
Abstract
Dyrk family enzymes are essential components of important signaling casades in the pathophysiology of cancer and Alzheimer’s disease. Especially, Dyrk2 biological expression levels regulate key signaling processes in these diseases. In the present work, a pharmacophore-based 3D-QSAR model was generated for a series of leucettine analogs possessing Dyrk2 inhibitory activity. Developed pharmacophore model contains four hydrogen bond acceptors (A) and one hydrophobic aromatic ring (R). These are crucial molecular fingerprints which predict binding efficacy of high affinity and low affinity ligands to the Dyrk2 enzyme. These pharmacophoric features point toward key structural requirements of leucettines for potent Dyrk2 inhibition. Furthermore, a biological correlation between pharmacophore hypothesis-based 3D-QSAR variables and functional fingerprints of leucettines responsible for the receptor binding was observed. Alignment of the developed model with Dyrk2 crystal structure indicated importance of A3 and A4 H-bond accetor sites, which are involved in the important interactions with Leu231A and Lys178A residues of the active site. Excellent statistical results of QSAR model such as good correlation coefficient (r 2 > 0.95), higher F value (F > 106), and excellent predictive power (Q 2 > 0.7) with low standard deviation (SD < 0.2) strongly suggest that the developed model is good for the future prediction of Dyrk2 inhibitory activity of new leucettine analogs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dual-specificity tyrosine-phosphorylation-regulated kinase (Dyrk) belongs to an evolutionarily conserved family of proteins known as Dyrk’s involved in functions generally related with cellular growth and development (Laguna et al., 2008). These kinases are over-expressed in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Pick disease (Park et al., 2009; Ferrer et al., 2005). Dyrk’s were well known as dual-specificity kinases because of their capacity to catalyze self-activation process through auto-phosphorylation of a single tyrosine residue in their activation loop (for example, Tyr321 in Dyrk1A) as well as the phosphorylation of serine and threonine residues in exogenous protein substrates (Lochhead et al., 2005). Dyrk2 plays important role during development of human adult testis. Dyrk2 is also over-expressed in adenocarcinomas of the esophagus and lung (Miller et al., 2003). Interestingly, in recurrent non-small cell lung cancer patients, high Dyrk2 expression is positive indicator of chemotherapy response (Yamashita et al., 2009). The precise role of Dyrk2 in cancer, however, needs further systematic experimentation. Dyrk2 was also implicated in developmental and cellular processes such as cellular proliferation, cytokinesis, regulation of protein and glycogen synthesis, and cellular differentiation (Yoshida, 2008). Dyrk2 also regulates p53 gene to induce apoptosis pathways in response to DNA damage or mutation (Taira et al., 2007). More recently Dyrk2’s role as a scaffold for an E3 ubiquitin ligase complex has been revealed. The scaffolding function is independent of its kinase activity, but the subsequent phosphorylation and degradation of the substrate katanin p60 is dependent on the catalytic activity of Dyrk2 (Maddika and Chen, 2009). Over the ensuing years, studies established the central role of Dyrk2 in regulation of body growth and normal brain development by phosphorylating numerous protein substrates including other kinases, as well as splicing, transcription, and translation factors. However, history of drug-discovery aiming Dyrk2 is very small. Extensive drug discovery efforts in last 3 years led to the development of novel Dyrk-2 inhibitors like indirubin-3′-monooxime derivative 1 (Filippakopoulos et al., 2010), harmine 2 (Cuny et al., 2012), acridine 3 (Cuny et al., 2010), and leucettine 4 (Debdab et al., 2011) (Fig. 1).

Chemical structures of known Dyrk2 enzyme inhibitors 1–4
In recent years, structure-based drug design and molecular docking are widely used tools in the drug discovery and development process. However, pharmacophore-based drug design has proved to be more efficient technique for identification of potential new drug-like candidates, as this method provides the information about key structural features in the form of an active pharmacophore which are essential for biological activity. In addition, the generated pharmacophore model could be employed as a novel searching tool for chemical databases to find potential lead candidates. As a part of our on-going drug discovery program based on marine scaffolds for cancer and Alzheimer (Bharate et al., 2012a, b, c; 2013a, b), in the present study, pharmacophore-based 3D-QSAR was carried out on a set of Dyrk2 inhibitory leucettines using pharmacophore alignment and scoring engine (PHASE) module of Schrodinger program to understand the relationship between biological activity and 3-dimensional molecular fingerprints of leucettines (Debdab et al., 2011).
