
Abstract
Anaplastic lymphoma kinase (ALK) is involved in many signaling mechanisms that lead to cell-cycle progression; overexpression of ALK has been found in many types of cancers. ALK is a recognized target for the development of small-molecule inhibitors for the treatment of cancer. In this study, a diverse set of 71 ALK inhibitors were aligned by three different methods (pharmacophore, docking-based, and rigid body (Distill) alignment) for the development of comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) models. The best 3D QSAR models were obtained, which used rigid body (Distill) alignment of test and training set molecules. CoMFA and CoMSIA models were found statistically significant with leave-one-out correlation coefficients (q 2) of 0.816 and 0.838, respectively; cross-validated coefficients (\(r_{\text{cv}}^{2}\)) of 0.812 and 0.837, respectively; and conventional coefficients (r 2) of 0.969 and 0.966, respectively. QSAR models were validated by a test set of 14 compounds giving satisfactory prediction of correlation coefficients (\(r_{\text{pred}}^{2}\)) of 0.910 and 0.904 for CoMFA and CoMSIA models, respectively. Based on the generated contour maps, we have designed 10 novel ALK inhibitors and predicted their activities. Finally, molecular docking study was performed for designed molecules. The designed compounds showed good potential to be used as ALK inhibitors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Anaplastic lymphoma kinase (ALK) is an enzyme, which belongs to insulin receptor superfamily. It is a tyrosine kinase-type receptor and formed by instructions of ALK gene (Cheng and Ott, 2010). ALK is 220-kDa receptor tyrosine kinase (RTK) identified in human as well as in drosophila and mouse, and discovered as fusion protein “nucleophosphamin-anaplastic lymphoma kinase” derived from chromosomal translocation. Owing to chromosomal translocation of ALK gene, the fusion ALK enzyme is synthesized, which has kinase activity and make its contribution to oncogenesis (Pulford et al., 2004). Structure of ALK is shared with three domains of RTKs: (1) intracellular, (2) transmembrane, and (3) extracellular. Level of ALK is seen decreased in healthy adult tissue compared with children (Zificsak et al., 2011). Physiological role of ALK is not well defined, but it is suggested that it contributes to the development of brain as well as to the proliferation of nerve cells and also affects the dopaminergic signaling (Moh et al., 2011). Translocation of ALK causes oncogenesis, which plays an important role as an oncogene in the development of anaplastic large cell lymphomas (ALCL) known as non-Hodgkin’s lymphoma (NHL), non-small cell lung cancer, and in inflammatory myofibroblastic tumors. Full-length ALK is usually expressed in various types of cancers such as breast cancer, neuroblastoma, Ewing sarcoma, retinoblastoma, and melanoma. The pathogenic role of ALK receptor has not been clearly understood but overexpression of ALK is seen in many cancers. Furthermore, ALK fuses with other proteins like nucleophosphamin (NPM), ALK lymphoma oligomerization on chromosome 17 (ALO17), TRK-fused gene (TFG), moesin (MSN), etc. and makes ALK-fusion proteins, which are also responsible for tumor growth (Palmer et al., 2009). Nucleophosmin-anaplastic lymphoma kinase protein (NPM-ALK) is a mutant form of ALK and seen in a majority of the cases of ALCL. In these cases, it is believed that ALK may give bases (purines and pyrimidines) in tumorigenesis via many signaling mechanisms that will lead to cell-cycle progression, durability, cell movement, and its shaping (Tripathy et al., 2011). Inhibition of ALK may reduce the growth of ALK-positive lymphoma cells. ALK is nowadays getting more attention as a therapeutic target for the treatment of cancer. Crizotinib is an ALK inhibitor, which is under clinical trials for its effectiveness and safety in anaplastic large cell lymphoma as well as in neuroblastoma (Shaw et al., 2011). More recently, ARIAD laboratories have synthesized a new compound named “AP-26133” which has better efficacy as an ALK inhibitor. Novel ALK inhibitors are necessary for improved kinase selectivity profile and to fight against the resistance mechanisms (Allwein et al., 2012). Structural study as well as characterization of ALK domain can help in identifying and the development of ALK inhibitors (Tartari et al., 2011). The present study was undertaken to explore key structural requirements of different chemical scaffolds as ALK inhibitors by utilizing comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) with three different (pharmacophore, docking-based, and rigid body) alignment methods. CoMFA is a versatile and powerful tool in rational drug design (Vyas and Ghate 2012a, b). CoMFA calculates steric and electrostatic fields surrounding the molecules and correlating the differences in these fields to inhibitory activity. In CoMSIA, similarity indices are calculated at regularly placed grid points for aligned molecules. CoMSIA calculates other molecular descriptors like hydrophobic fields, hydrogen-bond donor (HBD) and hydrogen-bond acceptor (HBA) fields (Klebe and Abraham, 1999). Contour maps of CoMFA and CoMSIA fields describe the “favorable” or “unfavorable” contributions of a region of interest surrounding the ligands to the target property (Vyas et al., 2013). Based on generated 3D QSAR models, we have designed 10 novel ALK inhibitors and predicted their activities. Docking study was performed for designed molecules to explore the binding mode of inhibitors in the active site of ALK. The aim was to explore combined use of docking study with 3D QSAR to design new compounds with improved potency against ALK.
