
Abstract
Gemini surfactants are promising molecules utilized as non-viral gene delivery vectors. However, little is known about their cellular uptake and distribution after they release their therapeutic cargo. Therefore, we quantitatively evaluated the cellular uptake and distribution of three gemini surfactants: unsubstituted (16-3-16), with pyridinium head groups (16(Py)-S-2-S-16(Py)) and substituted with a glycyl-lysine di-peptide (16-7N(GK)-16). We also assessed the relationship between cellular uptake and distribution of each gemini surfactant and its overall efficiency and toxicity. Epidermal keratinocytes PAM 212 were treated with gemini surfactant nanoparticles formulated with plasmid DNA and harvested at various time points to collect the enriched nuclear, mitochondrial, plasma membrane, and cytosolic fractions. Gemini surfactants were then extracted from each subcellular fraction and quantified using a validated flow injection analysis-tandem mass spectrometry (FIA-MS/MS) method. Mass spectrometry is superior to the use of fluorescent tags that alter the physicochemical properties and pharmacokinetics of the nanoparticles and can be cleaved from the gemini surfactant molecules within biological systems. Overall, a significantly higher cellular uptake was observed for 16-7N(GK)-16 (17.0%) compared with 16-3-6 (3.6%) and 16(Py)-S-2-S-16(Py) (1.4%), which explained the relatively higher transfection efficiency of 16-7N(GK)-16. Gemini surfactants 16-3-16 and 16(Py)-S-2-S-16(Py) displayed similar subcellular distribution patterns, with major accumulation in the nucleus, followed by the mitochondrion, cytosol, and plasma membrane. In contrast, 16-7N(GK)-16 was relatively evenly distributed across all four subcellular fractions. However, accumulation within the nucleus after 5 h of treatment was the highest for 16(Py)-S-2-S-16(Py) (50.3%), followed by 16-3-16 (41.8%) and then 16-7N(GK)-16 (33.4%), possibly leading to its relatively higher toxicity.

Graphical Abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gemini surfactants are a versatile family of lipids that have a general structure of two surfactant monomers chemically linked by a spacer [1]. In particular, cationic gemini surfactants possess dual positively charged hydrophilic head groups, a spacer region, and two hydrophobic tails (Fig. 1) [2]. They are promising vectors for non-viral gene delivery [3,4,5] as their structures enable them to bind and compact DNA, facilitating cellular entry for gene transfection [6,7]. Extensive research has been conducted to design and synthesize novel gemini surfactants with the aim of enhancing transfection efficiency while reducing toxicity. For example, the positively charged head groups were altered using various cationic moieties, such as di-quaternary amines and di-pyridines, to obtain effective compaction of DNA [2,8]. Furthermore, amino acid moieties have been incorporated into the spacer region to enhance the biocompatibility of gemini surfactants and thus increase their transfection efficiencies [9]. In addition, the formulation of gemini surfactant-based lipoplexes and their cellular uptake mechanisms have been well studied [10,11], and it has been found that endocytosis is the main pathway by which gemini surfactant nanoparticles are internalized by the cell [11].

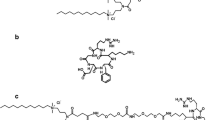
Schematic representation of the general structure of a gemini surfactant (a). The structures of gemini surfactants 16-3-16 (b), 16(Py)-S-2-S-16(Py) (c), and 16-7N(GK)-16 (d), showing their m/z values as well as the ions monitored during the FIA-MS/MS analysis
Despite their promise, gemini surfactants are still limited in their gene transfection efficiency [12,13], hindering their advancement from the experimental stage to clinical application. In addition, there are no clear explanations for the varying toxicities among different gemini surfactant structures. Therefore, a greater understanding of the mechanism of transfection and toxicity is required and will ultimately contribute to the development of more efficient and less toxic gemini surfactants. One factor that may be related to the overall efficiency and toxicity of gemini surfactants is their intracellular biological fate post-transfection; that is, how they are distributed at the subcellular and tissue levels. However, as of yet, their biological fate is poorly understood, and little is known about their cellular uptake, distribution, and metabolite formation upon transfection. Garnering such knowledge will contribute to the design and development of more effective gemini surfactants. In addition, an understanding of the cellular distribution of the delivery agents is crucial to achieve targeted delivery at the subcellular level [14]. Our research hypothesis is that the structure of a gemini surfactant significantly influences its cellular uptake and subsequent partitioning which in turn has profound consequences with respect to efficiency and toxicity. In fact, we have recently demonstrated the role of the molecular structure of gemini surfactants in determining their skin penetration efficiency [15].
Gemini surfactants designated as 16-3-16, 16(Py)-S-2-S-16(Py) and 16-7N(GK)-16) have been studied as non-viral gene delivery agents [16,17,18]. Structurally, 16-3-16 is a conventional m-s-m type gemini surfactant bearing two quaternary amines, linked by a 3-carbon spacer region (m is the number of carbon atoms in the tail and s is the number of carbon atoms in the spacer), while 16(Py)-S-2-S-(Py)16 is a pyrdinium-derived gemini surfactant containing two pyridines in the head groups, and 16-7N(GK)-16 bears a glycine-lysine di-peptide within the spacer region (Fig. 1), allowing for a better biocompatibility. In fact, these gemini surfactants have been successfully used for in vitro and in vivo gene delivery [16,17,18,19,20]. For instance, the 16-3-16 nanoparticles have shown great promise in the treatment of localized scleroderma, as transgene expression in animal models was significantly increased with treatment of the nanoparticles compared with naked DNA, showing the effectiveness of gemini surfactant-based gene delivery systems [6,20].
The selection of the three gemini surfactants (Fig. 1) was based on the variations in their molecular structures, transfection efficiency, and toxicity profiles [6,8,21]. Although these gemini surfactants belong to three different structural families that possess different head groups and spacer regions, they have the same number of carbon atoms in the tails (Fig. 1). We are currently conducting a wider assessment of the gemini surfactants in various families with respect to the relationship between cellular uptake, subcellular distribution, efficiency, and toxicity. As such, we chose these three structures as model compounds. Previously, we determined that the trend of cellular uptake and clearance of 16-3-16 and 16(Py)-S-2-S-(Py)16 nanoparticles were comparable in PAM 212 epidermal keratinocytes, which provides no explanation of the differential toxicities between the two compounds [22]. We, therefore, are testing the hypothesis that the relative efficiency and toxicity of these compounds is explained by a difference in their cellular uptake and subcellular distribution.
