
Abstract
A series of anilinoquinazoline derivatives with modification on the 2nd carbon of the aniline ring has been synthesized and characterized. The compounds have been tested for their in vitro antiproliferative activity against three NSCLC cell lines, including A549, H1650 and H1975. One of the products has demonstrated the highest IC50 value against A549 (17.60 ± 1.70 µM), surpassing the standard drug, gefitinib (34.32 ± 1.30 µM), another one has exhibited IC50 value against H1975 (9.75 ± 1.06 µM), surpassing gefitinib (31.12 ± 0.38 µM). The best performing derivatives in the antiproliferative assay have been selected for further in silico study for investigating their plausible binding mode in different EGFR kinases through molecular docking and molecular dynamics simulations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Quinazoline derivatives have demonstrated a broad spectrum of valuable biological activities, including antimicrobial [1], anti-inflammatory [2] and anticonvulsant [3]. Several derivatives of anilinoquinazoline, such as gefitinib and erlotinib, are widely used in treatment of non-small cell lung cancer (NSCLC) [4, 5]. Discovery of those anticancer agents has stimulated further attempts in derivativation of anilinoquinazoline scaffold and study of the products antitumor activity [6–9].
However, a limited attention has been payed to the derivatives with substituents on the 2nd carbon atom of the aniline ring of the anilinoquinazoline. Therefore, in this project, a series of anilinoquinazoline derivatives with an aromatic ring extended from an amide linker on the 2nd carbon of the aniline ring (Fig. 1) was synthesized and tested for antiproliferative activity against A549, H1650, and H1975 cell lines. Molecular docking and molecular dynamics simulations were involved for the insite in the mechanism of their cells proliferation inhibition.

General structure of the designed derivatives.
RESULTS AND DISCUSSION
Eight new anilinoquinazoline derivatives were synthesized according to the synthetic pathway outlined in Scheme 1. The commercially available 2-amino-4-chlorobenzoic acid (1) was reacted with formamide under MW irradiation to afford 7-chloroquinazolin-4(3H)-one (2). Initially the same process was carried out under conversion heating of 1 [10, 11]. The alternative MW irradiation [12] elevated the yield of 2 up to 90%.
Chlorination of 7-chloroquinazolin-4(3H)-one (2) using oxalyl chloride and DMF in chloroform produced 4,7-dichloroquinazoline (3) [13, 14], the following reaction of which with 2-amino-4-chlorobenzoic acid in THF targeted the 4-anilinoquinazoline scaffold. However, very low solubility of the precipitated product in common NMR solvents did not allow to characterize its structure. Instead, a series of aminobenzamide was prepared in situ from 2-amino-4-chlorobenzoic acid and different amines via CDI mediated coupling, before being reacted with 4,7-dichloroquinazoline (3) with formation of the target compounds 4a–4h (Scheme 1).

Synthetic pathway to compounds 4a–4h.
Antiproliferative study. The in vitro antiproliferative activity of the synthesized compounds was measured against three NSCLC cell lines: A549, H1650, and H1975 (Table 1). Gefitinib was used as the positive control.
The products demonstrated different selectivity on the cell lines. In comparison with gefitinib, seven compounds (except 4f) demonstrated better IC50 values on A549 cell line, and similarly, seven compounds (besides 4c) performed better on H1975 cell line. However, none of the compounds performed better than gefitinib on H1650 cell line.
Compounds 4a, 4g, and 4h were structurally different in term of the distance between the amido group and the phenyl ring. Specifically, there was one carbon atom separating the two groups in compound 4a, two carbons in compound 4g, and none in compound 4h. On A549 cell line, compound 4a (24.87 ± 1.74 µM) performed better than 4g (31.06 ± 0.28 µM) and 4h (33.64 ± 0.75 µM). On the other hand, on H1975 cell line, compound 4g (9.75 ± 1.06 µM) exhibited better results than 4a (23.83 ± 1.68 µM) and compound 4h (29.66 ± 0.85 µM). Compound 4f which contained pyridinyl ring instead of phenyl was determined to be of the lowest activity.
Molecular docking study. The molecular docking study was carried out for the most active products 4e and 4g interaction with EGFR kinase, which was chosen as the target protein [15, 16], and gefitinib and erlotinib were known as inhibitors of EGFR kinase [17].
