell survival were estimated by counting the number of surviving cells on a photograph of the cell monolayer and comparing it to untreated cells.
Transfections carried out with lipofectamine 2000 reagent were done according to the manufacturer’s specifications.
Cellular uptake of N-Rh-PE-labeled gemini lipoplexes
To determine the cellular uptake of lipoplex particles, lipoplexes were prepared with lipid vesicles containing 0.1% N-(lissamine rhodamine B sulphonyl)phosphatidylethanolamine (N-Rh-PE; Avanti Polar Lipids). N-Rh-PE-labeled lipoplexes were incubated with the cells exactly as described for the ‘Transfection in vitro’ protocol. After 4 h incubation at 37°C, the external fluorescence of non-internalized lipoplexes was quenched with a 0.4% trypan blue solution, thus allowing an accurate determination of genuinely internalized complexes. Rhodamine-positive cells were then sorted by FACS and the average fluorescence intensity per cell was measured.
Gel retardation assay for lipid/DNA interaction
A 0.5-μg plasmid was mixed with 12.5 nmol of gemini surfactant vesicles in 5 mM MES/HEPES/sodium acetate pH 7.4. The lipoplexes were then treated or not with Triton X-100 (1% final concentration). Subsequently, after addition of 3 μl of 30% glycerol, the samples were loaded on a 1% agarose gel containing 1.25 mM ethidium bromide. A voltage of 50 V was applied over the gel, immersed in a 1×TAE buffer, for 30 min. The DNA was then visualized by UV illumination.
Small-angle X-ray scattering (SAXS)
To determine the lipoplex structure, SAXS measurements of gemini lipoplexes were performed at 25°C using a NanoStar device (Brucker AXS and Anton Paar) with a ceramic fine-focus X-ray tube, operating in a point focus mode. The tube was powered with a Kristalloflex K760 generator at 35 kV and 40 mA. The primary beam was collimated using cross-coupled Göbel mirrors and a 0.1-mm pinhole providing a \({\text{CuK}}_{\alpha }\) radiation beam (wavelength λ=0.154 nm) with a full-width at half-maximum of about 0.2 mm in diameter at the sample position. The sample-detector distance was 0.24 m. The use of a Hi-Star position-sensitive area detector (Siemens AXS) allowed recording the scattering intensity in the q-range of 0.5 to 8.5 nm−1. The scattering vector q is defined as q=(4π/λ) sin(θ/2), where θ is the scattering angle. The measurements of the samples, prepared by mixing gemini liposomes (2.4 μmol) and plasmid DNA solution (100 μg, 8 to 1 molar charge ratio ±), were performed in flame-sealed quartz capillaries with a diameter of 1 mm. The measuring time was 9 h.
Determination and visualization of colloidal stability of liposomes and lipoplexes
Turbidity of liposome and lipoplexes, as a measure of their colloidal stability, was monitored as a function of time on a Perkin Elmer LAMBDA 25 UV/Vis spectrometer at a wavelength of 350 nm. The final concentration of lipids is 0.1 mM and the molar (±) charge ratio was 8 to 1. As a reference, SAINT-2/DOPE (1:1) lipoplexes at a 2.5 to 1 molar (±) charge ratio were used [21, 36]. The kinetics were measured as follows: vesicles were added to the buffer at t=1 min and plasmid DNA, in the case of lipoplexes, at t=5 min. To determine the effect of serum, lipoplexes were made in salt and incubated for 15 min at room temperature, after which serum was added to the final concentration of 10 and 50%. For each compound, the stability at five different conditions was tested, i.e., that of surfactant vesicles, prepared in MES/HEPES/sodium acetate buffer at pH 7 or in salt solution (HBS: 10 mM HEPES, 150 mM NaCl, pH 7), and that of lipoplexes, suspended in HBS with or without addition of 10 or 50% serum. In all cases, the initial kinetics of turbidity were monitored, which leveled off after approximately 15 min.
Lipoplexes samples examined by light microscopy were prepared as described for the turbidity measurements and incubated at room temperature for 24 h. When applicable, the final concentration of serum was 10%.
Transfection in vivo
All animal studies were performed after receiving approval of the Animal Experimentation Committee of the University of Groningen. Studies were performed on male Balb/c nude mice (8 to 10 weeks of age). For in vivo experiments, the plasmid DNA was freed from endotoxin, which was done by extraction, using the EndoFree Plasmid Maxi Kit from Qiagen. Some 200 μl of lipoplexes were injected via the penile vein while anesthetized, and the samples were prepared as follows: 40 μg of plasmid DNA were mixed with 0.5 μmol of gemini surfactant in a 20% sucrose solution. This sucrose solution was prepared in HBS at pH 7.4. Twenty-four hours after injection and 10 min before measurement, the mice were anaesthetized with 2.5% isoflurane gas in oxygen flow (1.5 l/min), and kept at 37°C body temperature. Ten minutes before imaging, mice were intraperitonealy injected with d-luciferin (150 mg/ml, Xenogen, Alameda, CA, USA) the substrate for luciferase. Luminescence emission was visualized by a cooled charged coupled device (CCD) camera, IVIS 100 system and Living Image Software (Xenogen, Alameda CA, USA).