To assess the cellular uptake and distribution of lipid-based nanoparticles, fluorescent tags have often been incorporated into their structures [14,23]. However, the use of fluorescent tags suffers from two main drawbacks. First, the addition of such structure-modified moieties alters the physicochemical properties and pharmacokinetics of the nanoparticles [24,25]. Second, the fluorescent tags can be cleaved from the gemini surfactant molecules within biological systems, confounding subsequent data interpretation. Such limitations motivated us to develop mass spectrometry (MS)-based methods to monitor the fate of gemini surfactant nanoparticles in cells [26,27]. The main advantage of MS is its capability to measure the original intact molecule with high selectivity and sensitivity [26]. Most recently, we developed and validated a simple flow injection analysis-tandem mass spectrometry (FIA-MS/MS) method that allows for the tracking of gemini surfactants at the subcellular level [28].
In the present study, the validated FIA-MS/MS method is applied to provide for the first time a quantitative assessment of the cellular uptake and subcellular distribution of three gemini surfactant gene delivery nanoparticles (Fig. 1) within PAM 212 cells. We found that variable cellular uptake of the three gemini surfactants explained the differences in transfection efficiency, and that accumulation of gemini surfactant in the nucleus may provide insights into the observed increased toxicity.
Materials and Methods
Materials
Gemini surfactants 16-3-16, 16(Py)-S-2-S-16(Py) and 6-7N(GK)-16 (Fig. 1) and their deuterated internal standards 16-3-16-D66, 16(Py)-S-2-S-16(Py)-D10, and 16-7N(GK)-16-D4 (Fig. S1, supporting information) were synthesized according to established protocols [16,19,29]. The neutral lipid 1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Chloroform, methanol, acetonitrile, formic acid, tissue culture flasks (75 cm2, 150 cm2), and petri dishes (150 cm2) were purchased from Fisher Scientific (Ottawa, ON, Canada). Ninety-six-well tissue culture plates were obtained from Falcon (BD Mississauga, ON, Canada). PAM 212 keratinocyte cells were kindly provided by Dr. S. Yuspa, National Cancer Institute, Bethesda, MD, USA. Fetal bovine serum albumin (FBS), antibiotic-antimycotic solution, and minimum essential media (MEM) were obtained from Sigma-Aldrich (Oakville, ON, Canada). The protease inhibitor cocktail was purchased from Invitrogen (Burlington, ON, Canada). The plasmid DNA (pGTmCMV.IFN-GFP) was constructed in-house as previously described [6]. The motorized homogenizer was purchased from Fisher Scientific (Toronto, ON, Canada).
Formulation
Gemini surfactants and internal standards were prepared at a concentration of 3 mM in aqueous solutions and stored at − 80 °C under darkness. DOPE vesicles were prepared freshly at a concentration of 1 mM in isotonic sucrose solution (9.25% w/v, pH = 9) based on an established protocol [5]. The plasmid DNA solution was prepared at 200 μg/mL in ultra-pure water and stored at − 80 °C.
The nano-lipoplex formulation (P/G/L) was prepared with plasmid DNA, gemini surfactant, and lipid DOPE as previously described [6] with a nitrogen (cationic) to phosphate (anionic) charge ratio (N/P) at 10 for 16-3-16 and 16(Py)-S-2-S-16(Py) and at 2.5 for 16-7N(GK)-16. Briefly, to prepare 1 mL of the P/G/L for 16-3-16 and 16(Py)-S-2-S-16(Py), 38 μL of 3 mM gemini surfactants was added to 38 μL of 200 μg/mL plasmid DNA, gently mixed by pipetting up and down several times, and incubated for 15 min at room temperature. Subsequently, 924 μL of 1 mM DOPE solution was added to the binary mixture, gently mixed with a pipette and incubated for 15 min at room temperature to produce the ternary P/G/L system (nanoparticles). To prepare 1 mL of the P/G/L for 16-7 N(GK)-16, 9.5 μL of 3 mM gemini surfactant was added to 38 μL of 200 μg/mL plasmid DNA, mixed and incubated for 15 min at room temperature, and 952.5 μL of 1 mM DOPE solution was then added, mixed, and incubated to generate the nanoparticles.
In Vitro Transfection
PAM 212 cells were cultured in MEM media supplemented with 10% (v/v) FBS and 1% (v/v) antibiotic-antimycotic solution in 75-cm2 tissue culture flasks in a humidified incubator at 37 °C at an atmosphere of 5% CO2. Upon reaching 80% confluence, cells were washed with phosphate-buffered saline (PBS, 8 mL), dissociated with a 5-min incubation in a Versene (10×, 3 mL) and Trypsin (10×, 0.3 mL) mixture and collected by centrifugation (250×g, 5 min, 4 °C). Twenty-four hours prior to transfection, three 96-well tissue culture plates were seeded with PAM 212 cells at a density of 2 × 104 cells/well. MEM was replaced with serum-free media 1 h prior to transfection. Cells were treated with 20 μL of the P/G/L nanoparticles per well and incubated for 5 h. The cells were then returned to supplemented MEM for further incubation, and the culture media were collected at 48 h for interferon-gamma (IFN-Ƴ) measurement.
An enzyme-linked immunosorbent assay (ELISA) was carried out to measure secreted IFN-Ƴ using flat-bottom 96-well plates (Immulon 2, Greiner Labortechnik, Germany) as per the BD Pharmingen protocol. An IFN-Ƴ standard curve was established using recombinant mouse IFN-Ƴ (BD Biosciences, Missisauga, ON, Canada) to allow for the concentration of secreted IFN-Ƴ in the cell media to be quantified. The experiments were conducted in three plates of quadruplicate wells.
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay
A 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was performed to determine the cytotoxicity of the gemini surfactants. PAM 212 cells were seeded in three 96-well cell culture plates at a density of 2 × 104 cells/well and treated with the P/G/L nanoparticles. Plates were incubated for 5 h at 37 °C with 5% CO2 in a humidified incubator and then the cell media were switched to supplemented MEM media. After 48 h of incubation, the cell media were removed and cell toxicity was evaluated by the determination of cell viability. Briefly, 100 μL of 0.5 mg/mL sterile MTT (Invitrogen, USA) in the supplemented media was added to each well and the plates were incubated for 3 h at 37 °C. The media were then removed, and 200 μL of dimethyl sulfoxide (DMSO) (spectroscopy grade, Sigma-Aldrich, ON, Canada) was added to each well to dissolve the formed purple formazan crystal. Subsequently, the plates were incubated at 37 °C for 10 min. Absorbance was measured at 550 nm using a microplate reader (BioTek® Microplate Synergy, HT, VT, USA). The experiments were conducted in three plates of quadruplicate wells, and the cytotoxicity of gemini surfactants reflects cell viability expressed as a percentage of the non-transfected cells (control).