Compound 4e and compound 4g were considered as ligands for docking in WT-EGFR and LRTM-EGFR, respectively (Table 2).
Compound 4e bound to WT-EGFR could be in the same orientation in the crystal structure of erlotinib in EGFR kinase [4]. The incorporated aromatic ring on the 2nd position of the aniline ring of compound 4e formed π–π interaction with PHE723. The aromatic ring also formed π-anion interaction with ASP855. π-Anion interaction with ASP855 had previously been observed in the docking of luteolin in EGFR kinase [18]. In LRTM-EGFR, compound 4g also formed hydrogen bonding with MET793, and its aromatic ring was involved in π- π interactions with PHE723.
The 3D diagrams of both docking results indicated that LYS745 and ASP855 residues were in close proximity to the amide group linker of compounds 4e and 4g. Despite the ability of the amide group to become a hydrogen donor and acceptor, such interactions were not observed.
Molecular dynamics simulations. The molecular dynamics simulations were carried out for the compounds 4e and 4g in the respective kinases for detecting possibile H-bonds formation with the residues that could contribute to their in vitro antiproliferative activity.
Stability and fluctuation of the systems were evaluated by the root-mean-square deviation (RMSD) of the backbone residues over 50 ns, that were calculated by using their respective energy minimized structures as a reference. Ten amino acid residues of both terminals of each kinase were opted out from RMSD calculations due to their highly fluctuating nature that might interfere with the interpretation of the results [19]. According to RMSD of the complexes the backbones of those were stabilized after 30 ns of simulations.
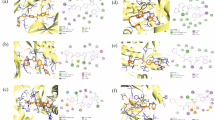
Hydrogen bond occupancy (Table 3 and Supplementary information). Though both compounds 4e and 4g contained the amide group on their aniline ring, compound 4g had a higher occupancy of the hydrogen bonding with ASP855 (23.95%), than 4e (8.72%). The 3D interaction diagram of LRTM-EGFR/compound 4g (Fig. 2a) supported the fact that presence of the bulky mutated arginine residue (ARG858) pushed ASP855 residue closer towards compound 4g, allowing it to form stronger hydrogen bonding with ASP855, whereas in WT-EGFR, ASP855 residue had more space to be flexible due to smaller LEU858 (Fig. 2b).

3D interaction diagrams of the lowest energy frame of (a) LTRM-EGFR/compound 4g, and (b) WT-EGFR/compound 4e.
All chemicals were obtained from various commercial sources, and used as received. TLC was performed using MERCK 25 TLC plates 20×20 cm silica gel 60 F254 precoated aluminum plates. The spots were visualized under the UV light. Ethyl acetate:hexane system was used as an eluent. Flash column chromatography was performed using silica gel (0.063–0.2 mm) as the stationary phase. Melting points were measured in glass capillaries on a MELTEMP II, Laboratory Device. NMR spectra were measured on a JEOL ECX-400 MHz using DMSO-d6 and CDCl3 as the solvents. Mass spectra were measured on an Agilent 6550 iFunnel ESI-Q-TOF LC-MS.
Synthesis of 7-chloroquinazolin-4(3H)-one (2). 2-amino-4-chlorobenzoic acid (0.5 g, 2.91 mmol) was stirred in 7.5 mL of formamide. The mixture was heated in a microwave oven at 300 kWatt, 190°C, for 30 min. The clear brown reaction mixture was cooled to room temperature. Ice cold water was added to the reaction mixture to induce precipitation. The precipitate was filtered, and the crude product was further purified using column chromatography (EA: hexane, 7 : 3) to give compound 2 as white powder. Yield 90%, mp 256–258°C. 1H NMR spectrum, δ, ppm: 7.49 d. d (1H, J = 8.71 Hz, J = 2.29 Hz, CH), 7.66 d (1H, J = 2.29 Hz, CH), 8.05 d (1H, J = 8.71 Hz, CH), 8.09 s (1H, CH), 12.35 s (1H, NH). 13C NMR spectrum, δC, ppm: 122.0, 126.9, 127.5, 128.5, 139.4, 147.4, 150.4, 160.7.
