
Abstract
Ischemia/reperfusion (IR) injury occurs in many organs and tissues, and contributes to morbidity and mortality worldwide. Melatonin, an endogenously produced indolamine, provides a strong defense against IR injury. Mitochondrion, an organelle for ATP production and a decider for cell fate, has been validated to be a crucial target for melatonin to exert its protection against IR injury. In this review, we first clarify the mechanisms underlying mitochondrial dysfunction during IR and melatonin’s protection of mitochondria under this condition. Thereafter, special focus is placed on the protective actions of melatonin against IR injury in brain, heart, liver, and others. Finally, we explore several potential future directions of research in this area. Collectively, the information compiled here will serve as a comprehensive reference for the actions of melatonin in IR injury identified to date and will hopefully aid in the design of future research and increase the potential of melatonin as a therapeutic agent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemia/reperfusion (IR) injury occurs when the blood supply to the tissue is blocked for minutes to hours (ischemia) and then restored (reperfusion) [1]. Ischemia elicits tissue anoxia which is the basis of ischemic injury and primes the tissue for subsequent reperfusion damage. IR injury affects many organs and tissues including brain [2, 3], heart [4, 5], liver [6, 7], lung [7, 8], kidney [9, 10], skeletal muscles [11, 12], testes tissue [13], and endothelial tissue [14] contributing to morbidity and mortality worldwide [15, 16]. Numerous efforts have attempted to search for proper agents for the treatment of IR injury every year. Notably, melatonin is the particularly promising one among various candidates.
Melatonin, an ancient molecular existing in various organism, is validated to be a potent antioxidant and exerts beneficial effects on many pathological conditions [17], including diabetes [18, 19], depression [20, 21], infection [22, 23], neurodegeneration [24, 25], and metabolic syndrome [26, 27]. The roles of melatonin on IR injury get much attention in recent years and multiple novel mechanisms have been revealed [28, 29]. Melatonin is highly concentrated in mitochondria and its roles on mitochondria have been widely explored in previous studies [30,31,32]. Mitochondrion, an organelle for ATP production and a decider for cell fate, has been verified to play crucial roles in IR injury and the protection of mitochondrion can inhibit IR injury in multiple organs [33,34,35]. Similar to studies about other pathophysiological processes, melatonin’s protective actions on IR injury are mainly achieved by inhibiting mitochondrial dysfunction [32, 36, 37]. Melatonin has been shown to ameliorate IR-induced disturbance in mitochondrial redox state, membrane structure, biogenesis, dynamics, and mitophagy and has attracted attention as an appealing therapeutic strategy [17, 30, 32].
The focus of this review is to summarize the latest research progress regarding the roles of melatonin in IR injury. First, we introduce the mechanisms underlying mitochondrial dysfunction in IR and melatonin’s protection of mitochondria under this condition. Thereafter, the protective effects of melatonin against IR injury in various organs and tissues, including brain, heart, liver, and others are presented. Finally, we explore several potential future directions of research in this area. Collectively, the information compiled here will serve as a comprehensive reference for the actions of melatonin in IR injury identified to date and will hopefully aid in the design of future research and increase the potential of melatonin as therapeutic agent.
Mitochondrial dysfunction induced by IR
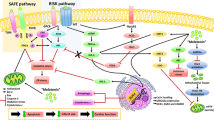
Mitochondrial dysfunction has been validated to be a crucial event in IR injury by numerous studies [38]. The period of ischemia primes the tissue for subsequent damage upon reperfusion which leads to a burst of free radical from mitochondria [39]. The excessive free radical directly causes oxidative damage to mitochondrial respiratory chain and metabolism enzymes further leading to more electron leakage and free radical production [40, 41]. Moreover, free radical is also validated to damage mitochondrial membrane structure [42] and increase mitochondrial permeability transition pore (MPTP) opening [43], resulting in loss of membrane potential and more free radical production [41]. The increased mitochondrial permeability also increases pro-apoptosis factors’ release to cytoplasm [44] (Fig. 1). Moreover, IR-induced damages also impair mitochondrial dynamics and mitophagy thereby affecting quality control of mitochondrial network [45, 46]. Eventually, mitochondrial dysfunction leads to increased apoptosis and exacerbates IR-induced injury in various organs and tissues.

The mechanisms underlying mitochondrial dysfunction in IR and melatonin’s protection of mitochondria under this condition. IR leads to electron leakage and excessive free radical production in mitochondria. The excessive free radical directly causes oxidative damage to mitochondrial respiratory chain and EME further leading to a burst of electron leakage and free radical production. Moreover, free radical also damages mitochondrial membrane structure (TOM complex reduction and mitochondrial membrane lipid peroxidation) and increases MPTP opening, resulting in membrane potential loss and pro-apoptosis factor release. Apart from directly scavenging free radical, melatonin also activates STAT3, a transcription factor for antioxidant enzymes, by activating SAFE pathway and JAK2. Melatonin activates AMPK–PGC-1α–SIRT3 axis to reduce mitochondrial oxidative stress and enhances its biogenesis. By activating PGC-1α, melatonin also upregulates TOM complex, the entry gate for the majority of precursor proteins that are imported into the mitochondria. As a result, melatonin exerts protective effects on diverse organs enduring IR injury. Red arrows damaging processes. Green arrows promotion or amelioration. Blue arrows inhibitory effects (AMPK adenosine monophosphate-activated protein kinase, EME energy metabolism enzymes, IR ischemia/reperfusion, JAK2 Janus kinase 2, MPTP mitochondrial permeability transition pore, SAFE survivor activating factor enhancement, SIRT3 silent information regulator 3, STAT3 signal transducer and activator of transcription 3, PGC-1α peroxisome proliferator-activated receptor-gamma coactivator-1α, TOM translocases in the outer membrane)
Melatonin protects mitochondria from IR injury
From the above brief clarification, we note that it is of great importance to break the vicious circle between free radical and mitochondrial injury during IR process. Notably, melatonin is an ideal candidate. First of all, as a potent-free radical scavenger, melatonin is highly concentrated in mitochondria, indicating its capacity to resist mitochondrial oxidative injury [32, 47]. Apart from directly scavenging free radical, melatonin also exerts antioxidant activity by upregulating antioxidant enzymes and downregulating pro-oxidant enzymes [48,49,50]. Besides, melatonin has been documented to upregulate the activity of all four complexes in respiratory chain under IR conditions [51, 52] which may reduce the production of free radical. At decreased level of oxidative stress, lipid peroxidation is repressed and the mitochondrial membrane structure is well preserved by melatonin [36]. Moreover, melatonin is also documented to regulate mitochondrial membrane permeability by modulating the translocases in the outer membrane (TOM) complex [28] and MPTP activity [53]. As a result, melatonin well preserves mitochondrial membrane potential and inhibits release of pro-apoptosis proteins including cytochrome c [54] and high-temperature requirement protein A 2 (HtrA2) [55]. Apart from above actions, melatonin was also validated to maintain a healthy mitochondrial network by regulating mitochondrial biogenesis [51], dynamics, and mitophagy [56]. Eventually, melatonin restores mitochondrial and organ function under IR conditions. Mechanistically, nuclear melatonin receptor RORα [57] and multiple pathways are revealed to be involved in the protection of melatonin, including silent information regulator 1 (SIRT1) [2], Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) [58], survivor activating factor enhancement (SAFE) [29], and adenosine monophosphate-activated protein kinase (AMPK) [51], which will be clarified later (Fig. 1). The activation or block of these targets could promote or repress the protective effect of melatonin on mitochondria.
