
Abstract
Mitochondria are dynamic organelles that supply energy required to drive key cellular processes, such as survival, proliferation, and migration. Critical to all of these processes are changes in mitochondrial architecture, a mechanical mechanism encompassing both fusion and fragmentation (fission) of the mitochondrial network. Changes to mitochondrial shape, size, and localization occur in a regulated manner to maintain energy and metabolic homeostasis, while deregulation of mitochondrial dynamics is associated with the onset of metabolic dysfunction and disease. In cancers, oncogenic signals that drive excessive proliferation, increase intracellular stress, and limit nutrient supply are all able to alter the bioenergetic and biosynthetic requirements of cancer cells. Consequently, mitochondrial function and shape rapidly adapt to these hostile conditions to support cancer cell proliferation and evade activation of cell death programs. In this review, we will discuss the molecular mechanisms governing mitochondrial dynamics and integrate recent insights into how changes in mitochondrial shape affect cellular migration, differentiation, apoptosis, and opportunities for the development of novel targeted cancer therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondria are double membrane organelles that consist of an outer mitochondrial membrane (OMM), inner membrane space (IMS), inner mitochondrial membrane (IMM), and matrix. The IMM has numerous folds called cristae and are sites for electron transport chain (ETC) assembly and oxidative phosphorylation (OXPHOS). Mitochondria also contain a genome (mitochondrial DNA; mtDNA) that exists in the matrix as thousands of copies of circular, double stranded DNA. mtDNA is comprised of 16,569 base pairs that encode for 13 protein ETC subunits, 22 transfer RNAs, and 2 ribosomal RNAs. Other proteins that function in mitochondria are encoded by the nuclear genome and contain a mitochondrial localization signal in their amino-terminus that allows for efficient delivery of the polypeptide to mitochondria. In addition to producing energy in the form of adenosine triphosphate (ATP), mitochondria also regulate biogenesis of iron-sulphur clusters, oxidation–reduction (redox) status, synthesize macromolecule precursors, and initiate apoptosis.
Mitochondria have a unique ability to regulate their morphology in response to various cellular stimuli. For example, during nutrient deprivation, mitochondria fuse together and create interconnected filamentous networks to share nutrient precursors, mtDNA, ETC components, and maintain OXPHOS. Conversely, mitochondrial fission produces smaller, fragmented mitochondria, which is important for mitochondrial movement to regions of high energy demand or to allow for equal mitochondrial distribution to daughter cells following mitosis [1]. Mitochondrial dysfunction has been associated with a number of degenerative diseases, such as Leigh’s disease and Parkinson’s disease [2], while disruption to mitochondrial dynamics has also been implicated in several neuropathies and cardiomyopathies [2]. Mitochondrial function and cellular metabolism in cancer have been an area of intense research over several decades; however, recently a number of studies have implicated a role changes in mitochondrial architecture during tumorigenesis.
Over the past decade, we have gained knowledge about how mitochondrial dynamic proteins are regulated at the transcriptional, translational, and post-translational levels as well as the cell-specific and intracellular contexts in which different mitochondrial morphologies are favoured. Nevertheless, there are a number of questions regarding how mitochondrial dynamics are regulated in cancers. For example, are mitochondrial dynamics regulated by oncogenic signaling? Do mitochondrial dynamics play a role in tumorgenic processes, such as differentiation and migration? What is the relationship between mitochondrial shape and the cell death machinery and what are the clinical opportunities for targeting mitochondrial dynamics proteins in cancer? We outline here some recent biochemical and cellular studies that have provided insights to these questions.
Mitochondrial dynamics machinery
Pioneering work by Lewis and Lewis over a century ago established that mitochondria constantly move and divide [3]. Since then, technological advances, such as mitochondrial-specific dyes (i.e., tetramethylrhoadamine, ethyl ester [TMRE]), and fluorescently-labelled proteins (i.e., green fluorescent-mito) [4, 5], have allowed researchers to depict a more accurate representation of fused and fragmented mitochondrial architecture and describe how these particular shapes directly relate to mitochondrial function. Mitochondrial dynamics is regulated by a number of highly conserved large guanosine triphosphatases (GTPases). Fusion of the OMM is mediated by mitofusin 1 and mitofusion 2 (Mfn1 and Mfn2), while IMM fusion is regulated by Optic Atrophy 1 (OPA1). On the other hand, mitochondrial fission is controlled by dynamin-related protein 1 (DRP1).
Outer mitochondrial membrane fusion
Mfn1 and 2 co-localize to the OMM and regulate mitochondrial fusion (Figs. 1a, 2a). Both mitofusins are broadly expressed in a range of tissues, although the relative levels of either protein can vary dramatically. For example, Mfn1 is ubiquitously expressed in most tissues, whereas Mfn2 has higher expression in skeletal muscle, brain, and heart, suggesting a dominant role for Mfn2-mediated fusion in these tissues. Generation of homozygous Mfn1 or Mfn2-null mice revealed an embryonic lethal phenotype [6]. However, while the mechanism underlying Mfn1 −/− lethality required further investigation, Mfn2 −/− mice died mid-gestation due to reduced trophoblast giant cells in the placenta and resulted in improper placental development, indicating that Mfn1 and Mfn2 are not functionally redundant [6, 7]. In humans, heterozygous missense mutations within Mfn2 are associated with an autosomal peripheral neuropathy called Charcot-Marie-Tooth hereditary neuropathy type 2 A (CMT) [8]. Electron micrographs of sural nerve specimens from CMT patients contain small, rounded mitochondria, suggesting that defects in mitochondrial fusion may contribute to CMT pathology [9, 10]. Collectively, these studies highlight the importance of mitofusins to embryogenesis and the development of specific tissues.

Dynamins are large GTPases that regulate mitochondrial fusion and fission. Schematic of Mfn1, Mfn2, OPA1, and DRP1 protein structures. a Amino-terminal region of Mfn1 and Mfn2 contain a GTPase domain. A centralized transmembrane (TM) domain enables insertion of the protein into the OMM. Flanking the TM region are two coiled–coiled (CC1, CC2) domains that permit homo- or hetero-dimerization of Mfn1 and Mfn2 proteins, and allow for fusion between adjacent mitochondria. b OPA1 consists of an amino-terminal mitochondrial localization signal, followed by two transmembrane domains, and a CC1 domain. The GTPase region is centrally located and is followed by a second CC domain (CC2), and GTPase effector domain (GED) at the carboxyl-terminus. OPA1 is proteolytically processed to produce long (L-OPA1) and short (S-OPA1) isoforms. c DRP1 consists of an amino-terminally located GTPase domain; followed by a middle domain that is involved in self-assembly and a variable CC region called “Insert B”. At the carboxyl-terminus is a GED, which is involved in intra-molecular interactions with the GTPase domain

Mechanisms of mitochondrial fusion and fission. a Fusion of the OMM is mediated by Mfn1 and Mfn2. The orientation of Mfn1 and Mfn2 domains suggests that the amino and carboxyl termini face the cytosol to facilitate interactions with other mitofusins on adjacent mitochondria, while the TM domain is embedded within the OMM and IMS. The mitofusin GTPase domain is required to pull the two opposing OMMs together resulting in bilayer fusion. Fusion of the OMM requires homo- or heterotypic interactions between Mfn1 and Mfn2, although heterotypic dimers (i.e., Mfn1:Mfn2) are more efficient at fusion compared to homotypic complexes. IMM fusion is coordinated by OPA1. L-OPA1 isoforms are anchored within the IMM and have their GTPase and GED exposed to the IMS. Proteolytic cleavage of L-OPA1 results in the generation of S-OPA1, allowing both to coordinate IMM fusion. OPA1 interacts with both Mfn1 and Mfn2 to form a bridge between the IMM and OMM, and is required for lipid mixing during fusion. b DRP1 regulates mitochondrial fission. Soluble, cytosolic DRP1 exists in the cytosol as dimers or trimers. Following activation via phosphorylation at Ser616, DRP1 translocates to the OMM where it binds to adaptor proteins (e.g., MFF, MiD49, MiD51, and FIS1). At the OMM, DRP1 undergoes conformational change, so that the middle domain and GED form a stalk-like structure. The DRP1 GTPase domain faces away from the OMM and connects with other DRP1 proteins on mitochondria. Once a DRP1 helix has completely spiralled around mitochondrion, GTP hydrolysis causes constriction of the helix and scission of the OMM and IMM. Actin polymerization also occurs at ER-mitochondrial junctions and facilitates migration of individual mitochondrion away from each other during fission
Genetic ablation of Mfn1 and/or Mfn2 significantly fragments the mitochondrial network and causes severe cellular defects, including disruption to mitochondrial membrane potential (ψ∆m), decreased respiration, and ATP production, which subsequently reduces cell proliferation [6, 7]. A consequence of fusion is the intermixing of matrix contents, including mitochondrial proteins, mtDNA, and nutrients, which promotes mitochondrial homogeneity and maintenance of OXPHOS, by diluting out any dysfunction proteins and mutated mtDNA [11, 12]. Cell fusion experiments between wild-type and Mfn1- or Mfn2-null cells revealed that mitochondrial fusion can only occur when mitofusins are present on opposing mitochondria, implying the formation of trans complexes during mitochondrial tethering [13, 14] (Fig. 2a). Interestingly, heterotypic dimers (i.e., Mfn1:Mfn2) are more efficient at fusion compared to homotypic complexes [14], while post-translational modifications can also regulate mitofusin activity [15–17] (Fig. 1a). The reason for the efficacy of heterotypic interactions may be related to the additional function Mfn2 plays in tethering mitochondria to the endoplasmic reticulum (ER) to regulate Ca2+ homeostasis [18, 19].