Methods
Dataset
The biological data used in the present study was imported from the published literature on Dyrk2 inhibitory leucettines L0–L41 (Debdab et al., 2011) (Table 1). The Dyrk2 inhibitory activity (IC50) values were converted to molar units and further to the negative logarithmic unit [\( {\text{pIC}}_{50} = - \log \left( {{\text{IC}}_{50} } \right) \)]. Compounds lacking well defined activity data were removed from the study. The data set of 36 compounds was then divided randomly into training and test set, respectively in such a way that both sets consisted of highly active, medium active and least active compounds. Training and test sets consisted of 26 and 10 compounds, respectively.

Pharmacophore-based QSAR study
A pharmacophore-based 3D-QSAR study was carried out using PHASE version 3.0.110 implemented in the Maestro 8.5.111 modeling package (Schrodinger, Inc., LLC, New York, USA) installed on a Intel Xenon processor-based HP xw6600 work station with Linux operating software. Structures of molecules were sketched, cleaned, prepared (using ligprep), minimized (using macromodel), and conformers were generated (using confogen) in Maestro under default settings. The active and inactive (−log IC50) threshold of ≥7.4 and <6.8, were applied to the training set and test set, respectively, which sorted 13 actives and 9 inactives. Redundancy was avoided in data set in terms of structural cluster or bioactivity (Dixon et al., 2006a, b; Bharate et al., 2012c). The important steps of common pharmacophore hypothesis development and 3D-QSAR model development using PHASE module were carried out in a default sequential order. Atom-based QSAR models were generated on the principle of identification and alignment of pharmacophoric features of the chemical structures, for all 16 generated hypotheses using the 26-member training set and a grid spacing of 1.0°A. The grid divides space into uniformly sized cubes (range 0.5–2.0°A), which are occupied by the atoms or pharmacophore sites that define each molecule. QSAR models containing one to four PLS factors were generated. Best 3D-QSAR model was selected based on the correlation coefficient values in training set molecules, which was further validated by predicting activities of 10 test set molecules. Three external test set predictors, namely Q 2, Pearson r, and RMSE were used to validate the developed model (Bharate et al., 2013b).
Structure-based validation of 3D-pharmacophore model
To validate the pharmacophore model using X-ray crystal structure-guided binding pattarn, the X-ray crystal structure of leucettine L41 bound to Dyrk2 was retrieved from RCSB (PDB: 4AZF), and minimized in Schrodinger protein preparation wizard and was compared to the developed pharmacophore hypothesis (Fig. 3).
Results and discussion
Pharmacophore-based 3D-QSAR study
A 3D-pharmacophore model for the set of 36 leucettines was developed using PHASE module of Schrodinger molecular modeling package. This 3D-QSAR approach involves the generation of a common pharmacophore hypothesis built feature definitions and creating sites to generate 28 variant combinations among given set of leucettines. These variants upon perceiving common pharmacophore generated 16 hypotheses. All hypotheses were scored for actives, inactives, volume, selectivity, and energy terms. Then the selection of best hypotheses was primarily dependent on best post-hoc survival score, which is a combination of active and inactive survival scores. Scores of different scoring parameters for best hypothesis AAAAR.29 are given in Table 2. Higher scoring hypothesis was further subjected to 3 and 4 factor PLS QSAR model development based on the alignment of the pharmacophoric features to the chemical fingerprints of leucettines. The validity of each model was predicted from calculated external (test set) correlation coefficient (Q 2). Among the two best models common pharmacophore hypothesis AAAAR.29 was considered optimal due to the superior correlation coefficient, cross correlation coefficient, stability, and F value of the hypothesis over AAAPR.23, and therefore selected for the 3D-QSAR model development. The stability of the model reflects the changes in the training set composition. The maximum value for stability can be 1; and the acceptable stability is >0.3. The stability statistics is often used to compare models generated through different hypotheses.