Protection of melatonin against IR injury in nervous system
The ischemic insult in the nervous system is accompanied by multiple physiopathological events, including reactive oxygen species (ROS) burst, Ca2+ dyshomeostasis, mitochondrial dysfunction, proinflammatory mediator release, excitotoxicity, and eventually programmed neuronal cell death [59, 60]. Melatonin was validated to be protective against ischemic injury in nervous system. In oxygen–glucose deprivation (OGD)-treated primary cerebrocortical neurons, melatonin inhibits loss of mitochondrial membrane potential, release of mitochondrial factors, and activation of caspase-1 and -3, thereby attenuating OGD-induced apoptosis [61]. After hypoxic exposure, the retinal ganglion cell showed mitochondrial dysfunction and increased oxidative stress. Melatonin treatment preserves mitochondrial function as indicated by a reduction in cytochrome c leakage into the cytosol [62]. The in vivo study revealed that melatonin decreases infarct size and improves neurological scores after permanent middle cerebral artery occlusion (MCAO) in mice, which is also associated with reduced cytochrome c release and caspase-3 activation in ischemic tissue [61].
Subsequent reperfusion exacerbates ischemia-induced injury in nervous system. The in vitro study revealed that melatonin application during reoxygenation inhibits the hypoxia/reoxygenation-induced loss of the mitochondrial membrane potential, release of mitochondrial cytochrome c and activation of caspase-3 [63]. Multiple in vivo studies also validated the protective role of melatonin after reperfusion. Fukaya and colleagues conducted a series of studies about the protective effect of melatonin on fetal brain IR injury. Their results showed that melatonin pre- or post-treatment effectively reverses IR-induced reductions in the respiratory control index (RCI) (a marker of mitochondrial respiratory activity) and in the ADP/oxygen ratio and also reduces the elevation in concentration of thiobarbituric acid-reactive substances in the mitochondria of fetal brain [37, 64, 65]. Melatonin also preserves mitochondrial function and ameliorates neuronal cell injury of newborn rats after hypoxia–ischemia/reperfusion [66, 67]. Similar protective effects were also observed in brain of adult animals. The mitochondrial complex I and IV activities were impaired with transient MCAO while melatonin administration restored them. Moreover, melatonin treatment after MCAO significantly inhibits inducible nitric oxide synthase (NOS) activity and attenuated expression of the inducible isoform, resulting in decreased total NOS activity and tissue nitrite levels [52]. Also in MCAO rats, melatonin regulates mitochondrial membrane permeability by inhibiting MPTP opening after IR, leading to decreased cytochrome c release and less caspase-3 activation in infarct area [53]. Our study on IR injury of adult mice brain revealed that melatonin confers a cerebral-protective effect through the activation of SIRT1 signaling which is associated with a well-preserved mitochondrial membrane potential, mitochondrial complex I activity, and mitochondrial cytochrome c level [2]. As a result, melatonin treatment diminishes the loss of neurons, decreases the infarct volume, lowers brain edema, and increases neurological scores after cerebral IR.
Apart from melatonin, melatonin’s precursor or metabolite also exerts neuroprotective effects under ischemia and IR conditions. N-Acetylserotonin, an immediate precursor of melatonin, inhibits cell death induced by OGD or H2O2 in primary cerebrocortical neurons, primary hippocampal neurons and organotypic hippocampal slice in vitro, meanwhile reduces hypoxia/ischemia injury in the MCAO mouse model of cerebral ischemia in vivo. Notably, the neuroprotective effects of N-acetylserotonin are also associated with its actions on MPTP opening, mitochondrial fragmentation, and subsequent pro-apoptosis factor release [68]. 6-Hydroxylmelatonin, a normal metabolite of melatonin in vivo, is shown to scavenge ROS, maintain mitochondrial transmembrane potential, and inhibit lactate dehydrogenase and cytochrome c release, and caspase-3 activity during IR [69]. However, unlike melatonin, melatonin’s precursor or metabolite cannot provide neuroprotection through the activation of melatonin receptors. Moreover, unlike free melatonin, the nanocapsulated melatonin is more slowly degraded by light and cleared by the circulating blood, which exhibits higher potential to rescue neuronal cells and mitochondria during cerebral IR insult [70].
Protection of melatonin against myocardial IR injury
Myocardial ischemia leads to cardiomyocyte anoxia, which is detrimental to the survival and function of these cells. In isoproterenol (ISO)-induced myocardial ischemia rat model, melatonin treatment reduces ischemia-induced mitochondrial dysfunction and rescues cardiac tissue [71]. The further study revealed that ISO induces myocardial ischemia and increases mitochondrial oxidative stress, leading to decreased activity of key enzymes of the Kreb’s cycle and the respiratory chain. Melatonin inhibits above changes and enhances the antioxidant enzymes activity, preserving mitochondrial redox potential [36]. The modulation of mitochondrial membrane permeability is also involved in melatonin’s protective effects during ischemia. The TOM complex located in the outer membrane of mitochondria is the entry gate for the majority of precursor proteins that are imported into the mitochondria [72]. TOM70 is an important receptor in TOM machinery and ischemic/hypoxic insult reduced TOM70 expression in cardiomyocytes which partially accounts for increased mitochondrial fragmentation and ROS overload. Melatonin was demonstrated to promote TOM70 expression by activating peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) and ameliorate ischemic injury which is absent in TOM70-deficient mice [28]. Moreover, in the long run, the improved mitochondrial function by melatonin is able to mitigate adverse left ventricle remodeling after myocardial infarction [73].
Ischemic injury is usually accompanied by subsequent reperfusion injury. Reperfusion was reported to significantly alter multiple mitochondrial parameters, including mitochondrial oxygen consumption rates, complex I and complex III activity, H2O2 production as well as the degree of lipid peroxidation [74]. Melatonin has been demonstrated to possess protective effect against myocardial IR injury through mitochondria-dependent mechanism. The STAT3 is a transcription factor of the manganese superoxide dismutase (MnSOD) gene and interacts with MnSOD protein to increase its antioxidant activity, which plays crucial roles in mitochondrial antioxidant defense [75, 76]. In cultured neonatal rat cardiomyocytes and isolated rat hearts, melatonin pretreatment attenuates IR-induced mitochondrial oxidative damage via the activation of the JAK2/STAT3 signaling pathway [58]. Moreover, melatonin can also activate mitochondrial STAT3 through SAFE pathway to reduced myocardial IR injury [29]. The regulation of mitochondrial membrane permeability is also involved in the protective actions of melatonin. The MPTP opening in the first few minutes of reperfusion is known to be a critical determinant of myocardial IR injury, contributing up to 50% of the final myocardial infarct size [77]. Also in isolated perfused rat hearts, melatonin desensitizes mitochondria from reperfused hearts to MPTP opening as demonstrated by their higher resistance to Ca2+, thereby improving the functional recovery and reducing myocardial injury after IR [4]. Moreover, when IR is combined with diabetes, melatonin preserves mitochondrial function by reducing mitochondrial oxidative stress and enhances its biogenesis, mainly by activating AMPK–PGC-1α–silent information regulator 3 (SIRT3) axis [51]. Apart from adult rat, melatonin was also documented to protect diabetic mother–offspring from myocardial IR injury. Diabetic mother–offspring exhibited augmented infarct size, cardiac dysfunction, and myocardial apoptosis in response to IR, in association with exaggerated activation of mitochondria- and endoplasmic reticulum (ER) stress-mediated apoptosis pathways and oxidative stress. The maternal melatonin application can improve the tolerance to myocardial IR injury in their offspring via restoring cardiac insulin receptor substrate 1/Akt signaling [78].
