
Abstract
Allosteric kinase inhibitors targets kinases with oncogenic driver mutations in malignancies as a potential new therapy strategy. EGFR inhibitors with a 4-oxo-chromane scaffold targeting the L858R/T790M/C797S mutation were identified, optimized, synthesized, and assessed for anticancer and EGFR enzyme inhibitory activity. Compounds 4i and 4l were shown to be very effective with IC50 values of 132 and 146 nM, respectively, and excellent selectivity in in silico study. Compound 4i showed substantial antioxidant activity at a concentration of 100 µM, with a DPPH radical scavenging value of 91.46%. The synthesized compounds 4i, 4k and 4l were found to be selective toward cancer cells since they did not exhibit cytotoxicity even at IC50 > 20 µM on normal cells. Compound potency was further assessed using in silico and in vitro biological evaluation.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is the world’s second biggest cause of death, accounting for around 22% of all deaths each year [1, 2]. Two kinds of lung cancers are non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Epidermal growth factor receptor (EGFR) has been connected to the signal transduction cascade that leads to the morphological and biochemical alterations occur during apoptosis [3]. The class of receptors that belong to the tyrosine kinase domain include EGFRs, which are found in the cell’s transmembrane area [4].
The EGFR enzyme is classified as (ErbB1, HER1), ErbB2 (HER2, neu in rodents), ErbB3 (HER3), and ErbB4 (HER4) based on structural similarities [5]. These receptors are in charge of cell division and proliferation in the body. EGFR is one of the most important therapeutic targets for NSCLC since it is involved in cancer cell proliferation, survival, adhesion, migration, and differentiation [6]. Overexpression or aberrant activation of EGFR and ErbB-2, according to the study, frequently leads to cell malignancy [5]. Targeted therapies work by specifically targeting a protein or enzyme that contains mutant or other genetically modified or changed cells that differ from normal tissue cells [7].
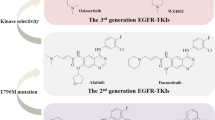
The first-generation EGFR inhibitors includes erlotinib and gefitinib, which are generally utilized in the early stages of cancer treatment (Fig. 1 consists of structures of standard EGFR inhibitors). Patients develop tumor resistance after a median response length, according to studies, owing to the emergence of the resistance mutation EGFRT790M [8]. The alterations found in wild-type enzymes necessitated the development of second-generation EGFR inhibitors. Both the wild-type and the mutant enzymes were effectively inhibited by these medicines, but the moieties were hazardous. After some time, similar changes occurred, as well as the emergence of a newly mutant residue, such as C797S [9]. As a result of many inconveniences and toxicity, these molecules have been replaced with molecules of the third generation. Exon 19 deletions, mutation in C797S, and mutation T790M were among the most recent developments of the third generation, with AZD9291 and rociletinib demonstrating considerable and increased selective mutation action [10]. Previously, our group designed and synthesized different anticancer derivatives containing various heterocyclic ring structures [11,12,13,14,15,16,17,18]. This helped us in the current work to design and synthesize 4-oxo-chromane derivatives.

Structures of standard EGFR inhibitors
Resistance emerged in the third generation, despite many generational improvements [19]. Wild and mutant enzymes were used to search for allosteric compounds [20]. The EAI001 was the first powerful (EGFR L858R/T790M IC50 of 0.024 mM) and selective (EGFR wild-type IC50 > 50 mM) mutant EGFR (EGFR allosteric-1 inhibitor). However, the individual’s efficacy against the L858R and T790M mutations was relatively moderate. Due to electronegative fluorine groups, EAI045 was shown to be highly potent and specific inhibitor for L858R/T790M mutations after medicinal-chemistry optimization [21].
In organic synthesis and creation of new lead compounds in drug design and discovery, the 4-oxo-chromane scaffold is an important intermediate and fascinating building block [22, 23]. The chroman-4-one/chromanone/oxo-chromane pharmacophore is a favored scaffold in medicinal research, consisting of two rings in which 2, 3-dihydro-γ-pyranone is fused with an aromatic benzene nucleus, and derivatization at the 2, 3, and 4-positions of the chromanone skeleton yields more effective flavonoids such as 3-benzylidene-chromanones, spirochromanone and hydrazone. This structural diversity of chromanone is essential in the pharmaceutical area since it has anticancer, antioxidant, anti-inflammatory, antiviral, antitubercular, antibacterial, antifungal, antiparasitic, anti-AchE, anticonvulsant, anti-HIV, and antileishmanial characteristics. Even though some derivatives of chromanone/oxo-chromane scaffolds, such as DSP-1053 (novel serotonin reuptake inhibitor and fast antidepressant), calanolide A (anti-HIV), Silibinin and chrysin (anticancer), taxifolin (antidiabetic), tetrazole (antidiabetic), troglitazone (antidiabetic), ormeloxifene (anticancer), and nebivolol (beta-blockers); nonetheless, there are fewer powerful chromanone/oxo-chromane analogs on the market. As a result, greater thought should be given to creating and manufacturing powerful synthetic chromanone/oxo-chromane analogs with superior therapeutic potential in the future [24].
Computational methodologies for developing new compounds are now commonly used due to the high prediction of effective compounds and proper screening of molecules. Scaffold hopping along with Molecular Docking is the most widely used computational strategy in drug design and discovery at the moment [25]. In structural molecular biology and computer-assisted drug design, molecular docking is an important technique [26]. The importance of halogen bonding and alkyl halide containing compounds and approved drugs were studied from literature [27, 28]. Bioactivity can be considerably altered by halogenation and the stereochemistry of halogen-bearing carbons. This can be accomplished by increasing lipophilicity, which results in improved pharmacokinetic qualities. Finally, a rising number of isolated halogenated natural compounds with distinct bioactivity indicate potential for future research.
A detailed synthetic scheme was designed, and novel compounds were submitted to molecular docking and ADMET experiments to understand the binding pocket and forecast potency. The most powerful compound was then subjected to molecular dynamics simulations after being evaluated in vitro on mutant cell lines.
