
Abstract
Mitochondrial structural and functional integrity defines the health of a cell by regulating cellular metabolism. Thus, mitochondria play an important role in both cell proliferation and cell death. Cancer cells are metabolically altered compared to normal cells for their ability to survive better and proliferate faster. Resistance to apoptosis is an important characteristic of cancer cells and given the contribution of mitochondria to apoptosis, it is imperative that mitochondria could behave differently in a tumor situation. The other feature associated with cancer cells is the Warburg effect, which engages a shift in metabolism. Although the Warburg effect often occurs in conjunction with dysfunctional mitochondria, the relationship between mitochondria, the Warburg effect, and cancer cell metabolism is not clearly decoded. Other than these changes, several mitochondrial gene mutations occur in cancer cells, mitochondrial biogenesis is affected and mitochondria see structural and functional variations. In cancer pharmacology, targeting mitochondria and mitochondria associated signaling pathways to reduce tumor proliferation is a growing field of interest. This chapter summarizes various changes in mitochondria in relevance to cancer, behavior of mitochondria during tumorigenesis, and the progress on using mitochondria as a therapeutic target for cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


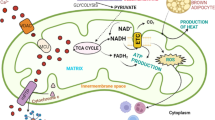
Keywords
1 Introduction
In routine clinical diagnosis of cancer, a glucose analogue (2-18 fluoro-2-deoxy-Glucose) is used to trace tumor tissue that would uptake more glucose than normal tissues due to its increased necessity for sugars. This technique is based on a hypothesis by Otto Warburg in the 1930s that cancer cells choose a different metabolic route than normal cells (Koppenol et al. 2011; Warburg 1956). This school of thought led to major studies on aerobic glycolysis in tumor cells where cells adapt to a glycolytic pathway to make adenosine triphosphate (ATP) instead of using the regular mitochondrial electron transport chain (ETC). Thus, cancer cells involve increased breakdown of glucose generating raw materials for the synthesis for other macromolecules, helping their rapid growth. Although tumor hypoxia is hypothesized to be a trigger (Gatenby and Gillies 2004), there are evidences where there is a metabolic shift to aerobic glycolysis in free availability of oxygen (Christofk et al. 2008). It is speculated that in cancer, there is a reprogramming of the cells driven by oncogenes into a proliferative metabolism, resembling an embryonic program (Vander Heiden et al. 2009) with upregulation of glycolytic enzymes (Christofk et al. 2008). Glycolysis under aerobic conditions makes cells acidic due to increased production of lactate as a result of glycolytic cycles, but this excess lactate is postulated to be a fuel for mitochondrial oxidative phosphorylation (Sonveaux et al. 2008). This phenomenon is proposed to be used by certain cancer cells in a reverse Warburg effect, where cancer cells induce Warburg effect in neighboring stromal cells and in turn receive lactate and pyruvate for oxidative phosphorylation (Pavlides et al. 2009). All these metabolic altercations point to an altered mitochondrial function in tumor cells that has led to years of research in this field with respect to ATP production and beyond (Boland et al. 2013). This chapter summarizes the important changes in mitochondria associated with onset and progression of cancer such as mitochondrial DNA (mtDNA) mutations, mitochondrial reactive oxygen species (ROS), mitochondrial mass regulation, and mitochondrial dynamics. The chapter also discusses and summarizes major cancer drug classes that target mitochondria (Table 1).
2 Mitochondrial Mutations in Cancer Cells
Each cell contains numerous mitochondria, and every mitochondrion has its own DNA (mtDNA) in multiple copies. Mammalian cells contain about 1000–10,000 copies of mtDNA which can replicate independent of cellular division (Lightowlers et al. 1997). Mitochondria can accumulate somatic mutations and lead to a heteroplasmic state where mitochondria with dissimilar DNA content co-exist. Mutations in mitochondrial DNA contribute to mitochondrial function, especially ROS production, and hence it is important to consider mitochondrial DNA as an important factor in tumorigenesis.