Materials and methods
Dataset
A dataset of 71 compounds (ALK inhibitors) consisting of 2-acyliminobenzimidazoles and piperidine carboxamides were collected from the literature (Lewis et al., 2012; Bryan et al., 2012). Chemical structures and activity data are shown in Table 1. ALK enzyme inhibitory IC50 (μM) values were converted to pIC50 and subsequently used as dependent variable for 3D QSAR study (Table 1).

Selection of training and test set
The total set of 71 inhibitors were divided into training set (57 compounds) for generating QSAR model and a test set (14 compounds) for validating the quality of the models. An ideal division of a training and test set will lead to dataset with resemblance of all the compounds of a test set in multidimensional descriptor space to a training set and resemblance of all representative compounds of a training set to a test set. Selection of a training and test set molecules was done by considering the fact that test set molecules represent a range of inhibitory activities similar to that of a training set. Thus, a test set was the true representative of the training set. This was achieved by arbitrarily setting aside 14 compounds as a test set with a regularly distributed biological data.
Computational details
QSAR modeling, calculations, and visualizations for CoMFA and CoMSIA analyses were performed using SYBYL X 1.3 software from Tripos Inc., St. Louis, MO, USA. Compound 66 was selected as template molecule because of its high inhibitory activity. The structures of all other compounds were constructed from the template molecule using “SKETCH” function in SYBYL, partial atomic charges were calculated by the Gasteiger-Huckel method. and energy minimization was performed using Tripos force field (Gasteiger and Marsili, 1980) with a distance-dependent dielectric and Powell conjugate gradient algorithm. The minimum gradient difference of 0.05 kcal/mol Å was set as a convergence criterion (Clark et al., 1989).
Alignment
The most crucial input for CoMFA and CoMSIA analyses is the alignment of the molecules that requires 3D structures of the analyzed molecules to be aligned according to a suitable conformational template. 3D QSAR models are often sensitive to a particular alignment scheme. Generally, the results of CoMFA and CoMSIA analyses depend upon the alignment method of molecules. The comparisons of different alignment techniques were reported (Vyas et al., 2012; Roy et al., 2008) in the literature for CoMFA and CoMSIA analyses. Therefore, three different alignment techniques were compared carefully in this work, to find the most efficient one for the present system.
Distill (align 1)
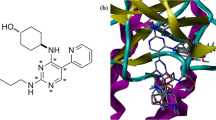
Rigid body alignment of molecules in a Mol2 database was performed using maximum common substructure (MCS) defined by Distill. Compound 66 was used as a template and all other compounds were aligned on the basis of the common structure. MCS represents a common core of all the structures used for the alignment. Distill generates MCS on the basic of a group of connected atoms common to a set of structures used for the alignment. A rigid alignment attempts to align molecules in a database to a template molecule on a common backbone or core (MCS). This core will typically have been produced by Distill. The minimum atom count in MCS fragments can be as small as 3. For the alignment of the molecules using Distill, first the core is looked for in all the molecule, if core may be found more than once, or there may be more than one mapping of the core atoms to the molecule atoms. In this case a single mapping is chosen. Finally, all the molecule are fit to the template using the best mapping of the core to the molecules and the database is updated with the molecule’s new orientation. Alignment of training and test set compounds using Distill module is shown in Fig. 1a.

