
Abstract
A simple and efficient protocol for the synthesis of novel 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) is described. Initially, p-anisaldehyde 1 was condensed (Mannich reaction) with acetone and ammonium acetate trihydrate afforded 2,6-bis(4-methoxyphenyl)piperidin-4-one 2. Then, methylation followed by oximation with hydroxylamine hydrochloride (NH2OH∙HCl) furnished a key scaffold 4. Further, to explore the enhanced biological properties of the piperidin-4-one core i.e. the key scaffold 4 was conjugated with substituted benzoyl chlorides in the presence of anhydrous K2CO3 as base to obtain novel 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) in excellent yields. The newly synthesized compounds were characterized by elemental analysis, IR, 1H NMR, 13C NMR and mass spectroscopic techniques, and screened for their in vitro antioxidant and antimicrobial activities. Most of the compounds exerted positive efficacy towards the biological assays performed. Among the synthesized analogues, compounds 4l and 4m exhibited promising antioxidant activity and on the other hand compounds 4b and 4d manifested persuasive antibacterial activity, whereas compound 4b displayed stupendous antifungal activity against A. flavus strain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Free radicals are naturally present in living systems; however, high amounts of free radicals can oxidise biomolecules, leading to tissue damage, cell death or degenerative processes, including aspects of ageing, cancer, cardiovascular diseases, arteriosclerosis, neural disorders, skin irritations and inflammation. Also, free radicals and lipid peroxides play an important role in oxidative stress (Gulçin, 2006). The harmful action of the free radicals can, however, be blocked by antioxidant substances, which scavenge the free radicals and detoxify the organism (Kumaran and Karunakaran, 2006). This had attracted a great deal of research interest in therapeutic antioxidant-based drugs formulations. Besides to this, the emerging resistance of microorganisms requires the careful use of existing antimicrobial drugs. However, there is a need for the design of novel antimicrobial agents, particularly for the treatment of the infections of hospitalized patients and protection of immune-suppressed or HIV-infected patients. A potential approach is the design of innovative drugs, with different mechanisms of action, in effort to avoid cross resistance (Pfeltz and Wilkinson, 2004). Heterocyclic compounds provide scaffolds on which pharmacophores can arrange to yield potent and selective drugs (Khan et al., 2005). Heterocyclic compounds specifically, piperidinone-based chemical entities with aryl substituents at C-2 and C-6 of the piperidinone ring have been documented as potent antioxidant and antimicrobial agents (Balasubramanian et al., 2005). Recently, oxime derivatives have attracted considerable attention in medicinal research due to their antiphytoviral, antitumor and antifungal bioactivities (Fig. 1) (Ravindra et al., 2009).

Structure of pharmacologically active oxime-derived drug
Motivated by the aforementioned findings and in continuation of our research interest on functionalization of new tricyclic and heterocyclic compounds (Vijay Kumar and Naik, 2010; Vijay Kumar et al., 2011; Nagaraja Naik et al., 2011; Rangaswamy et al., 2012) herein, we described the synthesis of novel 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) and evaluated their potential towards antioxidant and antimicrobial activity. The proposed general chemical motif of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters is depicted in Fig. 2.

General projected chemical motif of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters
Results and discussion
Chemistry
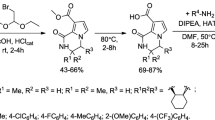
In the present work, four-step synthetic strategies were adapted for the synthesis of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q). p-anisaldehyde condensed (Mannich reaction) with acetone and ammonium acetate afforded 2,6-bis(4 methoxyphenyl)piperidin-4-one 2 in 70 % yield. Later, compound 2 was treated with iodomethane and anhydrous potassium carbonate (anh. K2CO3) in the presence of acetone to accomplish N-methylated product 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one 3. In the next step, oximation of compound 3 with NH2OH.HCl in the presence of sodium acetate trihydrate in absolute alcohol furnished key scaffold 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime 4. Further, a key scaffold 4 was conjugated with substituted benzoyl chlorides in the presence of t-BuOK as base afforded 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) in good yields. The synthesis of the target compounds were performed by the subsequent reaction pathways illustrated in Scheme 1. The formation of new compounds was confirmed by spectral and elemental analysis studies. The IR spectrum displayed the sharp bands that appeared at 1,608 and 3,617 cm−1 due to C=O and NH functions, respectively; on the other hand, 1H NMR spectrum revealed the signals resonated at δ 1.91 (s, 1H, NH), 4.10 (m, 2H, CH), 2.98 (m, 4H, CH2) and 6.94–7.10 (m, 8H, Ar–H), which confirms the formation of compound 2. IR spectrum of newly synthesized compounds 4(a–q) exhibited bands at 1691–1728 cm−1 due to the ester (–N–OCOR) group. 1H NMR spectrum of compounds 4(a–q) exhibited signal at δ 1.68–1.77 (s, 3H, N–CH3) which confirms N-methylation and also the absence of N–OH proton at δ 2.10 (s, 1H, N–OH) confirms the synthesis of piperidinone oxime esters.

Reaction protocol for the synthesis of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q)
Biological studies
In vitro antioxidant assays
The antioxidant potential of novel 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) was evaluated by means of different in vitro tests like 2,2′-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay (RSA), 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS•+) radical scavenging assay, ferric ion-reducing antioxidant power (FRAP) assay and cupric ion-reducing antioxidant capacity (CUPRAC) assay. Butylated hydroxy anisole (BHA) was used as an internal standard antioxidant.