The mitochondrial genome has been sequenced and characterized (Blanchard and Schmidt 1996; Grivell 1983), and several mutations in mtDNA have been associated with cancers in human tissues (Chatterjee et al. 2006). A list of major cancers and associated mtDNA mutations is provided in Table 2 (Abu-Amero et al. 2005; Fliss et al. 2000; Habano et al. 1999; Jeronimo et al. 2001; Jones et al. 2001; Maximo et al. 2002; Polyak et al. 1998; Sanchez-Cespedes et al. 2001; Canter et al. 2005; Parrella et al. 2001; Petros et al. 2005; Wong et al. 2003). Mitochondrial mutations (both homoplasmic and heteroplasmic) have been detected in body fluids of cancer patients (Chinnery et al. 2002; Fliss et al. 2000). Although normal subjects are reported to display age-associated accumulation of mitochondrial mutations (Cormio et al. 2005), mtDNA mutations are highly prevalent in cancer tissues. mtDNA coded enzymes contributing to mitochondrial oxidative phosphorylation (Luciakova and Kuzela 1992) are reported defective in tumor situations, which could possibly lead to deregulation of ROS production from the mitochondria. Thus, mtDNA mutations could contribute to solid tumors by favoring the Warburg phenomenon and by playing a role in apoptosis. These mutations can be traced along with tumors and thus could be used as markers for identifying types of tumors. However, it is not clear if mtDNA mutations themselves can drive tumor growth or merely provide an advantage to cancer cells. This is one key area awaiting extensive research in order to understand the mtDNA mutations and their relation to tumorigenesis.
3 Mitochondrial Reactive Oxygen Species
ROS are a by-product of the electron transport chain at the level of complex I and complex II/III where electrons escape (leak) the canonical pathway of electron transport, and are established to play a role in cellular signaling (Brand 2010; Chen et al. 2003). Reduced levels of antioxidants can also contribute to increased ROS levels in the cells (Hamanaka and Chandel 2010; Schumacker 2006). ROS can have both deleterious and favorable effects on cancer cells depending on the amount as well as rate of generation.
ROS can activate several signaling pathways promoting tumorigenesis and thus play a factor favoring the cancer cells. ROS stabilize hypoxia inducible factor (HIF)-α, an important protein for the survival of tumor cells in extremely hypoxic tumor environment (Jung et al. 2008). Intriguingly, antioxidant treatment of cancer cells can cause suppression of HIF1-α, again indicating the importance of ROS regulation in tumor microenvironment (Gao et al. 2009). Activation of the hypoxia pathway in cancer cells is also related to metabolic changes. HIF1-α induces the expression of glycolytic enzymes driving the cancer cells to adapt to an alternative ATP generation mechanism (Kim et al. 2006). The other way by which ROS regulate metabolism in cancer cells is by driving the activation of NRF2, a nuclear factor involved in increased production of anabolic enzymes (Mitsuishi et al. 2012). ROS also oxidize pyruvate kinase M2, which in turn drives a pentose pathway flux and increases glutathione levels promoting tumorigenesis (Anastasiou et al. 2011; Israelsen et al. 2013).
ROS regulate signaling pathways in cancer; the most studied one amongst them is the PI3Kinase pathway—a major growth-promoting signal in normal as well as cancer cells (Cantley 2002). The target of ROS within the context of cancer is the phosphatase PTEN, a negative regulator of the PI3Kinase pathway. ROS oxidize an active site cysteine on PTEN leading to the hyper-activation of the pathway (Lee et al. 2002; Leslie et al. 2003). ROS, on the other hand, can also inhibit the phosphatases of this pathway, namely PP2A and PTP1B, that negatively regulate Akt (Ostman et al. 2011), fostering the pathway’s activity and thus promoting cell survival and proliferation.
Another role of ROS in cancer cells is induction of oxidative DNA damage leading to the development and progression of tumor in several examples. Patients with increased oxidative damage are more likely to develop tumors (Hagen et al. 1994; Shimoda et al. 1994). The origin of ROS in tumor cells could often be oncogenes themselves (Irani et al. 1997) or increased activity of oxidases or peroxisome activity (Liou and Storz 2010). Mutations in mitochondrial DNA or mitochondrial dysfunction also cause increased levels of ROS. Heteroplasmic mitochondrial DNA mutations in ND (NADH Dehydrogenase) genes are shown to increase ROS production from the mitochondria (Larman et al. 2012). Tumor cells also have an elevated antioxidant response to balance the increased ROS, avoiding apoptosis (Liou and Storz 2010).