Alignment of training and test set compounds a rigid body alignment using Distill, b docking-based, c pharmacophore-based
Docking-based alignment (align 2)
The active conformation of all the compounds was achieved by molecular docking study. Docking experiments were performed using Surflex–Dock module of SYBYL X 1.3. The X-ray crystallographic structure of the ALK (PDB ID: 3L9P) (Lee, 2010) solved at 1.8 Å resolution was retrieved from the PDB databank. Each inhibitor was docked into the ALK using the flexible docking module implemented in Surflex–Dock (Jain, 1996). Active conformation was selected from the binding orientation in the active site of ALK and evaluated by consideration of binding free energy scores (Surflex–Dock score). Since for all compounds the best-docked geometries (active conformation) were in agreement with the crystallographic data of the ALK/glycerol complex (and thus already aligned) (Fig. 1b), they were directly submitted to QSAR study.
Pharmacophore-based alignment (align 3)
All the compounds (training and test sets) were selected to generate pharmacophore model using DISCOtech. All the compounds were aligned on some common features depending upon the position rotation and conformation. Generated pharmacophore model contains 1 donor site, 1 acceptor atom and 2 hydrophobic regions, which was then used for the alignment of the compounds in QSAR study (Fig. 1c).
These three different alignment methods yielded very good statistical results, but rigid body alignment of molecules by Distill (align 1) gave us the best results (Table 2) with a significant statistical value of q 2 and \(r_{\text{cv}}^{2}\) as compared to aligns 2 and 3, so further QSAR study was carried out using align 1 (Distill).
CoMFA model
In CoMFA analysis, steric and electrostatic potential energies were calculated using Tripos force field with a probe atom having a van der Waals radius of sp 3-hybridized carbon and a +1 charge to generate steric (Lennard-Jones 6–12 potential) field energies and electrostatic (Coulombic potential) fields with a distance-dependent dielectric at each lattice point. A lattice with 2 Å grid spacing extending at least 4 Å in each direction beyond the aligned molecules was used. The steric and electrostatic energy values were truncated at 30.0 kcal/mol. In order to reduce noise and improve efficiency, column filtering (minimum sigma) was set to 2.0 kcal/mol.
CoMSIA model
The CoMSIA similarity index descriptors were calculated using a dummy sp 3-hybridized carbon with +1 charge. The same lattice box used in CoMFA calculations was also applied to CoMSIA calculations with a grid spacing of 2 Å with a radius of 1.0 Å as implemented in SYBYL. Similarity indices were calculated between a probe and each atom of the molecules based on a Gaussian distance function. CoMSIA not only computes steric and electrostatic fields, but also calculates hydrophobic, HBD, and hydrogen-bond acceptor (HBA) fields. For the distance dependence between the probe atom and the molecule atoms, a Gaussian function was used. Because of the different shape of the Gaussian function, the similarity indices calculated at all grid points, both inside and outside the molecular surface.
Partial least square (PLS) analysis
CoMFA and CoMSIA models were derived using PLS regression analysis. Calculated CoMFA and CoMSIA descriptors were used as independent variables and ALK inhibitory activity (pIC50) as the dependent variable in the PLS analysis. PLS analysis was performed using the leave-one-out (LOO) and cross-validation (CV) methods for 3D QSAR analysis, which gives q 2 and \(r_{\text{cv}}^{2}\), respectively as a statistical index of predictive power. The non-cross-validated models were assessed by the conventional correlation coefficient (r 2), standard error of estimation (SEE), and F values. A 100-cycle bootstrap analysis was performed to assess the statistical confidence of the derived models. The mean correlation coefficient is represented as bootstrap r 2 (\(r_{\text{boot}}^{2}\)). The PLS analysis was then repeated with no validation using the optimal number of components to generate CoMFA and CoMSIA models (Cramer et al., 1988).
Predictive r 2 value
The predictive r 2 (\(r_{\text{pred}}^{2}\)) was based only on the molecules (14 compounds) not included in the training set and is defined as \(r_{\text{pred}}^{2}\) = SD − PRESS/SD where, SD is the sum of the squared deviations between the inhibitory activities of molecules in a test set and the mean inhibitory activity of a training set molecules, and PRESS is the sum of squared deviations between predicted and actual activity values for every molecule in a test set.
Analysis of the residuals
The training set was initially checked for outliers for 3D QSAR analysis. In general, if the residual of a compound between experimental pIC50 and predicted pIC50 values is greater than 1 logarithm unit, the compound is considered as outlier. Examination of the residuals from cross-validated predictions (Table 1) indicated that there is no outlier in 3D QSAR models.