DPPH radical scavenging activity
DPPH radical scavenging assay is a standard assay for antioxidant studies and offers a rapid technique for screening the radical scavenging activity of any compounds or extracts. RSA of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) was tested using an ethanolic solution of the ‘stable’ free radical, DPPH. Thus, antioxidant molecules can quench DPPH free radicals (i.e. by providing hydrogen atoms or by electron donation, conceivably via a free-radical attack on the DPPH molecule) and convert them to a colourless/bleached product (i.e. absorbance at 517 nm). Hence, the more rapidly the absorbance decreases, more potent the antioxidant activity of the compound. 50 % inhibition concentrations (IC50) for newly synthesized compounds were calculated and depicted in Table 1. Initially, 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime 4 exhibited certain degree of antioxidant activity. Further, to enhance the RSA, piperidin-4-one oxime 4 was coupled with different substituted benzoyl chlorides, and thus showed significant enhancement of activity. This indicates that electronic effects of substituents on phenyl ring attached to piperidin-4-one oxime play a vital role for enhanced antioxidant activity. Among the synthesized compounds 4l and 4m containing two and three –OH groups, respectively, on phenyl moiety displayed most potent DPPH radical scavenging capacity of about 13–15 folds compared to key scaffold 4 and indeed greater activity than the standard as well. Thus, free-radical scavenging capacity is primarily attributed to high reactivity of OH group (Heim et al., 2002). Whereas, compounds 4(i–k) possessing one hydroxyl group on the phenyl ring at different positions showed 7–10 folds increased activity. On the other hand, compounds 4(n–p) possessing electron-donating group –OCH3 on phenyl ring exhibited 3–5 folds increased DPPH activity. Whereas, compounds 4(b–g) holding electron-withdrawing groups (F, Br, Cl and NO2) registered the least activity compared to other analogues.
ABTS•+ radical scavenging assay
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) were subjected to ABTS•+ radical scavenging activity. Here the technique is based on the direct production of the blue/green ABTS•+ chromophore through the reaction between ABTS and potassium persulphate. The reduction of ABTS•+ concentration is directly related to the antioxidant capacity of the compounds being tested. In this assay, all the synthesized compounds displayed certain degree of ABTS•+ radical scavenging capacity. Compounds 4l and 4m bearing more hydroxyl groups on phenyl skeleton exhibited dominant activity among all the synthesized compounds and as well as the standard (Table 2). On the other hand, substitution of electron-withdrawing group like (F, Br, Cl and NO2) in compounds 4(b–g) does not showed appreciable enhanced antioxidant activity.
FRAP assay
The reducing capacity of compounds can be measured by the direct reduction of potassium ferricyanide (III) to potassium ferrocyanide (II) which leads to the formation of the intense Perl’s Prussian blue complex, which has a strong absorbance at 700 nm. Generally, increase in the absorbance of the reaction mixture indicates increase in the reducing capacity due to increase in the formation of the complex. The absorbance values of all the synthesized compounds are depicted in Table 2. Compounds 4(b–g) having electron-withdrawing groups (F, Cl, Br and NO2) on the phenyl ring at different positions displayed less absorbance values, while the compounds 4(i–k) possessing –OH group at ortho, meta and para position enhance the absorbance value reflecting higher but slightly lower activity than the standard. The introduction of benzoyl chlorides containing more number of hydroxy groups to compound 4 which led to compounds 4l and 4m resulted in higher ferric ion-reducing power than the standard.
CUPRAC assay
CUPRAC assay is based on the reduction of cupric (II) ion to cuprous (I) ion by antioxidants. The observed antioxidant potential for the ethanolic solutions of compounds can be related to the presence of various functional groups, such as hydroxyl and methoxy groups. The reducing power of all the tested compounds increased with increase in their concentration. Compounds 4l and 4m having the highest polyphenolic content revealed the highest absorption than the standard BHA and act as superior cupric ion reducer. Compounds 4(i–k) having single hydroxyl group and 4(n–p) bearing one or more methoxy group also exhibited the marked cupric ion-reducing ability but slightly less than compounds 4l and 4m. Rest of the compounds, 4a without any substitution, 4(b–h) possessing electron-withdrawing groups and 4q bearing methyl group, exhibit moderate antioxidant capacity (Table 2).
Antimicrobial studies
Antibacterial studies
The potentiality of the synthesized compounds as antimicrobials was appraised for their antibacterial studies against different strains of human pathogens namely Escherichia coli ATCC 25922 (Gram −Ve), Staphylococcus aureus ATCC 25923 (Gram +Ve) and Pseudomonas aeruginosa ATCC 27853 (Gram −Ve). The results obtained as zone of inhibition (mm) are presented in Table 3 and revealed that compounds 4b and 4d possessing electronegative fluoro (F) and chloro (Cl) group on benzoyl ester moiety at para-position showed excellent antibacterial activity against P. aeruginosa and E. coli at concentrations of 1,000 and 500 μg/mL compared to the standard drug streptomycin. The more lipophilic nature of piperidinone core along with the presence of electron-withdrawing groups like chloro/nitro in para-position of the phenyl rings may contribute for this enhanced antibacterial activity. Halogens like fluoro and chloro are very useful to modulate the electronic effects on phenyl ring and has strong inductive electron-attracting effects; moreover, these atoms may also influence the steric characteristics and the hydrophilic–hydrophobic balance of the molecules (Rossello et al., 2002). Compounds 4c and 4e having bromo and chloro group on benzoyl ester moiety exhibited appreciable activity. The replacement of fluoro group by nitro showed moderate activity, whereas introduction of electron-donating hydroxyl and methoxy groups in compounds 4(i–p) leads to marked decrease in activity.
Antifungal studies
Newly synthesized compounds 4(a–q) were also screened for their antifungal activity against three pathogenic fungal species namely Aspergillus flavus MTCC 3306, Candida albicans MTCC 3017 and Chrysosporium keratinophilum MTCC 2827. The compounds were dissolved in DMSO and antifungal activity was determined by well plate method at concentration of 1,000 and 500 μg/mL. The results are depicted in Table 4. Among the tested compounds, compound 4b possessing electronegative fluoro group on benzoyl ester moiety has emerged as active antifungal agent against A. flavus compared with the standard fluconazole. Whereas, other compounds showed weak to moderate antifungal activity against all the tested fungal strains compared to the standard.