As mentioned above, although ROS is a tumor-promoting signal, the levels of ROS clearly define if it is playing a deleterious effect for the cancer cells or an advantage. Given cancer cells carry high levels of ROS, they can be targeted to death by further elevating the levels of ROS using chemicals that produce ROS. Small molecules and alkaloids have been used to target cancer cells (Raj et al. 2011; Shaw et al. 2011; Trachootham et al. 2009). However, a major disadvantage of using ROS as a target for cancer cells stems from the fact that normal cells are also affected by increased ROS, especially in cells that utilize ROS as a physiological molecule (Sena and Chandel 2012; Nagaraj et al. 2012; Owusu-Ansah and Banerjee 2009). Moreover, not all cancer cells elevate ROS levels (Nagaraj et al. 2012; Shaw et al. 2011). Hence, ROS offers a tumor type specific therapeutic scope in cancer biology (discussed at the end of this chapter).
4 Mitochondrial Dynamics in Cancer
Mitochondria are dynamic organelles undergoing fusion and fission events constantly. The outer membrane consists of mitofusins, Mfn1 and Mfn2 and the inner membrane consists of Opa1, which facilitates fusion and fission in their respective membranes (Karbowski and Youle 2003). Drp1, a dynamin related GTPase is another protein required for mitochondrial fission, which forms rings where mitochondria pinch off from each other (Detmer and Chan 2007).
The shape of mitochondria changes throughout the cell cycle and apoptosis, which relates to their role in cancer (Van den Bogert et al. 1988). During G1-S phase there is an increased oxidative phosphorylation and the function of mitochondria to facilitate cell division. However during S-M phase, given the need of the cell to divide, the mitochondria become fragmented as they are distributed between the daughter cells (Margineantu et al. 2002). It is believed that the G1-S phase networking of mitochondria can regulate the cyclin E levels and thus is essential for cell cycle progression. This also involves membrane polarization and hyperfusion of mitochondria (Mitra et al. 2009). Although cyclin E expression is important for cell cycle progression, how this is driven by mitochondrial hyperfusion is not yet proved. However, ATP and ROS have been ruled out to be playing a major role (Qian et al. 2012). Glycolysis and glutaminolysis are noted to be increased around G1/S phase and which could be linked to the change in mitochondrial dynamics (Qian et al. 2012; Dang 2010). Thus, mitochondrial structural status can contribute to cell cycle progression and a deregulation would affect cell division.
Other than cell cycle, stress signals are documented to alter mitochondrial dynamics. Signaling pathways interact with mitochondrial dynamics and regulate the structure and function of the mitochondria in order to sustain stress. Drugs, UV, production of increased amounts of ATP, and higher rates of oxidative phosphorylation are all known to cause stress-induced hyperfusion to prevent apoptosis and mitophagy (Mitra et al. 2009; Rambold et al. 2011; Tondera et al. 2009). During glucose deprivation, cells switch to oxidative phosphorylation with increased mitochondrial fusion and cristae density as an adaptation for cell survival (Rossignol et al. 2004) in normal as well as tumor producing cells. Hence, such structural changes are relevant to cell survival and division. This was shown in an in vivo model Drosophila (Nagaraj et al. 2012), where the oncogene Yorkie (Yki) transcriptionally regulated mitochondrial proteins Opa1 and mitofusin causing increased mitochondrial fusion. This fusion was essential throughout proliferation mediated by Yorkie. This study was extended into human cells where Yap2 expression, a homologue of Yorkie showed hyperfusion. Mitochondrial fusion is also indirectly correlated with decreased levels of ROS (Nagaraj et al. 2012), a contradiction from the general notion that ROS are elevated in tumor cells. Given ROS can induce apoptosis, it can be postulated that mitochondrial fusion is pro-survival and thus helps cancer cell endurance.
Conversely, inhibiting mitochondrial fusion impedes cell growth and proliferation as well as oxidative metabolism (Bach et al. 2003). Hence, it is imperative that fusion promotes mitochondrial function by increasing the efficiency of oxidative phosphorylation as well as ATP production, and reducing ROS generation. Thus, fusion could also promote mitochondria related metabolic pathways such as the Kreb’s cycle, fatty acid oxidation, etc.
Mitochondrial fission, the opposite phenomenon to fusion, has opposite effects on metabolism. It is acknowledged to stunt growth and increase ROS (Nagaraj et al. 2012; Yarosh et al. 2008), which could be the result of compromised respiratory activity. Loss of Drp1, a protein required for fission is also shown to cause deregulation of cyclin E, leading to aberrant proliferation (Parker et al. 2015). Dysregulated Drp1 expression is linked to tumor cell fission (Rehman et al. 2012). Given fission increases ROS production, and the ability of ROS to regulate hypoxia and lactate production, it is an interesting problem to answer how fission contributes to the Warburg effect.