Inner mitochondrial membrane fusion
Similar to mitofusins, OPA1 expression is also essential for mammalian development as homozygous deletion of OPA1 in mice leads to the early embryonic lethality [20]. In addition, mutations in OPA1 frequently occur in dominant optic atrophy (DOA), where the retinal ganglion cells degenerate causing vision loss [21]. Myotubes from DOA patients have fragmented mitochondria, indicating that the disease phenotype manifests as a consequence of the role OPA1 play in coordinating IMM fusion and cristae remodelling [22].
OPA1 undergoes proteolytic processing to produce two distinct isoforms (Figs. 1b, 2a) that are critical to initiate fusion and maintain cristae junctions (Fig. 2a). Fusion of IMM and OMM is a temporally linked, multi-step process controlled by transmembrane adaptor proteins that span both membranes [23]. In yeast, the adaptor protein is Ugo1, and while a mammalian homolog has not yet been discovered, OPA1 interacts with Mfn1 and Mfn2 [24] (Fig. 2a), indicating that OPA1-adaptor complexes form bridges between the IMM and OMM, and thus facilitate lipid mixing during fusion.
Mitochondrial fission
DRP1-mediated mitochondrial fission is regulated by a range of post-translational modifications, including phosphorylation, ubiquitination, sumoylation, and nitrosylation [25–33] (Fig. 1c). Fission is a coordinated process that requires recruitment of cytosolic DRP1 to the OMM followed by self-assembly into spherical oligomers that wrap around and sever mitochondria (Fig. 2b). Genetically engineered mice ablated for Drp1 are embryonically lethal due to abnormal placental and cardiomyocyte development [34, 35]. Conditional brain Drp1 −/− mice had defective cerebellar development and died within 2 days of birth, while specific heart Drp1 −/− mice developed lethal cardiac dysfunction, which similar to the fusion proteins signifies the importance of mitochondrial dynamics to embryogenesis [35–38]. Despite the physiological defects exhibited in Drp1-null mice during development, Drp1 −/− mouse embryonic fibroblasts (MEFs) are still capable of undergoing cell division and partition their mitochondria to daughter cells. At the point of cytokinesis in Drp1-null cells, mitochondria undergo forced fragmentation at the mid-body and unequal distribution to daughter cells [39]. In addition, the rate of cell division following loss of DRP1 is dramatically lower compared to wild-type cells [35], suggesting that while DRP1 may function as a core component of mitochondrial fission, there appears to be some unidentified members that can complete this process. Indeed, recent findings from Lee et al. identified the DRP1-related protein, dynamin-2 (Dyn-2) as a fundamental component of the fission machinery in mammalian cells [40]. DRP1 oligomerization at the OMM constricts the mitochondrial membrane to a specific diameter that permits recruitment and assembly of Dyn-2, which then further drives membrane constriction and completes mitochondrial fission [40], suggesting that mitochondrial fission in Drp1 −/− MEFs may be mediated by Dyn-2.
Given the importance of mitochondrial dynamics to fundamental biological processes, such as development, a critical question to answer is do the mitochondrial dynamics machinery also play a significant role in the progression of diseases, such as cancer.
Oncogenic signaling and cancer metabolism
While tumors can contain hundreds of genomic mutations and chromosomal rearrangements, typically only two to eight genomic events cause the progression of cancer by providing specific growth advantages and evasion of cell death programs [41]. Some of these mutations occur in proto-oncogenes (i.e., RASG12V and B-RAFV600E), copy number amplification (i.e., MYC), or deletion of tumor suppressors (i.e., PTEN). However, overexpression of oncogenes in the background of functional tumor suppressors is not sufficient to induce cellular transformation and tumorigenesis, but instead arrests the cell cycle in a senescent-like state [42]. For example, benign melanocytic naevi that acquire B-RAFV600E mutations will undergo limited proliferation or attrition via oncogene-induced apoptosis before entering senescence [43–45]. Some naevi are able to overcome oncogene-induced senescence by acquiring additional somatic mutations (usually from exposure to ultraviolet radiation) in signaling pathways that control proliferation (i.e., NF1), cell cycle (i.e., CDKN2A), cell growth (i.e., PTEN), or apoptosis (i.e., TP53). Progression from benign to malignant lesions requires additional changes in cellular metabolism to support increased bioenergetic and biosynthetic demands that occur from excessive proliferation.
Over seven decades ago, Otto Warburg linked mitochondrial function to tumorigenesis by observing that cancer cells undergo aerobic glycolysis, which is the fermentation of glucose to lactate in the presence of oxygen [46, 47]. It is now becoming clear that many oncogenic and tumor suppressor networks converge on mitochondria and alter cellular metabolism to support excessive tumor cell proliferation [48]. Rapidly dividing cancer cells require three main metabolic adaptations: (1) increase ATP production to maintain energy demand, (2) increase biosynthesis of macromolecules, and (3) regulation of redox states. To facilitate these requirements, cancer cells frequently reprogram their metabolic circuitry with the Warburg effect being the best characterized metabolic phenotype. Under the Warburg effect, ATP production primarily occurs through glycolysis, which paradoxically offers a more rapid means of generating ATP, but overall is less efficient than OXPHOS in terms of total ATP molecules produced per molecule of glucose. Instead, glycolysis is favoured because of the effective shuttling of carbon into macromolecule biosynthetic pathways, such as the pentose phosphate pathway [49]. This is done by limiting pyruvate utilization by mitochondria via decreasing mitochondrial pyruvate carriers and/or reducing the activity of pyruvate kinase, which catalyzes the final step of glycolysis to produce pyruvate. As a consequence, glycolytic intermediates upstream of pyruvate accumulate and can be utilized in other anabolic processes.
The effect of oncogene and tumor suppressor networks on cancer metabolism has been extensively reviewed elsewhere [50–52]. In the following sections, we look at three of the most frequently mutated pathways in cancer (mitogen activated protein kinases (MAPK), phosphoinositide 3-kinase (PI3K)-AKT, and MYC) and how they affect mitochondrial shape and function.
Oncogenic signaling and regulation of mitochondrial dynamics
While the underlying mechanisms regulating mitochondrial dynamics in cancer still remain unknown, a number of recent studies have revealed that hyper-activated oncogenic pathways act as potent signals to remodel mitochondrial shape and metabolism during tumorigenesis. Oncogenic cancer metabolism is associated with decreased OXPHOS, ATP production, increased ROS, and glycolytic flux. As specific mitochondrial morphologies are associated with different energetic states of cells, it, therefore, stands to reason that oncogene-mediated metabolic reprogramming will induce changes in mitochondrial shape to support changing metabolism. Indeed, primary fibroblasts display fused mitochondria and rely on OXPHOS, while B-RAFV600E-driven melanoma cells contain a fragmented mitochondrial network and increased glycolytic metabolism [53]. Furthermore, changes in the expression of mitochondrial dynamics that promote mitochondrial fission have been discovered in many cancer patient samples, indicating a potential role in tumorigenesis. For example, OPA1 levels are decreased in 50% of hepatocellular carcinoma samples compared to patient-matched normal tissue [54]. Similarly, Mfn2 is downregulated in human gastric tumors [55], while sonic hedgehog signaling induces glycolysis in medulloblastomas in mice by decreasing the expression of Mfn1 and Mfn2 [56]. Conversely, DRP1 levels are upregulated in B-RAFV600E-positive nevi and melanoma, pancreatic, thyroid, and breast cancers [53, 55, 57–59].
The aforementioned studies indicate that loss of fusion components or gain of DRP1 to promote mitochondrial fragmentation frequently occurs in cancers, suggesting that a fragmented mitochondrial phenotype is essential to many tumors. A key question, therefore, is how oncogenic signaling might regulate mitochondrial dynamics to facilitate a fragmented mitochondrial network.
Oncogenic MAPK (RAS-RAF-ERK) signaling acts to promote mitochondrial fission
The mitogen activated protein kinases (MAPK) pathway is frequently mutated in many cancers with most of the activating mutations occurring in the small GTPase RAS or its downstream target serine/threonine kinase rapidly accelerated fibrosarcoma (RAF) [60]. Inactivation of pathway inhibitors, such as nuclear factor 1 (NF1), disabled homolog 2-interacting protein (DAB2IP), and RAS protein activator like 2 (RASAL2), have also been observed in cancers and act to further perpetuate RAS-RAF signaling [60]. RAS activates a downstream kinase cascade starting with RAF, followed by mitogen activated protein kinase kinase (MEK) and extracellular signal regulated kinases (ERK). Activated ERK phosphorylates a large number of substrates, including kinases and transcription factors, that execute various cellular programs related to cell cycle, proliferation, metabolism, and evasion from apoptosis.
Mitochondrial fission has previously been associated with upregulation of the MAPK pathway [61]. This association was confirmed by in vitro phosphorylation assays between ERK and DRP1 which determined that DRP1 is, indeed, an ERK substrate, and indicated that mitochondrial fission can proceed through the MAPK pathway [61, 62]. It was only until two recent studies demonstrated that the specific ERK phosphorylation site on DRP1 at serine residue 616 (DRP1 Ser616), resulting in DRP1 activation and mitochondrial fission [53, 58] (Fig. 3a). Oncogenic RASG12V or B-RAFV600E also increases DRP1 mRNA levels, which can be reversed by pharmacological inhibition of B-RAFV600E, MEK, and ERK (i.e., PLX-3042, GSK1120212, and PD0325901), respectively [53].