Size and Zeta-Potential Measurements
Size and zeta potential of the gemini surfactant-based nanoparticles were measured using a Zeta-sizer Nano ZS instrument (Malvern Instruments, Worcestershire, UK). The nanoparticles were prepared as described in “Formulation”. Three measurements were conducted for each sample. The reported results are the mean of triplicate measurements ± standard deviation.
Cell Treatment and Sample Collection
PAM 212 cells were cultured in 150 cm2 flasks until they reached 80% confluence. Cells were then washed with PBS (25 mL), dissociated with Versene (10×, 5 mL) and trypsin (10×, 0.5 mL) and collected by centrifugation (250×g, 5 min, 4 °C). Twenty-four hours prior to treatment, 8 × 106 cells were seeded in each petri dish (150 cm2). Cells were switched to serum-free media 1 h prior to transfection. Five hundred microliters of freshly prepared P/G/L nanoparticles were added to each dish in a drop-wise manner. Following 5 h of incubation, the cells were returned to supplemented MEM media for subsequent incubation steps. Triplicates of treated cell samples and one control (untreated cell) were trypsinized and collected at 2 h, 5 h, and 8 h, respectively. The collected cells were pelleted by centrifugation (250×g, 5 min, 4 °C), rinsed with PBS three times, resuspended in 500 μL of ice-cold hypotonic homogenization buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl [PH 7.5], and cOmplete™ protease inhibitor), and incubated on ice for 10 min.
Subcellular Fractionation Using Differential Centrifugation
Cells were gently homogenized on ice to break the plasma membrane and release subcellular organelles. The cell homogenates were then diluted with ice-cold hypertonic buffer (420 mM mannitol, 140 mM sucrose, 10 mM Tris–HCl [pH 7.5], and 2 mM EDTA [pH 7.5]) to a final volume of 1 mL and enriched nuclear, mitochondrial, plasma membrane and cytosolic fractions were isolated by differential centrifugation using an established protocol [30], with slight modifications (Fig. 2). Briefly, homogenates were first centrifuged at 1000×g for 10 min at 4 °C, and the S1 supernatant was transferred to a clean ice-cooled microcentrifuge tube while the P1 pellet was collected as the nuclear fraction (nuclei, unbroken cells and cell debris). The S1 supernatant was then subjected to further centrifugation at 15,000×g for 15 min at 4 °C, yielding the S2 supernatant and the P2 pellet which contained the mitochondrial fraction. The S2 supernatant was then centrifuged at 100,000×g for 60 min at 4 °C. The resultant S3 supernatant contained the cytosol and the P3 pellet (the plasma membrane along with microsomes, ER and Golgi). All collected fractions were kept on ice prior to being diluted to equal 950 μL volume with PBS and stored at − 80 °C. To verify the successful isolation and relative purity of enriched fractions, Western blot analysis was performed. Relevant experimental details and results are shown in the supporting information (see Appendix 2).

Schematic illustration of the homogenization and subcellular fractionation protocol. Differential centrifugation was used to isolate several subcellular fractions, including those enriched for nuclei, mitochondria, plasma membrane, and cytosol
Sample Preparation
As we previously described [28], subcellular fractions (950 μL) were lysed and spiked with 50 μL of internal standard and sample extractions were conducted using the Bligh/Dyer method [31]. Briefly, 3.75 mL of methanol-chloroform (2:1, v/v) was added per 1 mL of sample, followed by the addition of 1.25 mL of chloroform and 1.25 mL of water. At each step, samples and the extraction solvent were vortexed thoroughly. The final mixture was centrifuged at 2800×g for 10 min at room temperature to separate the aqueous and organic phases. The bottom organic phase (80% portion) was collected and dried under a N2 gas stream, followed by reconstitution in 200 μL of methanol. Finally, 150 μL of methanol solution was transferred into an HPLC vial for analysis.
FIA-MS/MS Analysis
The FIA-MS/MS analysis was performed on a quadrupole-linear ion trap (4000 QTRAP®) mass spectrometer with an electrospray ionization (ESI) source (AB Sciex, Concord, ON, Canada), coupled with an Agilent 1200 series HPLC (a quaternary pump, degasser and auto sampler) (Agilent Technologies, Mississauga, ON, Canada). As recently described [28], 3 μL of sample was injected into the ESI source at a flow rate of 0.5 mL/min with an acetonitrile-water mixture (98:2, v/v) containing 0.1% formic acid as the mobile phase. The instrument source temperature was set at 600 °C and the ion spray voltage was at 5500 V. Nitrogen gas was used for curtain gas at 30, nebulizer gas at 55, and heater gas at 50. Multiple reaction monitoring (MRM) in positive ion mode was used to monitor all analytes and internal standards. The monitored MRM transitions were as follows: 16-3-16 [M]2+ m/z 290.3 → 355.4, 86.1; 16-3-16-D66 [M]2+ m/z 323.5 → 388.6; 16(Py)-S-2-S-(Py)16 [M]2+ m/z 349.3 → 396.3, 203.1; 16(Py)-S-2-S-(Py)16-D10 [M]2+ m/z 354.3 → 401.3; and 16-7N(GK)-16 [M]2+ m/z 411.4 → 276.8, 268.3; 16-7N(GK)-16-D4 [M]2+ m/z 413.4 → 278.8 (Figs. 1 and S1). The compound-dependent parameters for analytes and internal standards were optimized as previously described [28]. A stable isotope dilution standard curve and three quality control samples (low, medium and high) were run along with the samples in each batch. The data acquisition time per sample was 2 min. Data acquisition and analysis was performed using AB Sciex Analyst software (version 1.6.0).
Ethidium Bromide Dye Exclusion Assay
The plasmid DNA (200 μg/mL) was complexed with the three gemini surfactants at various charge ratios in the presence or absence of DOPE on 96-well plates. Ethidium bromide was added to all samples at a final concentration of 1 μg/mL. The samples were then incubated for 10 min at room temperature. After that, fluorescence excitation was carried out at 530 nm and emission was measured at 590 nm using a microplate reader (BioTek Microplate Synergy HT, VT, USA). The relative fluorescence of the P/G/L and P/G complexes was expressed as a percentage of fluorescence of the pure plasmid DNA solution. Measurements were conducted in triplicate.