Synthesis of 4,7-dichloroquinazoline (3). Compound 2 (5 g, 27.69 mmol) and DMF (1 mL, 20% w/w) were added to 150 mL of chloroform. The reaction mixture was cooled in an ice-bath. Oxalyl chloride (4.8 mL, 55.38 mmol) was added dropwise to the reaction mixture which was left to warm up to room temperature upon stirring for 2 h. Then the reaction mixture was heated to 60oC and stirred overnight, upon completion of the process it was cooled in ice-bath. Then, 4.95 g of sodium hydrogen carbonate in 50 mL of cold water were added drop-wise to the reaction mixture. The mixture was separated, and the organic layer was dried using sodium sulphate and concentrated under vacuum. The crude product was purified by column chromatography (ethyl acetate: hexane, 1 : 1) to give the product 3 as white solid. Yield 80%, mp 140–142°C. 1H NMR spectrum, δ, ppm: 7.67 d. d (1H, J = 8.70 Hz, J = 1.84 Hz, CH), 8.07 d (1H, J = 1.84 Hz, CH), 8.21 d (1H, J = 8.70Hz, CH), 9.04 s (1H, CH). 13C NMR spectrum, δC, ppm: 122.6, 127.4, 128.0, 130.4, 141.6, 151.7, 154.8, 162.5.
Synthesis of compounds 4a–4g. Solution of amino-4-chlorobenzoic acid (0.30 g, 1.75 mmol) in 10 mL of THF was mixed with CDI (0.34 g, 2.10 mmol), and the mixture was stirred for 1 h, after which the desired amine was added to the mixture, and it was stirred overnight. Upon completion of the reaction (TLC), the mixture was filtered through silica, and the eluate was concentrated on a rotary evaporator. The crude intermediate and compound 3 (0.35 g, 1.75 mmol) were dissolved in 10 mL of THF at room temperature and stirred overnight. The resulting precipitate was filtered off. The crude product was stirred in isopropanol for 1 h and filtered to give the corresponding pure product.
N-benzyl-4-chloro-2-[(7-chloroquinazolin-4-yl)amino]benzamide (4a). Phenylmethanamine (0.16 mL, 1.50 mmol) was used as the reactant. Yellowish powder, yield 62%, mp 208–210°C. 1H NMR spectrum, δ, ppm: 4.40 d (2H, J = 5.88 Hz, CH2), 7.15 m (5H, C6H5), 7.44 d (1H, J = 8.55 Hz, CH), 7.89 d (2H, J = 8.55 Hz, CH), 7.98 s (1H, CH), 8.37 d (1H, J = 8.55 Hz, CH), 8.43 s (1H, CH), 8.84 s (1H, CH), 9.43 t (1H, J = 5.88 Hz, NH), 12.82 s (1H, NH). 13C NMR spectrum, δC, ppm: 43.2, 113.5, 122.3, 124.8, 125.2, 125.8, 126.0, 127.4, 127.8, 128.8, 129.7, 130.0, 136.5, 138.8, 139.2, 140.6, 143.5, 153.4, 159.1, 167.4. MS: m/z: 423.0769 [M + H]+.
N-(2-Bromobenzyl)-4-chloro-2-[(7-chloroquinazolin-4-yl)amino]benzamide (4b). (2-Bromophenyl)methanamine (0.19 mL, 1.50 mmol) was used as the reactant. Yellowish powder, yield 62%, mp 206–208°C. 1H NMR spectrum, δ, ppm: 4.44 d (2H, J = 2.28 Hz, CH2), 7.14 d. d (1H, J = 7.90 Hz, J = 7.34 Hz, CH), 7.23 d. d (1H, J = 7.90 Hz, J = 7.34 Hz, CH), 7.29 d (1H, J = 7.34 Hz, CH), 7.45 d (1H, J = 8.47 Hz, CH), 7.53 d (1H, J = 7.90 Hz, CH), 7.86 d (1H, J = 8.47 Hz, CH), 7.94 d (1H, J = 8.47 Hz, CH), 7.96 s (1H, CH), 8.33 d (1H, J = 8.47 Hz, CH), 8.49 s (1H, CH), 8.87 s (1H, CH), 9.44 s (1H, NH), 12.79 s (1H, NH). 13C NMR spectrum, δC, ppm: 43.8, 113.7, 122.9, 124.4, 124.6, 125.6, 125.9, 128.2, 129.4, 129.6, 131.0, 132.9, 136.7, 137.5, 139.2, 140.4, 144.2, 153.7, 158.9, 167.7. MS, m/z: 500.9893 [M + H]+.