Docking study
The Surflex–Dock module of SYBYL was used for molecular docking. The X-ray crystallographic structures of ALK (PDB ID: 3L9P) (Lee, 2010) solved at 1.8 Å resolution was retrieved from the PDB, and modified for docking calculations. Co-crystallized ligand was removed from the structure, water molecules were removed, H atoms were added, and side chains were fixed during protein preparation. Protein structure minimization was performed by applying Tripos force field, and partial atomic charges were calculated by Gasteiger-Huckel method.
Results and discussion
Results of the CoMFA analysis
The statistical parameters of standard CoMFA models constructed with steric and electrostatic fields are given in Table 2. The q 2, \(r_{\text{cv}}^{2}\), \(r_{\text{pred}}^{2}\), \(r_{\text{ncv}}^{2}\), F, and SEE values were computed as defined in SYBYL. PLS analysis showed a q 2 value of 0.816 and \(r_{\text{cv}}^{2}\) of 0.812. A non-cross-validated PLS analysis results in a conventional r 2 of 0.969, F = 403 and a standard error of estimation (SEE) of 0.218 with four components. In both steric and electrostatic field contributions, the former accounts for 0.742, while the latter contributes 0.258, indicating that steric field contributed the highest to the binding affinity. A high bootstrapped r 2 (0.978) value and low standard deviation (0.006) suggest a high degree of confidence in the analysis. The predicted and experimental pIC50 and residual values are listed in Table 1, and the correlation between the predicted and the experimental pIC50 of training and test set is depicted in Fig. 2a.

Plot of experimental versus predicted activities of training and test set compounds based on a CoMFA model, b CoMSIA model
Results of the CoMSIA analysis
CoMSIA offered steric, electrostatic, hydrophobic, HBD, and HBA fields’ information. Optimization of CoMSIA study was performed using the above fields. CoMSIA models were generated using steric, electrostatic, hydrophobic, HBD, and HBA fields in different combinations, and the results of this study are summarized in Table 3. CoMSIA models showed higher correlation and high predictive properties. In most of the models, steric and electrostatic fields were the common factors indicating the importance of these fields for the present series of molecules. We found that the CoMSIA descriptors such as steric, electrostatic, hydrophobic, HBD, and HBA fields played significant roles in the prediction of ALK inhibitory activity. These factors result in best CoMSIA models (Table 3). Statistically significant CoMSIA model gives q 2 of 0.838, \(r_{\text{cv}}^{2}\) of 0.837, r 2 of 0.966, F of 357, and SEE of 0.416 values with four components. The corresponding field contributions are 0.142 (steric), 0.153 (electrostatic), 0.228 (hydrophobic), 0.282 (HBD), and 0.195 (HBA). Plot of experimental and predicted pIC50 of training and test set is depicted in Fig. 2b.
Predictive power of CoMFA and CoMSIA models
The predictive abilities of 3D QSAR models were further validated using a test set of 14 compounds, not included in the model generation study. The predicted r 2 (\(r_{\text{pred}}^{2}\)) values of CoMFA and CoMSIA models are 0.910 and 0.904, respectively (Table 2). By comparison of experimental and predicted pIC50 values of a test set compounds, it is observed that CoMFA and CoMSIA models performed well in the predication of ALK inhibitory activity.