Result and discussion
Chemistry
A detailed synthetic protocol of compounds 4(a–l) is shown in Scheme 1. The complete multi-step reaction resulted high yields in each step. Synthesized compounds with their yields are shown in Table 1.

Synthesis of compounds 4(a–l). Reagents and conditions: a Thionyl chloride, Ethanol, 75–80 °C; b Potassium tert-butoxide, DCM, rt
As described in Scheme 1, substituted 4-oxo-chromane-2-carboxylic acid (1) was reacted in presence of thionyl chloride and ethanol at 75–80 °C to yield 4-oxo-chromane-2-carbonyl chloride (2). 4-oxo-chromane-2-carbonyl chloride (2) was stirred at rt with substituted benzhydryl amine in presence of potassium tert-butoxide and DCM to obtain final substituted 4-oxo-chromane derivatives 4(a–l).
Biological activity
In vitro cytotoxic activity
The synthesized compounds were tested in vitro against cancer cell lines. To test their toxicity, the cancer cell lines HCC827 with an EGFR-activating mutation (EGFR Del E746-A750), gefitinib-resistant NSCLC cell H1975 with L858R/T790M mutant EGFR, A549 over-expressing wild-type EGFR (WT-EGFR), and colon cancer cells HT-29 with a non-special gene type were used. BEAS-2B normal lung cell line was also used to check the toxicity of the synthesized compounds. The MTT test was utilized to assess their cytotoxic activities in vitro. Osimertinib was the standard drug utilized in this study. The results are presented in the form of IC50 values, as indicated in Table 2.
Here, we have tried to put forward the structure activity relationship of the novel synthesized compounds 4a–4l. In this novel series of 4-oxo-chromane we have substituted halogens on the chromane ring and also on phenyl ring and tried to study its effect on cytotoxic activity. Through literature survey we came to know about the purpose behind phenyl and chromane ring substitution taking into consideration already known drugs such as Afatinib, Osimertinib and Dacomitinib [29].
It was observed that substitution of halogen groups like Cl, F, I, Br were not in favor of good cytotoxic activity. But at the same time, it was observed that if the halogens were substituted on the phenyl ring, it gave compounds with good cytotoxic activity. Compounds with halogen substitution on phenyl ring are 4e, 4f, 4g, 4h, 4i, 4j, 4k and 4l. From the activity data as observed in Table 3 it was also clear that the substitution of halogen at the 3rd position of phenyl ring gave the compounds (4i, 4j, 4k and 4l) with good activity than those substituted at the 4th position of phenyl ring (4e, 4f, 4g and 4h). The type of halogen that is substituted at the 3rd position of the phenyl ring also plays an important role in cytotoxic activity. The effect of different halogen substitution on phenyl ring against cytotoxic activity goes in the decreasing order like Cl >I > F > Br.
The compounds 4i, 4k and 4l gave the most promising anticancer activity. These synthesized compounds were also tested on the BEAS-2B normal lung cell line. It was observed that the synthesized compound 4i has shown 96 times more selectivity toward BEAS-2B normal lung cell line when compared with the A-549 lung cancer cell line. The synthesized compounds 4i, 4k and 4l were found to be selective toward cancer cells since they did not exhibit cytotoxicity even at IC50 > 20 µM on normal cells.
In vitro enzymatic activity assay
The in vitro enzymatic inhibitory activity of the produced compounds 4i and 4l against EGFR L858R/T790M/C797S was investigated. The drug osimertinib was employed as a standard. Table 3 summarizes the findings. With IC50 values of 132 nM, the synthesized compound 4i showed substantial inhibitory action against the triple mutant EGFR L858R/T790M/C797S, and was also the most effective drug in anti-proliferative activities against cancer cells in vitro.
Western blot assay
To investigate the mechanism of action for cytotoxic activities, the synthesized compounds 4i and 4l were evaluated on the phosphorylation of EGFR and downstream signaling transduction in HCC827 cells using a western blot assay. HCC827 cells were given different doses of the produced compounds 4i and 4l, such as 1.00, 0.10, and 0.01 M for each molecule. The activity’s outcomes are depicted in Fig. 2.

Inhibition of EGFR autophosphorylation in HCC827 cells by western blot assay. STD standard drug Osimertinib
Apoptosis
Apoptosis, or programmed cell death, is a normal physiologic process that eliminates unwanted cells. One of the initial stages in apoptosis is the translocation of membrane phosphatidylserine (PS) from the inner side of the plasma membrane to the surface. PS binds to Annexin V, a Ca2+−dependent phospholipid-binding protein, with a high affinity. PS is found on the surface of apoptotic cells, and when it interacts with the tagged Annexin V in this assay, it fluoresces green. During early apoptosis, membrane asymmetry is eliminated, and PS translocate from the cytoplasmic side to the outer leaflet. Propidium iodide (PI), the counterstain employed in this experiment, can only traverse damaged membranes and intercalate into DNA. As a result, PI can identify the presence of red fluorescence in the late stages of apoptosis and necrosis.
From the results in Fig. 3, it is clear that compound 4i induced early apoptosis (34.6%) and late apoptosis (4.2%) in comparison with control (early apoptosis 1.5%, late apoptosis 1.8%).

Apoptosis study by Annexin V/FITC assay of compound 4i
DPPH radical scavenging activity
Free radicals have a key role in cancer, cardiovascular and auto-immune illnesses, as well as aging-related issues, leading to new medical approaches [30].
The scavenging capacity of the produced compounds was evaluated using the previously published DPPH (1,1-diphenyl-2-picrylhydrazyl) technique [31]. As a control, ascorbic acid was employed. As indicated in Table 4, the produced compounds 4i and 4l were evaluated at various concentrations such as 10, 50 and 100 µM. The chemicals studied, 4i and 4l, displayed good antioxidant properties. Compound 4i showed substantial antioxidant activity at a concentration of 100 µM, with a DPPH radical scavenging value of 91.46%.