Langmuir Studies
Langmuir trough was used to measure the monolayer surface area of the gemini surfactant head group. Surface pressure-mean molecular area isotherms were obtained using a KSV 2,000 L trough instrument (KSV Instruments Ltd., Helsinki, Finland). A stock solution of each gemini surfactant was prepared at 1 mM in chloroform, and 40 μL of each stock solution was added drop-wise on the sub-phase using a Hamilton syringe. The monolayer was left for a minimum of 10 min to allow chloroform to evaporate, and a constant rate compression of 20 mm/min was then applied on the monolayer molecules until collapse of the monolayer lipid. The ultra-pure water (Millipore, resistivity 18 MΩ· cm) was used as a sub-phase in the trough, and the sub-phase temperature was set at 22 °C. Triplicate measurements were collected for each gemini surfactant, and data collection was performed using the KSV software (KSV Instruments Ltd., Helsinki, Finland).
Statistical Analysis
Statistical analyses were performed by one-way analysis of variance (ANOVA) and Tukey’s multiple comparison tests using SPSS 25 software. Significant difference was established at the p < 0.05 level of significance. Results are expressed as the mean of triplicates ± standard deviation.
Results and Discussion
In Vitro Transfection Activity
These gemini surfactant nanoparticles have shown great promise in treating the localized scleroderma, a rare skin disease [6,20]; we are currently tuning the gemini surfactant nanoparticles to develop an effective, non-invasive topical gene delivery system for the treatment of the fibrotic skin conditions. As such, the epidermal keratinocyte, PAM 212, was used as the cell model in this study. To evaluate the efficiency of the gemini surfactants to mediate transfection in PAM 212 cells, the amount of secreted IFN-Ƴ was quantified 48 h post-transfection. As determined in previous work [6,16], the optimal N/P to obtain the best transfection efficiency for 16-3-16 is 10 and for 16-7N(GK)-16 is 2.5. We also determined that the optimal N/P for 16(Py)-S-2-S-16(Py) is 10 based on the assessment of various N/P ratios at 0.5, 1, 2.5, 5, 10, and 20 for transfection (data not shown). For the best comparison of transfection capability as well as for proper toxicity assessment, the in vitro transfection study was conducted at the optimal N/P of each gemini surfactant. It reflects the real conditions in which these compounds are used for in vitro gene transfer. At the optimal N/P of 10, the P/G/L of 16(Py)-S-2-S-(Py)16 resulted in a significantly lower level of IFN-Ƴ (1.19 ± 0.08 ng/2 × 104 cells) when compared with that of 16-3-16 (2.77 ± 0.13 ng/2 × 104 cells) (p < 0.05) (Fig. 3). In comparison, the P/G/L of 16-7N(GK)-16 at its optimal N/P of 2.5 yielded a IFN-Ƴ level (3.76 ± 0.27 ng/2 × 104 cells) [21] that is significantly higher than that of 16-3-16 (p < 0.05). In sum, the relative transfection efficiency of the three gemini surfactant nanoparticles was determined, with 16-7N(GK)-16 being the most effective and 16(Py)-S-2-S-(Py)16 the least effective.

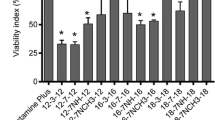
Transfection efficiencies of the P/G/Ls of 16-3-16, 16(Py)-S-2-S-(Py)16 and 16-7N(GK)-16 in PAM 212 cells. *16-7N(GK)-16 transfection was recently reported by our group, extracted from ref. [21]
Cytotoxicity
In the present study, the cytotoxicity of the gemini surfactants 16-3-16 and 16(Py)-S-2-S-(Py)16 was evaluated in PAM 212 cells. It was observed that cell viability was significantly higher upon treatment with the P/G/L of 16-3-16 (76%) compared with that of 16(Py)-S-2-S-(Py)16 (61%, p < 0.05) (Fig. 4), indicating that the former has significantly lower cytotoxicity than the latter for the tested PAM 212 cell line. However, 16-7N(GK)-16 was previously reported to have an even lower toxicity (89% cell viability) than 16-3-16 [21]. Hence, 16-7N(GK)-16 possesses the lowest cytotoxicity and 16(Py)-S-2-S-(Py)16 has the highest cytotoxicity among the three gemini surfactants.

Determination of Size and Zeta Potential
Size and zeta potential of the P/G/L nanoparticles were measured as they are important characteristics of the delivery systems, which can have an influence on their stability, cellular uptake, and cytotoxicity [32,33]. At the optimal N/P of 10, the P/G/L of 16-3-16 displayed a size of 131.9 ± 1.48 nm and positive zeta potential at 17.3 ± 1.38 mV, and the P/G/L of 16(Py)-S-2-S-(Py)16) showed a comparable size of 123.1 ± 0.76 nm and zeta potential at 23.3 ± 0.32 mV. Compared with the P/G/Ls of 16-3-16 and 16(Py)-S-2-S-(Py)16, the P/G/L of 16-7N(GK)-16 at its optimal N/P of 2.5 showed a similar zeta potential at 24 ± 2.00 mV but a bit smaller size of 80 ± 1.00 nm as reported in previous work [21]. Endocytosis has shown to be the main mechanism for the internalization of gemini surfactant-based nanoparticles [11,34]. In particular, clathrin and caeolae-mediated endocytosis are most common pathways for the cellular uptake of gemini surfactant nanoparticles. In fact, the internalization of amino acid-substituted gemini surfactant nanoparticles involves equally both clathrin and caeolae-mediated endocytosis [11]. While particles with smaller sizes of 60–80 nm typically undergo caveolae-mediated endocytosis for their internalization, particles with relatively larger size in the range of 120–200 nm often enter the cells via clathrin-mediated endocytosis [35,36]. As such, all three gemini surfactant nanoparticles have the particle sizes that are appropriate for their cellular uptake. However, the smaller size of the P/G/L of 16-7N(GK)-16 could be a contributing factor for its high efficiency in gene transfection, as nanoparticles with small size at 70 nm have been reported to display significantly higher transfection efficiency than large-sized nanoparticles at 200 nm [32].