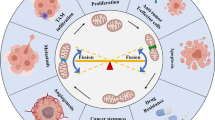
Role of mitochondrial dynamics in cancer processes. a, b Oncogenic signaling results in DRP1-dependent mitochondrial fragmentation. RAS and PI3K signaling up-regulates MYC, which subsequently promotes expression of pro-fusion and mitochondria biogenesis proteins. MYC gene amplification can also phenocopy these events in the absence of upstream stimuli. Mitochondrial shape also plays distinctive roles in regulating cellular metabolism. Fused mitochondria have increased oxidative metabolism, ATP production, and decreased ROS. Oncogenic signaling that fragments mitochondria increases glucose uptake, ROS, and decreases OXPHOS, which leads to a metabolic switch to glycolysis. Mitochondrial morphology is interchangeable between fused and fission states, and has implications for apoptosis, drug resistance, and clinical applications, such as biomarker discovery and targeted therapies. c Cell migration requires mitochondrial fission to enable movement of mitochondria to regions of the cell that have higher ATP requirements (i.e., lamellipodia). d Fused mitochondria are common in adult fibroblasts and stem cells, but mitochondrial network fragmentation is an initiating event following induction of pluripotency (i.e., iPCs) and cancer stem cells maintenance
Inhibition of MAPK signaling in B-RAF- or N-RAS-mutant melanomas promotes a metabolic shift towards OXPHOS, and increased mitochondrial biogenesis. This occurs through upregulation of melanocytic-specific transcription factor MITF, which, in turn, increases expression of the transcriptional coactivator peroxisome proliferator-activator receptor gamma coactivator-1 alpha (PGC-1α), a key mediator of mitochondrial biogenesis [53, 63, 64]. Increased PGC-1α levels have been observed in melanoma patient samples following administration of the B-RAFV600E inhibitor, PLX-4720. When dichotomized into high and low PGC-1α, it was found that high PGC-1α expression correlated with increased OXPHOS markers [63]. Another study also showed that inhibition of B-RAFV600E with PLX-3042 increased PGC-1α levels, which subsequently decreased the expression of numerous pro-metastatic genes [65]. While high PGC-1α expression has been associated with poor prognosis [63], Luo et al. revealed that low PGC-1α levels facilitated dissemination of tumor cells from the primary site [65]. Interestingly, PGC-1α expression increased in corresponding lung metastasises, which indicates that decreased mitochondrial mass and OXPHOS in primary melanoma supports a pro-metastatic program, while high PGC-1α and mitochondrial biogenesis promotes cell proliferation at metastatic sites [65]. Importantly, combining B-RAFV600E and OXPHOS inhibitors (i.e., oligomycin, TTFA) significantly decreased melanoma cell viability both in vitro and in mouse xenograft models, suggesting that combinatorial RAF-mitochondrial inhibition may be a novel strategy to treat cancers that revert back to oxidative metabolism. Curiously, little is known about how oncogenes directly regulate mitochondrial biogenesis and mitophagy to control cancer cell metabolism.
An alternative mechanism linking RAS signaling to mitochondrial fission was identified in HEK-293 and HeLa cells undergoing mitosis [66]. The GTPase RalA, an important downstream RAS substrate that is independent of the MAPK-ERK pathway [67, 68], and its effector RalBP1 actively promote mitochondrial fission by binding to Cdk1 and increasing DRP1 Ser616 phosphorylation during mitosis [66]. Collectively, oncogenic RAS promotes mitochondrial fragmentation through two separate pathways: ERK-DRP1 and RalA-RalBP1-Cdk1-DRP1, indicating that selective pressures within RAS-driven tumors promote fragmented mitochondria and force cellular metabolism towards glycolysis. In fact, the requirement for DRP1 is so strong that DRP1 expression is absolutely essential to facilitate oncogenic RASG12V-mediated cellular transformation [53]. DRP1-mediated fragmentation results in decreased ψ∆m, OXPHOS, and ATP production, suggesting that the requirement of DRP1 for RASG12V-induced transformation is due to metabolic reprogramming caused by mitochondrial fragmentation [53] (Fig. 3a). Meanwhile, Kashatus et al. demonstrated that knockdown of DRP1 in RASG12V-positive pancreatic xenografts significantly reduced tumor volume and progression [53, 58]. It should be mentioned that humans express four different RAS isoforms (H-RAS, N-RAS, and two K-RAS splice variants—K-RAS4A and K-RAS4B), all of which function in a similar manner at the plasma membrane [69]. Oncogenic RAS isoforms are distributed non-randomly across a range of tumors, indicating diversity between the RAS isoforms in different tissues [69]. For instance, H-RAS mutations are more frequently detected in melanoma and head and neck cancers, while K-RAS mutations are associated in lung, colorectal, and pancreatic tumors [69]. Despite these differences, it appears that RAS isoforms and tumor type (i.e., H-RAS, B-RAF in melanoma or K-RAS in pancreatic cancer) will result in DRP1 activation and mitochondrial fragmentation.
The fact that DRP1 is an important mediator of MAPK-driven tumorigenesis across multiple stages highlights two distinct impacts of DRP1 activity: (1) metabolic reprogramming during transformation and (2) requirement for equal mitochondrial distribution in rapidly proliferating cells. Together, these features offer a potential therapeutic window for cancer treatments that has yet to be exploited. It should also be noted that, recently, Mfn1 was discovered to be a substrate of ERK [70]. Specifically, ERK phosphorylated Mfn1 at tyrosine residue 562 (Tyr562), which is located within the first coiled-coil domain of Mfn1 [70]. Interestingly, either epidermal growth factor (EGF) stimulation or genetic engineering of cell lines to constitutively active MEK result in Mfn1 Tyr562 phosphorylation, followed by homotypic (Mfn1:Mfn1) interactions, and a fragmented mitochondrial phenotype [70]. However, the authors did not investigate these affects in DRP1-null cell lines, and, therefore, an effect of hyper-activated MAPK signaling will presumably feed onto DRP1 resulting in the same mitochondrial phenotype. Nevertheless, the fact that Mfn1 can be phosphorylated by ERK provides another layer of mitochondrial dynamic regulation, which, in the context of MAPK-driven cancers, may be physiologically relevant and should be further investigated.
PI3K-Akt signaling activates mitochondrial fission and promotes mitophagy
The PI3K pathway is composed of a number of lipid kinases that receives and transmits from growth factors, cytokines, and other extracellular stimuli to regulate cellular processes that include proliferation, survival, metabolism, and motility. Hyper-activation of PI3K signaling occurs in many solid tumors with somatic loss or epigenetic silencing of the PI3K inhibitor PTEN being the most common genetic alteration [71]. Somatic mutation, copy number gain, or amplification of the PI3K catalytic subunit alpha (PI3KCA) can be found in up to 40% of tumors, making it the second frequently mutated gene in this pathway [71].
Tumors with constitutive PI3K-AKT signaling actively increase glucose uptake to fuel glycolysis, which analogous to RAS-driven tumors, suggests that PI3K-Akt signaling fragments the mitochondrial network. Indeed, cells with oncogenic PI3K mutations contain a fragmented mitochondrial network that clusters around the nucleus [72] (Fig. 3a), while inhibition of PI3K signaling (e.g., PX-866 and GDC0941) rapidly fuses the mitochondrial network [72]. Although cancers upregulate nutrient transporters to continually fuel biosynthetic processes and rapid proliferation, nutrient and oxygen deprivation is a characteristic hurdle that many tumors face. As a means to overcome these challenges cancer cells rely on PI3K-AKT signaling to promote autophagy, a self-sustaining system that enables the cell to consume non-essential macromolecules to meet changing bioenergetic and biosynthetic needs. Mammalian target of rapamycin complex 1 (mTORC1), a downstream AKT target, is one of the best characterized regulators of autophagy. Under conditions where growth signals are abundant, PI3K activates mTORC1 via AKT to promote protein, lipid, and nucleic acid synthesis while inhibiting the autophagy machinery [73]. Conversely, mTORC1 can be inhibited through a number of mechanisms, including nutrient and oxygen deprivation, and the energy sensor adenosine monophosphate-activated protein kinase (AMPK), which is activated by a high AMP/ATP ratio. Inhibition of mTORC1 results in the release of its inhibitory control over autophagy, and allows the cell to maintain critical energy and macromolecule levels for proliferation and survival.
Mitophagy is a specialized form of autophagy where dysfunctional mitochondria are selected for degradation. Mitophagy can either proceed through two pathways; the PTEN-induced kinase 1 (PINK-1)-PARKIN E3 ubiquin ligase pathway [74] and the BNip3 pathway [75]. Consequently, the selective removal of damaged mitochondria also protects the cell from unwarranted release of pro-apoptotic mediators (i.e., cytochrome c, ROS) and reduces futile ATP usage [76, 77]. Although the mechanisms of how the cell distinguishes functional from dysfunctional mitochondria remain unclear, the loss of ψ∆m and mitochondrial fragmentation are events that precede mitophagy. Moreover, p53 inhibits PARKIN-mediated mitophagy in mouse heart and pancreatic β cells, suggesting a role for p53 in the regulation of energy metabolism, although this particular function has yet to be investigated in cancer cells [78, 79]. In the case of nutrient deprivation, healthy mitochondria are protected from mitophagy by fusing together and occurs through PKA-mediated inhibition of DRP1 [80]. In contrast, dysfunctional mitochondria are characterized by membrane depolarization, which causes the proteolytic cleavage and degradation of OPA1 as well as PARKIN-mediated degradation of Mfn1 and Mfn2 that consequently leads to mitochondrial fragmentation and turnover by mitophagy [81–83]. Interestingly, the PI3K inhibitors wortmannin and 3-methyladenine block mitophagy under nutrient deprivation conditions, suggesting that this pathway is required for mitophagy to proceed [84, 85]. How PI3K-Akt signaling may regulate mitochondrial dynamics under these conditions requires further investigation.
MYC overexpression promotes mitochondrial fusion and biogenesis
MYC is downstream of many signaling pathways that regulate cell growth, proliferation, and metabolism. The two pathways described above (MAPK and PI3K-AKT) are prime examples of upstream MYC effectors as they can stimulate MYC expression [60] (Fig. 3b). Non-cancerous cells tightly regulate MYC transcriptionally and post-transcriptionally by controlling the half-life of MYC mRNA and protein [86]. In addition, multiple cellular checkpoints are in place that can cause cell cycle arrest or death if MYC expression is deregulated [86]. The loss of these controls coupled with hyper-activation of growth promoting signals (i.e., oncogenic RAS, PI3K) increases MYC expression and can induce tumorigenesis. Equally, gene amplification that increase MYC copy number or chromosomal translocations that pair MYC with strong enhancers or promoters are frequent genetic alterations in cancers, thus severing the reliance of MYC from upstream stimuli.