In silico studies
Molecular docking with T790M and C797S mutated protein
The final derivatives’ molecular docking was performed using a mutant EGFR enzyme co-crystallized with EAI045 (PDB: 6P1L). This PDB was selected for docking to study EGFR mutations along with allosteric binding property of the designed ligands. The protein’s binding characteristics have been changed by the recently discovered C797S mutation, which has been coupled with the previously known T790M mutation. Compounds that interact with the ATP binding site have less efficacy due to the C797S mutation. To address these difficulties, a novel binding pocket in the form of an allosteric binding site was developed. The efficacy and market acceptability of EAI045 and other allosteric binding pocket interacting structures have been shown. The “Y” shaped compound’s pocket characteristics were likewise shown to be highly reliant on the shape configuration.
Compound 4i and 4l showed different hydrogen bonding interactions than EAI045, fitting properly into the binding pocket with higher docking score. Compounds 4e (−8.68) and 4h (−8.12) showed decreased docking scores, as they lacked proper pocket fitting and binding. Compound 4i (−10.64) and 4l (−10.52) exhibited hydrogen bonding interaction with amino acid residue LYS745, different from the interactions shown by EAI045. It can be observed that, more the number of hydrogen bond interactions between compound and amino acid residues, more is the binding affinity. Also, the hydrophobic interactions like π-Cation and π-π stacking, play an important role in determining the binding affinity and ultimately the docking score of a compound. The π-π stacking interaction with amino acid residue PHE 856 is seen with majority of the above synthesized compounds. All the compounds fit well into the allosteric binding site of C797S and T790M mutated EGFR protein (PDB ID: 6P1L). As illustrated in Fig. 4, molecular docking experiments indicated that molecules in various conformational states fit well into the pocket. The effectiveness of compound binding affinities was also discovered. Table 1 shows the aggregate docking scores as well as the structures.

Allosteric binding of designed compounds 4i, 4l and standard EAI045 over C797S and T790M mutated EGFR protein (PDB ID: 6P1L). Blue lines in the figures indicate π-π stacking interaction and pink lines indicate hydrogen bonding interaction
ADMET studies
Due to pharmacokinetic problems, a lot of compounds fail to become effective medicines and access the market. The physicochemical and pharmacokinetic characteristics of the synthesized derivatives were evaluated using the SwissADME website (http://www.swissadme.ch/index.php) in comparison to EAI045 [32]. The analysis of the pharmacokinetic parameters required for the ADMET research of compounds 4(a–l) is shown in Table 5.
Bioavailability radar was based on six physicochemical properties: size, polarity, insolubility, lipophilicity, flexibility, and in saturation. The optimal range for excellent oral bioavailability is shown by the middle pink hexagon area as shown in Fig. 5. The molecular weights (MW) of final compounds are well below 500 g/mol and MLOGP values well between 3.06 to 3.37, which are within the optimum range. The drug-likeness similarity of substances was assessed using the physicochemical characteristics of MW, total polar surface area (TPSA), and GI absorption. The use of in silico ADMET predictions in the drug development process assists in the creation of non-toxic new compounds with good oral human absorption. Lipinski’s rule-passing compounds have high oral absorption in humans [33, 34]. The partition coefficient (MLOGP) is used to determine how well a medication is absorbed and distributed in the body. The percent of GI absorption was monitored closely, suggesting that the chemicals were well absorbed.

Bioavailability radar plot of compounds 4i and 4l versus EAI045
All of the target compounds were tested for various pharmacokinetic characteristics and found to be within an acceptable range for human ingestion, indicating that they may be used as drug-like molecules [35,36,37]. The absence of hazardous or undesirable moiety in the molecule was also noticed via PAINS warnings, leaving the biological activity intact. The presence of a powerful chemical at the site of action was determined by bioavailability ratings. Overall, the drugs had acceptable pharmacokinetic characteristics for human usage, as well as respectable docking scores.
Molecular dynamic simulations
On the potent molecule 4i, molecular dynamic simulations were done to investigate the protein-ligand interactions. The movements of atoms and molecules were studied at this time. We used molecular dynamic simulations to assess the molecule’s and protein’s stability, which were interpreted using RMSD and root mean square fluctuation (RMSF). To immerse the system in a real-time virtual system of the body, the TIP3P water molecule set was employed. The entire system ran for 10 ns at 300 K temperature and 1.01325 bar pressure. A total of 1000 frames were used to analyze the dynamic simulations in depth. During the simulation, the root mean square deviation (RMSD) of the protein-ligand combination is shown in Fig. 6. Both protein and ligand were shown to be stable until the simulations were completed after the initial adjustment. For EGFR proteins, changes in the order 1–3 are regarded suitable. Any increase in the RMSD at the end of the runs indicates that the system is not balanced overall. During the simulations, the entire system was found to establish an equilibrium condition based on the acquired data. The ligand remained attached to the protein, regulating the system’s stability and binding affinity.

Time-dependent protein-ligand root mean square deviation (RMSD) plots (Angstrom)
Atoms in the protein RMSF break down local residue variations. Residues that contribute to complex structural fluctuations can be identified using RMSF (Fig. 7). RMSF values show that the prepared system had less variations. The peaks in the RMSF diagram reflect the volatility of an amino acid during the procedure. The identified protein’s low RMSF values also indicate that the system is in equilibrium.

Time-dependent protein root mean square fluctuation (RMSF) plots (Angstrom)
After determining ligand saturation, the capacity of the protein to bind to the ligand is investigated. Using ligand RMSF plots, these observations were observed. The ligand RMSF reveals how ligand fragments interact with proteins and have an entropic function in the binding process. Figure 8 depicts the ligand RMSF graph for all protein-ligand complexes. The system’s RMSF was found to be almost identical to the ligand plot. The ligand plot (compound 4i) looked to follow the protein RMSF curve perfectly. The saturation point of the ligand had been achieved, suggesting that the binding site was favorable for interaction.