4-Chloro-2-[(7-chloroquinazolin-4-yl)amino]-N-(4-fluorobenzyl)benzamide (4c). (4-Fluorophenyl)methanamine (0.17 mL, 1.50 mmol) was used as the reactant. White powder, yield 65%, mp 228–230°C. 1H NMR spectrum, δ, ppm: 4.36 d (2H, J = 5.64 Hz, CH2), 6.97 m (2H, CH), 7.23 m (2H, CH), 7.44 d (1H, J = 8.47 Hz, CH), 7.86 d (1H, J = 8.47 Hz, CH), 7.91 d (1H, J = 8.47 Hz, CH), 7.98 s (1H, CH), 8.38 m (2H, CH), 8.86 s (1H, CH), 9.43 s (1H, NH), 12.78 s (1H, NH). 13C NMR spectrum, δ, ppm: 42.5, 113.3, 115.3, 121.6, 125.0, 126.8, 129.7, 129.9, 131.0, 135.5, 136.4, 138.4, 140.8, 142.7, 153.2, 159.3, 160.5, 162.9, 167.2. MS: m/z: 441.0680 [M + H]+.
4-Chloro-2-[(7-chloroquinazolin-4-yl)amino]-N-(4-methoxybenzyl)benzamide (4d). (4-Methoxyphenyl)methanamine (0.20 mL, 1.50 mmol) was used as the reactant. White powder, yield 64%, mp 226–228°C. 1H NMR spectrum, δ, ppm: 3.77 s (3H, OCH3), 4.32 d (2H, J = 5.64 Hz, CH2), 6.70 d (2H, J = 7.90 Hz, CH), 7.11 d (2H, J = 7.90 Hz, CH), 7.43 d (1H, J = 8.47 Hz, CH), 7.85 d (1H, J = 8.47 Hz, CH), 7.89 d (1H, J = 8.47 Hz, CH), 7.97 s (1H, CH), 8.35 d (1H, J = 8.47 Hz, CH), 8.39 s (1H, CH), 8.86 s (1H, CH), 9.34 s (1H, NH), 12.81 s (1H, NH). 13C NMR spectrum, δ, ppm: 42.6, 55.5, 113.5, 114.1, 122.3, 124.5, 125.5, 125.8, 126.0, 129.2, 129.6, 130.9, 131.0, 131.2, 136.4, 138.8, 140.5, 153.5, 158.7, 159.1, 167.2. MS: m/z: 453.0861 [M + H]+.
4-Chloro-2-[(7-chloroquinazolin-4-yl)amino]-N-(4-methylbenzyl)benzamide (4e). p-Tolylmethanamine (0.20 mL, 1.50 mmol) was used as the reactant. White powder, yield 60%, mp 226–228°C. 1H NMR spectrum, δ, ppm: 2.19 s (3H, CH3), 4.33 d (2H, J = 5.64 Hz, CH2), 6.95 d (2H, J = 7.90 Hz, CH), 7.06 d (2H, J = 7.90 Hz, CH), 7.44 d. d (1H, J = 8.47 Hz, 2.26 Hz, CH), 7.85 d (1H, J = 8.47 Hz, CH), 7.89 d. d (1H, J = 9.03 Hz, 2.26 Hz, CH), 7.96 d (1H, J = 2.26 Hz, CH), 8.34 d (1H, J = 9.03 Hz, CH), 8.40 s (1H, CH), 8.85 s (1H, CH), 9.37 t (1H, J = 5.64 Hz, NH), 12.78 s (1H, NH). 13C NMR spectrum, δ, ppm: 21.2, 42.9, 113.5, 122.2, 124.7, 125. 6, 125.9, 126.1, 127.8, 129.3, 129.6, 130.9, 136.2, 136.4, 136.5, 138.6, 140.5, 143.4, 153.4, 159.1, 167.2. MS: m/z: 437.0919 [M + H]+.