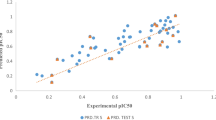
The robustness and reliability of the selected optimal model is justified by the fact that all statistical measures are significant. The model expresses 99 % variance exhibited by leucettines L0–L41, which is near to one and signifies a close agreement of fitting points on the regression line for the observed experimental and PHASE predicted activity, as depicted in Fig. 2.

Correlation graph between experimental and predicted Dyrk2 inhibitory activity of leucettines using pharmacophore-based QSAR model. a Training set. b Test set
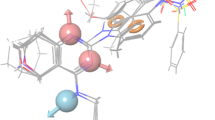
As shown in Table 3, the large value of F (F = 106.7) indicates a statistically significant regression model, which is also supported by the small value of statistical significance (P), an indication of a high degree of confidence. Further small value of standard deviation (SD = 0.16) of the regression and root-mean-square error (RMSE = 0.34) and higher value of the QSAR model stability (stability = 0.378) makes an obvious implication that the data used for model generation are best for the QSAR analysis. The Q 2 is more reliable and robust statistical parameter than r 2 because it is obtained by same set of test set molecules that were not involved in the model development. Therefore, validity of the developed model is expressed by higher correlation coefficient (Q 2 = 0.71) value, which was determined by multivariate method (PHASE User Manual, version 3.1). Figure 3a–c illustrates the comparative alignment of crystal structure of Dyrk2 (containing leucettine L41; Fig. 3a, c) (Tahtouh et al., 2012) with the pharmacophore model (Fig. 3b), which further validates our developed model. Upon visual overlay comparison of the pharmacophore model to the X-ray crystal structure of the leucettine series ligand L41 bound Dyrk2, it was observed that the H-bond acceptor feature of pharmacophore model, particularly the site A3 interacts with the NH of Leu-231A residue and site A4 interacts with the protonated amino group (NH3+) of Lys178A flanked side chain by strong H-bonding. The other two acceptor sites A1 and A2 were not clearly interacting with the receptor, and probably may not necessary; however, these sites were part of several active as well as inactive molecules and therefore these sites provides the opportunity to optimize the lead by lead-trimming process. Thus, the presence of multiple H-bond acceptor sites due to functionalities such as 1,3-dioxole, hydroxy, or methoxy (sites A2 and A3), lactam carbonyl (Site A4), and NH group of imidazolin-4-one scaffold (site A1) were identified to be important for Dyrk2 inhibition.

Comparative alignment of the developed pharmacophore model (AAAAR.29) with the Dyrk2 X-ray crystal structure (4AZF). a The structure-based interactions of the co-crystal ligand L41 with Dyrk2 enzyme; b developed pharmacophore PHASE hypothesis (AAAAR.29) superimposed on ligand L41 (in this figure LHS indicates left hand side; and RHS as right hand side of the pharmacophore); c molecular representation of the interactions of co-crystal ligand L41 with Dyrk2 active site residues
Similarly combined 3D-QSAR can be visualized from the Fig. 4. The blue/green and purple cubes in 3D-plots of the 3D-pharmacophore regions refer to ligand regions in which the specific feature is important for better activity, whereas the red cubes demonstrates that particular structural feature or functional group which is not essential for the activity or likely a reason for decreased activity. From 3D-QSAR model analysis (Fig. 4), it is observed that the plethora of the orange cubes near and around H-bond acceptor sites (A1–A4) positively contribute to Dyrk2 inhibition (Fig. 4b). The second significant factor which contributes to the Dyrk2 enzyme inhibition is the hydrophobic character, as visualized in Fig. 4c showing presence of large number of green cubes due to the presence of number of hydrophobic saturated alkyl (compounds L2, L3 and L4), cyclic alkyl (compounds L9 and L35), and aromatic phenyl rings (compounds L39 and L41). In the present study, it was observed that the 1,3-benzodioxole ring is an important feature of the pharmacophore hypothesis, which is also in agreement with the previously published SAR (Debdab et al., 2011). The 1,3-benzodioxole ring or its mimicry is crucial and optimal to interact with the Dyrk2 hinge region hydrophobic residues leucine residues (Leu230, Leu231, and Leu282) and Isoleucine residues (Ile155, Ile294, and Ile212). Replacement of the 1,3-benzodioxole ring causes complete loss of activity. For example, the most active ligand structure L41 carries 1,3-benzodioxole ring which fits complementary into the enzyme pocket and exhibits favorable interactions with Val222, Met240, Leu282A, Ala176A, and Ile294 residues. There exist few other hydrophobic interactions on the LHS (Fig. 3b) of the pharmacophore, which is not part of the pharmacophore hypothesis (e.g., electron-rich aromatic rings in case of compounds L36–L41). Although analogs L36–L41 possessing such interactions exhibited higher biological activity in the series, but in the structure-based interaction map (Fig. 3a, c), it is clear that this hydrophobic portion lies away from the active site residues, exhibiting minimal interactions.