Results
Structure of the sugar-based gemini surfactants
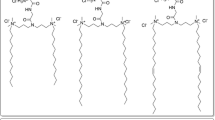
Five different sugar-based gemini surfactants were employed in this study. Their structural characteristics are shown in Fig. 1 and described in Table 1. Note that all compounds contain (unsaturated) oleoyl hydrocarbon tails. The head group is either a mannose or a glucose, which is connected via either an ethylene oxide (EO) spacer [–(EO)2–(CH2)2–] (GS1, GS2, and GS4) or a C6 aliphatic spacer [–(CH2)6–] (GS3 and GS5).

Structure of the gemini surfactants (GS) 1 to 5
The two amino moieties in the head groups of GS1, GS2, GS3, and GS5 are weak bases and are fully protonated at mild acidic pH, whereas the non-titratable amido linkage in GS4 gives rise to a net neutral charge. Previously, it has been shown [30, 31] that lipid vesicles prepared from gemini surfactants such as GS1, GS2, GS3, and GS5 adopt a micellar structure, whereas around pH 7, where these gemini surfactants are monoprotonated, the bilayer structure is maintained in aqueous solution. Surfactant-mediated gene delivery requires a membrane destabilization of the lipoplexes within endosomal compartments, which allows translocation of the gene into the cytosol via an as yet poorly defined mechanism [37]. To investigate whether such a destabilization could, thus, be triggered by a mild acidic pH within endosomes after endocytic internalization, subsequently giving rise to gene delivery and expression, we next determined the in vitro transfection efficiency of lipoplexes prepared from these gemini surfactants.
In vitro transfection mediated by the sugar-based gemini surfactants
Obviously, critical to transfection is the ability of the cationic vector to efficiently form complexes with plasmid DNA. Hence, this property was determined first for all five sugar-based gemini surfactants, using a gel retardation assay. Lipoplexes were prepared as described in Materials and methods and samples were subjected to electrophoresis on a 1% agarose gel. In this assay, non-entrapped DNA will migrate freely within the gel while complexed DNA will only do so after lysis of the lipoplex with detergent. As shown in Fig. 2a, for lipoplexes made with GS4, the plasmid DNA migrates into the gel, irrespective of the presence of detergent, whereas for all other complexes, migration of free plasmid was only apparent after lipoplex lysis.

Analysis of lipoplex assembly of gemini surfactants and cellular internalization of N-Rh-PE-labeled lipoplexes by CHO cells. a Lipoplexes were made at pH 7.4 with all five sugar-based gemini surfactants as indicated. The migration of free plasmid DNA with (+) or without (−) prior treatment of the lipoplexes with the Triton X-100 detergent was analyzed by agarose gel electrophoresis. Note that for all surfactants, except GS4, plasmid DNA did not migrate in the gel without treatment by a detergent. b N-Rh-PE-labeled lipoplexes were incubated with the cells. After 4 h, external fluorescence of non-internalized gemini lipoplexes was quenched with a trypan blue solution and the intracellular Rhodamine fluorescence as a reflection of internalized lipoplexes was measured by FACS
These data imply that GS4, which is neutral at pH 7.4, fails to engage in lipoplex assembly, in contrast to the mono-protonated GS1, GS2, GS3, and GS5 derivatives, which at similar conditions effectively complex DNA and thus form lipoplexes. The same patterns were found when lipoplexes, prepared at acidic pH 5.5 and 6.7, were analyzed, whereas at pH 8.5 (where the amino groups are no longer protonated), none of the gemini surfactants formed complexes with plasmid DNA (data not shown). These observations, thus, properly reflect the charge behavior of the gemini surfactants as primary parameter in lipoplex assembly, indicating that neither head group nor spacer properties interfered with this propensity.
Before transfection, we then determined the efficiency of interaction of either lipoplex with the cells, using N-Rh-PE labeled lipoplexes. After 4 h of incubation with the cells, non-bound lipoplexes were removed and the cells were washed with trypan blue to quench the fluorescence of attached complexes, non-quenched fluorescence thus representing genuinely internalized complexes. As shown in Fig. 2b, GS1, GS2, GS3, and GS5 lipoplexes are taken up efficiently by the cells to very similar extents. As expected, the neutral surfactant GS4, which also failed to form lipoplexes, was not significantly internalized by the cells. Moreover, fluorescent microscopy experiments were carried out in the presence of free sugars (0.25 mM mannose or glucose) and the extent of internalization was identical with or without sugars (not shown), implying that lipoplex charge rather than sugar specificity is the driving force in the interaction of these gemini lipoplexes with the cells.
Subsequently, CHO-K1 cells were transfected (at pH 7.0) in vitro, using a plasmid containing the reporter gene Green Fluorescent Protein (GFP). The transfection efficiency was measured by counting the percentage of GFP-positive cells by flow cytometry. To determine the optimal conditions for transfection, lipoplexes were prepared at various molar (±) charge ratios and cells were transfected according to the protocol as described in Materials and methods. In this manner, the highest efficiency was determined at a charge ratio of 8 to 1 (±), which was then used throughout this study, unless indicated otherwise. In addition, in contrast to observations made with other cationic systems in which the inclusion of the so-called helper lipid DOPE may improve transfection, an inhibition was observed in case of mixing the gemini surfactants with this lipid (not shown; see Discussion).