4-Chloro-2-[(7-chloroquinazolin-4-yl)amino]-N-(pyridin-2-ylmethyl)benzamide (4f). Pyridin-2-ylmethanamine (0.15 mL, 1.50 mmol) was used as the reactant. White powder, yield 57%, mp 214–216°C. 1H NMR spectrum, δ, ppm: 4.40 d (2H, J = 5.08 Hz, CH), 7.48 d. d (1H, J = 8.47 Hz, 1.70 Hz, CH), 7.65 d. d (1H, J = 5.64 Hz, 6.78 Hz, CH), 7.74 d (1H, J = 7.90 Hz, CH), 7.86 d. d (1H, J = 9.03 Hz, 1.69 Hz, CH), 7.97 (1H, d, J = 8.47 Hz, CH), 7.99 s (1H, CH), 8.18 d. d (1H, J = 6.78 Hz, 7.90 Hz, CH), 8.40 s (1H, CH), 8.42 d (1H, J = 9.03 Hz, CH), 8.65 d (1H, J = 5.64 Hz, CH), 8.87 s (1H, CH), 9.66 t (1H, J = 5.08 Hz, NH). 13C NMR spectrum, δ, ppm: 42.2, 113.3, 121.5, 125.1, 125.4 (CH, C-23), 125.8, 126.4, 126.6, 129.6, 131.3, 136.6, 138.3, 140.8, 142.8, 143.6, 144.7, 153.1, 155.1, 159.5, 167.8. MS: m/z: 424.0718 [M + H]+.
4-Chloro-2-[(7-chloroquinazolin-4-yl)amino]-N-phenethylbenzamide (4g). 2-Phenylethanamine (0.19 mL, 1.50 mmol) was used as the reactant. Yellowish powder, yield 61%, mp 216–218°C. 1H NMR spectrum, δ, ppm: 2.78 t (2H, J = 7.34 Hz, CH), 3.40 m (2H, CH), 7.07 m (1H, CH), 7.19 m (4H, CH), 7.38 d. d (1H, J = 8.47 Hz, J = 1.70 Hz, CH), 7.78 d (1H, J = 8.47 Hz, CH), 7.91 d. d (1H, J = 8.47 Hz, J = 1.70 Hz, CH), 7.96 s (1H, CH), 8.26 d (1H, J = 8.47 Hz, CH), 8.64 s (1H, CH), 8.90 s (1H, CH), 9.03 t (1H, J = 5.64 Hz, NH), 12.88 s (1H, NH). 13C NMR spectrum, δ, ppm: 35.2, 41.3, 114.0, 123.3, 123.5, 125.0, 125.6, 126.7, 127.3, 128.8, 129.2, 129.6, 130.7, 136.6, 139.4, 139.7, 140.3, 154.0, 158.6, 167.7. MS: m/z: 437.0911 [M + H]+.
4-Chloro-2-[(7-chloroquinazolin-4-yl)amino]-N-phenylbenzamide (4h). Aniline (0.14 mL, 1.50 mmol) was used as the reactant. Yellowish powder, yield 60%, mp 216–218°C. 1H NMR spectrum, δ, ppm: 7.03 t (1H, J = 7.34 Hz, CH), 7.25 d. d (2H, J = 7.90, 7.34 Hz, CH), 7.53 d (1H, J = 12.47 Hz, CH), 7.59 d (2H, J = 7.90 Hz, CH), 7.89 m (1H, CH), 7.91 m (1H, CH), 7.98 s (1H, CH), 8.12 s (1H, CH), 8.60 d (1H, J = 8.47 Hz, CH), 8.85 s (1H, CH), 10.66 s (1H, NH), 12.28 s (1H, NH). 13C NMR spectrum, δ, ppm: 113.2, 121.1, 121.4, 124.7, 125.4, 126.3, 126.7, 128.7, 129.2, 129.7, 131.5, 136.2, 137.7, 139.1, 140.9, 142.4, 152.9, 159.7, 165.6. MS: m/z: 409.0591 [M + H]+.