Conclusion
In wide search programme towards novel and efficient pharmacophore agents, we have reported a convenient four-step reaction protocol for the synthesis of novel series of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q) through the pathway involving Mannich reaction of p-anisaldehyde with acetone and ammonium acetate followed by N-methylation and oximation. Further, in order to improve the pharmacological activity of piperidin-4-one oxime core, esterification of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime 4 with substituted benzoyl chlorides was successfully done by employing a t-BuOK-assisted reaction. The newly synthesized compounds 4(a–q) were screened for their antioxidant and antimicrobial activity using assorted in vitro models. It is noteworthy that compounds 4l and 4m possessed dominant radical scavenging capacity than the standard because of having the highest phenolic content. The presence of hydroxyl group at para-position of phenyl rings is perhaps a greater determinant for antioxidant activity. Compounds 4(i–k), 4(n–p) having electron-donating groups like –OH and –OCH3 exhibit good antioxidant activity but slightly less than the standard. The compounds 4(b–e) having electron-withdrawing groups like fluoro, bromo and chloro demonstrated moderate activity. The rest of the compounds 4(f–h) and 4q exhibited slightly less to moderate activity. On the other hand antimicrobial study was also undertaken to appraise their antimicrobial potential against different strains of human pathogens. Compounds 4b and 4d showed excellent antibacterial activity against tested bacterial strains as compared to the standard drug. Among the screened samples compound 4b emerged as active antifungal agent against A. flavus. The compounds holding electron-donating hydroxyl and methoxy groups 4(i–p) showed marked decrease in antimicrobial activity. A careful observation of the results implies that the lipophilic nature of piperidinone skeleton and the electronic effects of the halogen substituent on the benzoyl ester moiety were significant towards increased antimicrobial activity. Our results prompt that, further studies on these compounds may be operating as a positive reinforce of the tendency for the construction of novel chemical entities with better pharmacological profiles than the standard drugs.
Experimental
Materials and methods
All the reagents used were purchased from commercial suppliers without further purification. Melting points were determined by using an open capillary method and are uncorrected. Thin-layer chromatography (TLC) was performed with aluminium sheets–Silica gel 60 F254 purchased from Merck. The compounds were purified by using column chromatography with silica gel (60–120 mesh) using hexane:ethylacetate (8:2) as eluent. IR: Nicolet 5700 FT–IR spectrophotometer; 1H NMR and 13C NMR spectra were acquired at 300 and 100 MHz, respectively, by using DMSO as a solvent for all the compounds. Mass spectra were obtained by Waters–Q–TOF ultima spectrometer. Micro analytical data were obtained by Elemental–Vario EL–III.
Synthesis of 2,6-bis(4-methoxyphenyl)piperidin-4-one (2)
The experimental procedure adopted for the condensations was as follows: a mixture of acetone (2 mmol), p-anisaldehyde (4 mmol) and ammonium acetate (2 mmol) in 10 ml of ethanol was heated until the acetate dissolved (Noller and Baliah, 1948) and a yellow colour developed. After cooling, the reaction mixture was taken in ether (20 ml) then addition of conc. HCl (1 ml) to the clear filtrate afforded the hydrochloride of the 2,6-bis(4-methoxyphenyl) piperidin-4-one. The free base was obtained by treating a suspension of the hydrochloride with aqueous ammonia, followed by dilution with water. The crude solid thus obtained was filtered. Crystallization from ethanol yielded the compound 2 (70 %).
Yield 70 % as brown solid, M.p 148–150 °C. IR (KBr) ν max (cm−1): 3030-2956 (Ar–CH), 1608 (C=O), 3617 (N–H). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.94–7.10 (m, 8H, Ar–H), 4.10 (m, 2H, CH), 3.81 (s, 6H, OCH3), 2.98 (m, 4H, CH2), 1.91 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 206.8, 158.0, 135.4, 127.3, 127.0, 126.9, 114.0, 114.0, 113.9, 113.9, 64.0, 55.6, 50.3, 50.1. Mass (m/z): (M+) 311.05. Anal.calcd. for C19H21NO3, C, 73.29; H, 6.80; N, 4.50; O, 15.41 found: C, 73.25; H, 6.84; N, 4.51, O, 15.41 %.
Synthesis of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one (3)
A mixture of 2,6-bis(4-methoxyphenyl)piperidin-4-one (2 mmol), potassium carbonate (K2CO3) (2 mmol) and iodomethane (2.5 mmol) in acetone was refluxed for 4 h. Progress of the reaction was monitored by TLC using hexane:ethylacetate (8:2) as solvent. After cooling, the solvent along with excess methyl iodide was stripped off from the reaction mixture under reduced pressure. Addition of distilled water (20 ml) followed by aqueous ammonia (5 ml) yielded a crude solid. The crude solid thus obtained was filtered and washed with distilled water. Crystallization from ethanol yielded the compound 3 (83 %).
Yield 83 % as brown solid, M.p 138–140 °C. IR (KBr) ν max (cm−1): 3092-2918 (Ar–CH), 1608 (C=O). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.79–6.90 (m, 6H, Ar–H), 3.83 (s, 6H, OCH3), 3.50 (m, 2H, CH), 2.96-2.70 (m, 4H, CH2), 1.73 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 206.8, 158.0, 135.6, 127.8, 127.0, 114.08, 113.5, 113.3, 63.8, 63.5, 55.8, 50.3, 55.0, 41.05. Mass (m/z): (M+) 325.10. Anal.calcd. for C20H24NO3, C, 73.82; H, 7.12; N, 4.30; O, 14.75 found: C, 73.80; H, 7.10; N, 4.28; O, 14.73 %.
Synthesis of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime (4)
To the ethanolic solution of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one (2 mmol) in a two-neck round-bottomed flask fitted with a reflux condenser, sodium acetate trihydrate (3 mmol) was added. The mixture was heated to boil for 15 min. Then, hydroxylamine hydrochloride (2.3 mmol) was added to the above mixture and was heated to reflux for further 2 h. After cooling, when the mixture was slowly poured into ice-cold water (50 ml) with constant stirring, the crude product was precipitated. Crystallization from absolute ethanol yielded the pure compound 4 (89 %).