Melatonin receptors play important roles in the cardiac protection of melatonin. Melatonin receptors include membrane melatonin receptor 1, melatonin receptor 2, and nuclear RZR/ROR receptors. In a study of hypoxia/reoxygenation model of H9c2 cells, melatonin receptor agonist Neu-p11 offers protection for mitochondria, inhibits cell apoptosis, and improves the morphology and rhythm of myocardial cells [79]. Melatonin could promote PGC-1α/TOM70 expression in ischemic myocardium but not when combined with melatonin receptor antagonist luzindole [28]. Moreover, RORα plays important roles in melatonin-exerted cardioprotection, in particular against myocardial IR injury. RORα deficiency promotes IR-induced mitochondrial impairments resulting in significantly increased myocardial infarct size, myocardial apoptosis and exacerbated contractile dysfunction [57].
Notably, cardiac mitochondria are considered as two distinct populations: subsarcolemmal mitochondria (SSM) located immediately underneath the plasma membrane and interfibrillar mitochondria (IFM) situated among the myofibrils [34]. SSM are more susceptible to calcium overload-mediated cytochrome c release and damage having a more rapid progression of ischemic injury than in IFM [34, 80]. However, this different response to ischemia and IR injury was neglected by above studies. Therefore, further studies can pay some attention to such issue for better clarification of melatonin’s roles.
Protection of melatonin against hepatic IR injury
Liver is one of the most frequently affected organs by IR injury and the protection by melatonin administration is highly evaluated [81]. Melatonin application after hepatic IR was shown to increase the energy charge and decrease the levels of plasma nitrite, tumor necrosis factor-α, aspartate aminotransferase, alanine aminotransferase, lipid peroxidation products, and inducible nitric oxide synthase, resulting in elevated 7-day survival rates in the end [82]. The maintenance of mitochondrial function is involved in such protective effects of melatonin. Melatonin was shown to restore mitochondria respiratory function as indicated by the preserved RCI, ADP/O and State 3 respiration [6]. In addition, melatonin treatment is able to decrease ROS production [83], increase mitochondrial glutathione peroxidase activity, and attenuate mitochondrial lipid peroxidation after IR [6]. Moreover, melatonin attenuates the extent of the mitochondrial permeability transition after hepatic IR as indicated by the decreased rate of mitochondrial swelling and cytochrome c release [54, 84]. Dynamin-related protein 1 (Drp1) is involved in mitochondrial outer membranes fission, a process that helps to maintain mitochondrial morphology and to reduce the accumulation of functional and structural defects in mitochondria [85]. In hepatic IR injury mouse model, melatonin was documented to ameliorate mitochondrial morphology and attenuate IR injury via restoring Drp1.
Protection of melatonin against IR injury in other organs and tissues
IR injury of skeletal muscles is a common pathophysiology during peripheral vascular injury and surgeries [11], which usually induces significant necrosis and apoptosis in the skeletal muscle cells. Mitochondrial dysfunction, such as the depolarization of mitochondrial membrane potential and the release of the proapoptotic protein, is induced by IR in skeletal muscle and melatonin significantly inhibited above changes [86]. Testicular IR injury is usually induced by torsion/detorsion, which causes an enhanced ROS formation and contributes to the pathophysiology of tissue damage [87, 88]. The melatonin treatment improves testicular histological appearance after IR, attenuates cell apoptosis, promotes cell proliferation, and increases testosterone in testis tissue, partially via the inhibition of mitochondrial degeneration [89]. Moreover, melatonin was also shown to protect placenta from IR injury. Maternally administered melatonin inhibits IR-induced changes in placental RCI and fetal growth restriction [90].
Different from other tissues, nitrosative stress is more common in endothelial tissue during OGD [91, 92]. OGD in endothelial cells was shown to promote peroxynitrite formation which further initiates the release of mitochondrial HtrA2 [55]. The mitochondrial protease HtrA2 is an acknowledged mitochondrial proapoptotic protein which participates in caspase-dependent apoptosis when released into the cytoplasm [93]. As a potent antioxidant, melatonin application provides significant protection against OGD-induced peroxynitrite formation and mitochondrial HtrA2 release, thereby attenuating ischemic-like injury in endothelial cells [55].
Tissue regeneration is a promising approach for IR injury treatment and stem cells are of great interest to achieve it [94, 95]. Apart from direct beneficial effect, melatonin was demonstrated to improve stem cell therapy efficacy on IR injuries [8]. Melatonin promotes the survival of engrafted mesenchymal stem cells under hypoxia and serum deprivation condition partially through the preservation of mitochondrial membrane potential [96]. In the rat model of small bowel IR injury, combined melatonin–adipose-derived mesenchymal stem cell treatment was shown to increase mitochondrial cytochrome c content, an indicator of mitochondrial integrity, in intestinal mucosal cells, offering beneficial effect against small bowel IR injury [97].
Further perspectives
Among the many recent findings on melatonin, the interaction between melatonin and other important cellular processes of IR injury and the regulatory roles of melatonin in kidney IR injury may hint at possible research opportunities.
We have discussed the free radical-induced mitochondrial damage and the melatonin’s protective effect under IR conditions. Actually, in addition to redox state, mitochondrial dynamics and mitophagy also play important roles in IR injury [45, 46]. The dynamic processes of mitochondrial fusion and fission allow for damaged mitochondria to be segregated and facilitate the equilibration of mitochondrial components such as DNA, proteins, and metabolites [45]. Melatonin’s roles on mitochondrial dynamics are drawing more attention in recent years and have been explored in multiple conditions, including cadmium-induced neurotoxicity [98], 1-methyl-4-phenylpyridinium-induced Parkinson’s disease model [99], lipotoxicity-mediated hepatic stellate cell activation [100], and methamphetamine-induced neurotoxicity [101]. Moreover, the free radical-damaged mitochondria can be selectively removed from the integrated network via an autophagy-related process, termed mitophagy. Melatonin exerts its roles on mitophagy under conditions including liver fibrosis [102], liver cancer [103], and traumatic brain injury [104]. Mitochondrial dynamics and mitophagy are very important for mitochondrial quality control while the roles of melatonin in such processes have not been well clarified under IR conditions. A recent study by Zhou and colleagues demonstrated that IR injury activates Drp1-dependent mitochondrial fission, which subsequently induces voltage-dependent anion channel 1 (VDAC1) oligomerization, hexokinase 2 (HK2) liberation, MPTP opening, PINK1/Parkin upregulation, and ultimately mitophagy-mediated cardiac microcirculation endothelial cell death. Melatonin activates AMPKα and inhibits mitochondrial fission–VDAC1–HK2–MPTP–mitophagy axis, thereby protecting cardiac microvasculature against IR injury [56]. However, the regulatory roles of melatonin on mitochondrial dynamics and mitophagy in cardiomyocytes, or in brain, kidney, lung, and liver have not been fully explored, which deserves much attention in the future.