Time-dependent ligand root mean square fluctuation (RMSF) plots (Angstrom)
Conclusion
Twelve new substituted 4-oxo-chromane derivatives targeting resistance in NSCLC were developed, optimized, and synthesized as small molecule L858R/T790M/C797S mutant EGFR inhibitors targeting resistance in NSCLC. Compounds 4i and 4l were shown to be effective in enzyme-based inhibition. The compounds suppressed the EGFR L858R/T790M/C797S with an IC50 values of 132 nM and 146 nM. The synthesized compounds 4i, 4k and 4l were found to be selective toward cancer cells since they did not exhibit cytotoxicity even at IC50 > 20 µM on normal cells. In addition, compound 4i clearly promoted early apoptosis (34.6%) and late apoptosis (4.2%) in contrast to control cells (early apoptosis 1.5%, late apoptosis 1.8%). It also showed substantial antioxidant activity at a concentration of 100 µM, with a DPPH radical scavenging value of 91.46%. According to molecular docking experiments, compounds in their conformational state fit well into the pocket of the T790M/C797S mutant (PDB ID: 6P1L) EGFR enzyme at the allosteric binding site. The potency of compound 4i was investigated using in silico studies along with in vitro biological evaluation.
Experimental
General procedures
Chemicals were purchased from commercial sources, viz. TCI and Avra Labs, and used without further purification. Progress of the reaction was monitored by thin-layer chromatography (TLC) on pre-coated silica gel F254 aluminum sheets (Merck), and visualization was done by UV light. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded on Bruker AVANCE II NMR spectrometer in Dimethylsulfoxide-d6 solution. Tetramethyl silane was used as an internal standard. Chemical shift values are given in ppm relative to TMS as an internal reference and coupling constants (J) in Hertz. The splitting pattern abbreviations are assigned as singlet (s), doublet (d), triplet (t), broad singlet (brs), double doublet (dd), and multiplet (m). High Resolution Mass were recorded on a XEVO G2-XS QTOF instrument. Elemental analysis was performed on Perkin-Elmer EAL-240 elemental analyzer.
Synthesis of substituted 4-oxo-chromane-2-carbonyl chloride (2)
In a 50 ml round bottom flask, 5 g of substituted 4-oxo-chromane-2-carboxylic acid was refluxed overnight with thionyl chloride in presence of ethanol as a solvent. The reaction was monitored by TLC. After completion of the reaction, the reaction mixture was allowed to stand at room temperature. The solvent was diluted with 20 ml ethyl acetate and washed with 10 ml water. The organic layer was extracted and treated with anhydrous Na2SO4 to remove water. The solvent was evaporated using a rotary vacuum evaporator to obtain the required intermediate. The obtained intermediates were purified by column chromatography using 20% EA:nH mobile phase and were used for further steps.
Synthesis of substituted 4-oxo-chromane derivatives 4(a-l)
Substituted 4-oxo-chromane-2-carbonyl chloride intermediates (2) were stirred at room temperature with substituted benzhydryl amines (3) in presence of potassium tert-butoxide and DCM as a solvent for 24 h. The reaction was monitored by TLC. After completion of reaction, product was isolated with layers of DCM-water. The organic layer was extracted and treated with anhydrous Na2SO4 to remove water. The solvent was evaporated on rotary vacuum evaporator. The obtained products were purified by column chromatography using 20% EA:nH mobile phase and were sent for HRMS, 1H and 13C NMR analysis. The diastereomers from 4e–4l were separated using column chromatography. The desired R configuration material was polar and was not easy to separate from the S configuration as the racemic mixtures do not impart color in the column separation. Still, the R configuration was separated from S with the help of a unique stain called p-anisaldehyde. This stain when applied to the TLC of the mixture imparted different colors to R and S configurations.
N-benzhydryl-7-chloro-4-oxochromane-3-carboxamide (4a)
Yellow solid; Yield: 89%; mp: 240–245 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.92 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.39–7.28 (m, 9H), 7.30–7.23 (m, 2H), 7.24 (dd, J = 8.5, 2.3 Hz, 1H), 7.05 (s, 0H), 5.89 (dp, J = 8.5, 1.0 Hz, 1H), 4.46–4.34 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 189.7, 171.1, 161.6, 142.6, 139.0, 129.9, 128.6, 127.5, 118.2, 115.8, 68.0, 58.5, 56.6; HRMS: m/z calcd: 391.10, found [M + H]+: 391.08; Anal. Calcd for C23H18ClNO3 (391.10): C, 70.50; H, 4.63; N, 3.57. Found: C, 70.41; H, 4.59; N, 3.53.
N-benzhydryl-7-bromo-4-oxochromane-3-carboxamide (4b)
Yellow solid; Yield: 90%; mp: 235–237 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.41–7.30 (m, 9H), 7.26 (ddt, J = 8.1, 6.5, 2.4 Hz, 2H), 5.89 (dp, J = 8.4, 1.0 Hz, 1H), 4.46–4.34 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 189.7, 171.1, 161.4, 142.6, 130.2, 128.6, 127.5, 125.6, 124.8, 119.2, 118.1, 68.0, 58.5, 56.6; HRMS: m/z calcd: 435.04, found [M + H]+: 436.31; Anal. Calcd for C23H18BrNO3 (435.04): C, 63.32; H, 4.16; N, 3.21. Found: C, 63.41; H, 4.20; N, 3.25.
N-benzhydryl-7-fluoro-4-oxochromane-3-carboxamide (4c)
Light-yellow solid; Yield: 91%; mp: 255–258 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.94 (dd, J = 8.4, 4.9 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.39–7.33 (m, 6H), 7.32 (s, 1H), 7.26 (ddt, J = 8.1, 6.5, 2.4 Hz, 2H), 7.00 (ddd, J = 10.4, 8.4, 2.2 Hz, 1H), 6.70 (dd, J = 12.1, 2.3 Hz, 1H), 5.89 (dp, J = 8.4, 0.9 Hz, 1H), 4.46–4.34 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 189.9, 171.1, 167.8, 165.8, 163.0, 162.9, 142.6, 130.15, 128.6, 127.9, 116.5, 109.7, 103.5, 68.0, 58.5, 56.6; HRMS: m/z calcd: 375.13, found [M + H]+: 375.40; Anal. Calcd for C23H18FNO3 (375.13): C, 73.59; H, 4.83; F, N, 3.73. Found: C, 73.50; H, 4.79; N, 3.69.