Cellular Uptake and Distribution of Gemini Surfactants
To determine the cellular uptake and distribution of the gemini surfactants in PAM 212 cells, fractions enriched for nuclei, mitochondria, plasma membrane, and cytosol were isolated by differential centrifugation, extracted and analyzed by the validated FIA-MS/MS method [28]. Differential centrifugation is an isolation technique that uses stepwise increases in centrifugal force to precipitate subcellular components based on their distinct density, size, and shape [37]. During the FIA-MS/MS analysis, the standard curve achieved a less than 15% deviation of the nominal value for each standard point other than the lower limit of quantification (LLOQ), which was 20%, and the quality control (QC) samples were accepted with a less than 15% deviation of the nominal values, as per FDA guidelines [38].
Cellular uptake of the three gemini surfactants, expressed as percentage of dose, was observed to increase rapidly over the course of a 5-h treatment in PAM 212 cells, reaching a maximum of 17.0% for 16-7N(GK)-16, 1.4% for 16-3-6, and 3.6% for 16(Py)-S-2-S-16(Py), followed by a gradual depletion after the removal of the nanoparticles from the media (Fig. 5a). A significantly higher cellular uptake was observed for the gemini surfactant 16-7N(GK)-16 compared with 16-3-16 and 16(Py)-S-2-S-16(Py) (Fig. 5a) (p < 0.05). It should be noted that the optimal N/P for 16-3-16 and 16(Py)-S-2-S-16(Py) was 10, whereas for the 16-7N(GK)-16 was 2.5 as previously determined in our lab [6,16], and all transfections were conducted with equal amounts of plasmid DNA in the P/G/L nanoparticles. Therefore, the higher cellular uptake of 16-7N(GK)-16 implies that there is a higher transfection efficiency with this gemini surfactant relative to 16-3-16 and 16(Py)-S-2-S-16(Py) and offers a mechanistic explanation as to why PAM 212 cells exposed to 16-7N(GK)-16 secrete greater amounts of IFN-Ƴ (Fig. 3).

The cellular uptake and distribution of gemini surfactants in PAM 212 cells. a Cellular uptake, normalized based on the dose, of three gemini surfactants. b–d Subcellular distribution, normalized based on the total cellular uptake, of 16-3-16, 16(Py)-S-2-S-(Py)16 and 16-7N(GK)-16. e Distribution percentage in nucleus. (Cyto—cytosol, Mito—mitochondria, Nuc—nucleus, and PM—plasma membrane), * indicates p < 0.05
While the gemini surfactant 16(Py)-S-2-S-16(Py) accumulated more significantly than 16-3-16 in PAM 212 after a 5-h treatment (p < 0.05), less IFN-Ƴ was produced, arguing that transfection efficiency is lower with 16(Py)-S-2-S-16(Py) than 16-3-16. The relatively higher toxicity of 16 (Py)-S-2-S-16 (Py) (Fig. 4), which caused greater cell death, might have contributed to reducing the overall gene transfection efficiency.
In terms of subcellular distribution, normalized for each surfactant to the total cellular uptake, the gemini surfactants 16-3-16 and 16(Py)-S-2-S-16(Py) exhibited comparable partitioning with significant accumulation in the nucleus, followed by mitochondria, cytosol, and plasma membrane (Fig. 5b, c) (p < 0.05). In contrast, the distribution of 16-7N(GK)-16 was relatively even across the four subcellular compartments (Fig. 5d). No significant difference was observed among the three gemini surfactants with respect to their relative distribution in the mitochondrial and cytosolic fractions. However, accumulation in the nucleus was the highest for 16(Py)-S-2-S-16(Py) (50.3%), followed by 16-3-16 (41.8%) and then 16-7N(GK)-16 (33.4%) (p < 0.05) at the 5-h duration of treatment (Fig. 5e), which correlates with the relative toxicity observed for the three gemini surfactants (Pearson’s correlation r = 0.9993). Even at the 2-h and 8-h time points post treatment, 16-7N(GK)-16 still displayed significantly lower accumulation in the nucleus relative to 16(Py)-S-2-S-16(Py) and 16-3-16 (p < 0.05). Although there is a difference in the accumulation among the three gemini surfactants at 2 h time point, this accumulation might not be informative as the cellular uptake are still in rapid progress at this stage. Surprisingly, 16(Py)-S-2-S-16(Py) showed a similar distribution percentage in the nucleus as 16-3-16 at the 8-h time point; however, the distribution quantity (in terms of absolute amount) of 16-3-16 is significantly less compared with that of 16(Py)-S-2-S-16(Py) (Table S1 in supporting information) as 16-3-16 has much lower cellular uptake in comparison with 16(Py)-S-2-S-16(Py) (Fig. 5a), resulting in less toxicity of 16-3-16.
Nuclear accumulation may result from either the entry of the gemini surfactants into the compartment or their association with the nuclear envelope, as it has been reported that lipoplexes can fuse with the membrane and release their DNA cargo into the nucleus [39]. However, it is currently not known that the entry of gemini surfactants into the nucleus is in the form of lipid molecules or lipoplexes. Although, the entry of lipoplexes is unlikely due to the small pore size of nuclear membrane, which can take place during cell mitosis. As such, it could be in either form or both.
The nucleus has crucial functions within a cell, ensuring the faithful storage and expression of genetic material essential to regulating cellular metabolism and growth [40]. Therefore, nuclear association or accumulation of gemini surfactants could impact outer membrane integrity and normal organelle function. As such, the higher cellular uptake and preferential accumulation of 16(Py)-S-2-S-16(Py) within the nucleus could provide a basis for its higher toxicity relative to 16-3-16 and 16-7N(GK)-16. Thus, to date, our findings provide the only quantitative distinction in the subcellular profiles of these three gemini surfactants, without the use of a florescent tag. The results may offer the first mechanistic insight into the various efficiencies and toxicities observed for the three promising gene delivery agents.
In addition, it was observed that 16-7N(GK)-16 has a significantly higher distribution within the plasma membrane compared with 16(Py)-S-2-S-16(Py) and 16-3-16 (Fig. 5b–d) (p < 0.05). However, this did not result in higher toxicity, as evidenced by the viability of PAM 212 cells treated with the 16-7N(GK)-16 nanoparticles (Fig. 4). In fact, 16-7N(GK)-16 displayed the lowest cytotoxicity among the three gemini surfactants in gene transfection. Stefanutti et al. [41] reported that the internalization of lipoplexes of DMPC, and a cationic gemini surfactant traversing cell membrane did not cause a significant biological damage to the cells. In addition, Marjan et al. [42] reported that although nanoparticle treatment disturbed membrane integrity, the cells were still alive and metabolically active during the transfection process. Therefore, accumulation in the plasma membrane does not appear to cause toxicity.