Yield 89 % as brown solid, M.p 243–244 °C. IR (KBr) ν max (cm−1): 3102-2956 (Ar–CH), 3411 (N–OH). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.94–7.10 (m, 8H, Ar–H), 1.75-1.48 (m, 4H, CH2), 1.72 (s, 3H, CH3), 3.30 (m, 2H, CH), 3.80 (s, 6H, OCH3), 2.12 (s, 1H, N–OH). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 159.5, 159.4, 156.5, 131.2, 131.1, 128.8, 128.2, 126.8, 126.0, 114.3, 114.2, 113.8, 113.5, 69.8, 69.5, 55.8, 55.1, 42.8, 37.5, 31.2. Mass (m/z): (M+) 340.28. Anal.calcd. for C20H24N2O3, C, 70.56; H, 7.11; N, 8.23; O, 14.10 found: C, 70.53; H, 7.09; N, 8.20; O, 14.08 %.
General procedure for the synthesis of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q)
To a stirring solution of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime (1 mmol) in N,N-dimethylformamide (DMF) at room temperature, potassium tert-butoxide (0.1 mmol) was added, after stirring for 25 min substituted benzoyl chlorides (0.9 mmol) was added in drop-wise fashion to the reaction mixture and the stirring was continued for further 8 h at room temperature. Progress of the reaction was monitored by TLC using hexane:ethyl acetate (8:2) mixture as mobile phase. Then, the reaction mixture was quenched with brine solution (5 ml). The organics was separated and the aqueous layer was extracted with diethyl ether (2 × 10 ml). The combined organics were dried over anhydrous sodium sulphate and concentrated under reduced pressure. Purification of the piperidinone oxime esters was accomplished by column chromatography over silica gel, eluting with a solvent system of hexane:ethyl acetate (80:20). The fractions containing products 4(a–q) were collected and concentrated under reduced pressure.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-benzoyl oxime (4a)
Yield 87.41 % as brown solid, M.p 229–231 °C. IR (KBr) ν max (cm−1): 3128-2978 (Ar–CH), 1692 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: δ 6.79–8.21 (m, 13H, Ar–H), 3.80 (s, 6H, OCH3), 3.31 (m, 2H, CH), 1.75-1.50 (m, 4H, CH2), 1.72 (s, 3H, N-CH3),. 13C NMR (100 MHz, DMSO- d 6 ) δ ppm: 165.6, 159.8, 158.5, 158.1, 133.5, 132.2, 132.8, 132.6, 131.5, 131.4, 128.9, 127.8, 127.3, 127.6, 125.5, 125.4, 114.8, 114.9, 113.6, 113.5, 68.8, 68.9, 55.8, 55.1, 42.1, 36.5. Mass (m/z): (M+) 444.28. Anal.calcd. for C27H28N2O4, C, 72.95; H, 6.35; N, 6.30; O, 14.40 found: C, 72.55; H, 6.34; N, 6.32; O, 14.41 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-fluorobenzoyl oxime (4b)
Yield 76.0 % as brown solid, M.p 173–175 °C. IR (KBr) ν max (cm−1): 3128-2917 (Ar–CH), 1695 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.19 (m, 12H, Ar–H), 3.84 (s, 6H, OCH3), 3.32 (m, 2H, CH), 1.73-1.47 (m, 4H, CH2), 1.68 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 168.6, 165.7, 159.5, 159.51, 156.5, 131.93, 131.25, 131.26, 128.82, 128.83, 127.5, 126.05, 126.04, 115.6, 115.3 114.5, 114.6, 113.66, 113.65, 69.8, 69.9, 55.5, 55.51, 43.8, 37.4, 32.85. Mass (m/z): (M+) 462.19. Anal.calcd. for C27H27FN2O4, C, 70.11; H, 5.88; F, 4.11; N, 6.06; O, 13.84 found: C, 70.09; H, 5.86; F, 4.14; N, 6.03; O, 13.82 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-bromobenzoyl oxime (4c)
Yield 75.93 % as off-white solid, M.p 215–217 °C. IR (KBr) ν max (cm−1): 3058-2914 (Ar–CH), 1701 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: δ 6.68–8.19 (m, 12H, Ar–H), 1.75-1.50 (m, 4H, CH2), 1.72 (s, 3H, N–CH3), 3.30 (m, 2H, CH), 3.83 (s, 6H, OCH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 165.5, 159.6, 159.4, 156.5, 139.2, 131.8, 131.6, 131.5, 131.1, 128.4, 128.3, 128.1, 126.5, 126.0, 114.3, 114.2, 113.8, 113.5, 69.8, 69.9, 55.2, 55.0, 42.8, 37.5, 31.8. Mass (m/z): (M+) 522.09. Anal.calcd. for C27H27BrN2O4, C, 61.96; H, 5.20; Br, 15.27; N, 5.35; O, 12.23 found C, 61.93; H, 5.18; Br, 15.24; N, 5.32; O, 12.25 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-chlorobenzoyl oxime (4d)
Yield 81.25 % as yellow solid, M.p 227–229 °C. IR (KBr) ν max (cm−1): 3069-2856 (Ar–CH), 1728 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–7.85 (m, 12H, Ar–H), 3.28 (m, 2H, CH), 3.81 (s, 6H, OCH3), 1.78-1.53 (m, 4H, CH2), 1.70 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 164.5 159.4, 158.8, 158.5, 147.4, 147.3, 146.8, 132.3, 131.7, 131.5, 121.7, 121.4, 115.1, 114.9, 112.4, 112.2, 110.5, 110.3, 108.8, 69.6, 69.8, 56.6, 56.7, 37.6, 31.6, 40.8, 30.2. Mass (m/z): (M+) 478.07. Anal.calcd. for C27H27ClN2O4, C, 67.71; H, 5.68; Cl, 7.40; N, 5.85; O, 13.36 found: C, 67.51; H, 5.18; Cl, 7.48; N, 5.25; O, 13.16 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-3-chlorobenzoyl oxime (4e)
