
Abstract
Anaplastic lymphoma kinase (ALK) is a particularly promising target for the development of small molecule anti-cancer drugs. In the present study, comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) were performed on 60 ALK inhibitors to build three-dimensional quantitative structure–activity relationship models. Both the ligand-based resultants of CoMFA (r 2 0.970, q 2 0.660) and CoMSIA (r 2 0.979, q 2 0.623) models exhibited good predictability. The resulting contour maps illustrated the regions where interactive fields may affect the activity. Molecular docking was then performed to explore the interactions between these inhibitors and the ALK-4DCE protein. A few key residues (His32, Gly31, Gly169, Asp170, Val35, Ala100, Pro160, Lys50, and Leu30) at the binding site of 4DCE were identified. Molecular dynamics simulation further verified the reliability. The information acquired in this work not only provides a better appreciation of interactions between these molecules and the ALK receptor but could also be applied to design more effective ALK inhibitors.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As a member of receptor tyrosine kinase, ALK (anaplastic lymphoma kinase) had attracted high clinical interest in the personalized treatment targeting the anti-cancer field [1], in particular, anaplastic large cell lymphoma (ALCL) [2], inflammatory myofibroblastic tumor [3], diffuse large B cell lymphoma (DLBCL) [4], renal cell carcinoma (RCC) [5], and non-small-cell lung cancer (NSCLC) [6,7,8,9].
The first ALK rearrangement was discovered as EML4-ALK fusion oncogene in NSCLC in 2007 [10]. Furthermore, ALK also fused with other proteins like nucleophosphamin (NPM) [11], ALK lymphoma oligomerization on chromosome 17 (ALO17) [12], TRK-fused gene (TFG) [13], moesin (MSN) [14] to form corresponding ALK-fusion proteins which are responsible for tumor growth [15]. Various ALK targeted drugs had been or are being tested in clinical trials and the first-in-class ALK inhibitor crizotinib was approved by FDA in 2011 for the ALK-positive NSCLC [16]. The second-generation ALK inhibitor ceritinib was approved by FDA in 2014 not only for its potent ALK inhibitory but also for its anti-drug-resistance to crizotinib [17, 18].
Other novel ALK inhibitors, including CH5424802 [19], AP26113 [20], NVP-TAE684 [21], LDK378, X-396 [22], and ASP3026 are also in Phase 1 and Phase 2 clinical trials displaying enhanced specificity [23] (Fig. 1). All these ALK inhibitors showed higher activity as well as enhanced significance in crizotinib-resistant ALK-positive NSCLC. In particular, the series of 2,4-diarylaminopyrimidine (DAAP) analogues showed high inhibitory activity against both c-Met and ALK kinases [24].

Structures of anaplastic lymphoma kinase inhibitors
Quantitative structure–activity relationship (QSAR) is one of the most widely used rational methods for drug design. In this method, the interactions between molecules and receptor depended on the difference of the molecular field around the compound. On the basis of quantitative molecular field parameters as variables, regression analysis of drug activity can reflect the interaction model between drugs and biological macromolecules, and then new drugs could be designed accordingly [25]. For instance, Vivek as well as the research group of Wang developed three-dimensional quantitative structure–activity relationship (3D-QSAR) models for different sets of compounds including 2-acyliminobenzimidazoles derivatives and piperidine carboxamides derivatives to understand chemical–biological interactions [15, 26]. We have investigated 2-acyliminobenzimidazoles derivatives as potent ALK inhibitors [27]. In the present work, 60 diarylaminopyrimidine (DAAP) derivatives reported [16, 24, 28] as potent and selective ALK inhibitors were collected as a dataset, which were studied using a combination of CoMFA, CoMSIA, molecular docking, and molecular dynamic simulation. The purpose of this study is to establish a reliable 3D-QSAR model by CoMFA and CoMSIA methods to elucidate the structural characteristics of some DAAP derivatives such as ALK inhibitors, which may provide valuable guidance in the rational synthesis of more effective inhibitors.