ER is an important intracellular membranous organelle which is responsible for protein folding and trafficking, lipid synthesis, and the maintenance of calcium homeostasis [105, 106]. ER stress, which is caused by a buildup of misfolded proteins, has been implicated in a series of pathophysiological processes [107]. Melatonin’s roles on ER stress have been studied in multiple conditions [106, 108,109,110,111,112], especially in myocardial [57, 78, 113] and cerebral [60, 114] IR injuries. Moreover, a previous study revealed that ER stress is able to promote mitochondrial damage under the condition of bacterial infection [115]. However, it has been not validated if melatonin exerts its beneficial roles on IR injury via modulating ER stress–mitochondrial damage axis in multiple organs and tissues.
Kidney IR injury occurs in multiple clinical conditions, being a great problem complicating the course and outcome [116]. Similarly, mitochondrial oxidative damage is a significant contributor to the early phases of kidney IR injury and mitochondria-targeted antioxidants were validated to be potential protectors for renal dysfunction caused by IR injury [117]. Melatonin has been demonstrated to preserve renal ultrastructural integrity after IR injury in the male rat, as indicated by decreased serum creatinine level, urine protein-to-creatinine ratio, podocyte injury score, kidney injury score, indicators of glomerular damage, renal tubular-damage, and glomerular integrity [9]. However, the mitochondrial protective effect has not been well investigated in melatonin’s protection of kidney IR injury, which deserves much attention.
Conclusion
Mitochondrial dysfunction is deeply involved in IR injury of various organs and tissues. Excessive free radical induced by ischemia and subsequent reperfusion directly damages multiple mitochondrial components including respiratory chain, metabolism enzymes, and mitochondrial membrane structure. Such damages result in mitochondrial malfunction, ATP shortage, and pro-apoptosis factor release. Moreover, the damaged mitochondria have impaired mitochondrial dynamics and mitophagy which are crucial for quality control of mitochondrial network [45, 46]. The above mitochondrial changes lead to increased apoptosis and exacerbate IR-induced injury in organs and tissues (Fig. 1).
Melatonin, an endogenous indolamine related to circadian rhythms, is a potent agent that could be for use in the treatment of IR injury. The application of melatonin ameliorates IR-induced disturbance in mitochondrial redox state, membrane structure, biogenesis, dynamics, and mitophagy and has attracted attention as an appealing therapeutic strategy. The impressive efficacy and safety of melatonin herald it as a promising agent for the treatment of IR injury. It also deserves our attention that the interaction between melatonin and other important cellular processes of IR injury and the regulatory roles of melatonin in kidney IR injury may hint at possible research opportunities in the future.
Change history
24 April 2018
In the original publication, affiliations were incorrectly published for the authors.
References
Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP (2016) A unifying mechanism for mitochondrial superoxide production during ischemia–reperfusion injury. Cell Metab 23:254–263. doi:10.1016/j.cmet.2015.12.009
Yang Y, Jiang S, Dong Y, Fan C, Zhao L, Yang X, Li J, Di S, Yue L, Liang G, Reiter RJ, Qu Y (2015) Melatonin prevents cell death and mitochondrial dysfunction via a SIRT1-dependent mechanism during ischemic-stroke in mice. J Pineal Res 58:61–70. doi:10.1111/jpi.12193
Ginsberg MD (2016) Expanding the concept of neuroprotection for acute ischemic stroke: the pivotal roles of reperfusion and the collateral circulation. Prog Neurobiol 145–146:46–77. doi:10.1016/j.pneurobio.2016.09.002
Petrosillo G, Colantuono G, Moro N, Ruggiero FM, Tiravanti E, Di Venosa N, Fiore T, Paradies G (2009) Melatonin protects against heart ischemia–reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol 297:H1487–H1493. doi:10.1152/ajpheart.00163.2009
Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, Zhou B, Cao Y, Ritterhoff J, Gu H, Raftery D, Sun H, Tian R (2017) Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia–reperfusion injury. Cell Metab 25:374–385. doi:10.1016/j.cmet.2016.11.005
Okatani Y, Wakatsuki A, Reiter RJ, Enzan H, Miyahara Y (2003) Protective effect of melatonin against mitochondrial injury induced by ischemia and reperfusion of rat liver. Eur J Pharmacol 469:145–152
Hu B, Guo Y, Garbacz WG, Jiang M, Xu M, Huang H, Tsung A, Billiar TR, Ramakrishnan SK, Shah YM, Lam KS, Huang M, Xie W (2015) Fatty acid binding protein-4 (FABP4) is a hypoxia inducible gene that sensitizes mice to liver ischemia/reperfusion injury. J Hepatol 63:855–862. doi:10.1016/j.jhep.2015.05.030
Yip HK, Chang YC, Wallace CG, Chang LT, Tsai TH, Chen YL, Chang HW, Leu S, Zhen YY, Tsai CY, Yeh KH, Sun CK, Yen CH (2013) Melatonin treatment improves adipose-derived mesenchymal stem cell therapy for acute lung ischemia–reperfusion injury. J Pineal Res 54:207–221. doi:10.1111/jpi.12020
Yip HK, Yang CC, Chen KH, Huang TH, Chen YL, Zhen YY, Sung PH, Chiang HJ, Sheu JJ, Chang CL, Chen CH, Chang HW, Chen YT (2015) Combined melatonin and exendin-4 therapy preserves renal ultrastructural integrity after ischemia–reperfusion injury in the male rat. J Pineal Res 59:434–447. doi:10.1111/jpi.12273
Block H, Herter JM, Rossaint J, Stadtmann A, Kliche S, Lowell CA, Zarbock A (2012) Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia–reperfusion injury. J Exp Med 209:407–421. doi:10.1084/jem.20111493
Zong H, Li X, Lin H, Hou C, Ma F (2017) Lipoxin A4 pretreatment mitigates skeletal muscle ischemia–reperfusion injury in rats. Am J Transl Res 9:1139–1150
Andreas M, Schmid AI, Keilani M, Doberer D, Bartko J, Crevenna R, Moser E, Wolzt M (2011) Effect of ischemic preconditioning in skeletal muscle measured by functional magnetic resonance imaging and spectroscopy: a randomized crossover trial. J Cardiovasc Magn Reson 13:32. doi:10.1186/1532-429x-13-32