Therefore, only the pure surfactants were examined for their transfection capacity. Fig. 3a shows the FACS analysis for untransfected cells (control) and cells transfected with GS1 or GS5. GFP-positive cells correspond to the cells in the area below the line marked by R2. Note that this analysis is made on living cells only, a first sorting allowing to separate cellular debris from normal cells. Fig. 3b summarizes the results for all surfactants. As anticipated, being incapable of effectively complexing DNA, GS4 only led to 0.8% of GFP-positive cells. In contrast, GS1, GS2, and GS3 displayed transfection efficiencies of 73.9, 71.2, and 68.9%, respectively, of GFP-positive cells. Interestingly, GS5 exhibited lower transfection efficiency than the other gemini surfactants, attaining a level of approximately 48% of GFP-positive cells, which is very similar as the transfection efficiency obtained with commercially available lipofectamine 2000 (LF2000).

Gemini lipoplexes efficiently transfect cells in vitro. a CHO-K1 cells were transfected with lipoplexes containing a plasmid coding for Green Fluorescent Protein (GFP) as described in the Materials and methods section. The percentage of GFP-positive cells as a measure of transfection efficiency was determined by FACS for all surfactants. FACS analysis of untransfected cells (Control) and cells transfected with GS1 or GS5 are presented here. The R2 area is marked for non-transfected cells (Control) and the fraction of GFP-transfected cells is determined from the shift into the R2 region (area underneath the line marked by R2). In b, the percentage of transfected cells for all surfactants (GS1, GS2, GS3, GS4, and GS5) is summarized. The transfection obtained with the commercially available Lipofectamine 2000 is shown as reference (LF2000). c Images of the cell monolayer were taken 48 h after transfection and control cell and cells transfected with GS2 and GS3 are shown (Control, GS2, and GS3, respectively)
Examination of the cell monolayer 48 h after transfection (Fig. 3c) revealed that the gemini surfactants affected cell survival to different extents. Commonly, more than 65% of the cells survived when the cells had been transfected with GS2. Cells transfected with GS1 showed a survival of approximately 40% of the cells. However, GS3 and GS5, both bearing aliphatic C6 spacers, showed the highest toxicity with 21 and 31%, respectively, of cell survival only. As a comparison, 52% of cells survived after transfection with lipofectamine 2000. Taken together, these data indicate that the transfection efficiency of the various sugar-based gemini surfactants did not correlate with head group specificity, as both GS1 and GS2 effectively transfected the cells, consistent with the interaction of the N-Rh-PE-labeled complexes with the cells. Also, there seems to be no structural preference, as no significant differences were seen in transfection efficiency between GS2, which contains an ethylene oxide spacer, and GS3, carrying the same head group, but a simple aliphatic C6 spacer. However, the nature of the spacer markedly affected cell survival, the ones containing an ethylene oxide spacer displaying a relatively low toxicity. The following experiments were, therefore, carried out with GS1 and GS2.
Colloidal stability of gemini surfactants lipoplexes
Lipoplexes that display a lamellar phase are colloidally more stable in an aqueous environment, i.e., they are less prone to aggregation than lipoplexes, which readily revert to an inverted hexagonal HII phase. Particularly in vivo, while circulating in the bloodstream, such properties are highly desirable, in that destabilization should preferably only take place once the complex has reached its site of destination. Previous work demonstrated that GS1 lipoplexes, formed at physiological pH, like lipid vesicles of the same compound, displays lamellar morphology with a d spacing (d=2π/q 001) of 59.8 Å [35].
To corroborate this observation for conditions that apply to the present work, we analyzed by small angle X-ray scattering (SAXS) the morphology of GS2 lipoplexes, charge ratio 8:1 at physiological pH. The spectrum obtained for GS2 lipoplexes is shown in Fig. 4 and reveals two diffraction peaks at q=0.1063 Å−1and q=0.2130 Å−1. The ratio between those q values of 1:2 is typical of a lamellar morphology. The calculated spacing between the two bilayers was d=59.1 Å. Consequently, both GS1 and GS2 lipoplexes have a lamellar morphology at physiological pH, when suspended in an aqueous environment.

GS2 lipoplexes display a lamellar \({\text{L}}_{\alpha }\) phase at physiological pH. GS2 lipoplexes were made at pH 7.3 as described in Materials and methods and examined by SAXS. The diffraction pattern revealed two peaks at q=0.1063 and 0.2130 (Å)−1 and the ratio of 1:2 between these two values is typical of a lamellar \({\text{L}}_{\alpha }\) phase. The bilayer spacing (d) was 59.1 Å
However, in the blood circulation, the complexes will be exposed to a higher ionic strength and serum proteins, which in turn may also deteriorate colloidal stability.