Results and discussion
CoMFA and CoMSIA statistical result
It is very necessary to make an initial inspection of the inhibitor molecules before establishing the 3D-QSAR models. Compound 50 was considered as an outlier in the CoMFA and CoMSIA models because the r 2 prediction of model was 0.400 on inclusion of this compound, while excluding this compound the r 2 prediction value increased to 0.983. Statistically, an r 2 value >0.3 of the predicted set is usually considered significant, while an r 2 value >0.5 is statistically more significant in CoMFA and CoMSIA studies [29]. The reason for this outlier may be the difference in structure or the different binding conformations, and the larger deviation between the actual and predicted pIC 50 values. Compound 50 and compounds 53 and 54 were very similar in structure, and the only difference was that the substituent at the nitrogen of azepane was a methyl, which might account for its outlier status since this molecule was the only compound with small volume group in this position.
Based on the internal research of the training set (44 molecules) and the external confirmation of the test set (16 molecules) the CoMFA and CoMSIA models were built. As shown in Table 1, the optimal CoMFA model resulted in a cross-validated q 2 of 0.660, a non-cross-validated correlation coefficient r 2 of 0.970, a standard error (SEE) value of 0.144, and F statistic value (F) of 167.010. For the CoMFA analysis, the q 2 value of 0.623, r 2 value of 0.979, SEE value of 0.120, and F statistic value of 241.162 were calculated, respectively.
For the CoMFA model, the contributions of the steric and electrostatic fields were calculated to be 43.8 and 56.2%, respectively; thus, the electrostatic field has more influence compared to the steric field. For the optimal CoMSIA model, five descriptor fields were considered including the steric, electrostatic, hydrophobic, hydrogen bond–donor, and hydrogen bond–acceptor. Their contributions were 10.6, 30.0, 19.2, 16.7, and 23.5%. Table 2 listed the actual and predicted pIC 50 values of the training and test set as well as the residues between them.
3D-QSAR contour maps
Through the superposition of the most active molecule 23 with the contour maps generated by CoMFA and CoMSIA, we explored the field effects on the target compounds in 3D space. These contour maps have great significance in explaining the relationship between molecular structure and biological activity because the regions displayed in 3D maps showed the influence of different substituents on the molecular activity.
CoMFA contour maps
The steric and electrostatic contour maps generated by the CoMFA model are shown in Fig. 2. The green polyhedrons represent bulk substituents which are beneficial to the potency, while yellow polyhedrons represent steric bulk groups that would decrease the activity (Fig. 2a).

CoMFA contour maps. Compound 23 is shown inside. a Steric field: favored (green) and disfavored (yellow). b Electrostatic field: electropositive (blue) and electronegative (red) (color figure online)
The medium yellow contour occurring at single side of the aromatic R1 ring indicated the compounds with bulk substituents at this site would decrease biological activity. The compound 38 (pIC 50 = 8.149) with cyclopropyl group at ortho-site of R1 aromatic ring possesses lower biological activity than the corresponding methyl and isopropyl group substituted compounds 37 (pIC 50 = 8.456) and 39 (pIC 50 = 8.886). In contrast, a big negative steric (yellow) and a big positive steric (green) emerged at the meta-site of the aromatic ring connected to R2. In addition, medium yellow contours appeared above of R2 position, which suggested that a bulky substituent in this region would decrease the biological activities of the molecule, such as 8 (pIC 50 = 8.745), (pIC 50 = 7.827), and 10 (pIC 50 = 7.294).
The electrostatic contour map of CoMFA is shown in Fig. 2b. The electrostatic field is represented by blue-colored and red-colored contours, in which the blue contours denote that the electropositive groups are favorable to the activity and the red regions indicate that the electronegative groups are positive to the activity. Three red contours at the site of R1 indicate that electronegative groups in this area are positive to the activity. More electronegative sulfonyl at R1 of compound 4 (pIC 50 = 8.569) leads to its high activity, while the less electronegative amide group at the same position of compound 3 (pIC 50 = 7.484), respectively, showed inactivity. As shown in the electrostatic contour maps, a big blue color contour encompassed the piperazine ring, which indicates that electropositive groups in this region would be beneficial to the activity. Small red color contours that appear near the methylene connecting the piperazine ring and benzene indicate that negatively charged substituents at this position also have a little influence on the activity.