Yield 73.50 % as brown solid, M.p 252–254 °C. IR (KBr) ν max (cm−1): 3030-2956 (Ar–CH), 1705 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–7.85 (m, 12H, Ar–H), 3.80 (s, 6H, OCH3), 1.73-1.48 (m, 4H, CH2), 1.75 (s, 3H, N–CH3), 3.31 (m, 2H, CH),. 13C NMR (100 MHz, DMSO- d 6 ) δ ppm: 164.5 159.4, 158.8, 158.5, 147.4, 147.3, 146.8, 132.3, 131.7, 131.5, 121.7, 121.4, 115.1, 114.9, 112.4, 112.2, 110.5, 110.3, 108.8, 69.6, 69.8, 56.6, 56.7, 37.6, 31.6, 40.8, 30.2. Mass (m/z): (M+) 478.19. Anal.calcd. for C27H27ClN2O4, C, 67.71; H, 5.68; Cl, 7.40; N, 5.85; O, 13.36 found: C, 67.54; H, 5.15; Cl, 7.44; N, 5.21; O, 13.16 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-nitrobenzoyl oxime (4f)
Yield 80.88 % as yellow solid, M.p 231–232 °C. IR (KBr) ν max (cm−1): 3028-2956 (Ar–CH), 1715 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–7.85 (m, 10H, Ar–H), 3.85 (s, 6H, OCH3), 3.30 (m, 2H, CH), 1.82-1.50 (m, 4H, CH2), 1.70 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 166.3, 161.3, 158.5, 147.9, 147.7, 145.8, 134.3, 133.4, 132.6, 131.9, 130.8, 130.1, 128.9,128.5, 127.4, 121.2, 120.5, 116.3, 115.8, 111.4,110.5, 67.8, 56.9, 55.8, 40.8, 37.9, 30.9. Mass (m/z): (M+) 489.13. Anal.calcd. for C27H27N4O6 C, 66.25; H, 5.56; N, 8.58; O, 19.61 found: C, 66.28; H, 5.54; N, 8.60; O, 19.60 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-2,4-dinitrobenzoyl oxime (4g)
Yield 86.54 % as yellow solid, M.p 238–240 °C. IR (KBr) ν max (cm−1): 3152-3010 (Ar–CH), 1711 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.94 (m, 9H, Ar–H), 3.79 (s, 6H, OCH3), 3.25 (m, 2H, CH), 1.78-1.54 (m, 4H, CH2), 1.76 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 165.3, 161.3, 154.7, 147.9, 147.7, 145.8, 134.3, 133.4, 132.6, 131.9, 130.8, 130.1, 128.9, 128.5, 127.4, 121.2, 120.5, 116.3, 115.8, 111.4,110.5, 68.6, 67.8, 56.9, 55.8, 38.9, 31.9. Mass (m/z): (M+) 534.08. Anal.calcd. for C27H26N4O8, C, 60.67; H, 4.90; N, 10.48; O, 23.95 found: C, 60.65; H, 4.91; N, 10.40; O, 23.94 %.
2-((2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-ylideneaminooxy)carbonyl)phenyl acetate (4h)
Yield 84.20 % as white solid, M.p 221–223 °C. IR (KBr) ν max (cm−1): 3069-2919 (Ar–CH), 1721 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.18 (m, 10H, Ar–H), 3.83 (s, 6H, OCH3), 3.30 (m, 2H, CH), 1.83-1.54 (m, 4H, CH2), 1.73 (s, 3H, N–CH3), 2.28 (s, 3H, COCH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 170.5, 165.3, 161.3, 159.7, 147.9, 147.7, 145.8, 134.3, 133.4, 132.6, 131.8, 130.5, 130.0, 128.7,128.4, 127.6, 121.0, 120.4, 116.7, 114.8, 111.4,110.5, 68.6, 65.8, 56.1, 55.8, 40.9, 31.5, 19.5. Mass (m/z): (M+) 502.31. Anal.calcd. for C29H30N2O6, C, 69.31; H, 6.02; N, 5.57; O, 19.10 found: C, 69.30; H, 6.04; N, 5.51; O, 19.11 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-2-hydroxybenzoyl oxime (4i)
Yield 79.35 % as brown solid, M.p 215–217 °C. IR (KBr) ν max (cm−1): 3081-2872 (Ar–CH), 1708 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.94 (m, 12H, Ar–H), 5.35 (s, 1H, OH), 3.80 (s, 6H, OCH3), 3.30 (m, 2H, CH), 1.72-1.49 (m, 4H, CH2), 1.68 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 168.2, 165.3, 161.3, 159.7, 147.9, 147.7, 145.8, 134.3, 133.4, 131.5, 130.0, 128.5, 128.4, 127.6, 121.2, 120.6, 116.7, 114.6, 111.4,110.5, 68.6, 65.8, 56.1, 55.0, 40.9, 38.7, 31.5. Mass (m/z): (M+) 460.16. Anal.calcd. for C27H28N2O5, C, 70.42; H, 6.13; N, 6.08; O, 17.37 found: C, 70.42; H, 6.13; N, 6.08; O, 17.37 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-3-hydroxybenzoyl oxime (4j)
Yield 69.56 % as brown solid, M.p 193–195 °C. IR (KBr) ν max (cm−1): 3162-2958 (Ar–CH), 1700 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.94 (m, 12H, Ar–H), 5.30 (s, 1H, OH), 3.81 (s, 6H, OCH3), 3.31 (m, 2H, CH), 1.80-1.50 (m, 4H, CH2), 1.73 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 165.4, 159.3, 147.8, 147.5, 145.0, 134.3, 143.4, 142.6, 141.9, 130.8, 130.1, 130.9,125.5, 121.4, 121.2, 115.5, 115.8, 112.2, 111.4,110.5, 68.6, 67.9, 55.9, 55.8, 40.8, 37.9, 30.9. Mass (m/z): (M+) 506.16. Anal.calcd. for C27H28N2O5, C, 70.42; H, 6.13; N, 6.08; O, 17.37 found: C, 70.41; H, 6.10; N, 6.04; O, 17.17 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-hydroxybenzoyl oxime (4k)