Dokmeci D, Kanter M, Inan M, Aydogdu N, Basaran UN, Yalcin O, Turan FN (2007) Protective effects of ibuprofen on testicular torsion/detorsion-induced ischemia/reperfusion injury in rats. Arch Toxicol 81:655–663. doi:10.1007/s00204-007-0189-2
Maessen MF, van Mil AC, Straathof Y, Riksen NP, Rongen GA, Hopman MT, Eijsvogels TM, Thijssen DH (2017) Impact of lifelong exercise training on endothelial ischemia–reperfusion and ischemic preconditioning in humans. Am J Physiol Regul Integr Comp Physiol. doi:10.1152/ajpregu.00466.2016
Ibanez B, Heusch G, Ovize M, Van de Werf F (2015) Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65:1454–1471. doi:10.1016/j.jacc.2015.02.032
Zimmerman MA, Tak E, Ehrentraut SF, Kaplan M, Giebler A, Weng T, Choi DS, Blackburn MR, Kam I, Eltzschig HK, Grenz A (2013) Equilibrative nucleoside transporter (ENT)-1-dependent elevation of extracellular adenosine protects the liver during ischemia and reperfusion. Hepatology 58:1766–1778. doi:10.1002/hep.26505
Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (2016) Melatonin as an antioxidant: under promises but over delivers. J Pineal Res 61:253–278. doi:10.1111/jpi.12360
Zephy D, Ahmad J (2015) Type 2 diabetes mellitus: role of melatonin and oxidative stress. Diabetes Metab Syndr 9:127–131. doi:10.1016/j.dsx.2014.09.018
Peschke E, Bahr I, Muhlbauer E (2015) Experimental and clinical aspects of melatonin and clock genes in diabetes. J Pineal Res 59:1–23. doi:10.1111/jpi.12240
Parry BL, Meliska CJ, Sorenson DL, Lopez AM, Martinez LF, Nowakowski S, Elliott JA, Hauger RL, Kripke DF (2008) Plasma melatonin circadian rhythm disturbances during pregnancy and postpartum in depressed women and women with personal or family histories of depression. Am J Psychiatry 165:1551–1558. doi:10.1176/appi.ajp.2008.08050709
Hickie IB, Rogers NL (2011) Novel melatonin-based therapies: potential advances in the treatment of major depression. Lancet 378:621–631. doi:10.1016/s0140-6736(11)60095-0
Ozdemir D, Uysal N, Tugyan K, Gonenc S, Acikgoz O, Aksu I, Ozkan H (2007) The effect of melatonin on endotoxemia-induced intestinal apoptosis and oxidative stress in infant rats. Intens Care Med 33:511–516. doi:10.1007/s00134-006-0492-z
Garcia JA, Volt H, Venegas C, Doerrier C, Escames G, Lopez LC, Acuna-Castroviejo D (2015) Disruption of the NF-kappaB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-alpha and blocks the septic response in mice. FASEB J 29:3863–3875. doi:10.1096/fj.15-273656
Deng Y, Jiao C, Mi C, Xu B, Li Y, Wang F, Liu W, Xu Z (2015) Melatonin inhibits manganese-induced motor dysfunction and neuronal loss in mice: involvement of oxidative stress and dopaminergic neurodegeneration. Mol Neurobiol 51:68–88. doi:10.1007/s12035-014-8789-3
Hardeland R, Cardinali DP, Brown GM, Pandi-Perumal SR (2015) Melatonin and brain inflammaging. Prog Neurobiol 127–128:46–63. doi:10.1016/j.pneurobio.2015.02.001
Cardinali DP, Hardeland R (2017) Inflammaging, metabolic syndrome and melatonin: a call for treatment studies. Neuroendocrinology 104:382–397. doi:10.1159/000446543
Demirtas CY, Pasaoglu OT, Bircan FS, Kantar S, Turkozkan N (2015) The investigation of melatonin effect on liver antioxidant and oxidant levels in fructose-mediated metabolic syndrome model. Eur Rev Med Pharmacol Sci 19:1915–1921
Pei HF, Hou JN, Wei FP, Xue Q, Zhang F, Peng CF, Yang Y, Tian Y, Feng J, Du J, He L, Li XC, Gao EH, Li Yang YJ (2017) Melatonin attenuates postmyocardial infarction injury via increasing Tom70 expression. J Pineal Res 62:e12371. doi:10.1111/jpi.12371
Nduhirabandi F, Lamont K, Albertyn Z, Opie LH, Lecour S (2016) Role of toll-like receptor 4 in melatonin-induced cardioprotection. J Pineal Res 60:39–47. doi:10.1111/jpi.12286
Venegas C, Garcia JA, Escames G, Ortiz F, Lopez A, Doerrier C, Garcia-Corzo L, Lopez LC, Reiter RJ, Acuna-Castroviejo D (2012) Extrapineal melatonin: analysis of its subcellular distribution and daily fluctuations. J Pineal Res 52:217–227. doi:10.1111/j.1600-079X.2011.00931.x
Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ (2013) Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin’s primary function and evolution in eukaryotes. J Pineal Res 54:127–138. doi:10.1111/jpi.12026
Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, Vriend J, Tan DX, Reiter RJ (2015) Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res 59:403–419. doi:10.1111/jpi.12267
Ertracht O, Malka A, Atar S, Binah O (2014) The mitochondria as a target for cardioprotection in acute myocardial ischemia. Pharmacol Ther 142:33–40. doi:10.1016/j.pharmthera.2013.11.003
Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL (2017) Mitochondrial dysfunction and myocardial ischemia–reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol 57:535–565. doi:10.1146/annurev-pharmtox-010715-103335
Brown DA, Sabbah HN, Shaikh SR (2013) Mitochondrial inner membrane lipids and proteins as targets for decreasing cardiac ischemia/reperfusion injury. Pharmacol Ther 140:258–266. doi:10.1016/j.pharmthera.2013.07.005
Mukherjee D, Ghosh AK, Dutta M, Mitra E, Mallick S, Saha B, Reiter RJ, Bandyopadhyay D (2015) Mechanisms of isoproterenol-induced cardiac mitochondrial damage: protective actions of melatonin. J Pineal Res 58:275–290. doi:10.1111/jpi.12213
Hamada F, Watanabe K, Wakatsuki A, Nagai R, Shinohara K, Hayashi Y, Imamura R, Fukaya T (2010) Therapeutic effects of maternal melatonin administration on ischemia/reperfusion-induced oxidative cerebral damage in neonatal rats. Neonatology 98:33–40. doi:10.1159/000264205
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. doi:10.1161/circresaha.116.305348
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion—from mechanism to translation. Nat Med 17:1391–1401. doi:10.1038/nm.2507
Musatov A, Robinson NC (2012) Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic Res 46:1313–1326. doi:10.3109/10715762.2012.717273
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950. doi:10.1152/physrev.00026.2013