N-benzhydryl-7-iodo-4-oxochromane-3-carboxamide (4d)
Yellow solid; Yield: 89%; mp: 260–263 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.80 (d, J = 7.9 Hz, 1H), 7.56–7.47 (m, 2H), 7.39–7.32 (m, 6H), 7.35–7.28 (m, 1H), 7.30–7.23 (m, 3H), 5.89 (dp, J = 8.4, 0.9 Hz, 1H), 4.46–4.34 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.3, 161.5, 142.5, 130.3, 129.6, 128.6, 127.5, 124.1, 116.8, 98.0, 69.0, 58.2, 57.6; HRMS: m/z calcd: 483.03, found [M + H]+: 483.31; Anal. Calcd for C23H18INO3 (483.03): C, 57.16; H, 3.75; N, 2.90. Found: C, 57.25; H, 3.79; N, 2.94.
N-((4-chlorophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4e)
Yellow-orange solid; Yield: 85%; mp: 245–250 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.39–7.23 (m, 9H), 7.20 (td, J = 7.9, 1.3 Hz, 1H), 7.06 (dd, J = 8.0, 1.2 Hz, 1H), 6.19–6.13 (m, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.1, 161.3, 142.7, 140.9, 136.1, 134.8, 128.6, 127.9, 121.1, 116.3, 68.0, 57.8, 56.6; HRMS: m/z calcd: 391.10, found [M + H]+: 391.85; Anal. Calcd for C23H18ClNO3 (391.10): C, 70.50; H, 4.63 N, 3.57. Found: C, 70.41; H, 4.59; N, 3.53.
N-((4-bromophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4f)
Yellow solid; Yield: 88%; mp: 283–285 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.53–7.47 (m, 3H), 7.39–7.32 (m, 4H), 7.35–7.27 (m, 4H), 7.30–7.23 (m, 1H), 7.20 (td, J = 7.9, 1.3 Hz, 1H), 7.06 (dd, J = 8.1, 1.2 Hz, 1H), 6.16 (dt, J = 8.5, 1.0 Hz, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.1, 161.3, 142.7, 141.4, 136.1, 129.0, 128.6, 127.5, 122.5, 121.7, 116.3, 68.0, 58.5, 56.6; HRMS: m/z calcd: 435.05, found [M + H]+: 436.31; Anal. Calcd for C23H18BrNO3 (435.05): C, 63.32; H, 4.16; N, 3.21. Found: C, 63.41; H, 4.20; N, 3.25.
N-((4-fluorophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4g)
Yellow solid; Yield: 91%; mp: 257–261 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.39–7.31 (m, 7H), 7.31–7.23 (m, 1H), 7.20 (td, J = 7.9, 1.3 Hz, 1H), 7.12–7.04 (m, 3H), 6.16 (dp, J = 8.4, 1.0 Hz, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.1, 163.0, 161.3, 142.6, 138.9, 136.1, 129.1, 128.8, 127.9, 121.7, 116.3, 115.9, 68.0, 58.2, 56.6; HRMS: m/z calcd: 375.13, found [M + H]+: 375.40; Anal. Calcd for C23H18FNO3 (375.13): C, 73.59; H, 4.83; N, 3.73. Found: C, 73.50; H, 4.79; N, 3.69.
N-((4-iodophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4h)
Light-yellow solid; Yield: 90%; mp: 235–237 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.60–7.54 (m, 2H), 7.50 (d, J = 8.4 Hz, 1H), 7.35 (dddd, J = 9.5, 8.5, 2.6, 1.5 Hz, 5H), 7.34–7.24 (m, 1H), 7.23 (d, J = 1.1 Hz, 1H), 7.24–7.17 (m, 2H), 7.06 (dd, J = 8.1, 1.3 Hz, 1H), 6.19–6.13 (m, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.8, 171.3, 161.7, 142.6, 140.7, 138.0, 136.1, 128.6, 127.9, 121.7, 116.3, 94.7, 69.1, 58.5, 57.6; HRMS: m/z calcd: 483.03, found [M + H]+: 483.31; Anal. Calcd for C23H18INO3 (483.03): C, 57.16; H, 3.75; N, 2.90. Found: C, 57.25; H, 3.79; N, 2.94.
N-((3-chlorophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4i)
Yellow solid; Yield: 93%; mp: 221–225 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.46–7.36 (m, 2H), 7.39–7.33 (m, 4H), 7.36–7.27 (m, 4H), 7.30–7.23 (m, 1H), 7.20 (td, J = 7.9, 1.3 Hz, 1H), 7.06 (dd, J = 8.0, 1.2 Hz, 1H), 6.16 (dt, J = 8.5, 1.1 Hz, 1H), 4.40 (dd, J = 12.2, 4.7 Hz, 1H), 4.34 (dd, J = 12.2, 4.9 Hz, 1H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.0, 161.3, 143.2, 140.9, 136.1, 130.6, 128.4, 127.5, 126.2, 122.5, 121.1, 116.3, 68.0, 58.6, 56.6; HRMS: m/z calcd: 391.10, found [M + H]+: 392.105; Anal. Calcd for C23H18ClNO3 (391.10): C, 70.50; H, 4.63; N, 3.57. Found: C, 70.59; H, 4.67; N, 3.61.
N-((3-bromophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4j)
Cream yellow solid; Yield: 90%; mp: 176–180 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.57 (td, J = 2.1, 1.0 Hz, 1H), 7.43–7.24 (m, 11H), 7.28–7.17 (m, 1H), 7.06 (dd, J = 8.1, 1.2 Hz, 1H), 6.15 (dt, J = 8.4, 0.9 Hz, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.1, 161.3, 143.1, 142.3, 136.1, 130.5, 129.7, 128.6, 127.5, 126.5, 124.9, 121.1, 116.3, 68.0, 58.2, 56.6; HRMS: m/z calcd: 435.05, found [M + H]+: 436.31; Anal. Calcd for C23H18BrNO3 (435.05): C, 63.32; H, 4.16; N, 3.21. Found: C, 63.23; H, 4.12; N, 3.17.