Ethidium Bromide Dye Exclusion Assay
To explore why unique gemini surfactants differentially accumulate within distinct subcellular compartments, an ethidium bromide dye exclusion assay was conducted to investigate their DNA binding and compaction capabilities. Gemini surfactants bind and compact plasmid DNA via electrostatic interactions to form nanosized particles, which hinder the penetration of ethidium bromide into the complexes. As a result, fluorescence is quenched due to the lack of intercalation between ethidium bromide and the base-pairs of DNA. The stronger the compaction of DNA by gemini surfactants, the more intense the fluorescence quenching in the complex. As shown in Fig. 6, the lowest fluorescence emission was observed at the N/P of 5, with 13.0% for 16-7N(GK)-16, 8.1% for 16-3-16, and 8.9% for 16(Py)-S-2-S-16(Py) in the absence of the helper lipid DOPE. The data indicate that 16-7N(GK)-16 has a significantly lower DNA compaction capability compared with the other two compounds (p < 0.05). In the presence of a helper lipid, the fluorescence values were increased to 45.7% for 16-7N(GK)-16, 23.6% for 16-3-16, and 28.3% for 16(Py)-S-2-S-16(Py), again showing 16-7N(GK)-16 is significantly less efficient at binding and compacting DNA relative to 16-3-16 and 16 (Py)-S-2-S-16(Py) (p < 0.05).

Ethidium bromide dye exclusion assay to evaluate the DNA binding and compaction capability of the gemini surfactants. A lower fluorescence indicates stronger DNA binding and compaction
However, the relatively lower DNA-binding capability of 16-7N(GK)-16 does not undermine its gene transfection capability, as evidenced in the transfection study, since gene transfection requires not only effective compaction of DNA for their protection and cellular entry but also efficient release from the complex into the nucleus for transgene expression. It is believed that the presence of glycyl-lysine moiety in the spacer offers conformational flexibility of the structure, which bestows 16-7N(GK)-16 with softened DNA binding properties [9,43]. Although 16-7N(GK)-16 has a relatively weaker DNA-binding ability than the other two compounds as indicated by its higher fluorescence emission of 13% compared with 8.1% for 16-3-16 and 8.9% for 16(Py)-S-2-S-16(Py), such binding provides the DNA with much needed protection against enzymatic degradation while also facilitating its intracellular release, thereby enhancing overall transfection efficiency [9,43]. This special binding capability may be caused by the overall interaction of multiple bonding forces, including hydrogen bonding and electrostatic interactions [44]. In addition, the various amine groups in the amino acids can allow for additional buffering capacity, which helps in the disruption of the endosomal membrane for the intracellular release of DNA and their nuclear transport [9].
Due to the weaker DNA binding of 16-7N(GK)-16, the encapsulated DNA could be released more efficiently from the lipoplexes into the cytoplasm to translocate into the nucleus for gene expression. As such, this led to a lesser accumulation of 16-7N(GK)-16 in the nucleus. Conversely, the lipoplexes formed with 16-3-16 and 16(Py)-S-2-S-16(Py) release the DNA less efficiently and potentially at a later stage due to their stronger association with DNA (Fig. 6), which could be one of the reasons for their lower transfection efficiency relative to 16-7N(GK)-16, as it has been reported that slow vector unpacking is linked to a decreased transfection efficiency [45,46]. In this case, the lipoplexes rather than free DNAs are more likely to be translocated into the nucleus. Consistent with this idea, we observed elevated accumulation of both 16-3-16 and 16(Py)-S-2-S-16(Py) in the nucleus (Fig. 5b and c).
Molecular Packing Parameter
In addition to their DNA compaction and binding properties, the molecular shape of gemini surfactants also have an great impact on the performance of the gene delivery system [16,47]. Therefore, to further understand the behavioral differences among the three gemini surfactants, structural differences in the formed aggregates was evaluated using the molecular packing parameter (P) [47]. The P was estimated based on the structures of gemini surfactants and the behavior of gemini surfactants at the air-water interface [48], and is defined as
where v is the volume of the hydrophobic tails, l is the length of the hydrocarbon tails, and ao is the head group area per molecule in aqueous solution. The v and l are the geometrical properties of gemini surfactants, which can be calculated from the chemical structures [49,50]. The ao is an equilibrium parameter dependent upon both the attractive forces of the hydrophobic chains and the repulsive forces of the head groups, which can be determined by the Langmuir studies. A specific P value can be linked to a particular geometrical shape [48]. Spherical micelles typically have a P value of less than 0.33; cylindrical micelles possess a P value between 0.33 and 0.50, whereas flexible bilayers (vesicles) usually have a P value between 0.5 and 1.0 [48].
As the three gemini surfactants have the same l and v of hydrophobic tails, the ao is the main determinant of the P value. The l was calculated to be 21.74 Å using the Avogardro software [51], and the v was calculated to be 918 Å3 with the Gaussian 09 software (revision B. 01) [52] (Table 1). Based on the Langmuir studies, 16-7N(GK)-16 showed the smallest a0 of 84 Å2 and thus the largest P of 0.51, indicating the aggregates formed by 16-7N(GK)-16 are typically flexible bilayers. 16-3-16 displayed an a0 of 116 Å2 and a P value of 0.36, which suggested the formation of aggregates shaped as cylindrical micelles (Table 1). This is in agreement with the literature [50,53] reporting that aggregates formed by m-3-m gemini surfactants tend to form cylindrical micelles. Similarly, 16(Py)-S-2-S-16(Py) had an a0 of 110 Å2 and a P value of 0.38, which argues it too forms cylindrical micelles in aqueous solution.
Although 16-3-16 and 16(Py)-S-2-S-16(Py) have unique structural head groups, their areas are comparable and enable them to have similar packing parameters and a preference towards forming cylindrical micelle aggregates (Table 1). The same aggregates would allow for a similar internalization process of the two gemini surfactant nanoparticles for gene delivery, which explained the similar trends observed for their uptake and subcellular distribution (Fig. 5b and c). Conversely, the substitution of a di-peptide in the spacer region provides 16-7N(GK)-16 with conformational flexibility [9]. It has a much smaller head group area and thus a flexible bilayer structure (Table 1), which allows for the formation of the inverted hexagonal phase of the lipoplex [47]. Such a conformation facilitates not only the destabilization of endosomal membrane to promote the cytoplasmic release of DNA but also the dissociation of DNA from the lipoplexes, thus resulting in enhanced gene transfection [54].