Tajeddine N (2016) How do reactive oxygen species and calcium trigger mitochondrial membrane permeabilisation? Biochim Biophys Acta 1860:1079–1088. doi:10.1016/j.bbagen.2016.02.013
Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, Hines KJ, Smith DJ, Eguchi A, Vallem S, Shaikh F, Cheung M, Leonard NJ, Stolakis RS, Wolfers MP, Ibetti J, Chuprun JK, Jog NR, Houser SR, Koch WJ, Elrod JW, Madesh M (2015) SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell 60:47–62. doi:10.1016/j.molcel.2015.08.009
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y (2017) Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc. doi:10.1161/jaha.116.005328
Anzell AR, Maizy R, Przyklenk K, Sanderson TH (2017) Mitochondrial quality control and disease: insights into ischemia–reperfusion injury. Mol Neurobiol. doi:10.1007/s12035-017-0503-9
Nan J, Zhu W, Rahman MS, Liu M, Li D, Su S, Zhang N, Hu X, Yu H, Gupta MP, Wang J (2017) Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta 1864:1260–1273. doi:10.1016/j.bbamcr.2017.03.006
Ma Z, Yang Y, Fan C, Han J, Wang D, Di S, Hu W, Liu D, Li X, Reiter RJ, Yan X (2016) Melatonin as a potential anticarcinogen for non-small-cell lung cancer. Oncotarget 7:46768–46784. doi:10.18632/oncotarget.8776
Brazao V, Santello FH, Colato RP, Mazotti TT, Tazinafo LF, Toldo MP, do Vale GT, Tirapelli CR, do Prado Jr JC (2017) Melatonin: antioxidant and modulatory properties in age-related changes during Trypanosoma cruzi infection. J Pineal Res. doi:10.1111/jpi.12409
Yu L, Sun Y, Cheng L, Jin Z, Yang Y, Zhai M, Pei H, Wang X, Zhang H, Meng Q, Zhang Y, Yu S, Duan W (2014) Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J Pineal Res 57:228–238. doi:10.1111/jpi.12161
Fischer TW, Kleszczynski K, Hardkop LH, Kruse N, Zillikens D (2013) Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVR-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2′-deoxyguanosine) in ex vivo human skin. J Pineal Res 54:303–312. doi:10.1111/jpi.12018
Yu L, Gong B, Duan W, Fan C, Zhang J, Li Z, Xue X, Xu Y, Meng D, Li B, Zhang M, Bin Z, Jin Z, Yu S, Yang Y, Wang H (2017) Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK-PGC-1alpha-SIRT3 signaling. Sci Rep 7:41337. doi:10.1038/srep41337
Nair SM, Rahman RM, Clarkson AN, Sutherland BA, Taurin S, Sammut IA, Appleton I (2011) Melatonin treatment following stroke induction modulates l-arginine metabolism. J Pineal Res 51:313–323. doi:10.1111/j.1600-079X.2011.00891.x
Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF (2004) Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J 18:869–871. doi:10.1096/fj.03-1031fje
Chen HH, Chen YT, Yang CC, Chen KH, Sung PH, Chiang HJ, Chen CH, Chua S, Chung SY, Chen YL, Huang TH, Kao GS, Chen SY, Lee MS, Yip HK (2016) Melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria against hepatic ischemia–reperfusion injury in rats through suppression of mitochondrial permeability transition. J Pineal Res 61:52–68. doi:10.1111/jpi.12326
Han F, Tao RR, Zhang GS, Lu YM, Liu LL, Chen YX, Lou YJ, Fukunaga K, Hong ZH (2011) Melatonin ameliorates ischemic-like injury-evoked nitrosative stress: involvement of HtrA2/PED pathways in endothelial cells. J Pineal Res 50:281–291. doi:10.1111/j.1600-079X.2010.00838.x
Jayaraman B, Smith AM, Fernandes JD, Frankel AD (2016) Oligomeric viral proteins: small in size, large in presence. Crit Rev Biochem Mol Biol 51:379–394. doi:10.1080/10409238.2016.1215406
He B, Zhao Y, Xu L, Gao L, Su Y, Lin N, Pu J (2016) The nuclear melatonin receptor RORalpha is a novel endogenous defender against myocardial ischemia/reperfusion injury. J Pineal Res 60:313–326. doi:10.1111/jpi.12312
Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, Wang N, Liang Z, Li Y, Chen W, Yi D, Yu S (2013) JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res 55:275–286. doi:10.1111/jpi.12070
Ramos E, Patino P, Reiter RJ, Gil-Martin E, Marco-Contelles J, Parada E, Los Rios C, Romero A, Egea J (2017) Ischemic brain injury: new insights on the protective role of melatonin. Free Radic Biol Med 104:32–53. doi:10.1016/j.freeradbiomed.2017.01.005
Feng D, Wang B, Wang L, Abraham N, Tao K, Huang L, Shi W, Dong Y, Qu Y (2017) Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res 62:e12395. doi:10.1111/jpi.12395
Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Sirianni AC, Zhu S, Day AL, Kristal BS, Friedlander RM (2009) Methazolamide and melatonin inhibit mitochondrial cytochrome C release and are neuroprotective in experimental models of ischemic injury. Stroke 40:1877–1885. doi:10.1161/strokeaha.108.540765
Kaur C, Sivakumar V, Robinson R, Foulds WS, Luu CD, Ling EA (2013) Neuroprotective effect of melatonin against hypoxia-induced retinal ganglion cell death in neonatal rats. J Pineal Res 54:190–206. doi:10.1111/jpi.12016
Han YX, Zhang SH, Wang XM, Wu JB (2006) Inhibition of mitochondria responsible for the anti-apoptotic effects of melatonin during ischemia–reperfusion. J Zhejiang Univ Sci B 7:142–147. doi:10.1631/jzus.2006.B0142
Wakatsuki A, Okatani Y, Shinohara K, Ikenoue N, Fukaya T (2001) Melatonin protects against ischemia/reperfusion-induced oxidative damage to mitochondria in fetal rat brain. J Pineal Res 31:167–172
Watanabe K, Wakatsuki A, Shinohara K, Ikenoue N, Yokota K, Fukaya T (2004) Maternally administered melatonin protects against ischemia and reperfusion-induced oxidative mitochondrial damage in premature fetal rat brain. J Pineal Res 37:276–280. doi:10.1111/j.1600-079X.2004.00167.x
Berger HR, Morken TS, Vettukattil R, Brubakk AM, Sonnewald U, Wideroe M (2016) No improvement of neuronal metabolism in the reperfusion phase with melatonin treatment after hypoxic-ischemic brain injury in the neonatal rat. J Neurochem 136:339–350. doi:10.1111/jnc.13420
Revuelta M, Arteaga O, Montalvo H, Alvarez A, Hilario E, Martinez-Ibarguen A (2016) Antioxidant treatments recover the alteration of auditory-evoked potentials and reduce morphological damage in the inferior colliculus after perinatal asphyxia in rat. Brain Pathol 26:186–198. doi:10.1111/bpa.12272