CoMFA contour maps
The contour maps of CoMFA denote the region in the space where the aligned molecules would favorably or unfavorably interact with ALK-binding site. Contour maps for the best CoMFA model are shown in Fig. 3. In the contour maps, the steric CoMFA contour plot of an active compound 66 is shown in Fig. 3a. The field energies at each lattice point were calculated as the scalar results of the coefficient and the standard deviation associated with a particular column of the data table (std*coeff), being always plotted as the percentages of the contribution of CoMFA equation. In this figure, the green color contours represent regions of high steric tolerance (80 % contribution), while the yellow color contours represent regions of low steric bulk tolerance (20 % contribution). The steric contour of CoMFA (Fig. 3a) model showed a large green color contour covering the amide linker between benzonitrile ring and imidazole ring of benzimidazole nucleus of template structure, which showed favorable bulky substitution. A second favorable steric contour was found near the piperidine ring indicating a favorable effect of steric bulk with high electron density of –N atom of piperidine ring. The active compounds of the dataset extended their bulky substitution into the sterically favored green color contour map of CoMFA and therefore exhibited good inhibitory activity. Difference between the activities of 40 (IC50 = 0.007 μM) and 41 (IC50 = 0.0016 nM) was due to the presence of more sterically favored ethyl piperidine ring in 41, whereas 40 does not have any substitution at this position. One steric unfavorable yellow color contour was observed near the carbonyl oxygen atom of amide linker between isopropyl group and cyclohexane ring. It suggested that bulky groups in these regions would decrease ALK inhibitory activity. CoMFA electrostatic contour map is shown in Fig. 3b. Regions where increased positive-charge is favorable for inhibitory activity are indicated in blue color (80 % contribution), while regions where increased negative charge is favorable for inhibitory activity are indicated in red color (20 % contribution). The electrostatic contour maps display a red color contour around the –C8 and –N1 atoms of benzimidazole ring, which indicates that a negatively charged group at –C8 position of benzimidazole ring would increase the activity. Negatively charged nitrogen atom is necessary for red colored favorable isopleths in proximity to this area. Nitrogen atoms in such a position usually can form H-bonds with amino acid residues of the binding site of ALK. Small blue color contours around the –NH of the amide linker between benzimidazole and benzonitrile ring system indicated that a positively charged hydrogen atom at this position would increase the activity.

CoMFA (std*coeff) contour maps. Compound 66 is shown inside the field, a contour maps of CoMFA steric map shown in green (80 % contribution) refer to sterically favored regions; yellow (20 % contribution) indicates disfavored areas, b contour maps of CoMFA electrostatic field. Electrostatic contour map is shown in red (20 % contribution) indicating regions where negatively charged substituents are favored, while blue contours (80 % contribution) refer to regions where negatively charged substituents are disfavored (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) (Color figure online)
CoMSIA contour maps
The CoMSIA contribution maps denote those areas within the specified region where the presence of a group with a particular physicochemical property will be favored or disfavored for good inhibitory activity. CoMSIA calculates both steric and electrostatic fields, as in CoMFA, but additionally uses hydrophobic, HBD, and HBA fields. The CoMSIA steric and electrostatic PLS contour maps were similarly placed as those for the CoMFA model. The contour plot of the CoMSIA hydrophobic, HBA, and HBD fields (std*coeff) are shown in Fig. 4. Favored and disfavored levels were fixed at 80 and 20 %, respectively. Compound 66 is overlaid in the maps once again. Figure 4a displayed the hydrophobic plot represented by yellow and gray color contours. In the CoMSIA hydrophobic map, two yellow color contours are present: one is distributed under the imidazole nitrogen of benzimidazole ring system, and other is found near the phenyl ring carrying the nitrile substitution, which indicated that hydrophobic groups in this area are beneficial to enhance ALK inhibitory activity. Compound 64 (IC50 = 0.002 μM) showed better activity because of the presence of yellow color contour at halogen (–F)-substituted phenyl ring, whereas 63 (IC50 = 0.006 μM) has unsubstituted pyridine at this position, which is less hydrophobic in nature and thus showed decrease in activity. The large gray color contour covering both the amide linker revealed the necessity of the hydrophilic amide groups on these positions to increase the activity. The graphical interpretation of the HBD interactions in the CoMSIA model is represented in Fig. 4b. Cyan color contours indicated the regions where HBD substituents on ligand are favored. In the HBD contour map, one large cyan color area was observed near the amide linker between benzimidazole and benzonitrile ring systems. Amide linker can form H-bonds with residues of ALK, which indicated the necessity of the –H atom at this position for high inhibitory activity. The graphical interpretation of the HBA interactions in the CoMSIA model is shown in Fig. 4c. Magenta color (80 % contribution) and red color (20 % contribution) contours represented the area where HBA are favored and disfavored, respectively. A large magenta color contour was observed near carbonyl oxygen of amide group. Carbonyl oxygen can act as HBA by attacking protons, indicating a favorable interaction of HBA group in this region to enhance inhibitory activity. A large red color contour was found near the benzimidazole ring, indicating that HBA substituents are disfavored at this position. However, in the present series, these substitutions are common in all compounds, and so to interpret the maps based on comparison of activity of compounds is very complex. Analyses of CoMFA and CoMSIA contour plots offered enough information to understand the importance of substituents at particular position for better activity.