Oncogenic MYC is also an activator of mitochondrial biogenesis by upregulating PGC-1β expression, thus coupling increased mitochondrial mass with rapid proliferation [51]. The involvement of MYC in mitochondrial biogenesis also suggests that it may play a role in regulating mitochondrial dynamics. MYC knockout MEFs have fragmented mitochondria, while re-expression of MYC promoted fusion by upregulation of OPA1 and Mfn2, although DRP1 and FIS1 levels were also increased [87] (Fig. 3b). Because this study did not investigate the status of DRP1 Ser616 and Ser637 phosphorylation, it, therefore, appears that a consequence of MYC-mediated mitochondrial biogenesis is increased fusion.
More recently, MYC signaling in triple-negative breast cancer cells induces mitochondrial fusion by upregulating phospholipase D Family member 6 (PLD6, which is also known as mitoPLD) [88]. Localization of PLD6 at the OMM facilitated cleavage of cardiolipin to phosphatidic acid, which is subsequently cleaved to diacylglycerol by the Lipin family of phosphatases [89]. A new report indicated that DRP1 GTPase activity is blocked following interactions with phosphatidic acid and mitoPLD on the OMM [90], implying that MYC is able to couple lipid metabolism at the OMM and mitochondrial dynamics.
Although tumorigenesis can be initiated via oncogenic MAPK or MYC signaling, it is interesting to note that both pathways result in different mitochondrial phenotypes (i.e., oncogenic MAPK induces mitochondrial fission, while MYC promotes fusion). These different mitochondrial shapes can be explained by the distinct mechanistic differences between MYC and MAPK signaling, whereby MYC is broadly responsible for gene expression, while MAPK signaling regulates protein activity by integrating plasma membrane receptor signals with multiple downstream kinase effector proteins. MYC controls the expression of genes involved in cell cycle progression, growth, and metabolism, including hundreds of genes that regulate mitochondrial mass and biogenesis [51]. Oncogenic MYC increases the biosynthetic, respiratory, and metabolic capacity of cancer cells to support rapid proliferation, which as mentioned above is coupled with mitochondrial fusion. In contrast, hyper-activation of MAPK signaling, either through increased receptor signaling or constitutively active RAS and B-RAF mutations, amplifies downstream kinase cascades that culminate in increased ERK activity, immediate activation of DRP1, inactivation of MFN1, and mitochondrial fission along with paralleled changes in gene transcription [53, 58, 70]. Although mitochondria display bioenergetic and structural plasticity in response to specific oncogenic stresses, understanding how these signaling pathways differentially influence mitochondrial shape and function to promote tumor progression requires further investigation.
Mitochondrial dynamics, movement, and metastasis
Tumor progression towards malignancy involves cancer cells generating the capacity to migrate to surrounding tissues and eventually metastasize to distal regions throughout the body. Cell migration is regulated by growth factors and cytokines that transmit signals through oncogenic pathways, such as MAPK and PI3K-AKT. These oncogenic signals can upregulate genes that promote changes in cell polarity, morphology, cytoskeletal dynamics, and cell adhesion that collectively can increase migratory capacity [60]. Concurrently, mitochondria actively migrate along cytoskeletal filaments to different cellular topographies that have high energetic demands. For example, cell migration requires formation of lamellipodia, an F-actin rich region at the leading edge of cells. Formation of F-actin polymers is a highly energetic process that requires extensive cytoskeletal reorganization, abundant ATP production, and Ca2+ buffering; hence, the requirement for mitochondria at lamellipodia is critical for cellular migration (Fig. 3c).
Access to microtubule-based motor proteins, such as dynein and kinesins, provides mitochondria with an appropriate scaffold that allows contact and movement along the cytoskeleton. Mitochondrial retrograde movement (towards the cell body) is regulated by dynein, whereas anterograde movement (away from the cell body) is dependent on kinesin. Besides microtubules, actin filaments and actin-based motors have also been implicated in mitochondrial movement over short distances. Two new studies have shown that actin polymerization occurs at ER-mitochondrial fission sites where the actin-nucleating protein Spire1C localizes and tethers mitochondria to the actin cytoskeleton [91]. Accumulation of mitochondria on actin filaments is closely followed by DRP1 localization and oligomerization on the OMM [92] (Fig. 2b). The fragmented mitochondria remain tethered to the actin cytoskeleton and are now able to be transported to other locations. The step-wise coordination of mitochondrial shape and cytoskeletal reorganization indicates that fragmentation and packaging of mitochondria as smaller parcels improve the efficiency of mitochondrial movement.
The spatial distribution of mitochondria in cancer cells and how mitochondrial dynamics regulates cellular migration has only recently been investigated. Several studies have demonstrated that mitochondrial fission is required to maintain the migratory and invasion potential of breast, thyroid, and glioblastoma cancer cells [59, 93, 94]. Breast cancer cells that migrated at a faster rate expressed higher total DRP1 and DRP1 Ser616 levels, while Mfn1 expression is lower compared to cancer cells with a low migratory capacity [93]. DRP1 knockdown or overexpression of Mfn1 or Mfn2 significantly decreased the migratory and invasive potential of cancer cells, suggesting that a highly fragmented mitochondrial network may be a selective pressure in tumors with a higher metastatic potential [59, 93]. It can, therefore, be hypothesized that cancers with high metastatic potential would contain high DRP1 activity as a means of maintaining a fragmented mitochondrial network. Indeed, increased DRP1 levels positively correlate with increased glioma tumor grade as well as invasive breast cancer and lymph node metastasis [93, 95], whereas decreased Mfn2 is associated with increased gastric tumor stage and decreased overall survival [93]. It is interesting that decrease Mfn2 levels would be associated with tumor progression as Mfn2 is involved in tethering mitochondria to the endoplasmic reticulum to promote Ca2+ homeostasis. Loss of Mfn2 would likely cause disruption to organelle contacts and decrease the capacity of mitochondria to buffer Ca2+. Oscillations in localized Ca2+ concentrations are considered important for cell movement, coupled with the fact that mitochondrial movement is halted in areas with high Ca2+ levels. This suggests that mitochondria with diminished capacity to sense and modulate Ca2+ levels may have inhibited movement to lamellipodia regions and promote cell migration. Further investigations are required to elucidate the underlying mechanisms that link Mfn2 with cell migration.
Similar to MAPK signaling, the PI3K-Akt pathway can also regulate cancer cell migration and invasion [96, 97]. Caino et al. recently investigated the underlying mechanisms of how PI3K antagonists paradoxically reactivate AKT and promote tumor progression. The authors found that PI3K inhibition increased the number, size, and persistence of lamellipodia in patient-derived glioblastoma spheroids [72]. Interestingly, inhibition of PI3K signaling induced mitochondrial elongation along the cytoskeleton towards lamellipodia, which contrasts with studies described above that stated mitochondrial fragmentation is required for cell migration [72]. This finding was also confirmed in A549 lung adenocarcinoma and LN299 glioblastoma cells, suggesting that the response to PI3K inhibition is not cell-type specific [72]. Knockdown of the mitofusins revealed that only Mfn1 was required for elongation of the mitochondrial network to lamellipodia to facilitate focal adhesion turnover and increased cellular migration [72]. Mitochondrial fusion increases respiration; however, the role respiration plays in cell migration remains controversial [98, 99]. A recent report proposed that localization of the inhibitor of apoptosis protein survivin to mitochondria promoted prostate and breast cancer cell migration and invasion in vivo and in vitro by stabilizing complex II of the ETC, supporting mitochondrial trafficking to lamellipodia and promoting focal adhesion turnover [100]. Mitochondrial fusion increases ETC assembly, oxidative metabolism, and ATP production, suggesting that enhanced OXPHOS function plays an active role in cell migration. Indeed, Caino and colleagues determined that combined inhibition of PI3K and mitochondrial ETC activity diminished mitochondrial positioning at lamellipodia and cell invasion [72]. These findings indicate that Mfn1-mediated mitochondrial fusion and increased ATP production are important to support cell migration.
Mitochondrial determinants in stem cells and cell differentiation
In the sections above, the role of mitochondrial dynamics has been described in processes of cell proliferation, metabolic reprogramming, and migration. However, new information relating both mitochondrial function and architecture to stem cell biology is beginning to offer novel insights into how mitochondria maintain quiescent cell populations throughout ageing and cancer [101].
Somatic stem cells (SSCs) are rare, undifferentiated cells that exist within different tissues and give rise to functionally mature progeny to ensure tissue homeostasis [102]. The morphology of mitochondria in SSCs is a fused, elongated network containing electron-dense matrix with numerous cristae folds [103]. This is in stark contrast to the mitochondrial network from embryonic stem cells, which form immature, small puncta with poorly developed cristae [102]. Recent studies have found that mitochondrial fusion and oxidative metabolism are essential in maintaining the SSC niche. For example, IMM fusion mediated by OPA1 is critical for regulating tight cristae junctions and the proximity of ETC complexes to each other in memory T cells compared to effector T cells [104], while Mfn2 specifically maintains populations of haematopoietic stem cells with lymphoid pluripotency [105]. Likewise, depletion of Mfn1, Mfn2, or OPA1 impaired neural stem cell renewal [106]. These studies suggest that SSCs contain fused mitochondrial networks as a means of maintaining OXPHOS and ATP supply, while keeping ROS levels to a minimum. In agreement, the transcription factor forkhead box O3a (Foxo3a) is a critical regulator of mitochondrial biogenesis, OXPHOS, and redox status in hematopoietic stem cells [107]. Loss of Foxo3a resulted in increased mitochondrial fragmentation, metabolic switch to glycolysis, and decreased pluripotency, indicating that regulation of mitochondrial shape is important to maintaining the stem cell niche [107]. In contrast, mitochondrial fragmentation is a key early marker of inducible pluripotent stem cell (iPCs) reprogramming [108]. iPC technology allows researchers to artificially reprogram adult cells, such as fibroblasts, to an embryonic stem cell-like state. The mitochondrial fragmentation observed in iPCs was driven by decreased Mfn1 and Mfn2 expression and accompanied by decreased OXPHOS, increased glycolysis, and lactate production [108] (Fig. 3d). Moreover, mitochondrial fragmentation in iPCs also corresponded to activation of MAPK signaling, which contributed to glycolysis by upregulating glucose transporters and HIF-1α, a known regulator metabolism and cell reprogramming [108].