Zhou H, Wang J, Jiang J, Stavrovskaya IG, Li M, Li W, Wu Q, Zhang X, Luo C, Zhou S, Sirianni AC, Sarkar S, Kristal BS, Friedlander RM, Wang X (2014) N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J Neurosci 34:2967–2978. doi:10.1523/jneurosci.1948-13.2014
Duan Q, Wang Z, Lu T, Chen J, Wang X (2006) Comparison of 6-hydroxylmelatonin or melatonin in protecting neurons against ischemia/reperfusion-mediated injury. J Pineal Res 41:351–357. doi:10.1111/j.1600-079X.2006.00374.x
Sarkar S, Mukherjee A, Das N, Swarnakar S (2017) Protective roles of nanomelatonin in cerebral ischemia–reperfusion of aged brain: matrix metalloproteinases as regulators. Exp Gerontol 92:13–22. doi:10.1016/j.exger.2017.03.009
Mukherjee D, Ghosh AK, Bandyopadhyay A, Basu A, Datta S, Pattari SK, Reiter RJ, Bandyopadhyay D (2012) Melatonin protects against isoproterenol-induced alterations in cardiac mitochondrial energy-metabolizing enzymes, apoptotic proteins, and assists in complete recovery from myocardial injury in rats. J Pineal Res 53:166–179
Harner M, Neupert W, Deponte M (2011) Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. EMBO J 30:3232–3241. doi:10.1038/emboj.2011.235
Hu J, Zhang L, Yang Y, Guo Y, Fan Y, Zhang M, Man W, Gao E, Hu W, Reiter RJ, Wang H, Sun D (2017) Melatonin alleviates postinfarction cardiac remodeling and dysfunction by inhibiting Mst1. J Pineal Res 62:e12368. doi:10.1111/jpi.12368
Petrosillo G, Di Venosa N, Pistolese M, Casanova G, Tiravanti E, Colantuono G, Federici A, Paradies G, Ruggiero FM (2006) Protective effect of melatonin against mitochondrial dysfunction associated with cardiac ischemia–reperfusion: role of cardiolipin. FASEB J 20:269–276. doi:10.1096/fj.05-4692com
Luo S, Gu X, Ma F, Liu C, Shen Y, Ge R, Zhu Y (2016) ZYZ451 protects cardiomyocytes from hypoxia-induced apoptosis via enhancing MnSOD and STAT3 interaction. Free Radic Biol Med 92:1–14. doi:10.1016/j.freeradbiomed.2015.12.026
Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH (2009) Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci 29:7003–7014. doi:10.1523/jneurosci.1110-09.2009
Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ (2015) The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 78:23–34. doi:10.1016/j.yjmcc.2014.11.005
Gao L, Zhao YC, Liang Y, Lin XH, Tan YJ, Wu DD, Li XZ, Ye BZ, Kong FQ, Sheng JZ, Huang HF (2016) The impaired myocardial ischemic tolerance in adult offspring of diabetic pregnancy is restored by maternal melatonin treatment. J Pineal Res 61:340–352. doi:10.1111/jpi.12351
Yu J, Wei J, Ji L, Hong X (2014) Exploration on mechanism of a new type of melatonin receptor agonist Neu-p11 in hypoxia-reoxygenation injury of myocardial cells. Cell Biochem Biophys 70:999–1003. doi:10.1007/s12013-014-0009-2
Palmer JW, Tandler B, Hoppel CL (1986) Heterogeneous response of subsarcolemmal heart mitochondria to calcium. Am J Physiol 250:H741–H748
Li Y, Yang Y, Feng Y, Yan J, Fan C, Jiang S, Qu Y (2014) A review of melatonin in hepatic ischemia/reperfusion injury and clinical liver disease. Ann Med 46:503–511. doi:10.3109/07853890.2014.934275
Rodriguez-Reynoso S, Leal C, Portilla E, Olivares N, Muniz J (2001) Effect of exogenous melatonin on hepatic energetic status during ischemia/reperfusion: possible role of tumor necrosis factor-alpha and nitric oxide. J Surg Res 100:141–149. doi:10.1006/jsre.2001.6185
Freitas I, Bertone V, Guarnaschelli C, Ferrigno A, Boncompagni E, Rizzo V, Reiter RJ, Barni S, Vairetti M (2006) In situ demonstration of improvement of liver mitochondria function by melatonin after cold ischemia. In Vivo 20:229–237
Kim SH, Lee SM (2008) Cytoprotective effects of melatonin against necrosis and apoptosis induced by ischemia/reperfusion injury in rat liver. J Pineal Res 44:165–171. doi:10.1111/j.1600-079X.2007.00504.x
Zhang Y, Gao X, Garavito RM (2011) Biochemical characterization of human dynamin-like protein 1. J Biochem 150:627–633. doi:10.1093/jb/mvr102
Wang WZ, Fang XH, Stephenson LL, Zhang X, Khiabani KT, Zamboni WA (2011) Melatonin attenuates I/R-induced mitochondrial dysfunction in skeletal muscle. J Surg Res 171:108–113. doi:10.1016/j.jss.2010.01.019
Filho DW, Torres MA, Bordin AL, Crezcynski-Pasa TB, Boveris A (2004) Spermatic cord torsion, reactive oxygen and nitrogen species and ischemia–reperfusion injury. Mol Aspects Med 25:199–210. doi:10.1016/j.mam.2004.02.020
Okur MH, Arslan S, Aydogdu B, Zeytun H, Basuguy E, Arslan MS, Ibiloglu I, Kaplan I (2017) Protective effect of cordycepin on experimental testicular ischemia/reperfusion injury in rats. J Invest Surg. doi:10.1080/08941939.2016.1246629
Kanter M (2010) Protective effects of melatonin on testicular torsion/detorsion-induced ischemia–reperfusion injury in rats. Exp Mol Pathol 89:314–320. doi:10.1016/j.yexmp.2010.07.006
Nagai R, Watanabe K, Wakatsuki A, Hamada F, Shinohara K, Hayashi Y, Imamura R, Fukaya T (2008) Melatonin preserves fetal growth in rats by protecting against ischemia/reperfusion-induced oxidative/nitrosative mitochondrial damage in the placenta. J Pineal Res 45:271–276. doi:10.1111/j.1600-079X.2008.00586.x
Tao RR, Huang JY, Shao XJ, Ye WF, Tian Y, Liao MH, Fukunaga K, Lou YJ, Han F, Lu YM (2013) Ischemic injury promotes Keap1 nitration and disturbance of antioxidative responses in endothelial cells: a potential vasoprotective effect of melatonin. J Pineal Res 54:271–281. doi:10.1111/jpi.12009
Han F, Chen YX, Lu YM, Huang JY, Zhang GS, Tao RR, Ji YL, Liao MH, Fukunaga K, Qin ZH (2011) Regulation of the ischemia-induced autophagy-lysosome processes by nitrosative stress in endothelial cells. J Pineal Res 51:124–135. doi:10.1111/j.1600-079X.2011.00869.x
Wang K, Yuan Y, Liu X, Lau WB, Zuo L, Wang X, Ma L, Jiao K, Shang J, Wang W, Ma X, Liu H (2016) Cardiac specific overexpression of mitochondrial Omi/HtrA2 induces myocardial apoptosis and cardiac dysfunction. Sci Rep 6:37927. doi:10.1038/srep37927
Preda MB, Ronningen T, Burlacu A, Simionescu M, Moskaug JO, Valen G (2014) Remote transplantation of mesenchymal stem cells protects the heart against ischemia–reperfusion injury. Stem Cells 32:2123–2134. doi:10.1002/stem.1687