CoMSIA (std*coeff) contour maps. Compound 66 is shown inside the field. Hydrophobic field (a), HBD field (b), and HBA field (c). Yellow and gray contours indicate regions where hydrophobic groups favored and disfavored the activity, respectively. Cyan contour represent areas where HBD is favored. Magenta and red contours represent areas where HBA is favored and disfavored, respectively. Favored and disfavored levels were fixed at 80 and 20 %, respectively (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) (Color figure online)
Designing of the novel ALK inhibitors
Based on the results drawn from the 3D QSAR analysis, we have designed 10 (1–10d) novel potent ALK inhibitors considering the structural requirement for inhibition ALK, and predicted ALK inhibitory activity of the designed compounds (Table 4). The compounds were designed considering the substituents found to be significant with the contour maps analyses like (1) sterically favored bulky group (–R) attached to cyclohexane ring system; (2) negatively charged group at –C8 position of benzimidazole ring (R2); (3) the presence of amide spacer between benzimidazole nucleus and substituted phenyl ring system, which will act as HBA/HBD; (4) –R1-substituted phenyl ring is responsible for aromatic hydrophobic interaction with ALK; and (5) the presence of alkoxy groups (–OMe/–OEt/–OPh) on piperidine ring system which will have hydrophobic interactions with the active site of ALK (Fig. 5).

Hypothetical interactions model of new designed ALK inhibitors with ALK enzyme
Molecular docking analysis
To study the binding modes of the designed molecules with the ALK, we performed molecular docking experiments into the ligand-binding site of ALK. The Surflex–Dock uses an empirically derived scoring function that is based on the binding affinities of protein–ligand complexes and on their X-ray structures. The protomol is a unique and important factor of the docking algorithm and is a computational representation of assumed ligands that interact with the binding site. The co-crystal structure of human ALK was retrieved from the protein data bank. After running Surflex–Dock, the scores of the active docked conformers were ranked in a molecular spread sheet. We selected the best total score conformers and speculated regarding the detailed binding patterns in the cavity. A total dock score—the total Surflex–Dock score expressed as −log (K d) to represent binding affinities which include hydrophobic, polar, repulsive, entropic, and salvation—is given in Table 4.
Binding pose of designed ALK inhibitor (8d) in active site of ALK
Docking results suggested that compound 8d has the highest good docking score of 8.048. The overall binding of 8d is illustrated in Fig. 6. Compound 8d formed two H-bonds with ALK. The oxygen and –F atom of trifluoromethoxy group substituted on cyclohexane ring formed H-bond with hydrogen atom of –NH of Asp1203 (Asp1203NH···OCF3, 1.93 Å; Asp1203NH···FF2CO, 2.47 Å). Docking studies showed that most of the compounds have a common binding mode and occupied in the vicinity of active site of ALK.

Docking interactions of designed compound 8d (ball-and-stick with color by atom) in the active site of ALK using Surflex–Dock module. The labeled protein residues are in capped stick model with color by atom. Hydrogen bonds are colored in yellow lines with distance in Å (For interpretation of the references to color in the citation of this figure, the reader is referred to the web version of the article.) (Color figure online)
Conclusions
ALK is an attractive target for small-molecule drug discovery in cancer therapy because of its key role in tumor biology. In this study, we described 3D QSAR analysis as a rational strategy to design potent ALK inhibitors, using three different alignment methods. CoMFA and CoMSIA models with good predictive capabilities were developed in this study, which were found satisfactory according to the statistical results as well as the contour maps analyses, and used for prediction of ALK inhibitory activity of the designed compounds. Overall, the predictive power of CoMFA model appeared to be better than that of CoMSIA model. 3D QSAR models discussed in this study were exploited to design novel ALK inhibitors with higher selectivity and efficacy for ALK inhibition. Finally, docking study was performed with the designed compound. The designed compounds showed good potential to be used as ALK inhibitors.