In vitro antiproliferative study. The in vitro antiproliferation activity of the synthesized derivatives was tested using the MTT assay. Gefitinib was used as the positive control. Three NSCLC cell lines, namely A549, H1650, and H1975 which were obtained from AddexBio Ltd. USA. DMEM was used as the media for A549 cell line, while RPMI 1640 was used as the media for H1650 and H1975 cell lines. The cells were cultured in a cell incubator at 37°C, where the humidified atmosphere of 5% CO2 was maintained. The cells were plated into 96-wells microplate at a density of 104 cells per well after which the cells were incubated over a period of 24 h. The treatment of the test compounds onto the cells was executed using 100 µL of nine working concentrations (3.91–100 µM). The working solutions were prepared in 0.5% v/v DMSO in respective media. For negative control, the cells were treated with 100 µL of 0.5% v/v DMSO in media. The cells were incubated for another 72 h. Next, 20 µL of MTT reagent (5 mg/mL) were added to each well. After the MTT treatment, the cells were incubated for another 3 h. Then, the supernatant was removed carefully, after which 100 µL of DMSO was added to dissolve the MTT formazan crystals. The absorbance of the formazan product was read on a microplate reader at 570 nm, with 620 nm as the background wavelength (Infinite 200, Tecan, Männedorf, Switzerland). The percentage of the viable cells in each well was calculated from the absorbance of the respective well over that of the negative control well. The IC50 values of the test compounds were determined from dose-response curves using Prism 8.0.2, software for Windows (GraphPad Software Inc., La Jolla, CA, USA). The results were expressed as mean ±standard deviation of triplicates [20].
Molecular docking. A workstation with four Intel Core 2 Duo E6850 3.00 GHz microprocessors, 8 GigaBytes of Random-Access Memory (RAM) and an Ubuntu 18.04 Linux operating system was used. Blind docking was performed on WT-EGFR and LRTM-EGFR using AutoDock Vina software [21]. The models for the kinases were obtained from the Protein Data Bank (PDB ID: 1M17 and 5EDR, respectively) [4, 22].
The present ligands and water molecules were removed from the kinases models using Biovia Discovery Studio Visualiser 4.5 software [23]. Then, AutoDockTools 1.5.6 software was used to add all hydrogen atoms, merging nonpolar hydrogen atoms, checking and repairing missing atoms, adding Gasteiger charges, assigning AD4 atom types to the protein structure, and finally saving the protein structure in .pdbqt format for further docking [24, 25]. A grid box of the kinase structure was then set using AutoDockTools 1.5.6 software with a grid spacing of 1.0 Å, dimensions of 126×126×126 points along the x, y and z axes, and centered on the kinase and covered the whole kinase for blind docking [26].
The structure of the ligands to be run in the molecular docking were constructed using ChemBio 3D Ultra 12.0, and their energy were minimized using MM2 energy minimization calculation [27]. Then, AutoDockTools 1.5.6 software was used to add flexibility to flexible bonds on the ligands and save the ligands in .pdbqt format as input files for molecular docking [24].
Molecular dynamics simulations. The MD simulations were carried out using GROMACS 2019 [28]. GROMACS “pdb2gmx” module with AMBER force field ff14SB [29] were used to prepare the receptor files for the simulations. Antechamber Python Parser Interface (ACPYPE) [30] software was used to prepare the ligand files, after which Generalized AMBER Force Field (GAFF) [31, 32] and AM1-BCC charge models were applied to the files. The receptor-ligand complex was suspended at the center of a TIP3P water box [33], where the edge of box was set to be at least 10 Å away from the complex. Explicit solvent model was used to solvate the system, after which the system was neutralized by adding counter ions prior to the energy minimization stage. The energy of the solvated system was minimized using the steepest descent algorithm, and followed by the conjugated gradient algorithm. After the energy minimization, equilibrium simulation was carried out with constant temperature and pressure at 300 K and 1 atm, respectively. All MD simulations during the production stage was carried out for 50 ns, while a trajectory was captured for every 20 ps. For RMSD calculation, the analysis of the collected trajectories was carried out using the GROMACS ‘rms’ module. For the hydrogen bond occupancy calculations, the trajectories collected from GROMACS MD simulation were converted into AMBER netcdf format (.nc) with cpptraj [34] module in AmberTools. The converted trajectories were then used for hydrogen bond occupancy analysis with cpptraj’s hbond module.
CONCLUSIONS
Eight new anilinoquinazoline with an aromatic ring at the 2nd carbon of the aniline ring with amide group linker have been synthesized and tested for their in vitro antiproliferative activity against A549, H1650 and H1975 cell lines. The results have indicated that seven derivatives have demonstrated performance higher than that of gefitinib in A549 and H1975 cell lines, while none of the derivatives have performed better than the standard drug in H1650 cell line. Compound 4e has demonstrated the best antiproliferative activity in A549 cell line (17.60 ± 1.70 µM), while compound 4g has demonstrated the highest antiproliferative activity in H1975 cell line (9.75 ± 1.06 µM). The molecular docking study against EGFR kinases has proposed that both compounds have maintained the hydrogen bonding interaction with MET793 as observed in anilinoquinazoline derivatives, while forming the additional π–π stacking interaction with PHE723. Further in silico study by the molecular dynamics simulations has indicated that in WT-EGFR, compound 4e could have interacted with ASP855 through the hydrogen bonding. In LRTM-EGFR, compound 4g also could have form hydrogen bonding with ASP855. This study demonstrates that the incorporation of both amide group and phenyl ring on the 2nd carbon of the aniline ring increases the potency of the anilinoquinazoline derivatives against certain NCSLC cells. In conclusion, compound 4e and compound 4g can be considered as the potential leads for further development of antitumor agents.