N-((3-fluorophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4k)
Light-yellow solid; Yield: 85%; mp: 260–263 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.45 (td, J = 7.6, 4.9 Hz, 1H), 7.40–7.30 (m, 6H), 7.30–7.23 (m, 1H), 7.26–7.12 (m, 2H), 7.09–7.01 (m, 2H), 6.15 (dp, J = 8.5, 1.0 Hz, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.1, 171.0, 164.2, 162.2, 161.3, 143.4, 141.5, 136.1, 130.5, 128.8, 127.9, 123.2, 121.7, 116.3, 115.8, 114.9, 68.0, 58.3, 56.6; HRMS: m/z calcd: 375.13, found [M + H]+: 375.31; Anal. Calcd for C23H18FNO3 (375.13): C, 73.59; H, 4.83; N, 3.73. Found: C, 73.68; H, 4.87; N, 3.77.
N-((3-iodophenyl) (phenyl)methyl)-4-oxochromane-3-carboxamide (4l)
Yellow solid; Yield: 87%; mp: 247–251 °C. 1H NMR (500 MHz, Chloroform-d) δ 7.90 (dd, J = 8.2, 1.6 Hz, 1H), 7.70 (td, J = 2.3, 1.1 Hz, 1H), 7.61 (ddd, J = 7.3, 2.3, 1.2 Hz, 1H), 7.40–7.33 (m, 5H), 7.36–7.28 (m, 3H), 7.30–7.24 (m, 1H), 7.27–7.21 (m, 1H), 7.20 (td, J = 7.9, 1.3 Hz, 1H), 7.06 (dd, J = 8.1, 1.2 Hz, 1H), 6.15 (dq, J = 8.4, 1.0 Hz, 1H), 4.43–4.32 (m, 2H), 4.29 (t, J = 4.8 Hz, 1H). 13C NMR (125 MHz, Common NMR Solvents) δ 190.8, 171.35, 161.7, 144.8, 143.0, 138.3, 137.9, 136.1, 131.2, 128.6, 127.1, 121.2, 116.3, 92.6, 69.1, 59.4, 57.6; HRMS: m/z calcd: 483.03, found [M + H]+: 483.31; Anal. Calcd for C23H18INO3 (483.03): C, 57.16; H, 3.75; N, 2.90. Found: C, 57.25; H, 3.79; N, 2.94.
In silico methodology
All computational studies were performed using AutoDock Vina. The molecular docking tool, AutoDock Vina, was used for docking studies of all compounds on the EGFR enzyme. SwissADME tool was used to determine ADMET studies. Molecular dynamic simulations were carried out using Desmond.
Molecular docking
To investigate the binding pocket, residue interactions and predicted binding affinity of the enumerated compounds, molecular docking protocol was carried out using AutoDock tools. Through Chimera modeler, the protein was fetched from the open source PDB libraries. Explicit hydrogen bonds were added followed by removal of water molecules. Using Gastegier charges, ions were removed and finally the protein was minimized using Amber94 force field. The total number of loops were set to 1000 to ensure maximum minimization of the protein. The minimization was terminated when the energy converged or the root mean square deviation (RMSD) reached a maximum cut-off of 0.30 Å. Ramachandran plot was set to check the total number of disallowed residues. Through AutoDock 4.0 GUI, the minimized PDB was converted to PDBQT file followed by generation of grid. To perform flexible docking, torsions were chosen in the selected residues and saved as flex.PDBQT. The configuration file, which was used to outline the grid area as well as the protein and ligand input, was defined and saved as a text document. All the ligands were prepared using Gastegier charges and saved as PDBQT file. The files saved in the fold were called through command line under vina environment to start the docking process. The results for each input ligand were saved as a text file consisting of docking scores and RMSD values of each ligand conformation which was used for analysis of data. The output file was then converted into Maestro readable extension for image saving [38,39,40,41].
ADMET studies
The ADMET results were generated using SwissADME [38, 42].
Molecular dynamic simulations
To understand the stability and interactions affinity of the ligand-bound protein, the most potent compound 6a, screened based on in silico and in vitro results were subjected to molecular dynamics simulations. The ligand-bound protein was immersed in a TIP3P orthorhombic box of 10 Å_10Å_10 Å and system was then neutralized by the system-built option in the protocol by adding 0.15 M NaCl in a buffer. Then, steepest-descent and conjugate-gradient minimization methods were utilized to a satisfactory convergence of 0.1 kcal/mol-Å energy gradient. NPT ensemble class with a temperature of 300 K and a pressure of 1.01325 bar was used. Using VIPARR facility of DESMOND, the best suitable AMBER99 force field parameters were generated for duplex while the OPLS2005 force field was used for ligands [19, 43].
In vitro methodology
In vitro enzymatic activity assay
The assay was performed as reported in literature [38, 43, 44]. All of the enzymatic reactions were conducted at 30 °C for 40 min. The 50 µl reaction mixture contains 40 mM Tris, pH 7.4, 10 mM MgCl2, 0.1 mg/ml BSA, 1 mM DTT, 10 µM ATP, 25 ng kinase and the 0.2 mg/ml enzyme-substrate (Poly (Glu, Tyr)). The compounds were diluted in 10% DMSO and 5 µl of the dilution was added to a 50 µl reaction so that the final concentration of DMSO is 1% in all of the reactions. The assay was performed by using the Kinase-Glo Plus luminescence kinase assay kit. It measures kinase activity by quantitating the amount of ATP remaining in the solution following a kinase reaction. The luminescent signal from the assay is correlated with the amount of ATP present and is inversely correlated with the amount of kinase activity. The IC50 values were calculated using nonlinear regression with normalized dose-response fit using GraphPad Prism 5.0 Software.