References
Allwein SP, Roemmele RC, Haley JJ, Mowrey DR, Petrillo DE, Reif JJ, Gingrich DE, Bakale RP (2012) Development and scale-up of an optimized route to the ALK inhibitor CEP-28122. Org Process Res Dev 16:148–155
Bryan MC, Whittington DA, Doherty EM, Falsey JR, Cheng AC, Emkey R, Brake RL, Lewis RT (2012) Rapid development of piperidine carboxamides as potent and selective anaplastic lymphoma kinase inhibitors. J Med Chem 55:1698–1705
Cheng M, Ott GR (2010) Anaplastic lymphoma kinase as a therapeutic target in anaplastic large cell lymphoma, non-small cell lung cancer and neuroblastoma. Anti-Cancer Agents Med Chem 10:236–249
Clark M, Cramer RD, Opdenbosch NV (1989) Validation of the general purpose tripos 5.2 force field. J Comput Chem 10:982–1012
Cramer RD III, Bunce JD, Patterson DE (1988) Crossvalidation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies. Quant Struct Act Relat 7:18–25
Gasteiger J, Marsili M (1980) Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 36:3219–3228
Jain AN (1996) Scoring non-covalent protein-ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided-Mol Des 10:427–440
Klebe G, Abraham U (1999) Comparative molecular similarity index analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J Comput Aided Mol Des 13:1–10
Lee C (2010) Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J 430:425–437
Lewis RT, Bode CM, Choquette DM, Potashman M, Romero K, Stellwagen JC, Teffera Y, Moore E, Whittington DA, Chen H, Epstein LF, Emkey R, Andrews PS, Yu VL, Saffran DC, Xu M, Drew A, Merkel P, Szilvassy S, Brake RL (2012) The discovery and optimization of a novel class of potent, selective, and orally bioavailable anaplastic lymphoma kinase (ALK) inhibitors with potential utility for the treatment of cancer. J Med Chem 55:6523–6540
Moh K, Saito N, Okabe T, Masuda Y, Furuya T, Nagano T (2011) Virtual screening and further development of novel ALK inhibitors. Bioorg Med Chem 19:3086–3095
Palmer RH, Vernersson E, Grabbe C, Hallberg B (2009) Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 15:345–361
Pulford K, Morris SW, Turturro F (2004) Anaplastic lymphoma kinase proteins in growth control and cancer. J Cell Physiol 199:330–358
Roy KK, Dixit A, Saxena AK (2008) An investigation of structurally diverse carbamates for acetylcholinesterase (AChE) inhibition using 3D-QSAR analysis. J Mol Graphics Model 27:197–208
Shaw AT, Yasothan U, Kirkpatrick P (2011) Crizotinib. Nature Rev Drug Discov 10:897–898
Tartari CJ, Scapozza L, Gambacorti-Passerini C (2011) The ALK gene an attractive target for inhibitor development. Curr Top Med Chem 11:1406–1419
Tripathy R, McHugh RJ, Ghose AK, Ott GR, Angeles TS, Albom MS, Huang Z, Aimone LD, Cheng M, Dorsey BD (2011) Pyrazolone-based anaplastic lymphoma kinase (ALK) inhibitors: control of selectivity by a benzyloxy group. Bioorg Med Chem Lett 21:7261–7264
Vyas VK, Ghate M (2012a) 2D and 3D QSAR study on amino nicotinic acid and isonicotinic acid derivatives as potential inhibitors of dihydroorotate dehydrogenase (DHODH). Med Chem Res 21:3021–3034
Vyas VK, Ghate M (2012b) CoMFA and CoMSIA studies on aryl carboxylic acid amide derivatives as dihydroorotate dehydrogenase (DHODH) inhibitors. Cur Comp Aided-Drug Des 8:271–282
Vyas VK, Bhatt HG, Patel PK, Jalu J, Chintha C, Gupta N, Ghate M (2012) CoMFA and CoMSIA studies on C-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents. SAR QSAR Environ Res. doi:10.1080/1062936X.2012.751553
Vyas VK, Ghate M, Parikh H (2013) 3D QSAR studies on 5-(2-methylbenzimidazol-1-yl)-N-alkylthiophene-2-carboxamide derivatives as P. falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. Med Chem Res. doi:10.1007/s00044-012-0216-6
Zificsak CA, Theroff JP, Aimone LD, Angeles TS, Albom MS, Cheng M, Mesaros EF, Ott GR, Quail MR, Underiner TL, Wan W, Dorsey BD (2011) Methanesulfonamido-cyclohexylamine derivatives of 2,4-diaminopyrimidine as potent ALK inhibitors. Bioorg Med Chem Lett 21:3877–3880
Acknowledgments
The authors would like to thank Nirma University, Ahmedabad, India for providing the necessary facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vyas, V.K., Patel, A., Gupta, N. et al. Design of novel anaplastic lymphoma kinase (ALK) inhibitors based on predictive 3D QSAR models using different alignment strategies. Med Chem Res 23, 603–617 (2014). https://doi.org/10.1007/s00044-013-0662-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0662-9