Mitochondrial fission also promotes membrane depolarization, cytochrome c release, and apoptosis. Drp1 promotes Bax oligomerization, possibly allowing fragmented mitochondria to form Bax/Bak openings (Brooks et al. 2007; Montessuit et al. 2010). Other classes of apoptotic proteins, such as Bcl2 family members (Bcl-XL), promote mitochondrial fission (Li H 2008) and are overexpressed in tumors (Kelly and Strasser 2011). These are predicted to promote a Warburg shift by reducing oxidative phosphorylation. However, their mechanistic connection to mitochondrial activity and metabolic changes is to be explored.
Mitochondrial dynamics (both fusion and fission) contribute to tumor cell proliferation by different mechanisms. However, if these changes can act as cause for tumor progression or if they are a consequence of tumor growth is yet to be understood.
5 Mitochondrial Content in Cancer
Mitochondrial content is decided by two factors in cancer: mitochondrial biogenesis and mitophagy. Signaling pathways, especially the oncogenes, control mitochondrial biogenesis, as established in cell culture systems. However, there is no clear evidence of altered mitochondrial biogenesis in tumors. Yet, given the established roles of oncogenes such as Myc in mitochondrial biogenesis (Li et al. 2005; Morrish and Hockenbery 2014), it is vital to understand the importance of mitochondrial amounts in tumor cells.
Mitochondrial biogenesis involves nuclear factors as well as mitochondrial genes; most of the mitochondrial proteins are synthesized by the nuclear factor and imported to mitochondria, while some enzymes in the ETC are encoded by the mitochondrial genes themselves (Chacinska et al. 2009). It is an essential process for the cells, and the mitochondrial content of the cells depends on the nutrient availability, dividing status and physiological (and/or pathophysiological) state of the cells (Wenz 2013). Mitochondrial biogenesis is controlled by peroxisome-proliferator activator receptor-alpha and gamma (PPARα, PPARγ), nuclear respiratory factor 1 (NRF1), nuclear respiratory factor 2 (NRF2), and also estrogen related receptors (ERR) α, β, γ (Scarpulla et al. 2012). PPAR-γ co-activator 1-alpha (PGC-1α), with its partners (PGC-1β, and PRC-PGC related co-activator) termed as the master regulator of mitochondrial biogenesis. It forms a protein complex, controlling and maintaining the expression of mitochondrial biogenesis factors, antioxidants, and several metabolic genes (Dominy et al. 2010).
The role of mitochondrial biogenesis in cancer comes to picture with the discoveries of oncogenes regulating mitochondrial biogenesis through the PGC family. The most well-known oncogene that triggers mitochondrial biogenesis is c-Myc, which control biogenesis through PGC-1β (Zhang et al. 2007). However, tumor related genes such as HIF-1α negatively regulate mitochondrial biogenesis by inhibiting c-Myc (Zhang et al. 2007). PGC-1α activity is upregulated in a subset of melanoma where oxidative phosphorylation dependency is seen (Vazquez et al. 2013). PGC-1β and its targets are upregulated in ALT (Alternative Lengthening of Telomeres) positive tumors (Hu et al. 2012). p53, the tumor suppressor protein negatively regulates mitochondrial biogenesis, again indicating a relation between cancer and mitochondrial function (Sahin et al. 2011).
Taken together, it is not clear whether mitochondrial biogenesis is beneficial for tumor cells or is inhibiting rapid proliferation. Not all tumor cells carry the same metabolic profiles. Thus, tumor mitochondrial biogenesis needs to be studied in specification to the oncogenes involved and the nature of the tumors.