CoMSIA contour maps
The CoMSIA-generated maps indicate that the presence of a group with a special physicochemical property in the designated area would be beneficial or detrimental to good inhibitory activity. The CoMSIA not only calculated both steric and electrostatic fields the same way as the CoMFA, but also covered hydrophobic, H-bond donor (HBD), and H-bond acceptor (HBA) fields. Favorable and unfavorable contributions were fixed at 80 and 20%, respectively. Once again, we choose the most active compound 23 to analyze the effects of the five force fields.
Figure 3a displays the steric plot represented by yellow and green color contours. The whole area of R2 is covered by the yellow color contour, which shows that compounds with bulk substituents in this area would decrease biological activity. The difference between the activities of 1 (pIC 50 = 6.553) and 3 (pIC 50 = 7.484) is due to the presence of small volume of imide in 3, whereas 1 has a sterically more demanding substitution at this position. The second adverse steric contour was discovered near the R1 ring indicating the adverse effect of steric bulk, while there are also two green contours at the opposite sites of the yellow polyhedral suggesting that bulky groups were acceptable at this position.

CoMSIA contour maps. Compound 23 is shown inside. a Steric field: favored (green) and disfavored (yellow). b Electrostatic field: electropositive (blue) and electronegative (red). c Hydrophobic field: favored (yellow) and disfavored (gray). d Hydrogen bond donor field: favored (cyan) and disfavored (purple). e Hydrogen bond acceptor field: favored (magenta) and disfavored (red) (color figure online)
Figure 3b shows the influence of the electrostatic field in the CoMSIA model. The red contour overlapping the amide at the meta-site of the R2 aromatic means that the electronegative groups in this region could improve the inhibition. The large blue contour encompassing the piperazine ring denotes that the electropositive substituents in this area have a positive effect on the molecular activity. For example, the activity of 50 (pIC 50 = 6.112) with methyl was less potent than compound 52 (pIC 50 = 6.750), which possesses an ester at this position.
In Fig. 3c, yellow and gray color contours represent the effect of hydrophobicity on the molecular activity. One yellow color contour can be seen covering the methylene and ketone located in the meta-site of the R2 ring, which suggests that hydrophobic groups in this region contribute to the enhancement of the inhibition. But an equal volume of gray color contour also can be seen near the piperazine ring. In general, both of the hydrophobic favored and hydrophilic contours emerge at the same area indicating that the two groups are in equilibrium in this region. For example, both values of 51 (pIC 50 = 6.767) and 52 (pIC 50 = 6.750) are basically the same.
Hydrogen bond donor (HBD) groups represented by cyan (favorable) and purple (unfavorable) contour maps are shown in Fig. 3d. From the contour map of the HBD, a large cyan color contour can be seen in the vicinity of the piperazine ring. For example, in the most active molecule 23, there is an imine between R1 and piperazine which could form H-bonds with residues of the protein, indicating that a hydrogen atom in this position is favorable to the activity of molecule. The alignment of the blue polyhedron has a small purple contour, which indicates that hydrogen bond acceptor groups have little effect on the molecular activity in this position.
Figure 3e illustrates the effects of a hydrogen bond acceptor (HBA) in the CoMSIA model. Magenta color (80% contribution) and red color (20% contribution) contours, respectively, representing the HBA are favorable or unfavorable to the biological activity. One large volume of magenta color contour is discovered near the R1 ring indicating that the presence of sulfonic groups in this area could act as HBA attacking protons, which further showed that HBA in this position was conductive to improving inhibitory activity. Another magenta color contour emerges below the piperazine ring, while there are two red contours at the opposite position, which suggests a balance between H-bond donor and H-bond acceptor in the same region.
In summary, the structural characteristics for better inhibitory activities from the above-mentioned contour analysis of CoMFA and CoMSIA models are:
-
1.