To examine this effect, GS1 and GS2 lipid vesicles and lipoplexes, respectively, were incubated in buffer of physiological ionic strength and in 10 or 50% serum. As a measure of colloidal stability, the initial kinetics (20 min) in turbidity change were determined at pH 7 (Fig. 5a). GS1 and GS2 were compared to lipid vesicles and lipoplexes, prepared from the dialkyl pyridinium surfactant SAINT-2/DOPE (1:1). This system has been shown to transform from a lamellar \({\text{L}}_{\alpha }\) phase to an inverted hexagonal phase HII [36, 38], when transferred from water to a physiological salt solution, a feature accompanied by a dramatic enhancement in turbidity (Fig. 5a, tracks VI and VII; b, +NaCl and pDNA). Interestingly, in contrast to a pronounced aggregation of SAINT-2/DOPE liposomes and lipoplexes, GS1 and GS2 liposomes and lipoplexes, when transferred from a buffer of low ionic strength to a HBS solution, exhibited a negligible increase in turbidity when placed in HBS solution [Fig. 5a (I, II, and III); b (−NaCl, +NaCl, and pDNA)]. This reflects the ability of the HII forming SAINT-2/DOPE lipoplexes to rapidly cluster [39], while, at the same conditions, gemini lipoplexes do not aggregate. However, in the presence of serum, GS1 and GS2 lipoplexes display a distinct degree of aggregation, which, remarkably, appeared to be more pronounced in 10% serum than in 50%, as reflected by an increase of turbidity [Fig. 5a (IV), b (10 and 50% serum)], although to a lesser extent than that observed for SAINT-2/DOPE complexes.

Effect of salt and serum on lipoplexes colloidal stability. a Turbidity changes at 350 nm for GS1 and SAINT-2/DOPE liposomes and lipoplexes was monitored as a function of time. Traces of the kinetics for GS1 (I, II, III, and IV) and SAINT-2/DOPE (V, VI, VII, and VIII) are shown and arrows indicate the time where liposomes and plasmid DNA were added. Kinetichs for GS2, similar to the one of GS1, are not presented here. b The maximum turbidity values reached for GS1, GS2, and SAINT-2/DOPE liposomes and lipoplexes are summarized. “−NaCl” and “+NaCl” refer to the lipids without plasmid DNA with or without salt, respectively. The legend “pDNA” indicates lipoplexes in salt solution (HBS); “10% Serum” and “50% Serum” refer to lipoplexes that were made in HBS and which were then incubated in the presence of either 10 or 50% serum. The maximum turbidity values are indicated on the histogram bars
The long-term effect of serum on the lipoplexes was analyzed after 24 h by light microscopy (Fig. 6). It should be noted, however, that at none of our transfection conditions, the complexes were actually exposed to these extreme conditions per se, but such extended incubation conditions may be of help to further define the colloidal stability of the complexes. Interestingly, after these extended incubation periods, SAINT-2/DOPE lipoplexes may grow, in the presence of salt only, to giant complexes of up to 10 μm (Fig. 6d). Such clustering can be precluded by ‘coating’ of the lipoplexes with serum proteins, as after serum addition, the size of the lipoplexes stabilizes around 0.5 to 2 μm (Fig. 6c, c.f. also [40]).

Effect of salt and serum on lipoplexes colloidal stability after 24 h. The lipoplexes were prepared as in Fig. 5. a GS1 lipoplexes made in HBS and 10% serum was added. b GS2 lipoplexes in HBS, 10% serum was added. c SAINT-2/DOPE lipoplexes in HBS, 10% serum added. d SAINT-2/DOPE lipoplexes in HBS no serum added. The bar represents 40 μm
As shown above for the turbidity measurements, the behavior of GS1 and GS2 lipoplexes is opposite to that of SAINT-2/DOPE lipoplexes. In salt, the gemini lipoplexes do not aggregate, and after 24 h, no significant clustering of particles could be observed. In 10% serum, however, the gemini lipoplexes also aggregate after 24 h, leading to particles of around 2 μm (Fig. 6a,b), implying long-term changes that appear clearly different from those seen after relatively short time intervals that are more reminiscent of physiologically relevant conditions. Thus, after such shorter time intervals the gemini surfactants show a superior ‘stability’ over systems like those of the HII phase adopting SAINT-2/DOPE system (Fig. 5a). This relative stability could, thus, be exploited for in vivo purposes in that the low level of aggregation, especially at higher serum concentration, might allow the gemini surfactant complexes to circulate for prolonged time intervals rather than to accumulate readily in pulmonary capillaries, shortly after administration.
Transfection in vivo using GS1 and GS2 lipoplexes
To investigate the potential of GS1 and GS2 lipoplexes for in vivo transfection purposes, a plasmid was used that encodes the firefly luciferase enzyme, which emits light in presence of its substrate luciferin, oxygen, ATP and magnesium [41, 42], thus allowing in situ detection of gene expression by luminescence imaging. Three groups of three nude mice each were transfected using GS1 lipoplexes, GS2 lipoplexes or naked plasmid DNA. Lipoplexes were injected intravenously in the penile vein; each mouse received 0.5 μmol of lipids and 40 μg of plasmid DNA or only naked plasmid DNA. Consecutive to the injection of lipoplexes and naked DNA, the mice experienced a mild discomfort, as shown by a reduced mobility and the tendency to nest. Nevertheless, after 24 h, all mice recovered perfectly and none of the animals died after injection of either lipoplexes or naked DNA.