Conclusions
The cellular uptake and distribution of the gemini surfactants 16-3-16, 16(Py)-S-2-S-16(Py) and 16-7N(GK)-16 as gene delivery agents in PAM 212 cells was evaluated by analyzing subcellular fractions collected by differential centrifugation using a validated FIA-MS/MS method. The three gemini surfactants varied with respect to their uptake and subcellular distribution profiles, with 16-7N(GK)-16 exhibiting greater uptake and a higher transfection efficiency. Preferential nuclear accumulation or association of 16(Py)-S-2-S-16(Py) may explain its relatively higher toxicity. DNA binding and molecular packing experiments provided explanations to the different cellular behaviors of the gemini surfactants. Overall, the results presented herein demonstrate the general applicability of the combined differential centrifugation and MS approach for assessing the uptake and subcellular distribution of gemini surfactants and emphasize that it is superior to a fluorescence-labeling method as it does not require any structural modifications. We are currently investigating the metabolite formation of the three structures, which may provide additional insight into their relative efficiencies and toxicities. In the future, it may be worthwhile isolating more cellular organelles, such as the endosomes and lysosomes to better understand the cellular trafficking of lipid-based gene delivery agents.
References
Menger FM, Keiper JS. Gemini surfactants. Angew Chem Int Ed. 2000;39(11):1906–20.
Wettig SD, Verrall RE, Foldvari M. Gemini surfactants: a new family of building blocks for non-viral gene delivery systems. Curr Gene Ther. 2008;8(1):9–23.
Donkuru M, Badea I, Wettig S, Verrall R, Elsabahy M, Foldvari M. Advancing nonviral gene delivery: lipid-and surfactant-based nanoparticle design strategies. Nanomedicine. 2010;5(7):1103–27.
Bombelli C, Giansanti L, Luciani P, Mancini G. Gemini surfactant based carriers in gene and drug delivery. Curr Med Chem. 2009;16(2):171–83.
Makhlouf A, Hajdu I, Badea I. Gemini surfactant-based systems for drug and gene delivery, in Organic Materials as Smart Nanocarriers for Drug Delivery: Published by William Andrew, Applied Science Publisher. 2018. p. 561–600.
Badea I, Verrall R, Baca-Estrada M, Tikoo S, Rosenberg A, Kumar P, et al. In vivo cutaneous interferon-gamma gene delivery using novel dicationic (gemini) surfactant-plasmid complexes. J Gene Med. 2005;7(9):1200–14.
Grueso E, Kuliszewska E, Roldan E, Perez-Tejeda P, Prado-Gotor R, Brecker L. DNA conformational changes induced by cationic gemini surfactants: the key to switching DNA compact structures into elongated forms. RSC Adv. 2015;5(37):29433–46.
Van Der Woude I, et al. Novel pyridinium surfactants for efficient, nontoxic in vitro gene delivery. Proc Natl Acad Sci. 1997;94(4):1160–5.
Singh J, Yang P, Michel D, E. Verrall R, Foldvari M, Badea I. Amino acid-substituted gemini surfactant-based nanoparticles as safe and versatile gene delivery agents. Curr Drug Deliv. 2011;8(3):299–306.
Mohammed-Saeid W, Michel D, el-Aneed A, Verrall RE, Low NH, Badea I. Development of lyophilized gemini surfactant-based gene delivery systems: influence of lyophilization on the structure, activity and stability of the lipoplexes. J Pharm Pharm Sci. 2012;15(4):548–67.
Singh J, Michel D, Chitanda JM, Verrall RE, Badea I. Evaluation of cellular uptake and intracellular trafficking as determining factors of gene expression for amino acid-substituted gemini surfactant-based DNA nanoparticles. J Nanobiotechnol. 2012;10:7.
Ramamoorth M, Narvekar A. Non viral vectors in gene therapy-an overview. J Clin Diagn Res. 2015;9(1):GE01–6.
Al-Dosari MS, Gao XJTAj. Nonviral gene delivery: principle, limitations, and recent progress. AAPS J. 2009;11(4):671.
Savić R, et al. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science. 2003;300(5619):615–8.
Al-Dulaymi M, et al. The development of simple flow injection analysis tandem mass spectrometric methods for the cutaneous determination of peptide-modified cationic gemini surfactants used as gene delivery vectors. J Pharm Biomed Anal. 2018;159:536–47.
Al-Dulaymi MA, et al. Di-peptide-modified gemini surfactants as gene delivery vectors: exploring the role of the alkyl tail in their physicochemical behavior and biological activity. AAPS J. 2016;18(5):1168–81.
Singh J, Michel D, Getson HM, Chitanda JM, Verrall RE, Badea I. Development of amino acid substituted gemini surfactant-based mucoadhesive gene delivery systems for potential use as noninvasive vaginal genetic vaccination. Nanomedicine. 2015;10(3):405–17.
Badea I, Virtanen C, Verrall RE, Rosenberg A, Foldvari M. Effect of topical interferon-γ gene therapy using gemini nanoparticles on pathophysiological markers of cutaneous scleroderma in Tsk/+ mice. Gene Ther. 2012;19(10):978–87.
Bhadani A, Singh S. Novel gemini pyridinium surfactants: synthesis and study of their surface activity, DNA binding, and cytotoxicity. Langmuir. 2009;25(19):11703–12.
Badea I, Wettig S, Verrall R, Foldvari M. Topical non-invasive gene delivery using gemini nanoparticles in interferon-gamma-deficient mice. Eur J Pharm Biopharm. 2007;65(3):414–22.
Al-Dulaymi M, et al. Molecular engineering as an approach to modulate gene delivery efficiency of peptide-modified gemini surfactants. Bioconjug Chem. 2018;29(10):3293–308.
Donkuru M, Michel D, Awad H, Katselis G, el-Aneed A. Hydrophilic interaction liquid chromatography-tandem mass spectrometry quantitative method for the cellular analysis of varying structures of gemini surfactants designed as nanomaterial drug carriers. J Chromatogr A. 2016;1446:114–24.
Wang C, Li X, Wettig SD, Badea I, Foldvari M, Verrall RE. Investigation of complexes formed by interaction of cationic gemini surfactants with deoxyribonucleic acid. Phys Chem Chem Phys. 2007;9(13):1616–28.
Wettig SD, Deubry R, Akbar J, Kaur T, Wang H, Sheinin T, et al. Thermodynamic investigation of the binding of dissymmetric pyrenyl-gemini surfactants to DNA. Phys Chem Chem Phys. 2010;12(18):4821–6.
Gemeinhart RA, Luo D, Saltzman WM. Cellular fate of a modular DNA delivery system mediated by silica nanoparticles. Biotechnol Prog. 2005;21(2):532–7.