R1: medium-sized and electronegative substituents at R1 site, and hydrogen bond acceptor (favorable).
-
2.
R2: bulk substituents, electropositive and hydrophobic substitutes at meta-site of R2 aromatic ring, and hydrogen bond donor near the piperazine ring (favorable).
Docking analysis
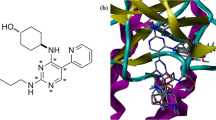
Through molecular docking, we found that the activity of molecules is related to the free energy changes in the process of binding with the protein. The most active inhibitor 23 was selected to dock with the ALK-4DCE protein and the results explain the interaction mechanism between the ligand and the receptor, which is shown in Fig. 6. The benzene ring among the common skeleton formed a π–alkyl interaction with Pro160 (2.45 Å). On the other hand, the benzene ring at R2 forms a π–σ interaction with Gly31 (2.42 Å). Alkyl hydrophobic interactions form between the piperazine ring and Val35 (5.23 Å) and Lys50 (4.73 Å), which coincides with the hydrophobic contour map depicted in Fig. 3e. There is a large yellow contour around the benzene ring indicating that the introduction of hydrophobic groups in this region is beneficial for the inhibitory activity. According to the docking results, the large pocket composed of Asp170, Gly169, Lys50 as displayed in Fig. 4 is sufficient for medium bulky substituents. The green contour generated by the COMFA model at this position also verified this conclusion. However, large bulky substituents in this position would lead to steric hindrance with the surrounding amino acids, which would lead to a reduction in activity. Therefore, the introduction of too large groups in this position is detrimental, which is consistent with the steric contour map in the COMSIA model of Fig. 2b. The results of molecular docking show that most of the inhibitors have a similar binding pattern at the active site of ALK.

Docking result of the representative ligand 23 into the binding site of the ALK protein. Ligands and the important residues for binding interaction are depicted by stick and line models

a Plot of the root-mean-square deviation (RMSD) of docked complex versus the MD simulation time in the MD-simulated structures. b View of superimposed backbone atoms of the lowest-energy structure of the MD simulation (blue) and the initial structure (green) for the 23/4DCE complex
Molecular dynamics simulation
In order to further verify the models of 3D-QSAR and molecular docking, we applied MD simulations to establish a more reliable mechanism to illustrate interactions between ligand and receptor. The basic theory of MD simulations is given by the molecular system initial motion state and the natural motion of the molecules in the phase space. A 15 ns simulation was run to obtain a stable conformation of ligand–receptor complex in this study was shown in Fig. 5a. The RMSDs of the trajectory with respect to the initial structure ranged from 2.5 to 3.0 Å. After 2 ns, the RMSDs of the complex reached about 5.3 Å and maintained a similar value in the following simulation, which indicated that the docked complex could reach metastable conformation after 2 ns of simulation. A superposition of the lowest-energy structure extracted from the MD simulation (blue) and the initial structure (green) for the 23–4DCE complex were shown in Fig. 5b. Through the analysis of the interactions between 23 and the receptor after MD simulation, we explored the similarities and differences between molecular docking and MD simulation. Figure 6 shows the lowest-energy structure extracted from the MD simulation, from which we can see that it mainly forms three hydrogen bonds between the ligand and the receptor. The amide oxygen at the meta position of the benzene ring forms a hydrogen bond with NH of His32 (–C=O···HN–, 2.45 Å). The oxygen atom of the sulfonic group acts as a hydrogen bond acceptor to form a H-bond with NH of Gly31 (–C=O···HN–, 1.77 Å), which is consistent with the H-bond acceptor contour depicted in Fig. 3e. There is a large magenta contour near the R1 position, which indicates that H-bond acceptor groups in this area are favorable for the inhibitory activity. Residues Gly31, Val35, and His32 form hydrophobic contacts with the ligands, which are beneficial for the inhibitory activity. On the other hand, the benzene ring at the R2 forms a π-stacking bond with His32 which further strengthens the correlation between the inhibitory and the receptor.