The other process that regulated mitochondrial mass in cancer cells is mitochondrial autophagy. Autophagy is activated in a situation of nutrient deprivation to the cells, and mitophagy is a specific process where mitochondria are targeted for degradation to provide nutrient for cell survival (Rabinowitz and White 2010). Mitochondrial fusion and fission events control mitophagy, where fission facilitates mitophagy by marking them for degradation and fusion keeps the mitochondria healthy, protecting them from mitophagy (Twig et al. 2008). Mitophagy is regulated by two pathways postulated to be tumor suppressors: the Parkin pathway and the BNIP3/NIX pathway (Youle and Narendra 2011; Zhang and Ney 2009). Parkin promotes mitochondrial turnover via fission and assists in mitochondrial transport via microtubules (Narendra et al. 2010; Narendra et al. 2008). Parkin, although is a gene associated with Parkinson’s syndrome (Youle and Narendra 2011), is also identified as a tumor suppressor gene in several cancers, and Parkin mutant mice are susceptible to tumors (Cesari et al. 2003; Fujiwara et al. 2008). Parkin promotes oxidative metabolism as a p53 target while inhibiting the development of Warburg effect (Zhang et al. 2011). Although direct evidences of Parkin involved in a tumor scenario via mitophagy are unclear, it can be postulated that Parkin maintains healthy mitochondria balancing metabolism in cells and thus can cause deregulation of metabolism in cancer cells upon its mutation.
BNIP3 and NIX are redox-sensing hypoxia inducible genes promoting mitophagy (Zhang and Ney 2009). These proteins directly interact with the autophagy protein LC3 as adaptors targeting mitochondria for degradation (Hanna et al. 2012). They reduce mitochondrial mass in hypoxic condition by inducing mitophagy helping the cells regulate excessive ROS production in a state of hypoxia (Tracy et al. 2007). BNIP3 and NIX are characterized as dysregulated in several tumors. In certain malignant tumors, the expression of these genes decreases, whereas pre-malignant stages see an increase (Okami et al. 2004; Tan et al. 2007; Sowter et al. 2003). Loss of BNIP3 promotes tumors in mouse models, and hence it is considered to be a tumor suppressor (Chourasia and Macleod 2015). Similar to Parkin, BNIP3 and NIX proteins are considered to control mitochondrial turnover. Although it is established that Parkin and BNIP3/NIX pathways control mitophagy in tumor scenarios, a definitive mechanism for the effect of mitophagy in cancer is yet to emerge. Mitophagy is also directly related to aging as cancer mutations accumulate with age and the development of tumor progresses. Hence mitophagy and aging work in tandem in cancer situations. However, targeting mitophagy offers an advantage in tumor therapy over apoptosis and general autophagy due to specificity.
Along with the above discussed major mitochondrial changes, retrograde signals from mitochondria to the nucleus (Wallace 2012), and oncogenic control of mitochondria by oncogenes, such as Myc and KRas, and tumors suppressors, such as p53 and RB (Sherr and McCormick 2002; Vousden and Prives 2009; Dang 2010), also bring about mitochondrial changes in tumor situations. Although it is now established that mitochondrial changes occur in cancer cells, how these changes affect tumor growth and how they can be manipulated to achieve cancer therapy still require specific research.
6 Targeting Mitochondria for Cancer Therapy
Given the multilevel involvement of mitochondria in cancer, researchers have used several ways to target mitochondria for cancer therapy.
Mitochondrial DNA is targeted by drugs that reduce the copy number of mitochondrial DNA or by inhibiting replication. Vitamin K3 (menadione) (Sasaki et al. 2008) inhibits DNA polymerase gamma, which is important for replication of mitochondrial DNA. Parkinsonian toxin 1-methyl-4-phenylpyridinium reduces the copy number of mtDNA by destabilizing the structure (Umeda et al. 2000; Neuzil et al. 2007).
Dysregulated ROS production from mitochondria in cancer is another target for therapy. However, the human trials have not had great success (Bjelakovic and Gluud 2007) as they fail to inhibit the mitochondrial generated ROS. Another problem with the antioxidant drugs is that normal cells such as immune cells also produce ROS for physiological functions. Nevertheless, another approach in ROS based therapeutic experiments is using the ability of cancer cells to produce more antioxidants and ROS. Hence, inhibition of antioxidants in cancer cells could lead to excessive ROS in cancer cells and cause selective killing of cancer cells (Raj et al. 2011; Glasauer et al. 2014).
The Bcl-2 family of proteins consists of pro and anti-survival factors (Youle and Strasser 2008). Cell death is favored in the absence of pro-survival factors. Mimetic drugs that target BH3 domains of these proteins, the domain that interacts with Bax/Bak proteins (Youle and Strasser 2008), are used to target tumors. Drugs such as ABT-263, Gossypol, antimycin A, alpha-tocopheryl succinates are some examples of Bcl-2 family targets that induce mitochondria mediated cell death, partly due to their interaction with the BH3 domain (Kang et al. 2010; Neuzil et al. 2007).