Western blot assay
HCC827 cells (5 × 105/well) were seeded in 6-well plates overnight. The cell was exposed to 1 μM synthesized analogs for 1 h at 37 °C and then either immediately treated with media containing EGF (20 ng/ml) for 15 min or thoroughly washed with fresh medium 10 times for 5 h before EGF treatment. Whole-cell lysates were prepared and total protein concentrations were determined. Proteins were extracted with lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 0.5% deoxycholic acid, 0.02% sodium azide, 1% NP-40, 2.0 μg/ml aprotinin, 1 mM phenylmethylsulfonylfluoride). The lysates were centrifuged at 13,000 rpm for 30 min at 40 °C. Equivalent amounts of proteins were loaded on SDS–PAGE gels for electrophoresis and were subjected to transfer onto PVDF membranes. Appropriate antibodies to EGFR and p-EGFR from Cell signaling Technology (Danvers, MA) and anti-β-actin from Santa Cruz Biotech (Santa Cruz, CA) were used. Proteins were visualized with peroxidase-coupled secondary antibody from Southern Biotech (Birmingham, UK), using an ECL-plus kit from Amersham Biosciences (UK) for detection.
MTT assay
Virtually screened compounds had been screened against HCC827, H1975, A549, and HT-29 cells by standard MTT assay. These cell lines were cultured in 10% fetal bovine serum. In exact 4 × 103 cells have been suspended in MEM medium and plated into the 96-well plate in 5% CO2 for 24 h. The synthesized compounds were added to the culture medium and cells cultured maintained for 72 h. Freshly prepared MTT was added to each well and incubated with cells at 37 °C for 4 h. MTT reduction was quantified by measurement of absorbance at 492 nm using a multimode reader (Synergy Mx, BioTek).
Apoptosis
Apoptosis was determined by staining the cells with Annexin V-Fluorescein isothiocyanate (FITC) and counterstaining with PI using the Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Diego, CA) according to the manufacturer’s instructions. Briefly, 4 × 106 cell/T 75 flasks were exposed to compound at its IC50 concentration for 24 and 48 h. The cells then were collected by trypsinization and 0.5 × 106 cells were washed twice with phosphate-buffered saline and stained with 5 µl Annexin V-FITC and 5 µl PI in 1x binding buffer for 15 min at room temperature in the dark. Analyses were performed using BD LSR Fortessa) with FACS Diva version 6.2 software [33, 38, 39].
DPPH (1,1-diphenyl-2-picrylhydrazyl) assay
The reported DPPH method was applied to assess the scavenging ability of the compounds [26]. The compounds were tested in the range of 0–25 µg/ml in methanol. To 2.5 ml of compound in three different concentrations, 1 ml of 0.3 mM DPPH ethanol solution was added. Then 1 ml of methanol was added to the solution and allowed to react for 30 min in the dark at room temperature. The change in the absorbance was read at 518 nm. The blank was comprised of 2.5 ml of test compound and 1 ml methanol, while the mixture of 1 ml DPPH and 2.5 ml of methanol served as negative control. The percentage antioxidant activity was calculated as follows:
where AB: absorption of blank sample, AA: absorption of test samples. The percentage inhibition value was calculated and compared with that of Ascorbic acid as a reference.
Abbreviations
- 1. Cys797 to Ser797 (C797S):
-
Cysteine to serine
- 2. T790M:
-
Threonine 790 methionine
- 3. EGFR:
-
Epidermal growth factor receptor
- 4. PDB:
-
Protein Data Bank
- 5. ADMET:
-
Absorption, distribution, metabolism, excretion and toxicity
- 6. MDS:
-
Molecular dynamic simulations
- 7. Mol MW:
-
Molecular weight
- 8. NSCLC:
-
Non-small cell lung cancer
- 9. MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
References
Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med. 2016;10:383–8.
Sung H, Ferlay J, Siegel RL, Laversannek M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2021;71:209–49.
Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015;5:a006098–a006119.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34.
Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566–84.
Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. 2017;9:52–97.
Paul MK, Mukhopadhyay AK. Tyrosine kinase—role and significance in Cancer. Int J Med Sci. 2004;1:101–15.
Hirsch FR, Varella-Garcia M, Cappuzzo F. Predictive value of EGFR and HER2 overexpression in advanced non-small-cell lung cancer. Oncogene. 2009;28:S32–7.
Riely GJ. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:S146–9.
Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–9.
Dofe VS, Sarkate AP, Shaikh ZM, Jadhav CK, Nipte AS, Gill CH. Ultrasound-assisted synthesis of novel pyrazole and pyrimidine derivatives as antimicrobial agents. J Heterocycl Chem. 2018;55:756–63.
Chate AV, Kamdi SP, Bhagat AN, Jadhav CK, Nipte AS, Sarkate AP, et al. Design, synthesis and SAR study of novel Spiro [Pyrimido[5,4-b]Quinoline-10,50-Pyrrolo[2,3-d]Pyrimidine] derivatives as promising anticancer agents. J Heterocycl Chem. 2018;55:764–9.
Dofe VS, Sarkate AP, Shaikh ZM, Gill CH. Ultrasound mediated synthesis of novel 1,2,3-triazole-based pyrazole and pyrimidine derivatives as antimicrobial agents. J Heterocycl Chem. 2017;55:3195–201.
Dofe VS, Sarkate AP, Lokwani DK, Shinde DB, Kathwate SH, Gill CH. Novel O-Alkylated chromones as antimicrobial agents: ultrasound mediated synthesis, molecular docking and ADME prediction. J Heterocycl Chem. 2017;54:2678–85.
Bhosle MR, Wahul DB, Bondle GM, Sarkate AP, Tiwari SV. An efficient multi-component synthesis and in vitro anticancer activity of dihydropyranochromene and chromenopyrimidine-2,5-diones. Synth Commun. 2018;48:2046–60.
Bhosle MR, Andil P, Wahul DB, Bondle GM, Sarkate AP, Tiwari SV. Straightforward multicomponent synthesis of pyrano[2,3-d]pyrimidine-2,4,7-triones in β-cyclodextrin cavity and evaluation of their anticancer activity. J Iran Chem Soc. 2019;16:1553–61.