REFERENCES
Raghavendra, N.M., Thampi, P., Gurubasavarajaswamy, P.M., and Sriram, D., Chem. Pharm. Bull., 2007, vol. 55, p. 1615. https://doi.org/10.1248/cpb.55.1615
Manivannan, E. and Chaturvedi, S.C., Bioorg. Med. Chem., 2011, vol. 19, p. 4520. https://doi.org/10.1016/j.bmc.2011.06.019
Georgey, H., Abdel-Gawad, N., and Abbas, S., Molecules, 2008, vol. 13, p. 2557. https://doi.org/10.3390/molecules13102557
Stamos, J., Sliwkowski, M.X., and Eigenbrot, C., J. Biol. Chem., 2002, vol. 277, p. 46265. https://doi.org/10.1074/jbc.M207135200
Yun, C.H., Boggon, T.J., Li, Y., Woo, M.S., Greulich, H., Meyerson, M., and Eck, M.J., Cancer Cell, 2007, vol. 11, p. 217. https://doi.org/10.1016/j.ccr.2006.12.017
Yang, Z., Gu, J.-M., Ma, Q.-Y., Xue, N., Shi, X.-W., Wang, L., Zhang, K., Wang, Y.-B., Cao, D.-Y., Guo, R., and Xing, R.-J., Future Med. Chem., 2019, vol. 11, p. 2821. https://doi.org/10.4155/fmc-2019-0220
Cheng, W., Wang, S., Yang, Z., Tian, X., and Hu, Y., Drug Des. Devel. Ther., 2019, vol. 13, p. 3079. https://doi.org/10.2147/DDDT.S209481
Wei, H., Duan, Y., Gou, W., Cui, J., Ning, H., Li, D., Qin, Y., Liu, Q., and Li, Y., Eur. J. Med. Chem., 2019, vol. 181, p. 111552. https://doi.org/10.1016/j.ejmech.2019.07.055
Hassan, H.M.A., Denetiu, I., Khan, S.A., Rehan, M., Sakkaf, K., and Gauthaman, K., Med. Chem. Res., 2019, vol. 28, p. 1766. https://doi.org/10.1007/s00044-019-02413-6
Baumann, M. and Baxendale, I.R., Beilstein J. Org. Chem., 2013, vol. 9, p. 2265. https://doi.org/10.3762/bjoc.9.265
Davoodnia, A., Asian J. Chem., 2010, vol. 22, p. 1591.