The tumor initiating cell or cancer stem cell (CSC) hypothesis states that the presence of cancer cells with stem cell-like properties is responsible for tumor growth, cellular heterogeneity within cancers, and treatment resistance. Normal SSCs and CSCs share many properties, including self-renewal while maintaining an undifferentiated state and expression of similar cell surface markers [109]. However, very few studies have compared the metabolic profiles of CSCs to either SSCs or differentiated cells. Several reports have suggested that CSCs from ovarian, breast, and colon cancers are more glycolytic than differentiated cells based on increased glucose uptake, lactate production, and expression of glycolytic enzymes, which indicate that these cells have fragmented mitochondria [110–112]. Conversely, CSCs with fused mitochondrial networks rely more on OXPHOS as they have increased ψ∆m, enhanced oxygen consumption rates, and increased mitochondrial biogenesis through expression of PGC-1α [113, 114]. Moreover, knockdown of PGC-1α reduced the stemness properties of breast CSCs, suggesting that maintenance of mitochondrial populations in CSCs is critical for pluripotency [115].
A recent report investigated if mitochondrial shape influences the metabolic growth of brain tumor initiating cells (BTICs) [116]. These cells have similar properties to normal neural stem cells, in that they share cell-autonomous regulatory pathways that control continual proliferation and differentiation, but differ in their metabolic features. For example, BTICs increase glycolytic flux and glucose uptake by upregulating GLUT3 [116]. Moreover, it was found that BTICs tend to have higher DRP1 Ser616 and reduced Ser637 levels compared to non-BTICs, while survival of BTICs depended on the expression of DRP1, indicating that the coupling of glycolysis to mitochondrial fragmentation is essential in these neuronal CSCs [116].
The distinction between fused and fragmented mitochondrial networks across different CSCs may be reflected in the specific tissues where they reside, nutrient, and oxygen supply or if they carry different oncogenic mutations. The utilization of iPCs technology may provide clues as to why different CSCs have heterogeneous mitochondrial morphologies. The switch from OXPHOS to glycolysis following induction of pluripotency was partially attributed to activation of MAPK signaling [108]. Prieto et al. demonstrated that ERK-mediated DRP1 Ser616 phosphorylation was responsible for the early wave of mitochondrial fragmentation during cellular reprogramming [117]. However, once pluripotency was reached the mitochondrial network quickly refused, indicating a desired return to oxidative metabolism and quiescence, indicating plasticity in mitochondrial shape during the intermediary steps of cell reprogramming [117]. Oxidative metabolism may be favoured by quiescent cells as a means of maintaining mitochondrial ψ∆m, ATP production, and mtDNA content. Therefore, CSCs that display different mitochondrial morphologies may simply reflect various energetic states of CSC differentiation. For instance, CSCs that require upregulation of macromolecule biosynthetic pathways during differentiation will fragment their mitochondrial network and switch cellular metabolism to glycolysis, while quiescent CSCs will retain fused mitochondria and OXPHOS. The plasticity of metabolic profiles in CSCs and their relative dependence on mitochondrial dynamics may offer new therapeutic avenues for cancer treatments, particularly as CSCs display extreme resistance to most conventional cancer treatments [118, 119].
Mitochondrial dynamics and apoptosis
One aspect of oncogenic and tumor suppressor signaling pathways is their ability to regulate cellular sensitivity to mitochondrial-dependent apoptosis by converging on the the B-cell chronic lymphatic leukemia/lymphoma (BCL-2) family of pro- and anti-apoptotic proteins. The underlying mechanisms of transcriptional, translational, and post-translational regulation of the BCL-2 family in cancers have extensively been reviewed [120–125]. Here, we will discuss how the mitochondrial dynamics machinery intersects with the BCL-2 family to regulate apoptosis.
The mitochondrial-dependent or intrinsic pathway of apoptosis is activated as a result of intracellular cell stress or damage (i.e., nutrient deprivation, DNA damage), which engages the BCL-2 family of pro-apoptotic proteins, including BCL-2 antagonist killer 1 (BAK), and BCL-2 associated × protein (BAX), which cooperates to form pores in the OMM. Pore formation, also referred to as mitochondrial outer membrane permeabilization (MOMP), allows the release of pro-apoptotic factors (i.e., cytochrome c) that interact with adaptor protein apoptotic protease activating factor 1 (APAF-1) and trigger recruitment and activation of cysteine-aspartic proteases (caspases). Caspase-dependent cleavage of numerous substrates is the final stage of apoptosis, which results in the efficient packaging and elimination of targeted cells.
One of the most salient morphological features of apoptosis is the fragmentation of the mitochondrial network. While a number of early studies indicated mitochondrial fragmentation and clustering at perinuclear region to occur just prior to cytochrome c release, suggesting that regulated mitochondrial fission may be responsible for apoptosis [5, 126–128]. This is supported by the fact that DRP1 is heavily recruited to the OMM, while BAX translocation to mitochondria co-localizes with DRP1 at fission sites, implying that DRP1 marks regions of the OMM where MOMP will occur [5]. However, given that Drp1 −/− MEFs are still able to undergo mitosis and apoptosis indicates that DRP1-dependent mitochondrial fragmentation is not necessary for intrinsic apoptosis to proceed. Instead, initiation of apoptosis involves regions of mitochondria where the ER wraps around and marks sites for division. These ER-mitochondrial contact sites, known as ER-associated mitochondrial division (ERMD), are important for phospholipid synthesis and Ca2+ signaling [18], and serve as “hot-spots” for DRP1 recruitment and fission. Given the co-localization between DRP1 and BAX at the OMM, the ER-mitochondrial interface may also represent a membrane microenvironment that is critical for BAX oligomerization and MOMP.
Non-apoptotic, soluble BAX associates with Mfn2 at ER-mitochondrial junctions and promotes mitochondrial fusion [14]. Increased mitochondrial fission during apoptosis is thought to be a consequence of decreased soluble, inactivated BAX coupled with increased membrane inserted, and oligomerized BAX at the OMM. Bax −/− Bak −/− cells have fragmented mitochondria, which fuse upon re-expression of either BAX or BAK [129]. Moreover, BAX re-expression in Bax −/− Bak −/− cells reorganised Mfn2 OMM localisation to ER-mitochondrial junctions and increased Mfn2 GTPase activity [129]. In this context, the non-apoptotic functions of BAX are to promote ER-mitochondrial tethering and Ca2+ homeostatsis by increasing Mfn2 activity. Conversely, Mfn2 can be considered an anti-apoptotic effector by sequestering soluble BAX from the cytosol and preventing its activation. Indeed, overexpression of a dominant active Mfn2 mutant protected against staurosporine-induced apoptosis, while reciprocal Mfn2 knockdown enhanced apoptosis [130, 131]. It would be interestingly for future studies to determine if mitochondrial dynamics are involved in the formation and regulation of ERMDs during tumorigenesis.
Sphigolipid metabolites derived from ER and mitochondrial membranes promote BAX and MOMP [132, 133], suggesting that the shuttling of lipid effector molecules through ERMD sites where BAX is localized may increase the likelihood of MOMP. In this context, changes in mitochondrial dynamics can either positively or negatively regulate BAX activity. Montessuit and colleagues showed that DRP1 promoted BAX oligomerization through a process of membrane tethering and hemifusion [134]. This process was independent of the DRP1 GTPase domain, but instead relied on the positively charged arginine residue at amino acid 247 (DRP1 Arg247) to interact with negatively charged cardiolipin phospholipids. Expression of DRP1 R247A mutants significantly decreased the interaction between DRP1 and cardiolipin in liposomes and impaired BAX oligomerization [134]. As previously mentioned, the interaction between DRP1 and cardiolipin inhibits mitochondrial fragmentation [90], but instead results in membrane tethering and hemifusion, which may provide the appropriate membrane curvature and lipid mixing considered to be important for recruitment of activated BAX and MOMP [134]. Indeed, activated BAX is localized to additional membranes, such as the Golgi, where following cell stress (i.e., DNA damage), it rapidly disassociates from anti-apoptotic proteins and retro-translocates to the OMM [135, 136].
In addition to OMM microenvironment and lipid composition, a recent report demonstrated that modification of mitochondrial membrane curvature through dynamics proteins can regulate BAX-mediated MOMP [137]. By comparing mitochondrial networks between Mfn1 −/− and Mfn2 −/− MEFs, it was demonstrated that Mfn1 −/− cells contain hyper-fragmented mitochondria and were intrinsically resistant to ER stressors, whereas mitochondria from Mfn2 −/− MEFs were short, swollen tubular structures and retained sensitivity to induction of apoptosis [137]. The hyper-fragmented mitochondrial phenotype in Mfn1 −/− cells was mediated by DRP1, and either genetic or pharmacological DRP1 inhibition resulted in fusion of the mitochondrial network and re-sensitization to apoptotic stimuli [137]. It was found that hyper-fragmented mitochondria from Mfn1 −/− cells were resistant to BAX accumulation on the OMM, thus preventing MOMP. Following a series of biochemical and in cellulo experiments, it was demonstrated that mitochondrial shape and membrane curvature were primarily involved regulating the release and insertion of the carboxyl terminal tail of BAX in the OMM [137]. Collectively, these studies indicate that fission per se is not required for apoptosis to proceed, but rather remodelling the OMM to facilitate efficient BAX oligomerization and MOMP. Deregulation of mitochondrial dynamics proteins in cancers, therefore, represents a new means of evasion from cell death programs and development of drug resistance.