Li GH, Luo B, Lv YX, Zheng F, Wang L, Wei MX, Li XY, Zhang L, Wang JN, Chen SY, Tang JM, He X (2016) Dual effects of VEGF-B on activating cardiomyocytes and cardiac stem cells to protect the heart against short- and long-term ischemia–reperfusion injury. J Transl Med 14:116. doi:10.1186/s12967-016-0847-3
Wang F, Zhou H, Du Z, Chen X, Zhu F, Wang Z, Zhang Y, Lin L, Qian M, Zhang X, Li X, Hao A (2015) Cytoprotective effect of melatonin against hypoxia/serum deprivation-induced cell death of bone marrow mesenchymal stem cells in vitro. Eur J Pharmacol 748:157–165. doi:10.1016/j.ejphar.2014.09.033
Chang CL, Sung PH, Sun CK, Chen CH, Chiang HJ, Huang TH, Chen YL, Zhen YY, Chai HT, Chung SY, Tong MS, Chang HW, Chen HH, Yip HK (2015) Protective effect of melatonin-supported adipose-derived mesenchymal stem cells against small bowel ischemia–reperfusion injury in rat. J Pineal Res 59:206–220. doi:10.1111/jpi.12251
Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang H, Tang J, Li H, Feng M, Deng P, Guo P, Tian L, Xie J, He M, Lu Y, Zhong M, Zhang Y, Wang W, Reiter RJ, Yu Z, Zhou Z (2016) Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J Pineal Res 60:291–302. doi:10.1111/jpi.12310
Chuang JI, Pan IL, Hsieh CY, Huang CY, Chen PC, Shin JW (2016) Melatonin prevents the dynamin-related protein 1-dependent mitochondrial fission and oxidative insult in the cortical neurons after 1-methyl-4-phenylpyridinium treatment. J Pineal Res 61:230–240. doi:10.1111/jpi.12343
Das N, Mandala A, Naaz S, Giri S, Jain M, Bandyopadhyay D, Reiter RJ, Roy SS (2017) Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J Pineal Res 62:e12404. doi:10.1111/jpi.12404
Parameyong A, Charngkaew K, Govitrapong P, Chetsawang B (2013) Melatonin attenuates methamphetamine-induced disturbances in mitochondrial dynamics and degeneration in neuroblastoma SH-SY5Y cells. J Pineal Res 55:313–323. doi:10.1111/jpi.12078
Kang JW, Hong JM, Lee SM (2016) Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res 60:383–393. doi:10.1111/jpi.12319
Prieto-Dominguez N, Ordonez R, Fernandez A, Mendez-Blanco C, Baulies A, Garcia-Ruiz C, Fernandez-Checa JC, Mauriz JL, Gonzalez-Gallego J (2016) Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J Pineal Res 61:396–407. doi:10.1111/jpi.12358
Lin C, Chao H, Li Z, Xu X, Liu Y, Hou L, Liu N, Ji J (2016) Melatonin attenuates traumatic brain injury-induced inflammation: a possible role for mitophagy. J Pineal Res 61:177–186. doi:10.1111/jpi.12337
Hu W, Ma Z, Di S, Jiang S, Li Y, Fan C, Yang Y, Wang D (2016) Snapshot: implications for melatonin in endoplasmic reticulum homeostasis. Br J Pharmacol 173:3431–3442. doi:10.1111/bph.13651
de Luxan-Delgado B, Potes Y, Rubio-Gonzalez A, Caballero B, Solano JJ, Fernandez-Fernandez M, Bermudez M, Rodrigues Moreira Guimaraes M, Vega-Naredo I, Boga JA, Coto-Montes A (2016) Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin-deficient mice. J Pineal Res 61:108–123. doi:10.1111/jpi.12333
Oakes SA, Papa FR (2015) The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 10:173–194. doi:10.1146/annurev-pathol-012513-104649
Park JH, Shim HM, Na AY, Bae KC, Bae JH, Im SS, Cho HC, Song DK (2014) Melatonin prevents pancreatic beta-cell loss due to glucotoxicity: the relationship between oxidative stress and endoplasmic reticulum stress. J Pineal Res 56:143–153. doi:10.1111/jpi.12106
San-Miguel B, Crespo I, Sanchez DI, Gonzalez-Fernandez B, Ortiz de Urbina JJ, Tunon MJ, Gonzalez-Gallego J (2015) Melatonin inhibits autophagy and endoplasmic reticulum stress in mice with carbon tetrachloride-induced fibrosis. J Pineal Res 59:151–162. doi:10.1111/jpi.12247
Wu SM, Lin WY, Shen CC, Pan HC, Keh-Bin W, Chen YC, Jan YJ, Lai DW, Tang SC, Tien HR, Chiu CS, Tsai TC, Lai YL, Sheu ML (2016) Melatonin set out to ER stress signaling thwarts epithelial mesenchymal transition and peritoneal dissemination via calpain-mediated C/EBPbeta and NFkappaB cleavage. J Pineal Res 60:142–154. doi:10.1111/jpi.12295
Hosseinzadeh A, Kamrava SK, Joghataei MT, Darabi R, Shakeri-Zadeh A, Shahriari M, Reiter RJ, Ghaznavi H, Mehrzadi S (2016) Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J Pineal Res 61:411–425. doi:10.1111/jpi.12362
Fernandez A, Ordonez R, Reiter RJ, Gonzalez-Gallego J, Mauriz JL (2015) Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res 59:292–307. doi:10.1111/jpi.12264
Yu L, Liang H, Dong X, Zhao G, Jin Z, Zhai M, Yang Y, Chen W, Liu J, Yi W, Yang J, Yi D, Duan W, Yu S (2015) Reduced silent information regulator 1 signaling exacerbates myocardial ischemia–reperfusion injury in type 2 diabetic rats and the protective effect of melatonin. J Pineal Res 59:376–390. doi:10.1111/jpi.12269
Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proietti F, Balduini W (2014) Melatonin reduces endoplasmic reticulum stress and preserves sirtuin 1 expression in neuronal cells of newborn rats after hypoxia–ischemia. J Pineal Res 57:192–199. doi:10.1111/jpi.12156
Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, Yin XM, O’Riordan MX (2015) Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 43:451–462. doi:10.1016/j.immuni.2015.08.008
Kezic A, Spasojevic I, Lezaic V, Bajcetic M (2016) Mitochondria-targeted antioxidants: future perspectives in kidney ischemia reperfusion injury. Oxid Med Cell Longev 2016:2950503. doi:10.1155/2016/2950503
Dare AJ, Bolton EA, Pettigrew GJ, Bradley JA, Saeb-Parsy K, Murphy MP (2015) Protection against renal ischemia–reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol 5:163–168. doi:10.1016/j.redox.2015.04.008
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81500263) and China Postdoctoral Science Foundation (2016T90973 and 2015M572681).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ma, Z., Xin, Z., Di, W. et al. Melatonin and mitochondrial function during ischemia/reperfusion injury. Cell. Mol. Life Sci. 74, 3989–3998 (2017). https://doi.org/10.1007/s00018-017-2618-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2618-6