Ouahrouch, A., Taourirte, M., Engels, J.W., Benjelloun, S., and Lazrek, H.B., Molecules., 2014, vol. 19, p. 3638. https://doi.org/10.3390/molecules19033638
Shen, C., Wang, L., Wen, M., Shen, H., Jin, J., and Zhang, P., Ind. Eng. Chem. Res., 2016, vol. 55, p. 3177. https://doi.org/10.1021/acs.iecr.5b04452
Min, J., Guo, K., Suryadevara, P.K., Zhu, F., Holbrook, G., Chen, Y., Feau, C., Young, B.M., Lemoff, A., Connelly, M.C., Kastan, M.B., and Guy, R.K., J. Med. Chem., 2016, vol. 59, p. 559. https://doi.org/10.1021/acs.jmedchem.5b01092
Nicholson, R.I., Gee, J.M., and Harper, M.E., Eur. J. Cancer, 2001, vol. 37, p. S9-15. https://doi.org/10.1016/s0959-8049(01)00231-3
Harari, P.M., Endocr. Relat. Cancer, 2004, vol. 11, p. 689. https://doi.org/10.1677/erc.1.00600
Blackledge, G. and Averbuch, S., Br. J. Cancer, 2004, vol. 90, p. 566. https://doi.org/10.1038/sj.bjc.6601550
Ambrose, G.O., Afees, O.J., Nwamaka, N.C., Simon, N., Oluwaseun, A.A., Soyinka, T., Oluwaseun, A.S., and Bankole, S., Bioinformation, 2018, vol. 14, p. 241. https://doi.org/10.6026/97320630014241
Tiwari, G. and Mohanty, D., PLoS One, 2013, vol. 8, p. e71340. https://doi.org/10.1371/journal.pone.0071340
Nazarbahjat, N., Ariffin, A., Abdullah, Z., Abdulla, M.A., Shia, J.K.S., and Leong, K.H., Med. Chem. Res., 2016, vol. 25, p. 2015. https://doi.org/10.1007/s00044-016-1660-5
Trott, O. and Olson, A.J., J. Comput. Chem., 2010, vol. 31, p. 455. https://doi.org/10.1002/jcc.21334
Hanan, E.J., Baumgardner, M., Bryan, M.C., Chen, Y., Eigenbrot, C., Fan, P., Gu, X.-H., La, H., Malek, S., Purkey, H.E., Schaefer, G., Schmidt, S., Sideris, S., Yen, I., Yu, C., and Heffron, T.P., Bioorg. Med. Chem. Lett., 2016, vol. 26, p. 534. https://doi.org/10.1016/j.bmcl.2015.11.078
BIOVIA, D.S., Discovery Studio Visualizer. 2016, Dassault Systèmes, San Diego.
Heh, C.H., Othman, R., Buckle, M.J.C., Sharifuddin, Y., Yusof, R., and Rahman, N.A., Chem. Biol. Drug Des., 2013, vol. 82, p. 1. https://doi.org/10.1111/cbdd.12122
Morris, G.M., Huey, R., Lindstrom, W., Sanner, M.F., Belew, R.K., Goodsell, D.S., and Olson, A.J., J. Comput. Chem., 2009, vol. 30, p. 2785. https://doi.org/10.1002/jcc.21256
Lim, S.K., Othman, R., Yusof, R., and Heh, C.H., Curr. Comput. Aided Drug. Des., 2017, vol. 13, p. 160. https://doi.org/10.2174/1573409912666161130122622
Cousins, K.R., J. Am. Chem. Soc., 2011, vol. 133, p. 8388. https://doi.org/10.1021/ja204075s
Abraham, M.J., Murtola, T., Schulz, R., Páll, S., Smith, J.C., Hess, B., and Lindahl, E., SoftwareX, 2015, vol. 1, p. 19. https://doi.org/10.1016/j.softx.2015.06.001
Maier, J.A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K.E., and Simmerling, C., J. Chem. Theory Comput., 2015, vol. 11, p. 3696. https://doi.org/10.1021/acs.jctc.5b00255
Sousa da Silva, A.W. and Vranken, W.F., BMC Res. Notes, 2012, vol. 5, p. 367. https://doi.org/10.1186/1756-0500-5-367
Wang, J., Wang, W., Kollman, P.A., and Case, D.A., J. Mol. Graph. Model., 2006, vol. 25, p. 247. https://doi.org/10.1016/j.jmgm.2005.12.005
Wang, J., Wolf, R.M., Caldwell, J.W., Kollman, P.A., and Case, D.A., J. Comput. Chem., 2004, vol. 25, p. 1157. https://doi.org/10.1002/jcc.20035
Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., and Klein, M.L., 1983, vol. 79. https://doi.org/10.1063/1.445869
Roe, D.R. and Cheatham, T.E., J. Chem. Theory Comput., 2013, vol. 9, p. 3084. https://doi.org/10.1021/ct400341p
Funding
This work was financially supported by the Ministry of Higher Education Malaysia, Fundamental Research Grant (FRGS/1/2019/SKK09/UM/02/1) and the University of Malaya Research Programme (RP035-17AFR).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare no conflict of interest in this work.
Supplementary information
Rights and permissions
About this article

Cite this article
Abdulwahab, M.K., Dzulkeflee, R., Han, T.K. et al. Synthesis, In Vitro Antiproliferative Activity, and In Silico Studies of New Anilinoquinazoline Derivatives as Potential AntitumorAgents. Russ J Gen Chem 90, 2410–2418 (2020). https://doi.org/10.1134/S1070363220120294
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363220120294