Translation of mitochondrial dynamics from the bench to bedside
Given the importance of mitochondria in multiple aspects of tumorigenesis, targeting mitochondrial function and more specifically mitochondrial dynamics has been proposed as an effective strategy to induce apoptosis in cancer [52, 138]. To the best of our knowledge, no specific inhibitors target mitofusins or OPA1, which may, in part, be due to their overlapping functions to fuse mitochondria. There is, however, a small molecule (hydrazone M1) that actively promotes mitochondrial fusion in Mfn1 −/− or Mfn2 −/− MEFs, but not in Mfn1 −/−; Mfn2 −/− double knockout or OPA1 −/− cells [139]. Mechanistically, hydrazone M1 treatment increases the expression of ATP synthase subunit α and β, while oligomycin-mediated inhibition of ATPase synthase blocked the pro-fusion function of M1 [139]. These findings indicate that increase ATP synthase activity and subsequent ATP production are important for fusion, although the underlying mechanism remains unclear. Nevertheless, it would be curious to determine if pharmacological induction of mitochondrial fusion has any anti-tumor properties. Conversely, two pharmacological inhibitors against DRP1 have been developed. Mitochondrial division inhibitor (mDIVI-1) came out of a yeast screen of 23,000 compounds that inhibited DRP1 GTPase activity and self-assembly in mammalian cells [140]. Years later, another group developed a specific peptide (P110) that interfered with DRP1-Fis1 interactions and decreased DRP1 GTPase activity in neurons [141].
By inhibiting DRP1, both mDIVI-1 and P110 enforce mitochondrial fusion, increase ψ∆m, enhance ATP production, decrease ROS, and protect against apoptosis in neuron and cardiomyocytes. These cytoprotective properties may be therapeutically effective against cardiovascular and neurodegenerative disorders like ischemic heart disease and Parkinson disease [140–142]. Of the two drugs, only mDIVI-1 has extensively been studied in a cancer setting, and in contrast to the cytoprotective effects, mDIVI-1 possesses cytotoxic properties across a wide range of neoplasms [142]. Given that DRP1 is upregulated in many cancers and is required for oncogenic transformation, indicates that cancer cells may be exquisitely sensitive to DRP1 inhibition. This hypothesis has thus far held true as pharmacological and genetic inhibition of DRP1 decreased the in vitro and in vivo growth of glioblastomas, melanoma, hepatocellular carcinoma, and mesothelioma [57, 116, 143, 144].
Besides directly inhibiting DRP1, mDIVI-1 also has off-target effects unrelated to mitochondrial function, such as interfering with DNA replication and impairing mitotic spindle assembly that together result in G2/M cell cycle arrest [142]. Furthermore, mDIVI-1 synergizes with cisplatin to induce apoptosis in Drp1-null cells, highlighting the ability of mDIVI-1 to kill cells in the absence of DRP1 [145]. Importantly though, mDIVI-1 does not affect cell survival or proliferation in non-transformed fibroblasts and epithelial cells, suggesting that it specifically acts upon cancer cells. Nonetheless, the pharmacokinetics and direct targets of mDIVI-1 remain poorly defined, particularly as the effects of this drug are dependent on cell and disease type [142]. Therefore, to define the therapeutic potential for DRP1 inhibitors in clinical trials, there is an urgent need to further elucidate the pharmacodynamics and pharmacokinetic properties of mDIVI-1 as well as to develop new chemical screens to identify more specific and potent DRP1 inhibitors.
Mitochondrial dynamics as biomarkers
Biomarkers that dichotomize tumors into categories that predict prognosis and therapeutic responses play an important role within the clinical setting. The clinical utility of mitochondrial dynamics as biomarkers for cancer progression is only in its infancy and requires substantial future efforts. However, early work indicates that upregulation of DRP1 may be predictive of breast cancer progression and metastasis [54], while an increased DRP1 Ser616 to Ser637 phosphorylation ratio (meaning increased DRP1 activity) predicts cisplatin resistance and relapse in lung adenocarcinoma patients [146]. In addition, DRP1 Ser616 phosphorylation status dichotomized wild-type B-RAF from B-RAFV600E-positive dysplastic nevi and melanoma [53, 57], while another study found DRP1 Ser616 positively correlated with ERK phosphorylation in human pancreatic adenocarcinoma [58], indicating that DRP1 Ser616 status significantly relates to oncogenic MAPK signaling may be useful in determining which lesions may develop into cancer. Finally, DRP1 Ser616 was significantly expressed in BTICs, while evaluation of total DRP1 and DRP1 S616 in normal brain and glioblastoma specimens demonstrated strong positive correlation between DRP1 Ser616 and cancer progression [116]. These findings suggest that DRP1 Ser616 status is a contributing factor to cancer and may be a useful biomarker to predict cancer progression and response to treatment.
Concluding remarks and future directions
Our knowledge of mitochondrial biology in tumorigenesis remains rudimentary. However, significant efforts in recent years have illuminated the area of mitochondrial dynamics as critical for cancer progression and survival. Outlined in this review, we have summarized the importance of individual mitochondrial dynamics components across a range of biological processes that are essential for cancers. Oncogenic MAPK and PI3K-AKT cause DRP1-dependent fragmentation of the mitochondrial network, which, in turn, may be a key component of metabolic reprogramming during tumorigenesis. Maintaining mitochondria in a fragmented state is also important as tumors progress and migrate away from primary tissues. Similarly, CSCs that change their mitochondrial networks from fused to fragmented states are essential during cellular differentiation and tumor growth. These alterations in mitochondrial shape influence the sensitivity of cancer cells to engage the pro-apoptotic machinery and may contribute to increased resistance to chemotherapy.
While mitochondrial dynamics have now been implicated in multiple cancer processes, we still know very little about the mechanisms that connect mitochondrial architecture and metabolism to different stages of tumor progression. Moving forward, it will be important to find answers to the following questions. Although DRP1-mediated mitochondrial fragmentation occurs in many cancers, do the fusion mechanics play a role in tumorigenesis and response to cellular stress? What role do tumor suppressors, such as p53, play in regulating mitochondrial dynamics and what are the regulatory signals that control mitochondria in cancer stem cells? How do mitochondrial dynamics regulate metabolic heterogenity and cell migration, and are these processes mechanistically linked? Are mitochondrial dynamics proteins viable therapeutic targets for cancer treatment and can their expression and activity status be used as predictive diagnostic and/or prognostic biomarkers? As our understanding of mitochondrial dynamics expands, we anticipate learning more about mitochondrial biology and its role in cancer as well as how this ancient organelle interacts with other cellular compartments, including the ER and nucleus. New discoveries in these areas have implications for cancer, and will also impact upon our knowledge of fundamental cell biology.
References
Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. TEM 27(2):105–117. doi:10.1016/j.tem.2015.12.001
Archer SL (2013) Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med 369(23):2236–2251. doi:10.1056/NEJMra1215233
Lewis MR, Lewis WH (1915) Mitochondria (and other cytoplasmic structures) in tissue cultures. Am J Anat 17(3):339–401. doi:10.1002/aja.1000170304
Jakobs S (2006) High resolution imaging of live mitochondria. Biochim Biophys Acta 1763(5–6):561–575. doi:10.1016/j.bbamcr.2006.04.004
Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ (2002) Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159(6):931–938. doi:10.1083/jcb.200209124
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160(2):189–200. doi:10.1083/jcb.200211046
Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280(28):26185–26192. doi:10.1074/jbc.M503062200
Feely SM, Laura M, Siskind CE, Sottile S, Davis M, Gibbons VS, Reilly MM, Shy ME (2011) MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 76(20):1690–1696. doi:10.1212/WNL.0b013e31821a441e
Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, Jordanova A, Nelis E, De Vriendt E, Van Hul M, Seeman P, Mazanec R, Saifi GM, Szigeti K, Mancias P, Butler IJ, Kochanski A, Ryniewicz B, De Bleecker J, Van den Bergh P, Verellen C, Van Coster R, Goemans N, Auer-Grumbach M, Robberecht W, Milic Rasic V, Nevo Y, Tournev I, Guergueltcheva V, Roelens F, Vieregge P, Vinci P, Moreno MT, Christen HJ, Shy ME, Lupski JR, Vance JM, De Jonghe P, Timmerman V (2006) MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain J Neurol 129(Pt 8):2093–2102. doi:10.1093/brain/awl126
Vallat JM, Ouvrier RA, Pollard JD, Magdelaine C, Zhu D, Nicholson GA, Grew S, Ryan MM, Funalot B (2008) Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations. J Neuropathol Exp Neurol 67(11):1097–1102. doi:10.1097/NEN.0b013e31818b6cbc
Chan DC (2006) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22:79–99. doi:10.1146/annurev.cellbio.22.010305.104638
Chen H, Chan DC (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet 18(R2):R169–R176. doi:10.1093/hmg/ddp326
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305(5685):858–862. doi:10.1126/science.1099793
Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J (2011) The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell 41(2):150–160. doi:10.1016/j.molcel.2010.11.030
Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM (2012) Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell 47(4):547–557. doi:10.1016/j.molcel.2012.05.041
Park YY, Nguyen OT, Kang H, Cho H (2014) MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis 5:e1172. doi:10.1038/cddis.2014.142
Park YY, Cho H (2012) Mitofusin 1 is degraded at G2/M phase through ubiquitylation by MARCH5. Cell Div 7(1):25. doi:10.1186/1747-1028-7-25
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362. doi:10.1126/science.1207385
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernandez-Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd, Scorrano L (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci USA 113(40):11249–11254. doi:10.1073/pnas.1606786113
Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M (2007) Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet 16(11):1307–1318. doi:10.1093/hmg/ddm079
Ranieri M, Del Bo R, Bordoni A, Ronchi D, Colombo I, Riboldi G, Cosi A, Servida M, Magri F, Moggio M, Bresolin N, Comi GP, Corti S (2012) Optic atrophy plus phenotype due to mutations in the OPA1 gene: two more Italian families. J Neurol Sci 315(1–2):146–149. doi:10.1016/j.jns.2011.12.002
Belenguer P, Pellegrini L (2013) The dynamin GTPase OPA1: more than mitochondria? Biochim Biophys Acta 1833(1):176–183. doi:10.1016/j.bbamcr.2012.08.004
Hoppins S, Nunnari J (2009) The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta 1793(1):20–26. doi:10.1016/j.bbamcr.2008.07.005
Guillery O, Malka F, Landes T, Guillou E, Blackstone C, Lombes A, Belenguer P, Arnoult D, Rojo M (2008) Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 100(5):315–325. doi:10.1042/BC20070110
Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10(7):748–754. doi:10.1038/embor.2009.86
Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM (2015) MAPL SUMOylation of Drp1 stabilizes an ER/Mitochondrial platform required for cell death. Mol Cell 59(6):941–955. doi:10.1016/j.molcel.2015.08.001
Di Bacco A, Ouyang J, Lee HY, Catic A, Ploegh H, Gill G (2006) The SUMO-specific protease SENP5 is required for cell division. Mol Cell Biol 26(12):4489–4498. doi:10.1128/MCB.02301-05
Zunino R, Braschi E, Xu L, McBride HM (2009) Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem 284(26):17783–17795. doi:10.1074/jbc.M901902200
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324(5923):102–105. doi:10.1126/science.1171091
Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, Masliah E, Schwarzenbacher R, Bossy-Wetzel E (2010) S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J Alzheimer’s Dis JAD 20(Suppl 2):S513–S526. doi:10.3233/JAD-2010-100552
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105(41):15803–15808. doi:10.1073/pnas.0808249105
Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286(13):11649–11658. doi:10.1074/jbc.M110.144238
Karbowski M, Neutzner A, Youle RJ (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178(1):71–84. doi:10.1083/jcb.200611064
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11(8):958–966. doi:10.1038/ncb1907
Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H (2009) The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186(6):805–816. doi:10.1083/jcb.200903065
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116(2):264–278. doi:10.1161/CIRCRESAHA.116.303356
Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33(23):2798–2813. doi:10.15252/embj.201488658
Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21(2):273–285. doi:10.1016/j.cmet.2014.12.011
Otera H, Ishihara N, Mihara K (2013) New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 1833(5):1256–1268. doi:10.1016/j.bbamcr.2013.02.002
Lee JE, Westrate LM, Wu H, Page C, Voeltz GK (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540(7631):139–143. doi:10.1038/nature20555
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339(6127):1546–1558. doi:10.1126/science.1235122
Courtois-Cox S, Jones SL, Cichowski K (2008) Many roads lead to oncogene-induced senescence. Oncogene 27(20):2801–2809. doi:10.1038/sj.onc.1210950
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434(7035):907–913. doi:10.1038/nature03485
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436(7051):720–724. doi:10.1038/nature03890
Shain AH, Bastian BC (2016) From melanocytes to melanomas. Nat Rev Cancer 16(6):345–358. doi:10.1038/nrc.2016.37
Warburg O, Wind F, Negelein E (1927) The Metabolism of Tumors in the Body. J Gen Physiol 8(6):519–530
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Hay N (2016) Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer 16(10):635–649. doi:10.1038/nrc.2016.77
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12(10):685–698. doi:10.1038/nrc3365
Vyas S, Zaganjor E, Haigis MC (2016) Mitochondria and cancer. Cell 166(3):555–566. doi:10.1016/j.cell.2016.07.002
Zong WX, Rabinowitz JD, White E (2016) Mitochondria and cancer. Mol Cell 61(5):667–676. doi:10.1016/j.molcel.2016.02.011
Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE (2015) Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 57(3):521–536. doi:10.1016/j.molcel.2015.01.003
Zhao X, Tian C, Puszyk WM, Ogunwobi OO, Cao M, Wang T, Cabrera R, Nelson DR, Liu C (2013) OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab Investig J Tech Methods Pathol 93 (1):8–19. doi:10.1038/labinvest.2012.144
Zhang GE, Jin HL, Lin XK, Chen C, Liu XS, Zhang Q, Yu JR (2013) Anti-tumor effects of Mfn2 in gastric cancer. Int J Mol Sci 14(7):13005–13021. doi:10.3390/ijms140713005
Malhotra A, Dey A, Prasad N, Kenney AM (2016) Sonic Hedgehog signaling drives mitochondrial fragmentation by suppressing mitofusins in cerebellar granule neuron precursors and medulloblastoma. Mol Cancer Res MCR 14(1):114–124. doi:10.1158/1541-7786.MCR-15-0278
Wieder SY, Serasinghe MN, Sung JC, Choi DC, Birge MB, Yao JL, Bernstein E, Celebi JT, Chipuk JE (2015) Activation of the mitochondrial fragmentation protein DRP1 correlates with BRAF(V600E) Melanoma. J Invest Dermatol 135(10):2544–2547. doi:10.1038/jid.2015.196
Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57(3):537–551. doi:10.1016/j.molcel.2015.01.002
Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, Scorrano L, Maximo V, Campello S (2015) Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PloS one 10(3):e0122308. doi:10.1371/journal.pone.0122308
Sever R, Brugge JS (2015) Signal transduction in cancer. Cold Spring Harbor Perspect Med. doi:10.1101/cshperspect.a006098
Yu T, Jhun BS, Yoon Y (2011) High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal 14(3):425–437. doi:10.1089/ars.2010.3284
Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, Zhang H, Yu H, Swerdlow RH, Chen JX, Yan SS (2014) Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta 1842(2):220–231. doi:10.1016/j.bbadis.2013.11.009
Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR (2013) Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer cell 23(3):302–315. doi:10.1016/j.ccr.2013.02.003
Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V, Zhang J, Patel L, Pereira CG, Broom BM, Mills GB, Ram P, Smith PD, Wargo JA, Long GV, Davies MA (2014) Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res 74(23):7037–7047. doi:10.1158/0008-5472.CAN-14-1392
Luo C, Lim J-H, Lee Y, Granter SR, Thomas A, Vazquez F, Widlund HR, Puigserver P (2016) A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 537 (7620):422–426. doi:10.1038/nature19347. http://www.nature.com/nature/journal/v537/n7620/abs/nature19347.html#supplementary-information
Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM (2011) RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 13(9):1108–1115. doi:10.1038/ncb2310
Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM (2005) Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer cell 7(6):533–545. doi:10.1016/j.ccr.2005.04.030
Shields JM, Pruitt K, McFall A, Shaub A, Der CJ (2000) Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol 10(4):147–154
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D (2011) RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11(11):761–774. doi:10.1038/nrc3106
Pyakurel A, Savoia C, Hess D, Scorrano L (2015) Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell 58(2):244–254. doi:10.1016/j.molcel.2015.02.021
Thorpe LM, Yuzugullu H, Zhao JJ (2015) PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15(1):7–24. doi:10.1038/nrc3860
Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, Webster MR, Weeraratna AT, Bosari S, Languino LR, Altieri DC (2015) PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci USA 112(28):8638–8643. doi:10.1073/pnas.1500722112
Dibble CC, Manning BD (2013) Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15(6):555–564. doi:10.1038/ncb2763
Jin SM, Youle RJ (2012) PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 125(Pt 4):795–799. doi:10.1242/jcs.093849
Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16(9):1145–1163. doi:10.15252/embr.201540759
Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 288(2):1099–1113. doi:10.1074/jbc.M112.399345
Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A (2009) Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 106(8):2770–2775. doi:10.1073/pnas.0807694106
Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S (2013) Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nature communications 4:2308. doi:10.1038/ncomms3308
Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Ikeda K, Ueyama T, Ogata T, Matoba S (2014) Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc Natl Acad Sci USA 111(8):3116–3121. doi:10.1073/pnas.1318951111
Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13(5):589–598. doi:10.1038/ncb2220
Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187(7):959–966. doi:10.1083/jcb.200906083
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191(7):1367–1380. doi:10.1083/jcb.201007013
Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PloS one 5(4):e10054. doi:10.1371/journal.pone.0010054
Kim I, Lemasters JJ (2011) Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxidants redox signaling 14(10):1919–1928. doi:10.1089/ars.2010.3768
Lemasters JJ (2014) Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox biology 2:749–754. doi:10.1016/j.redox.2014.06.004
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV (2015) MYC, metabolism, and cancer. Cancer Discov 5(10):1024–1039. doi:10.1158/2159-8290.CD-15-0507
Graves JA, Wang Y, Sims-Lucas S, Cherok E, Rothermund K, Branca MF, Elster J, Beer-Stolz D, Van Houten B, Vockley J, Prochownik EV (2012) Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PloS One 7(5):e37699. doi:10.1371/journal.pone.0037699
von Eyss B, Jaenicke LA, Kortlever RM, Royla N, Wiese KE, Letschert S, McDuffus LA, Sauer M, Rosenwald A, Evan GI, Kempa S, Eilers M (2015) A MYC-driven change in mitochondrial dynamics limits YAP/TAZ function in mammary epithelial cells and breast cancer. Cancer cell 28(6):743–757. doi:10.1016/j.ccell.2015.10.013
Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA (2011) piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell 20(3):376–387. doi:10.1016/j.devcel.2011.01.004
Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R, Iijima M, Sesaki H (2016) Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol Cell 63(6):1034–1043. doi:10.1016/j.molcel.2016.08.013
Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, Spudich J, Lippincott-Schwartz J (2015) A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife. doi:10.7554/eLife.08828
Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 4:e11553. doi:10.7554/eLife.11553
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32(40):4814–4824. doi:10.1038/onc.2012.494
Wan YY, Zhang JF, Yang ZJ, Jiang LP, Wei YF, Lai QN, Wang JB, Xin HB, Han XJ (2014) Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol Rep 32(2):619–626. doi:10.3892/or.2014.3235
Yin M, Lu Q, Liu X, Wang T, Liu Y, Chen L (2016) Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ ROCK1 pathway. Biochem Biophys Res Commun 478(2):663–668. doi:10.1016/j.bbrc.2016.08.003
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274. doi:10.1016/j.cell.2007.06.009
Zhou H, Huang S (2011) Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci 12 (1):30–42
LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R (2014) PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16(10):992–1003, 1001–1015. doi:10.1038/ncb3039
Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G, Zarif JC, Epstein T, Gatenby R, McCartney A, Elisseeff JH, Mooney SM, An SS, Pienta KJ (2015) Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6(1):130–143. doi:10.18632/oncotarget.2766
Rivadeneira DB, Caino MC, Seo JH, Angelin A, Wallace DC, Languino LR, Altieri DC (2015) Survivin promotes oxidative phosphorylation, subcellular mitochondrial repositioning, and tumor cell invasion. Sci Signal 8(389):ra80. doi:10.1126/scisignal.aab1624
Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC (2013) Mitochondrial regulation in pluripotent stem cells. Cell Metab 18(3):325–332. doi:10.1016/j.cmet.2013.06.005
Wanet A, Arnould T, Najimi M, Renard P (2015) Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev 24(17):1957–1971. doi:10.1089/scd.2015.0117
Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CAt, Ramalho-Santos J, Van Houten B, Schatten G (2011) Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PloS one 6(6):e20914. doi:10.1371/journal.pone.0020914
Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, Chen Q, Huang SC, O’Neill CM, Edelson BT, Pearce EJ, Sesaki H, Huber TB, Rambold AS, Pearce EL (2016) Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166(1):63–76. doi:10.1016/j.cell.2016.05.035
Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW (2016) Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529(7587):528–531. doi:10.1038/nature16500
Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, Park DS, Slack RS (2016) Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell stem cell 19(2):232–247. doi:10.1016/j.stem.2016.04.015
Rimmele P, Liang R, Bigarella CL, Kocabas F, Xie J, Serasinghe MN, Chipuk J, Sadek H, Zhang CC, Ghaffari S (2015) Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep 16(9):1164–1176. doi:10.15252/embr.201439704
Son MJ, Kwon Y, Son MY, Seol B, Choi HS, Ryu SW, Choi C, Cho YS (2015) Mitofusins deficiency elicits mitochondrial metabolic reprogramming to pluripotency. Cell Death Differ 22(12):1957–1969. doi:10.1038/cdd.2015.43
Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414(6859):105–111. doi:10.1038/35102167
Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, De Cola A, Scavo E, Carollo R, D’Agostino D, Forli F, D’Aguanno S, Todaro M, Stassi G, Di Ilio C, De Laurenzi V, Urbani A (2014) Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis 5:e1336. doi:10.1038/cddis.2014.285
Emmink BL, Verheem A, Van Houdt WJ, Steller EJ, Govaert KM, Pham TV, Piersma SR, Borel Rinkes IH, Jimenez CR, Kranenburg O (2013) The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J Proteom 91:84–96. doi:10.1016/j.jprot.2013.06.027
Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, Wang X, Huss WJ, Lele SB, Morrison CD, Odunsi K (2014) Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PloS One 9(1):e84941. doi:10.1371/journal.pone.0084941
Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, Rasola A, Frasson C, Nardo G, Zulato E, Nicoletto MO, Manicone M, Indraccolo S, Amadori A (2014) Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 5(12):4305–4319. doi:10.18632/oncotarget.2010
Sancho P, Barneda D, Heeschen C (2016) Hallmarks of cancer stem cell metabolism. Br J Cancer 114(12):1305–1312. doi:10.1038/bjc.2016.152
De Luca A, Fiorillo M, Peiris-Pages M, Ozsvari B, Smith DL, Sanchez-Alvarez R, Martinez-Outschoorn UE, Cappello AR, Pezzi V, Lisanti MP, Sotgia F (2015) Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 6(17):14777–14795. doi:10.18632/oncotarget.4401
Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN (2015) Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18(4):501–510. doi:10.1038/nn.3960
Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, Raya A, Lopez-Garcia C, Torres J (2016) Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun 7:11124. doi:10.1038/ncomms11124
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444(7120):756–760. doi:10.1038/nature05236
Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R (2006) Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 13(7):1238–1241. doi:10.1038/sj.cdd.4401872
Delbridge AR, Strasser A (2015) The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ 22(7):1071–1080. doi:10.1038/cdd.2015.50
Delbridge AR, Grabow S, Strasser A, Vaux DL (2016) Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nature reviews Cancer 16(2):99–109. doi:10.1038/nrc.2015.17
Luna-Vargas MP, Chipuk JE (2016) Physiological and Pharmacological Control of BAK, BAX, and Beyond. Trends Cell Biol. doi:10.1016/j.tcb.2016.07.002
Elkholi R, Renault TT, Serasinghe MN, Chipuk JE (2014) Putting the pieces together: How is the mitochondrial pathway of apoptosis regulated in cancer and chemotherapy? Cancer Metabol 2:16. doi:10.1186/2049-3002-2-16
Anvekar RA, Asciolla JJ, Missert DJ, Chipuk JE (2011) Born to be alive: a role for the BCL-2 family in melanoma tumor cell survival, apoptosis, and treatment. Front Oncol 1 (34). doi:10.3389/fonc.2011.00034
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR (2010) The BCL-2 family reunion. Mol Cell 37(3):299–310. doi:10.1016/j.molcel.2010.01.025
Mancini M, Anderson BO, Caldwell E, Sedghinasab M, Paty PB, Hockenbery DM (1997) Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J Cell Biol 138(2):449–469
Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R (2001) The Ca2 + concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 20(11):2690–2701. doi:10.1093/emboj/20.11.2690
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1(4):515–525
Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ (2006) Role of Bax and Bak in mitochondrial morphogenesis. Nature 443(7112):658–662. doi:10.1038/nature05111
Sugioka R, Shimizu S, Tsujimoto Y (2004) Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279(50):52726–52734. doi:10.1074/jbc.M408910200
Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H (2005) Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem 280(26):25060–25070. doi:10.1074/jbc.M501599200
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148(5):988–1000. doi:10.1016/j.cell.2012.01.038
Lee H, Rotolo JA, Mesicek J, Penate-Medina T, Rimner A, Liao WC, Yin X, Ragupathi G, Ehleiter D, Gulbins E, Zhai D, Reed JC, Haimovitz-Friedman A, Fuks Z, Kolesnick R (2011) Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PloS one 6(6):e19783. doi:10.1371/journal.pone.0019783
Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, Martinou JC (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142(6):889–901. doi:10.1016/j.cell.2010.08.017
Dumitru R, Gama V, Fagan BM, Bower JJ, Swahari V, Pevny LH, Deshmukh M (2012) Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell 46(5):573–583. doi:10.1016/j.molcel.2012.04.002
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, Neutzner A, Tjandra N, Youle RJ (2011) Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145(1):104–116. doi:10.1016/j.cell.2011.02.034
Renault TT, Floros KV, Elkholi R, Corrigan KA, Kushnareva Y, Wieder SY, Lindtner C, Serasinghe MN, Asciolla JJ, Buettner C, Newmeyer DD, Chipuk JE (2015) Mitochondrial shape governs BAX-induced membrane permeabilization and apoptosis. Mol Cell 57(1):69–82. doi:10.1016/j.molcel.2014.10.028
Weinberg SE, Chandel NS (2015) Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 11(1):9–15. doi:10.1038/nchembio.1712
Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, Yang P, Schultz PG (2012) A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem 51(37):9302–9305. doi:10.1002/anie.201204589
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14(2):193–204. doi:10.1016/j.devcel.2007.11.019
Qi X, Qvit N, Su YC, Mochly-Rosen D (2013) A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126(Pt 3):789–802. doi:10.1242/jcs.114439
Rosdah AA, J KH, Delbridge LM, Dusting GJ, Lim SY (2016) Mitochondrial fission—a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect 4 (3):e00235. doi:10.1002/prp2.235
Zhan L, Cao H, Wang G, Lyu Y, Sun X, An J, Wu Z, Huang Q, Liu B, Xing J (2016) Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget. doi:10.18632/oncotarget.11339
Lennon FE, Cianci GC, Kanteti R, Riehm JJ, Arif Q, Poroyko VA, Lupovitch E, Vigneswaran W, Husain A, Chen P, Liao JK, Sattler M, Kindler HL, Salgia R (2016) Unique fractal evaluation and therapeutic implications of mitochondrial morphology in malignant mesothelioma. Sci Rep 6:24578. doi:10.1038/srep24578
Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, Llambi F, Green DR, Van Houten B (2014) Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 5(12):4180–4194. doi:10.18632/oncotarget.1944
Chiang YY, Chen SL, Hsiao YT, Huang CH, Lin TY, Chiang IP, Hsu WH, Chow KC (2009) Nuclear expression of dynamin-related protein 1 in lung adenocarcinomas. Mod Pathol Off J US Can Acad Pathol Inc 22(9):1139–1150. doi:10.1038/modpathol.2009.83
Acknowledgements
This work was supported by: NIH Grants CA157740 (J. E. C.) and CA206005 (J. E. C.); the JJR Foundation, the William A. Spivak Fund, the Fridolin Charitable Trust, an American Cancer Society Research Scholar Award, a Leukemia and Lymphoma Society Career Development Award, and an Irma T. Hirschl/Monique Weill-Caulier Trust Research Award. This work was also supported in part by two research Grants (5FY1174 and 1FY13416) from the March of Dimes Foundation, and the Developmental Research Pilot Project Program within the Department of Oncological Sciences at the Icahn School of Medicine at Mount Sinai.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Trotta, A.P., Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 74, 1999–2017 (2017). https://doi.org/10.1007/s00018-016-2451-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2451-3