Expression of luciferase was examined 24 h after injection of the lipoplexes using a cooled sensitive CCD camera for detection of bioluminescence. As shown in Fig. 7, administration of both GS1 (panel GS1) and GS2 (panel GS2) lipoplexes suggested expression of luciferase in the living mice. For some mice a local expression at the site of injection around the penile vein (Fig. 7; regions c and e; panel GS1 and GS2) can be observed. However, the expression of luciferase is mainly located in the lower abdominal region of the mice (Fig. 7; regions a, b, and d; panel GS1 and GS2).

In vivo transfection mediated by GS1 and GS2. Male nude mice were injected with GS1 or GS2 lipoplexes or with naked plasmid DNA. The plasmid codes for the luciferase enzyme and its activity is visualized by luminescence after injection of luciferin using a cooled charged coupled device (CCD) camera
Ex-vivo analysis of liver and spleen for luciferase activity confirmed gene expression, mediated by both formulations, in these organs, and in case of GS1 a relatively enhanced expression was observed in the spleen (data not shown). Consistent with the bioluminescence image, neither GS1 nor GS2 lipoplexes led to significant gene expression in the lungs. Some expression is found in the mouth of the animal (Fig. 7; region f; panel GS1 and GS2), which likely results from the animal licking the injection site. Note, however, the absence of oral expression in mice treated with naked DNA (Fig. 7; panel pDNA).
Discussion
The purpose of this work was to investigate the transfection capacity of sugar-based gemini surfactants, which are capable of undergoing a pH-triggered structural change, i.e., from a lamellar phase at physiological pH to a non-lamellar phase at mild acid pH. It was reasoned that this property would convey colloidal stability to gemini lipoplexes before cellular uptake, while exerting destabilizing properties necessary for gene delivery only after internalization within mild acidic endosomal compartments. By the same token, such a colloidal stability could likely be exploited in vivo, avoiding massive aggregation of these cationic lipids while in the circulation, which would also preclude pulmonary capture, as often observed for these systems [13–16].
Our data are consistent with this concept in that effective transfection of those gemini surfactants capable of forming DNA-bound lipoplexes was observed in vitro. Interestingly, significant transfection was similarly observed in vivo, showing gene expression in organs other than lungs, as directly visualized by bioluminescence imaging. Thus, the present systems should allow the possibility of better defining potential correlations between mechanism(s) and transfection efficiency as obtained in vitro vs in vivo. Given the apparent colloidal stability of these particles in the circulation, it should also become possible to exploit them for targeting purposes.
This study shows that the sugar-based gemini surfactants GS1, GS2, GS3, and GS5 can efficiently form complexes with plasmid DNA and mediate transfection in vitro. The effect of the head group, glucose vs mannose, or the effect of the spacer, C6-alkyl vs ethylene oxide, does not appear to modulate the level of transfection to a significant extent. Indeed GS1, GS2, and GS3 all show a transfection efficiency of approximately 70%. GS5 showed a somewhat lower level of transfection (approximately 50%). This could suggest that the combination of a C6-alkyl spacer and a reduced glucose as head group are less favorable in bringing about transfection, although the cellular uptake of all gemini lipoplexes was very similar, as determined by marking the complexes with a fluorescent lipid analogue N-Rh-PE, allowing to distinguish binding from genuine internalization of the lipoplexes.
Aside from a less efficient transfection, GS5 also showed a relatively high toxicity, which appears to be related to the nature of the spacer, as a similar negative effect on cell survival was seen for GS3, which in turn shares the aliphatic C6 spacer with GS5. Thus, GS1 and GS2, both containing an ethylene oxide spacer, appear to be the geminis of choice, as they displayed both a low toxicity and relatively high transfection efficiency. Apparently the nature of the head group, i.e., glucose vs mannose, exerts little effect on lipoplex assembly, cell association, and eventual transfection efficiency.
The molecular shape of the monoprotonated gemini surfactant is such that it conveys a higher colloidal stability than similar complexes formed by different cationic amphiphiles. The preferred morphology of the gemini lipoplex at neutral pH is lamellar (Fig. 4). In contrast, cationic amphiphiles are often characterized by a packing parameter (relatively small head group area vs relative large hydrocarbon tail area) that leads to the formation of lipoplexes organized in inverted phases [9]. The HII phase has been widely recognized as promoting transfection in vitro [21, 36, 37, 43]; however, as noted, such a colloidal destabilization before reaching the target site could prove disadvantageous, particularly upon their administration in vivo. As lipoplexes with a lamellar organization are less prone to aggregation than lipoplexes that have adopted an inverted hexagonal phase, such a property would preclude them from accumulating in the lungs, as commonly seen for cationic delivery vehicles [15, 44, 45].
Indeed, both GS1 and GS2 lipoplexes are in a lamellar phase at physiological pH, and, after injection in vivo, show no transfection in the lungs. In this context, it is relevant to note that in very recent work, applying a novel and highly sensitive assay to monitor lipid phase changes [46], we have determined that upon acidification, GS1 and GS2 lipoplexes undergo a lamellar to a HI non-inverted micellar transition and that this transition is impeded when the HII (i.e., inverted) phase preferring DOPE is included [33] (Wasungu et al., submitted for publication). These data, thus, entirely explain the observation that DOPE inhibits rather than promotes transfection of gemini complexes.