Plot of the MD-simulated structure of the binding site with the ligand. Compound 23 in the complex is in the active site of the ALK enzyme. Active site amino acid residues are represented as sticks; the inhibitor is shown as stick and ball model
Conclusion
QSAR and molecular dynamics were applied to analyze and explore characteristics of DAAP analogues as ALK inhibitors. The CoMFA and CoMSIA models nicely explained the intermolecular interactions between the inhibitors and the surrounding environment. Docking and molecular dynamics studies demonstrated that hydrogen bond formed between the inhibitors and ALK-4DCE protein play an important role in activity of the inhibitors. In addition, the MD simulation results are consistent with the results of QSAR models and molecular docking in terms of the reliability and stability of the derived models. Some key residues (His32, Gly31, Gly169, Asp170, Val35, Ala100, Pro160, Lys50, and Leu30) and three hydrogen bonds (His32, Gly31, and Leu30) were discovered in the binding site, which indicated that the model could provide guidance for further research in the development of new ALK inhibitors.
Materials and methods
Dataset and biological activity
Sixty DAAP analogues involved in this work were reported by Ao Zhang and co-workers [16, 24, 28]. The range of IC 50 values for these compounds was 0.7–775 nM. The bioactivities of the derivatives were expressed as pIC 50 (= −log IC 50) values. The samples were divided into a training set of 44 molecules for model generation and a test set of 15 molecules for model validation at a ratio of 3:1. The structures and activity values of each molecule used in the study are shown in Table 3. The test molecules were selected randomly such that the dataset showed high structural diversity and a wide range of activities [30].

Molecular modeling and alignment procedure
CoMFA and CoMSIA models were all performed using the SYBYL-X 2.0 software. All molecules were loaded with Gasteiger–Hückel charges and optimized by using the Tripos force field [31] with Powell energy gradient algorithms at a convergence criterion of 0.02 kJ/mol Å and a maximum of 1000 iterations [32]. Table 3 lists the common scaffold of the samples, various substituents, and the IC 50 value of each molecule. Molecular alignment was the most critical step in the establishment of the CoMFA and CoMSIA models, which needed to analyze the three-dimensional structure of the samples to find a suitable conformational template for alignment [33]. Since the molecules share a common structure, it was assumed that each molecule binds into the active site of protein in a similar way. In this context, we adopted the rigid body alignment rule. Compound 23, which had the highest pIC 50 (9.155), was selected as the template molecule of DAAP derivatives. In the end, the program automatically superposed all the molecules and then the database was updated to a new molecular library with new orientation [32]. Alignment of training and test set compounds is shown in Fig. 7. The common substructure is depicted in bold.

Molecular alignments of all compounds in the dataset. Compound 23 was used as the template for alignment
CoMFA and CoMSIA
In order to build a reliable 3D-QSAR model, the partial least squares (PLS) method was carried out based on the above alignment of molecules [34]. To find the best models, we calculated various parameters which were used to evaluate and analyze the robustness and predictive ability of these models, including the internal validations of LOO cross-validated q 2, non-cross-validated coefficient r 2, standard error of estimate (SEE), and F statistic values. According to these statistical results, the final models were established. The statistical results of the CoMFA and CoMSIA models are summarized in Table 1. We used the default settings in SYBYL in the optimization process of the CoMFA and CoMSIA descriptors [35]. Figure 8 showed a linear relationship between the predicted and true values calculated by the CoMFA and CoMSIA models.

Plots of predicted versus actual pIC 50 values for all the molecules based on CoMFA (a) and CoMSIA models (b)
Molecular docking
Molecular docking is an important computational chemistry tool with clear and intuitive definition. The structure and binding energy of protein–ligand complexes can be found in the case of known protein and ligand space structures [36]. We applied the SYBYL-X 2.0, which is based on a prototype to explore more information on the binding mode of ligand and ALK protein. The crystal structure of the ALK protein complex was obtained from the RCSB Protein Data Bank (PDB entry code: 4DCE). After the extraction of ligands, removal of water molecules, and hydrogenation, a prototype was generated by using the ligand extraction method for molecular docking. The energy minimization of the protein structure was performed by applying the Tripos force field, and partial atomic charges were calculated by means of the Gasteiger–Hückel method. The protein interaction of the ligand was visualized by using Discovery Studio Visualizer 2.5 (Accelrys Software Inc.), which provided a molecular modeling environment for both small molecule and macromolecule.