Deshmukh TR, Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Shingate BB. New amide linked dimeric 1,2,3-triazoles bearing aryloxy scaffolds as a potent antiproliferative agents and EGFR tyrosine kinase phosphorylation inhibitors. Bioorg Med Chem Lett. 2019;29:1–8.
Lokwani DK, Sarkate AP, Shinde DB. 3D-QSAR and docking studies of benzoyl urea derivatives as tubulin-binding agents for antiproliferative activity. Med Chem Res. 2013;22:1415–25.
Karnik KS, Sarkate AP, Lokwani DK, Narula IS, Burra PVLS, Wakte PS. Development of triple mutant T790M/C797S allosteric EGFR inhibitors: a computational approach. J Biomol Struct Dyn. 2021;39:5376–98.
Minari R, Bordi P, Tiseo M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: review on emerged mechanisms of resistance. Transl Lung Cancer Res. 2016;5:695–708.
Varkondi E, Schäfer E, Bökönyi G, Gyökeres T, Schwab R. Comparison of ELISA-based tyrosine kinase assays for screening EGFR inhibitors. J Recept Signal Transduct Res. 2005;25:45–56.
Emami S, Ghanbarimasir Z. Recent advances of chroman-4-one derivatives: synthetic approaches and bioactivities. Eur J Med Chem. 2015;93:539–63.
Rawat P, Verma SM. Design and synthesis of chroman derivatives with dual anti-breast cancer and antiepileptic activities. Drug Des Devel Ther. 2016;10:2779–88.
Kamboj S, Singh R. Chromanone-A prerogative therapeutic scaffold: an overview. Arab J Sci Eng. 2022;47:75–111.
Li R, Stumpfe D, Vogt M, Geppert H, Bajorath J. Development of a method to consistently quantify the structural distance between scaffolds and to assess scaffold hopping potential. J Chem Inf Model. 2011;51:2507–14.
Morris GM, Lim-Wilby M. Molecular docking. In: Kukol A, editors. Molecular modeling of proteins. Methods molecular biology™, Vol. 443. Totowa, NJ: Human Press; 2008.
Gál B, Bucher C, Burns NZ. Chiral alkyl halides: underexplored motifs in medicine. Mar Drugs. 2016;14:206–17.
Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56:1363–88.
Patel H, Pawara R, Ansari A, Surana S. Recent updates on third generation EGFR inhibitors and emergence of fourth generation EGFR inhibitors to combat C797S resistance. Eur J Med Chem. 2017;142:32–47.
Youssif BGM, Abdelrahman MH, Abdelazeem AH, Abdelgawad MA, Ibrahim HM, Salem OIA, et al. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a]indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur J Med Chem. 2018;146:260–73.
Blois MS. Antioxidant determinations by the use of a stable free radical. Nature. 1958;181:1199–200.
Sivanandan S, Jain K, Plakkal N, Bahl M, Sahoo T, Mukherjee S, et al. Issues, challenges, and the way forward in conducting clinical trials among neonates: investigators’ perspective. J Perinatol. 2019;3:20–30.
Angelis ID, Turco L. Caco-2 cells as a model for intestinal absorption. Curr Protoc Toxicol. 2011;47:20–6.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17.
Lokwani DK, Azad R, Sarkate AP, Reddanna P, Shinde DB. Structure based library design (SBLD) for new 1,4-dihydropyrimidine scaffold as simultaneous COX-1/COX-2 and 5-LOX inhibitors. Bioorg Med Chem. 2015;23:4533–43.
Bhosle MR, Khillare LD, Mali JR, Sarkate AP, Lokwani DK, Tiwari SV. DIPEAc promoted one-pot synthesis of dihydropyrido[2,3-d:6,5-d′]dipyrimidinetetraone and pyrimido[4,5-d]pyrimidine derivatives as potent tyrosinase inhibitors and anticancer agents: in vitro screening, molecular docking and ADMET predictions. N J Chem. 2018;42:18621–32.
Tiwari SV, Seijas JA, Vazquez-Tato MP, Sarkate AP, Karnik KS, Nikalje APG. Ionic liquid-promoted synthesis of novel chromone-pyrimidine coupled derivatives, antimicrobial analysis, enzyme assay, docking study and toxicity study. Molecules. 2018;23:1–23.
Karnik KS, Sarkate AP, Tiwari SV, Azad R, Wakte PS. Free energy perturbation guided synthesis with biological evaluation of substituted quinoline derivatives as small molecule L858R/T790M/C797S mutant EGFR inhibitors targeting resistance in non-small cell lung cancer (NSCLC). Bioorg Chem. 2021;115:105226–38.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–61.
Forli S, Huey R, Pique ME, Sanner M, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11:905–19.
Seeliger D, Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24:417–22.
SwissADME. 2021. http://www.swissadme.ch.
Karnik KS, Sarkate AP, Tiwari SV, Azad R, Burra PVLS, Wakte PS. Computational and synthetic approach with biological evaluation of substituted quinoline derivatives as small molecule L858R/T790M/C797S triple mutant EGFR inhibitors targeting resistance in non-small cell lung cancer (NSCLC). Bioorg Chem. 2021;107:104612–28.
Kashem MA, Nelson RM, Yingling JD, Pullen SS, Prokopowicz AS, Jones JW, et al. Three mechanistically distinct kinase assays compared: measurement of intrinsic ATPase activity identified the most comprehensive set of ITK inhibitors. J Biomol Screen. 2007;12:70–83.
Acknowledgements
The authors KSK and PSW are thankful to Indian Council of Medical Research (ICMR) for providing funding in support of Senior Research Fellowship with Ref No: 3/2/2/33/2018/Online OncoFship/NCD-III. The authors are thankful to The Head, Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad 431 004 (MS), India, for providing the laboratory facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Karnik, K.S., Sarkate, A.P., Tiwari, S.V. et al. Design, synthesis, biological evaluation and in silico studies of EGFR inhibitors based on 4-oxo-chromane scaffold targeting resistance in non-small cell lung cancer (NSCLC). Med Chem Res 31, 1500–1516 (2022). https://doi.org/10.1007/s00044-022-02929-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02929-4