3D-Pharmacophore regions around developed QSAR model with most active ligand. a Combined effect (blue cubes are favored areas contributing for increase in activity, whereas red cubes indicate unfavorable areas contributing for decrease in activity); b hydrogen-bond donor (orange cubes are favored areas contributing for increase in activity, whereas red cubes indicate unfavorable areas contributing for decrease in activity); c hydrophobic region (green cubes are favored areas contributing for increase in activity, whereas red cubes indicate unfavorable areas contributing for decrease in activity); d electron-withdrawing effects (blue cubes are favored areas contributing for increase in activity, whereas red cubes indicate unfavorable areas contributing for decrease in activity) (Color figure online)
Conclusion
A atom-based pharmacophoric 3D-QSAR model was successfully developed for a set of Dyrk2 inhibitory leucettine class of compounds. The derived model exhibits good fitness with the experimental data, with a maximum correlation coefficient of 0.95 and having hydrophobic and H-bond acceptor features as crucial parameters for enzyme inhibition. Furthermore, external validation coefficient also known as Q 2 > 0.7, reflects good predictive power of the model. Robustness of the model was indicated by the higher degree of closeness between experimental and predicted activity. Developed pharmacophore model was efficiently compared to the reference molecule L41 crystal structure bound to Dyrk2 and importance of pharmacophoric features in hypothesis discussed in contrast to crystal structure, which validates the hypothesis. Therefore, the developed 3D-QSAR model could be employed to predict Dyrk2 inhibitory activity of natural and synthetic leucettines. This developed model will be useful to identify new potent Dyrk2 inhibitors by virtual screening of compound libraries. This will further add in designing better molecules with enhanced Dyrk2 inhibitory activity.
Abbreviations
- 3D-QSAR:
-
3-Dimensional quantitative structure–activity relationship
- Dyrk1A:
-
Dual-specificity tyrosine-phosphorylated and regulated kinase 1a
- Dyrk2:
-
Dual-specificity tyrosine-phosphorylated and regulated kinase 2
- PLS:
-
Partial least square
- RMSD:
-
Relative mean square deviation
- RMSE:
-
Root-mean-square error
- SD:
-
Standard deviation
- r 2 :
-
Correlation coefficient
- Q 2 :
-
Correlation coefficient for test set
References
Bharate SB, Yadav RR, Battula S, Vishwakarma RA (2012a) Meridianins: marine-derived potent kinase inhibitors. Mini Rev Med Chem 12:618–631
Bharate SB, Manda S, Mupparapu N, Battini N, Vishwakarma RA (2012b) Chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini Rev Med Chem 12:650–664
Bharate SB, Manda S, Joshi P, Singh B, Vishwakarma RA (2012c) Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. Med Chem Commun 3:1098–1103
Bharate SB, Yadav RR, Vishwakarma RA (2013a) QSAR and pharmacophore study of Dyrk1A inhibitory meridianin analogs as potential agents for treatment of neurodegenerative diseases. Med Chem 9:152–161
Bharate SB, Singh B, Bharate JB, Jain SK, Meena S, Vishwakarma RA (2013b) QSAR and pharmacophore modeling of N-acetyl-2-aminobenzothiazole class of phosphoinositide-3-kinase-alpha inhibitors. Med Chem Res 22:890–899
Cuny GD, Robin M, Ulyanova NP, Patnaik D, Pique V, Casano G, Liu J-F, Lin X, Xian J, Glicksman MA, Stein RL, Higgins JMG (2010) Structure–activity relationship study of acridine analogs as haspin and DYRK2 kinase inhibitors. Bioorg Med Chem Lett 20:3491–3494