References
Achary R, Yun JI, Park CM (2016) Bioorg Med Chem 24:207
Le Beau MM, Bitter MA, Larson RA (1989) Leukemia 3:866
Gleason BC, Hornick JL (2008) J Clin Pathol 61:428
Lee HW, Kim K, Kim W, Ko YH (2008) Hematol Oncol 26:108
Debelenko LV, Raimondi SC, Daw N (2011) Mod Pathol 24:430
Soda M, Choi YL, Enomoto M (2007) Nature 448:561
Caren H, Abel F, Kogner P, Martinsson T (2008) Biochem J 416:153
Mosse YP, Laudenslager M, Longo L (2008) Nature 455:930
Ren H, Tan ZP, Zhu X (2012) Cancer Res 72:3312
Katayama R, Friboulet L, Koike S (2014) Clin Cancer Res 20:5686
Choudhari R, Minero VG, Menotti M (2016) Blood 127:1297
Moritake H, Shimonodan H, Marutsuka K (2011) Am J Hematol 86:75
Wynne C, Lazzari E, Smith S (2014) PLoS One 9:e101503
Tort F, Pinyol M, Pulford K (2001) Lab Invest 81:419
Vyas VK, Patel A, Gupta N, Ghate M (2014) Med Chem Res 23:603
Liu ZQ, Yue XH, Song ZL (2014) Eur J Med Chem 86:438
Marsilje TH, Pei W, Chen B (2013) J Med Chem 56:5675
Christensen JG, Zou HY, Arango ME (2007) Mol Cancer Ther 6:3314
Kinoshita K, Asoh K, Furuichi N (2012) Bioorg Med Chem 20:1271
Katayama R, Khan TM, Benes C (2011) Proc Natl Acad Sci USA 108:7535
Galkin AV, Melnick JS, Kim S (2007) Proc Natl Acad Sci USA 104:270
Lovly CM, Heuckmann JM, Thomas RK (2011) Cancer Res 71:4920
Ke ZP, Lu T, Liu HC (2014) J Mol Struct 1067:127
Song ZL, Yang YH, Liu ZQ (2015) J Med Chem 58:197
Cheng LP, Huang XY, Wang Z, Kai ZP, Wu FH (2014) Monatsh Chem 145:1213
Wang P, Cai J, Chen JQ (2014) Med Chem Res 23:2576
Lv QQ, Wang ZH, Wang ZH, Wu FH (2015) Lett Drug Des Discov 12:219
Liu ZQ, Ai J, Peng X, Song ZL (2014) ACS Med Chem Lett 5:304
Boehm M, Stuerzebecher J, Klebe G (1999) J Med Chem 42:458
Liu J, Li Y, Zhang HX, Zhang SW (2012) J Mol Model 18:991
Clark M, Cramer RD III, Van ON (1989) J Comput Chem 10:982
Wang FF, Yang W, Shi YH, Le GW (2015) J Biomol Struct Dyn 33:1929
Thaimattam R, Daga PR, Banerjee R, Iqbal J (2005) Bioorg Med Chem 13:4704
Liu JL, Wang FF, Ma Z, Wang X (2011) Int J Mol Sci 12:946
Xu C, Ren YJ (2015) Bioorg Med Chem Lett 25:4522
Kirkpatrick P (2004) Nat Rev Drug Discov 3:299
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21172148, 21472126, and 21502117) and Shanghai Municipal Education Commission (Plateau Discipline Construction Program).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, DD., Wu, FL., Wang, ZH. et al. Identification of 2,4-diarylaminopyrimidine analogues as ALK inhibitors by using 3D-QSAR, molecular docking, and molecular dynamics simulations. Monatsh Chem 148, 1711–1725 (2017). https://doi.org/10.1007/s00706-017-1999-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-017-1999-4