Donkuru M, Michel D, Awad H, Katselis G, el-Aneed A. Hydrophilic interaction liquid chromatography–tandem mass spectrometry quantitative method for the cellular analysis of varying structures of gemini surfactants designed as nanomaterial drug carriers. J Chromatogr A. 2016;1446:114–24.
Buse J, Badea I, Verrall RE, el-Aneed A. A general liquid chromatography tandem mass spectrometry method for the quantitative determination of diquaternary ammonium gemini surfactant drug delivery agents in mouse keratinocytes’ cellular lysate. J Chromatogr A. 2013;1294:98–105.
Jin W, Badea I, Leary SC, el-Aneed A. The determination of gemini surfactants used as gene delivery agents in cellular matrix using validated tandem mass spectrometric method. J Pharm Biomed Anal. 2019;164:164–72.
Donkuru M, Wettig SD, Verrall RE, Badea I, Foldvari M. Designing pH-sensitive gemini nanoparticles for non-viral gene delivery into keratinocytes. J Mater Chem. 2012;22(13):6232–44.
Rangel R, Dobroff AS, Guzman-Rojas L, Salmeron CC, Gelovani JG, Sidman RL, et al. Targeting mammalian organelles with internalizing phage (iPhage) libraries. Nat Protoc. 2013;8(10):1916–39.
Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–7.
Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244(1):105–15.
Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577.
Cardoso AM, Morais CM, Cruz AR, Cardoso AL, Silva SG, do Vale ML, et al. Gemini surfactants mediate efficient mitochondrial gene delivery and expression. Mol Pharm. 2015;12(3):716–30.
El-Sayed A, Harashima H. Endocytosis of gene delivery vectors: from clathrin-dependent to lipid raft-mediated endocytosis. Mol Ther. 2013;21(6):1118–30.
Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44.
Graham JM, Rickwood D. Subcellular fractionation: a practical approach. Published by Oxford University Press Inc, New York, 1997.
FDA. Guidance for Industry: bioanalytical method validation. US: Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM); 2013.
Kamiya H, Fujimura Y, Matsuoka I, Harashima H. Visualization of intracellular trafficking of exogenous DNA delivered by cationic liposomes. Biochem Biophys Res Commun. 2002;298(4):591–7.
Alberts B, et al. Essential cell biology: Published by Garland Science, Taylor & Francis Group, LLC. New York, USA and Abingdon, UK, 2014.
Stefanutti E, et al. Cationic liposomes formulated with DMPC and a gemini surfactant traverse the cell membrane without causing a significant bio-damage. Biochim Biophys Acta Biomembr. 2014;1838(10):2646–55.
Gharagozloo M, Rafiee A, Chen DW, Foldvari M. A flow cytometric approach to study the mechanism of gene delivery to cells by gemini-lipid nanoparticles: an implication for cell membrane nanoporation. J Nanobiotechnol. 2015;13(1):62.
Yang P, Singh J, Wettig S, Foldvari M, Verrall RE, Badea I. Enhanced gene expression in epithelial cells transfected with amino acid-substituted gemini nanoparticles. Eur J Pharm Biopharm. 2010;75(3):311–20.
Colombo G, Soto P, Gazit E. Peptide self-assembly at the nanoscale: a challenging target for computational and experimental biotechnology. Trends Biotechnol. 2007;25(5):211–8.
Lin AJ, Slack NL, Ahmad A, George CX, Samuel CE, Safinya CR. Three-dimensional imaging of lipid gene-carriers: membrane charge density controls universal transfection behavior in lamellar cationic liposome-DNA complexes. Biophys J. 2003;84(5):3307–16.
Schaffer DV, Fidelman NA, Dan N, Lauffenburger DA. Vector unpacking as a potential barrier for receptor-mediated polyplex gene delivery. Biotechnol Bioeng. 2000;67(5):598–606.
Smisterová J, et al. Molecular shape of the cationic lipid controls the structure of cationic lipid/dioleylphosphatidylethanolamine-DNA complexes and the efficiency of gene delivery. J Biol Chem. 2001;276(50):47615–22.
Israelachvili JN, Mitchell DJ, Ninham BW. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J Chem Soc, Faraday Trans 2. 1976;72:1525–68.
Koenig BW, Gawrisch K. Specific volumes of unsaturated phosphatidylcholines in the liquid crystalline lamellar phase. Biochim Biophys Acta Biomembr. 2005;1715(1):65–70.
Wang H, Wettig SD. Synthesis and aggregation properties of dissymmetric phytanyl-gemini surfactants for use as improved DNA transfection vectors. Phys Chem Chem Phys. 2011;13(2):637–42.
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminf. 2012;4(1):17.
Frisch M, et al. Gaussian 09, Revision D. 01. Wallingford: Gaussian, Inc.; 2013.
Wettig S, Verrall R. Thermodynamic studies of aqueous m-s-m gemini surfactant systems. J Colloid Interface Sci. 2001;235(2):310–6.
Zuhorn IS, Bakowsky U, Polushkin E, Visser WH, Stuart MCA, Engberts JBFN, et al. Nonbilayer phase of lipoplex–membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol Ther. 2005;11(5):801–10.
Acknowledgments
The authors would like to thank Dr. Jackson M. Chitanda for the synthesis of gemini surfactants and standards, and Ms. Deborah Michael for her technical assistance on the operation of 4000 QTRAP® mass spectrometry system. We also thank Dr. Waleed Mohammed-Saeid for the training in the cell transfection and purification of plasmid DNA. Dr. George Katselis and David Schneberger are greatly appreciated for the training in Western blotting assay. We acknowledge Dr. Matthew F. Paige’s lab for collecting the data on the Langmuir trough system. Dr. McDonald Donkuru is acknowledged for his initial help and discussions regarding the subcellular fractionation.
Funding
A postgraduate scholarship for Wei Jin is provided by the Natural Sciences and Engineering Research Council of Canada (NSERC). Funding to purchase the 4000 QTRAP® instrument was obtained through the Canada Foundation for Innovation-Leaders Opportunity Fund. The research was supported by a NSERC discovery grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 176 kb)
Rights and permissions
About this article

Cite this article
Jin, W., Al-Dulaymi, M., Badea, I. et al. Cellular Uptake and Distribution of Gemini Surfactant Nanoparticles Used as Gene Delivery Agents. AAPS J 21, 98 (2019). https://doi.org/10.1208/s12248-019-0367-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-019-0367-1