Yield 76.24 % as brown solid, M.p 233–235 °C. IR (KBr) ν max (cm−1): 3079-2952 (Ar–CH), 1713 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.94 (m, 12H, Ar–H), 5.35 (s, 1H, OH), 3.85 (s, 6H, OCH3), 3.33 (m, 2H, CH), 1.79-1.53 (m, 4H, CH2), 1.77 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d6) δ ppm: 168.5,166.2, 162.2, 148.3, 147.3, 147.2, 146.5, 134.0, 133.8, 132.6, 130.9, 130.5, 130.2, 127.0, 121.3, 120.2, 116.3, 115.8, 112.4, 110.5, 68.2, 67.8, 56.0, 55.8, 40.2, 38.1, 30.5. Mass (m/z): (M+) 506.19. Anal.calcd. for C27H28N2O5, C, 70.42; H, 6.13; N, 6.08; O, 17.37 found: C, 70.44; H, 6.11; N, 6.01; O, 17.14 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-3,5-dihydroxybenzoyl oxime (4l)
Yield 75.39 % as brown solid, M.p 261–263 °C. IR (KBr) ν max (cm−1): 3120-2917 (Ar–CH), 1716 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.10 (m, 11H, Ar–H), 5.34 (s, 2H, OH), 3.80 (s, 6H, OCH3), 3.28 (m, 2H, CH), 1.82-1.57 (m, 4H, CH2), 1.72 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 165.4, 159.3, 147.8, 147.5, 145.0, 134.3, 143.4, 142.6, 141.9, 130.8, 130.1, 130.9, 125.5, 121.4, 121.2, 115.5, 115.8, 112.2, 111.4, 110.5, 68.6, 67.9, 55.9, 55.8, 40.8, 37.9, 30.9. Mass (m/z): (M+) 476.28. Anal.calcd. for C27H28N2O6, C, 68.05; H, 5.92; N, 5.88; O, 20.15 found: C, 68.04; H, 5.91; N, 5.85; O, 20.16 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-3,4,5-trihydroxybenzoyl oxime (4m)
Yield 87.17 % as brown solid, M.p 245–247 °C. IR (KBr) ν max (cm−1): 3101-2972 (Ar–CH), 1691 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.94–7.10 (m, 10H, Ar–H), 5.32 (s, 3H, OH), 3.84 (s, 9H, OCH3), 3.30 (m, 2H, CH), 1.71-1.52 (m, 4H, CH2), 1.68 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 165.4, 159.3, 147.8, 147.5, 145.0, 134.3, 143.4, 142.6, 141.9, 130.8, 130.1, 130.9, 125.5, 121.4, 121.2, 115.5, 115.8, 112.2, 111.4, 110.5, 68.6, 67.9, 55.9, 55.8, 40.8, 37.9, 30.9. Mass (m/z): (M+) 492.24. Anal.calcd. for C27H28N2O7 C, 65.84; H, 5.73; N, 5.69; O, 22.74 found: C, 65.81; H, 5.75; N, 5.67; O, 22.72 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-methoxybenzoyl oxime (4n)
Yield 81.34 % as white solid, M.p 212–214 °C. IR (KBr) ν max (cm−1): 3178-2911 (Ar–CH), 1689 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.94–7.10 (m, 12H, Ar–H), 3.81 (s, 9H, OCH3), 3.30 (m, 2H, CH), 1.73-1.50 (m, 4H, CH2), 1.70 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 168.1, 164.7, 160.8, 158.1, 146.0, 147.1, 147.0, 146.9, 133.7, 131.8, 131.7, 122.0, 118.8, 115.7, 114.2, 112.9, 111.7, 106.8, 105.0, 101.8, 69.8, 69.4, 56.1, 56.0, 55.2, 55.4, 40.5, 37.4. Mass (m/z): (M+) 474.20. Anal.calcd. for C28H30N2O5, C, 65.84; H, 5.73; N, 5.69; O, 22.74 found: C, 65.81; H, 5.75; N, 5.67; O, 22.72 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-2,4-dimethoxybenzoyl oxime (4o)
Yield 74.50 % as brown solid, M.p 219–221 °C. IR (KBr) ν max (cm−1): 3205-2950 (Ar–CH), 1711 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.12 (m, 10H, Ar–H), 3.80 (s, 12H, OCH3), 3.30 (m, 2H, CH), 1.72-1.53 (m, 4H, CH2), 1.69 (s, 3H, N–CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 168.5, 164.1, 160.5, 159.1, 146.9, 147.3, 147.4, 147.1, 133.6, 131.5, 131.2, 121.0, 119.8, 115.6, 115.2, 112.5, 111.8, 105.8, 105.0, 101.8, 69.5, 69.4, 56.3, 56.0, 55.7, 55.4, 40.5, 37.4, 31.2. Mass (m/z): (M+) 504.33. Anal.calcd. for C29H32N2O6, C, 69.03; H, 6.39; N, 5.55; O, 19.03 found: C, 69.05; H, 6.34; N, 5.53; O, 19.01 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-3,4,5-trimethoxybenzoyl oxime (4p)
Yield 76.36 % as brown solid, M.p 247–249 °C. IR (KBr) ν max (cm−1): 3058-2977 (Ar–CH), 1702 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–8.13 (m, 10H, Ar–H), 1.77-1.49 (m, 4H, CH2), 1.69 (s, 3H, N–CH3), 3.30 (m, 2H, CH), 3.78 (s, 15H, OCH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm: 168.5, 164.1, 160.5, 159.1, 146.9, 147.3, 147.4, 147.1, 133.6, 131.5, 131.2, 121.0, 119.8, 115.6, 115.2, 112.5, 111.8, 105.8, 105.0, 101.8, 69.5, 69.4, 56.3, 56.1, 56.0, 55.7, 55.4, 40.5, 37.4, 31.2. Mass (m/z): (M+) 534.14. Anal.calcd. for C30H34N2O7, C, 67.40; H, 6.41; N, 5.24; O, 20.95 found: C, 67.36; H, 6.31; N, 5.20; O, 20.98 %.