The sensitivity to serum is an important issue when applying cationic lipids for in vivo transfection. Two kinds of effects of serum have been reported. One of these effects is on the colloidal stability of lipoplexes. Indeed, the presence of serum can cause aggregation and size increase of the particles, which leads to pulmonary accumulation [9, 13, 47]. The second effect is a destabilization of the lipoplexes structure and lipid/DNA interaction, which may result in the destruction of the complexes [9]. This sensitivity to serum can vary according to the type of cationic lipids and the type of helper lipids. Thus, SAINT-2/DOPE-mediated lipofection has been reported to be serum resistant [48]. In the case of the widely used cationic lipid DOTAP, it was shown to be sensitive to destabilization by serum proteins when used in combination with DOPE but not when used in combination with cholesterol [13]. Likewise, the use of cholesterol as helper lipid with DOTAP has also been shown to increase the delivery of plasmid DNA to cells in vitro [49]. For DOTAP/DOPE, the hexagonal structure of DOPE has been suggested to facilitate penetration of serum proteins into the complex, thus causing their disruption [9]. Clearly, in case of sugar-based gemini surfactants, the lamellar organization of lipoplexes not only provides an improved colloidal stability but would also preclude complex disruption by penetration of serum proteins.
In this study, we observed that serum added to gemini surfactants lipoplexes provoked a mild aggregation (Fig. 5b). As these lipoplexes have been shown to be lamellar, this aggregation is not likely the result of hydrophobic interactions between inverted hexagonal particles (as noted above, a non-inverted structure is maintained, also after exposure to mild acidic pH) but rather the consequence of charge neutralization at the surface of those lamellar lipoplexes. Interestingly, the extent of lipoplex aggregation was inversely related to the serum concentration, the aggregation being diminished at a higher serum concentration. This effect has been reported before for DOTAP/cholesterol lipoplexes [9], and was proposed to be related to the possibility that a low concentration may cause bridging of adjacent lipoplexes, whereas an enhanced concentration may uniformly coat lipoplexes, thereby preventing clustering.
A similar phenomenom could be at stake for the gemini surfactants, as observed in the present study (Fig. 5b). Consistent with this observation is the conspicuous absence of extensive accumulation of lipoplexes in pulmonary capillaries, which suggests that in vivo massive clustering of gemini lipoplexes did not occur. This potential of avoiding ‘preliminary capture’ could thus be further exploited in developing devices for specific targeting of gemini lipoplexes. Together, these data would also argue strongly against the possibility that the lamellar phase, as determined in vitro, would be converted to a non-lamellar phase by serum proteins after injection into the circulation. Moreover, such structural changes in circulating lipo- or polyplexes have not been reported thus far, although further work will be required to firmly exclude such a possibility.
Interestingly, when compared to the absence of oral expression in mice treated with naked DNA (Fig. 7, panel pDNA), and yet the presence of expression on the injection site (Fig. 7; regions b, c, and d, panel pDNA), it seems that the gemini surfactants can play a protective role for the DNA when present in the oral tract. We will, therefore, further examine the interesting option of using the sugar-based gemini surfactants for oral gene therapy.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- lipolexes:
-
Complexes of DNA and cationic lipids
- PEG:
-
Poly(ethylene glycol)
- GFP:
-
Green fluorescent protein
- GS:
-
Gemini surfactant
- DOPE:
-
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOTAP:
-
N-[1-(2,3-dioleyl)propyl]-N,N,N-trimethylammonim chloride
- Saint-2:
-
N-methyl-4-(dioleyl)methylpyridinium
- N-Rh-PE:
-
N-(lissamine rhodamine B sulphonyl)phosphatidylethanolamine
- FACS:
-
fluorescence-activated cell sorting
- SAXS:
-
Small angle X-ray scattering
- HEPES:
-
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- MES:
-
2-[N-morpholino]ethanesulfonic acid
- HBS solution:
-
HEPES buffered saline solution
- CHO cells:
-
Chinese hamster ovarian cells
- AU:
-
Arbitrary unit
References
Felgner PL (1996) Improvements in cationic liposomes for in vivo gene transfer. Hum Gene Ther 7(15):1791–1793
Romano G, Claudio PP, Kaiser HE, Giordano A (1998) Recent advances, prospects and problems in designing new strategies for oligonucleotide and gene delivery in therapy. In Vivo 12(1):59–67