Cuny GD, Ulyanova NP, Patnaik D, Liu J-F, Lin X, Auerbach K, Ray SS, Xian J, Glicksman MA, Stein RL, Higgins JM (2012) Structure–activity relationship study of beta-carboline derivatives as haspin kinase inhibitors. Bioorg Med Chem Lett 22:2015–2019
Debdab M, Carreaux F, Renault S, Soundararajan M, Fedorov O, Filippakopoulos P, Lozach O, Babault L, Tahtouh T, Baratte B, Ogawa Y, Hagiwara M, Eisenreich A, Rauch U, Knapp S, Meijer L, Bazureau JP (2011) Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: modulation of alternative pre-RNA splicing. J Med Chem 54:4172–4186
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA (2006a) PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 20:647–671
Dixon SL, Smondyrev AM, Rao SN (2006b) PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug Des 67:370–372
Ferrer I, Barrachina M, Puig B, Lagran MMD, Marti E, Avila J, Dierssen M (2005) Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol Dis 20:392–400
Filippakopoulos P, Myrianthopoulos V, Kritsanida M, Magiatis P, Skaltsounis AL, Krojer T, Gileadi O, Hapka E, Fedorov O, Berridge G, Wang J, Shrestha L, Vollmar M, Von Delft F, Arrowsmith CH, Edwards A, Weigelt J, Bountra C, Mikros E, Knapp S (2010) Crystal structure of dual-specificity tyrosine phosphorylation regulated kinase 2 (Dyrk2) in complex with an indirubin ligand. RCSB. doi:10.2210/pdb3kvw/pdb
Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernández E, Fotaki V, Delabar JM, de la Luna S, de la Villa P, Arbonés ML (2008) The protein kinase DYRK1A regulates caspase-9-mediated apotposis during retina development. Dev Cell 15:841–853
Lochhead PA, Sibbet G, Morrice N, Cleghon V (2005) Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 121:925–936
Maddika S, Chen J (2009) Protein kinase Dyrk2 is a scaffold that facilitates assembly of an E3 ligase. Nat Cell Biol 11:409–419
Miller CT, Aggarwal S, Lin TK, Dagenais SL, Contreras JI, Orringer MB, Glover TW, Beer DG, Lin L (2003) Amplification and overexpression of the dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 2 (DYRK2) gene in esophageal and lung adenocarcinomas. Cancer Res 63:4136–4143
Park J, Song WJ, Chung KC (2009) Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol Life Sci 66:3235–3240
Tahtouh T, Elkins JM, Filippakopoulos P, Soundararajan M, Burgy G, Durieu E, Cochet C, Schmid RS, Lo DC, Delhommel F, Oberholzer AE, Pearl LH, Carreaux F, Bazureau JP, Knapp S, Meijer L (2012) Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J Med Chem 55:9312–9330
Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K (2007) Dyrk2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 25:725–738
Yamashita S, Chujo M, Moroga T, Anami K, Tokuishi K, Miyawaki M, Kawano Y, Takeno S, Yamamoto S, Kawahara K (2009) Dyrk2 expression may be a predictive marker for chemotherapy in non-small cell lung cancer. Anticancer Res 29:2753–2757
Yoshida K (2008) Role for Dyrk family kinases on regulation of apoptosis. Biochem Pharmacol 76:1389–1394
Acknowledgments
Authors are thankful to Schrodinger Inc. for providing license Schrodinger molecular modeling software.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bahl, A., Joshi, P., Bharate, S.B. et al. Pharmacophore modeling and 3D-QSAR studies of leucettines as potent Dyrk2 inhibitors. Med Chem Res 23, 1925–1933 (2014). https://doi.org/10.1007/s00044-013-0767-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0767-1