2,6-Bis(4-methoxyphenyl)-1-methylpiperidin-4-one O-4-methylbenzoyl oxime (4q)
Yield 81.55 % as off-white solid, M.p 216–218 °C. IR (KBr) ν max (cm−1): 3030-2917 (Ar–CH), 1698 (NOCOR). 1H NMR (300 MHz, DMSO-d 6 ) δ ppm: 6.68–7.60 (m, 10H, Ar–H), 7.48 (m, 1H, C6H5CH), 6.31 (d,1H, COCH), 1.75 (s, 3H, N–CH3), 1.69–1.55 (m, 4H, CH2), 3.30 (m, 2H, CH), 3.82 (s, 6H, OCH3), 2.13 (s, 3H, C6H5CH3). 13C NMR (100 MHz, DMSO-d 6 ) δ ppm 171.8, 159.5, 147.9, 147.6, 147.5, 146.8, 146.1, 136.1, 131.6, 130.8, 128.6, 128.5, 127.9, 127.6, 126.9, 121.3, 121.5, 115.5, 114.5, 112.0, 111.5, 69.8, 68.9, 56.5, 56.4, 41.6, 31.5, 21.3. Mass (m/z): (M+) 458.26. Anal.calcd. for C28H30N2O4, C, 73.34; H, 6.59; N, 6.11; O, 13.96 found: C, 73.31; H, 6.55; N, 6.09; O, 13.94 %.
Antioxidant evaluation
2,2′-Diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay (RSA)
The evaluation of antioxidant activity of newly synthesized compounds was done by DPPH radical scavenging assay (Blois, 1958). Internal standard BHA and the synthesized compounds 4(a–q) of different concentrations were prepared in distilled ethanol, 1 mL of each compound solutions having different concentrations (10, 25, 50, 100, 200 and 500 μM) were taken in different test tubes, 4 mL of 0.1 mM ethanol solution of DPPH was added and shaken vigorously. The tubes were then incubated in the dark room at RT for 20 min. A DPPH blank was prepared without compound and ethanol was used for the baseline correction. Changes (decrease) in the absorbance at 517 nm were measured using a UV–visible spectrophotometer and the remaining DPPH was calculated. The percent decrease in the absorbance was recorded for each concentration and percent quenching of DPPH was calculated on the basis of the observed decreased in absorbance of the radical. The radical scavenging activity was expressed as the inhibition percentage and was calculated using the formula:
where Ao is the absorbance of the control (blank, without compound) and A1 is the absorbance of the compound.
2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS•+) radical scavenging assay
The ABTS•+ cation was produced by the reaction between 7 mmol ABTS in H2O and 2.45 mmol potassium persulphate, stored in the dark at room temperature for 12 h (Re et al., 1999). Before usage, the ABTS•+ solution was diluted to get an absorbance of 0.700 ± 0.025 at 734 nm with phosphate buffer (0.1 M, pH 7.4). Then, 1 ml of ABTS•+ solution was added to the compounds 4(a–q) solution in ethanol at different concentrations (10, 25, 50, 100, 200 and 500 μM/ml). After 30 min, the percentage inhibition at 734 nm was calculated for each concentration relative to a blank absorbance (ethanol). The scavenging capability of ABTS•+ radical was calculated using the following equation:
where, Ac is the absorbance of the initial concentration of the ABTS•+ and As is the absorbance of the remaining concentration of ABTS•+ in the presence of the compounds.
Ferric ion-reducing antioxidant power (FRAP) assay
The reducing power of the synthesized compounds was determined according to the method of Oyaizu (Oyaizu, 1986). The compounds having 10 μM were mixed with 2.5 mL of phosphate buffer (0.2 M, pH 6.6) and 2.5 mL of 1 % potassium ferric cyanide and then incubated at 50 °C for 20 min. To this mixture 2.5 mL of 10 % trichloroacetic acid was added and the mixture was centrifuged at 3000 rpm for 20 min. The upper layer (2.5 mL) was mixed with 2.5 mL of deionised water and 0.5 mL of 0.1 % ferric chloride and the absorbance was measured at 700 nm using a spectrophotometer (Shimadzu 160A). Increases of absorbance of the reaction mixture indicate higher reducing power. Mean values from three independent samples were calculated for each compounds.
Cupric ion-reducing antioxidant capacity (CUPRAC) assay
In order to determine the cupric ions (Cu2+)-reducing ability of 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters 4(a–q), the method of Huang was used (Huang and Kuo, 2000). For the assay, 0.25 mL CuCl2 solution (0.01 M), 0.25 mL ethanolic neocuproine solution (7.5 × 10−3 M) and 0.25 mL CH3COONH4 buffer solution (1 M) were added to a test tube, followed by mixing of compounds at 10 μM. Then, total volume was adjusted to 2 mL with distilled water and the solution was mixed well. The tubes were stoppered and kept at room temperature. Absorbance was measured at 450 nm against a reagent blank 30 min later. Increased absorbance of the reaction mixture indicates increased reduction capability.
Antimicrobial studies
Antibacterial studies
The antibacterial activities of newly synthesized compounds 4(a–q) were determined by well plate method in Mueller–Hinton Agar (Arthington-Skaggs et al., 2000). The in vitro antibacterial activity was carried out against 24-h-old cultures of bacterial strains. In this work, E. coli ATCC 25922 (Gram −Ve), Staphylococcus aureus ATCC 25923 (Gram +Ve) and Pseudomonas aeruginosa ATCC 27853 (Gram −Ve) were used to investigate the activity. The test compounds were dissolved in dimethyl sulphoxide (DMSO) at concentration of 1,000 and 500 μg/mL. Twenty millilitres of sterilized agar media was poured into each pre-sterilized Petri dish. Excess of suspension was decanted and plates were dried by placing in an incubator at 37 °C for an hour. About 60 ml of 24-h-old culture suspension was poured and neatly swabbed with the pre-sterilized cotton swabs. Six millimetre diameter wells were then punched carefully using a sterile cork borer and 30 ml of test solutions of different concentrations were added into each labelled well. The plates were incubated for 24 h at 37 °C. The inhibition zone that appeared after 24 h around the well in each plate was measured as zone of inhibition in mm. Experiments were triplicates and standard deviation was calculated.