Felgner PL (1997) Nonviral strategies for gene therapy. Sci Am 276(6):102–106
van der Woude I, Wagenaar A, Meekel AA, ter Beest MB, Ruiters MH, Engberts JB, Hoekstra D (1997) Novel pyridinium surfactants for efficient, nontoxic in vitro gene delivery. Proc Natl Acad Sci U S A 94(4):1160–1165
Shi F, Nomden A, Oberle V, Engberts JB, Hoekstra D (2001) Efficient cationic lipid-mediated delivery of antisense oligonucleotides into eukaryotic cells: down-regulation of the corticotropin-releasing factor receptor. Nucleic Acids Res 29(10):2079–2087
Garcia-Chaumont C, Seksek O, Grzybowska J, Borowski E, Bolard J (2000) Delivery systems for antisense oligonucleotides. Pharmacol Ther 87(2-3):255–277
Chien PY, Wang J, Carbonaro D, Lei S, Miller B, Sheikh S, Ali SM, Ahmad MU, Ahmad I (2005) Novel cationic cardiolipin analogue-based liposome for efficient DNA and small interfering RNA delivery in vitro and in vivo. Cancer Gene Ther 12(3):321–328
Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J (2001) Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem 276(37):35103–35110
Simberg D, Weisman S, Talmon Y, Barenholz Y (2004) DOTAP (and other cationic lipids): chemistry, biophysics, and transfection. Crit Rev Ther Drug Carr Syst 21(4):257–317
Simeoni F, Morris MC, Heitz F, Divita G (2003) Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res 31(11):2717–2724
Mountain A (2000) Gene therapy: the first decade. Trends Biotechnol 18(3):119–128
de Lima MC, Simoes S, Pires P, Faneca H, Duzgunes N (2001) Cationic lipid-DNA complexes in gene delivery: from biophysics to biological applications. Adv Drug Deliv Rev 47(2–3):277–294
Simberg D, Weisman S, Talmon Y, Faerman A, Shoshani T, Barenholz Y (2003) The role of organ vascularization and lipoplex-serum initial contact in intravenous murine lipofection. J Biol Chem 278(41):39858–39865
Mahato RI, Anwer K, Tagliaferri F, Meaney C, Leonard P, Wadhwa MS, Logan M, French M, Rolland A (1998) Biodistribution and gene expression of lipid/plasmid complexes after systemic administration. Hum Gene Ther 9(14):2083–2099
Niven R, Pearlman R, Wedeking T, Mackeigan J, Noker P, Simpson-Herren L, Smith JG (1998) Biodistribution of radiolabeled lipid-DNA complexes and DNA in mice. J Pharm Sci 87(11):1292–1299
Li S, Tseng WC, Stolz DB, Wu SP, Watkins SC, Huang L (1999) Dynamic changes in the characteristics of cationic lipidic vectors after exposure to mouse serum: implications for intravenous lipofection. Gene Ther 6(4):585–594
Li S, Rizzo MA, Bhattacharya S, Huang L (1998) Characterization of cationic lipid-protamine-DNA (LPD) complexes for intravenous gene delivery. Gene Ther 5(7):930–937
Tam P, Monck M, Lee D, Ludkovski O, Leng EC, Clow K, Stark H, Scherrer P, Graham RW, Cullis PR (2000) Stabilized plasmid-lipid particles for systemic gene therapy. Gene Ther 7(21):1867–1874
Bartsch M, Weeke-Klimp AH, Hoenselaar EP, Stuart MC, Meijer DK, Scherphof GL, Kamps JA (2004) Stabilized lipid coated lipoplexes for the delivery of antisense oligonucleotides to liver endothelial cells in vitro and in vivo. J Drug Target 12(9–10):613–621
Holland JW, Cullis PR, Madden TD (1996) Poly(ethylene glycol)-lipid conjugates promote bilayer formation in mixtures of non-bilayer-forming lipids. Biochemistry 35(8):2610–2617
Shi F, Wasungu L, Nomden A, Stuart MCA, Polushkin E, Engberts JBFN, Hoekstra D (2002) Interference of polyethylene glycol-lipid analogues with cationic lipid-mediated delivery of oligonucleotides; role of lipid exchangeability and non-lamellar transitions. Biochem J 366:333–341
Martin-Herranz A, Ahmad A, Evans HM, Ewert K, Schulze U, Safinya CR (2004) Surface functionalized cationic lipid-DNA complexes for gene delivery: PEGylated lamellar complexes exhibit distinct DNA-DNA interaction regimes. Biophys J 86(2):1160–1168
Woodle MC (1998) Controlling liposome blood clearance by surface-grafted polymers. Adv Drug Deliv Rev 32(1–2):139–152
Song LY, Ahkong QF, Rong Q, Wang Z, Ansell S, Hope MJ, Mui B (2002) Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim Biophys Acta 1558(1):1–13
Bergstrand N, Arfvidsson MC, Kim JM, Thompson DH, Edwards K (2003) Interactions between pH-sensitive liposomes and model membranes. Biophys Chem 104(1):361–379
Li W, Huang Z, MacKay JA, Grube S, Szoka FC Jr (2005) Low-pH-sensitive poly(ethylene glycol) (PEG)-stabilized plasmid nanolipoparticles: effects of PEG chain length, lipid composition and assembly conditions on gene delivery. J Gene Med 7(1):67–79
Guo X, MacKay JA, Szoka FC Jr (2003) Mechanism of pH-triggered collapse of phosphatidylethanolamine liposomes stabilized by an ortho ester polyethyleneglycol lipid. Biophys J 84(3):1784–1795
Ambegia E, Ansell S, Cullis P, Heyes J, Palmer L, MacLachlan I (2005) Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim Biophys Acta 1669(2):155–163
Rejman J, Wagenaar A, Engberts JB, Hoekstra D (2004) Characterization and transfection properties of lipoplexes stabilized with novel exchangeable polyethylene glycol-lipid conjugates. Biochim Biophys Acta 1660(1-2):41–52
Johnsson M, Wagenaar A, Stuart MCA, Engberts JBFN (2003) Sugar-based gemini surfactants with pH-dependent aggregation behavior: vesicle-to-micelle transition, critical micelle concentration, and vesicle surface charge reversal. Langmuir 19(11):4609–4618
Johnsson M, Wagenaar A, Engberts JBFN (2003) Sugar-based gemini surfactant with a vesicle-to-micelle transition at acidic pH and a reversible vesicle flocculation near neutral pH. J Am Chem Soc 125(3):757–760
Johnsson M, Engberts JBFN (2004) Novel sugar-based gemini surfactants: aggregation properties in aqueous solution. J Phys Org Chem 17(11):934–944
Scarzello M, Klijn JE, Wagenaar A, Stuart MCA, Hulst R, Engberts JBFN (2006) pH-dependent aggregation properties of mixtures of sugar-based gemini surfactants with phospholipids and single-tailed surfactants. Langmuir 22(6):2558–2568
Fielden ML, Perrin C, Kremer A, Bergsma M, Stuart MCA, Camilleri P, Engberts JBFN (2001) Sugar-based tertiary amino gemini surfactants with a vesicle-to-micelle transition in the endosomal pH range mediate efficient transfection in vitro. Eur J Biochem 268(5):1269–1279
Bell PC, Bergsma M, Dolbnya IP, Bras W, Stuart MCA, Rowan AE, Feiters MC, Engberts JBFN (2003) Transfection mediated by gemini surfactants: engineered escape from the endosomal compartment. J Am Chem Soc 125(6):1551–1558
Smisterova J, Wagenaar A, Stuart MCA, Polushkin E, ten Brinke G, Hulst R, Engberts JBFN, Hoekstra D (2001) Molecular shape of the cationic lipid controls the structure of cationic lipid/dioleylphosphatidylethanolamine-DNA complexes and the efficiency of gene delivery. J Biol Chem 276(50):47615–47622
Zuhorn IS, Bakowsky U, Polushkin E, Visser WH, Stuart MCA, Engberts JBFN, Hoekstra D (2005) Nonbilayer phase of lipoplex-membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol Ther 11(5):801–810
Scarzello M, Chupin V, Wagenaar A, Stuart MCA, Engberts JBFN, Hulst R (2005) Polymorphism of pyridinium amphiphiles for gene delivery: influence of ionic strength, helper lipid content, and plasmid DNA complexation. Biophys J 88(3):2104–2113
Oberle V, Bakowsky U, Zuhorn IS, Hoekstra D (2000) Lipoplex formation under equilibrium conditions reveals a three-step mechanism. Biophys J 79(3):1447–1454
Zuhorn IS, Visser WH, Bakowsky U, Engberts JB, Hoekstra D (2002) Interference of serum with lipoplex-cell interaction: modulation of intracellular processing. Biochim Biophys Acta 1560(1–2):25–36
McElroy WD, DeLuca MA (1983) Firefly and bacterial luminescence: basic science and applications. J Appl Biochem 5(3):197–209
Wilson T, Hastings JW (1998) Bioluminescence. Annu Rev Cell Dev Biol 14:197–230
Scarzello M, Smisterova J, Wagenaar A, Stuart MC, Hoekstra D, Engberts JB, Hulst R (2005) Sunfish cationic amphiphiles: toward an adaptative lipoplex morphology. J Am Chem Soc 127(29):10420–10429
Audouy SA, de Leij LF, Hoekstra D, Molema G (2002) In vivo characteristics of cationic liposomes as delivery vectors for gene therapy. Pharm Res 19(11):1599–1605
Iyer M, Berenji M, Templeton NS, Gambhir SS (2002) Noninvasive imaging of cationic lipid-mediated delivery of optical and PET reporter genes in living mice. Mol Ther 6(4):555–562
Stuart MCA, van de Pas JC, Engberts JBFN (2005) The use of Nile Red to monitor the aggregation behavior in ternary surfactant-water-organic solvent systems. J Phys Org Chem 18(9):929–934
Li W, Ishida T, Okada Y, Oku N, Kiwada H (2005) Increased gene expression by cationic liposomes (TFL-3) in lung metastases following intravenous injection. Biol Pharm Bull 28(4):701-706
Audouy S, Molema G, de Leij L, Hoekstra D (2000) Serum as a modulator of lipoplex-mediated gene transfection: dependence of amphiphile, cell type and complex stability. J Gene Med 2(6):465-476
Crook K, Stevenson BJ, Dubouchet M, Porteous DJ (1998) Inclusion of cholesterol in DOTAP transfection complexes increases the delivery of DNA to cells in vitro in the presence of serum. Gene Ther 5(1):137-143
Acknowledgements
This work was supported by a grant from The Netherlands Organization for Scientific Research (NWO)/NDRF Innovative Drug Research (940-70-001). The authors would like to thank Dr. Marc Stuart and Dr. Jaap Klijn for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wasungu, L., Scarzello, M., van Dam, G. et al. Transfection mediated by pH-sensitive sugar-based gemini surfactants; potential for in vivo gene therapy applications. J Mol Med 84, 774–784 (2006). https://doi.org/10.1007/s00109-006-0067-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-006-0067-z