Antifungal studies
Antifungal studies of newly synthesized compounds 4(a–q) were carried out against Aspergillus flavus MTCC 3306, Candida albicans MTCC 3017 and Chrysosporium keratinophilum MTCC 2827 were determined by well plate (Lowry et al., 1970). Sabourands agar media was prepared by dissolving peptone (10 g), d-glucose (40 g) and agar (20 g) in distilled water (1,000 mL) and adjusting the pH to 5.7. Normal saline was used to make a suspension of spore of fungal strains for lawning. A loopful of particular fungal strain was transferred to 3 mL saline to get a suspension of the corresponding species. Twenty millilitres of agar media was poured into each Petri dish. Excess of suspension was decanted and plates were dried by placing in incubator at 37 °C for 1 h. Using sterile cork borer punched carefully, wells were made on these seeded agar plates and different concentrations of the test compounds in DMSO were added into each labelled well. A control was also prepared for the plates in the same way using solvent DMSO. The Petri dishes were prepared in triplicate and maintained at 25 °C for 72 h. Antifungal activity was determined by measuring the diameter of inhibition zone. Activity of each compound was compared with fluconazole as the standard. Zones of inhibition were determined for compounds 4(a–q).
Statistical analysis
Assays were carried out in triplicate for 3–5 separate experiments. The amount of compound needed to inhibit DPPH free radical and ABTS•+ cation by 50 % (IC50) was graphically estimated using a linear regression algorithm. The antimicrobial assays were carried out in triplicate and standard deviation was calculated.
References
Arthington-Skaggs A, Motley M, Warnock DW, Morrison CJ (2000) Comparative evaluation of PASCO and national committee for clinical D.W. laboratory standards M27-A broth microdilution methods for antifungal drug susceptibility testing of yeasts. J Clin Microbiol 38:2254–2260
Balasubramanian S, Ramalingan C, Aridoss G, Kabilan S (2005) Synthesis and study of antibacterial and antifungal activities of novel 8-methyl-7,9-diaryl-1,2,4,8-tetraazaspiro[4.5]decan-3-thiones. Eur J Med Chem 40:694–700
Blois MS (1958) Antioxidant determinations by the use of a stable free radical. Nature 181:1199
Gulçin I (2006) Antioxidant and antiradical activities of l-carnitine. Life Sci 78:803–811
Heim KE, Tagliaferro AR, Bobilya DJ (2002) Flavanoid antioxidants: chemistry, metabolism and structure- activity relationships. J Nut Biochem 13:572–584
Huang S, Kuo JC (2000) Concentrations and antioxidative activity of anserine and carnosine in poultry meat extracts treated with demineralization and papain. Proc Natl Sci Counc ROC 24:193–201
Khan MW, Alam MJ, Rashid MA, Chowdhury R (2005) A new structural alternative in benzo[b]furans for antimicrobial activity. Bioorg Med Chem 13:4796–4805
Kumaran A, Karunakaran RJ (2006) Antioxidant and free radical scavenging activity of an aqueous extract of Coleus aromaticus. Food Chem 97:109–114
Lowry DJM, Jaqua MJ, Selepak ST (1970) Detailed Methodology and implementation of automated dilution microtechnique for semi automated susceptibility testing. J Appl Microbiol 20:46–53
Naik Nagaraja, Vijay Kumar H, Harini ST (2011) Synthesis and antioxidant evaluation of novel indole-3-acetic acid analogues. Eur J Chem 2:337–341
Noller CR, Baliah V (1948) The preparation of some piperidine derivatives by the mannich reaction. J Am Chem Soc 70:3853–3855
Oyaizu M (1986) Studies on product of browning reaction prepared from glucose amine. Jpn J Nutr 44:307–315
Pfeltz RF, Wilkinson BJ (2004) The escalating challenge of vancomycin resistance in Staphylococcus aureus. Curr Drug Targets Infect Disord 4:273–294
Rangaswamy J, Vijay Kumar H, Harini ST, Naik Nagaraja (2012) Synthesis of benzofuran based 1,3,5-substituted pyrazole derivatives: as a new class of potent antioxidants and antimicrobials-A novel accost to amend biocompatibility. Bioorg Med Chem Lett 22:4773–4777
Ravindra RK, Sudha SB, Ravindranath A, Imthiyaz AK (2009) Synthesis and evaluation of benzophenone oximes derivatized with sydnone as inhibitors of secretory phospholipase A2 with anti-inflammatory activity. Chem Pharm Bull 57:16–21
Re R, Pellergini N, Proteggenete A, Pannala A, Yang M, Rice Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radical Biol Med 26:1231–1237
Rossello A, Bertini S, Lapucci A, Macchia M, Martinelli A, Rapposelli S, Herreros E, Macchia B (2002) Synthesis, antifungal activity, and molecular modeling studies of new inverted oxime ethers of oxiconazole. J Med Chem 45:4903–4912
Vijay Kumar H, Naik Nagaraja (2010) Synthesis and antioxidant properties of some novel 5H-dibenz[b, f]azepinederivatives in different in vitro model systems. Eur J Med Chem 45:2–10
Vijay Kumar H, Kishor Kumar C, Naik Nagaraja (2011) Synthesis of novel 3-chloro-1-(5H-dibenz[b, f] azepine-5yl)propan-1-one derivatives with antioxidant activity. Med Chem Res 20:101–108
Acknowledgments
The authors are thankful to NMR Research Center, Indian Institute of Science, Bangalore for providing spectral data.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Harini, S.T., Kumar, H.V., Peethambar, S.K. et al. Novel 2,6-bis(4-methoxyphenyl)-1-methylpiperidin-4-one oxime esters: synthesis and a new insight into their antioxidant and antimicrobial potential. Med Chem Res 23, 1887–1898 (2014). https://doi.org/10.1007/s00044-013-0793-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0793-z