Given ATP generation is essential for normal cells as well as tumor cells (Zu and Guppy 2004), mitochondrial bioenergetic targeting drugs need to be specifically targeted to tumor cells. Poorly perfused tumors which make ATP (Rumsey et al. 1990) are an apt target for drugs that inhibit ATP production given it would cause cell death in such poorly perfused tumors only. The antidiabetic drug, metformin, a biguanide, is thought to be such a candidate to target ATP production without affecting the normal tissue. Metformin decreases hepatic gluconeogenesis and brings down insulin levels (Bailey and Turner 1996). Metformin has many effects on cancer cells. It decreases blood glucose and insulin levels, and inhibits the growth of tumors that are insulin dependent (Pollak 2014). It acts on the mitochondrial complex I (El-Mir et al. 2000; Owen et al. 2000). It also impairs glycolysis by decreasing the activity of enzyme hexokinase 2, an important enzyme to carry out glycolysis (Salani et al. 2013). Thus, metformin is believed to inhibit tumor growth by lowering glucose supply, acting on complex I and thereby reducing ATP production. The dosages and complete mechanism of action for this drug are still under investigation in clinical trials. Another drug similar to metformin is phenformin, a biguanide that also inhibits mitochondrial complex I (Birsoy et al. 2014). It has higher affinity to mitochondria and recently shown to work better than metformin in breast tumors (Appleyard et al. 2012). Although lactose acidosis is a drawback of phenformin, there are evidences that the drug can be used in combination with BRAF inhibitors to control melanomas effectively (Yuan et al. 2013). Hence, phenformin also makes an attractive candidate for clinical trials.
One of the classes of drugs also tried on inhibiting ATP production is VLX600, an ETC inhibitor. This is shown to reduce colon cancer tumor growth (Zhang et al. 2014) at experimental levels. Mitochondrial protein translation is targeted by certain drugs such as tigecycline that reduces the expression of 13 subunits of ETC. This drug has been efficient on leukemic cells that survive primarily on mitochondrial ATP production (Skrtic et al. 2011). Mitochondrial chaperones, such as heat shock proteins, are targeted by drugs such as Gamitrinib, which are modified to accumulate in mitochondria and reduce the activity of HSP90 and ATPase-1, thus reducing energy production of mitochondria.
Another target in mitochondrial cancer drugs is biosynthetic pathways. The glutamine addicted tumors are formed mostly due to Myc and Kras (Gaglio et al. 2011). Such tumors can be targeted by using inhibitors of glumatinases that use glutamine in their reaction to continue the tricarboxylic acid cycle. Glutaminase inhibitors such as bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide) or compound 968 (Le et al. 2012) already attenuate tumor growth. Mitophagy also produces raw materials for the mitochondrial TCA cycle as discussed in the review before. Several autophagy inhibitors are on trials and chloroquine is one such drug (Balic et al. 2014). Targeting mitophagy specifically would be a safer option for such treatments given the toxicity associated with autophagy drugs.
Several mitochondria related protein specific inhibitors, such as hexokinase inhibitors (Ben Sahra et al. 2010; Chen et al. 2009; Mathupala et al. 2006), VDAC, and ANT inhibitors, are also reportedly used as mitochondrial targets in cancers (Belzacq et al. 2001; Don et al. 2003).
In conclusion, although there are several attempts to target mitochondria in cancer, the efficiency has been low due to the lack of complete understanding of the role of mitochondria in cancer. Mitochondria are complex organelles and they undergo drastic structural and functional changes during tumor development. As every cancer is defined by its own oncogenic signals, and every signal will have a different effect on mitochondria, there is a tremendous difference in how mitochondria are affected in each scenario. Like the saying—one glove does not fit all, there is a need to understand specific changes in mitochondria in specific cancers and target processes with one or a combination of drugs effectively.
References
Abu-Amero KK, Alzahrani AS, Zou M, Shi Y (2005) High frequency of somatic mitochondrial DNA mutations in human thyroid carcinomas and complex I respiratory defect in thyroid cancer cell lines. Oncogene 24:1455–1460
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334:1278–1283
Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, Fleming S, Alessi DR, Thompson AM (2012) Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer 106:1117–1122
Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR et al (2003) Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 278:17190–17197
Bailey CJ, Turner RC (1996) Metformin. N Engl J Med 334:574–579
Balic A, Sorensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J, Erkan M et al (2014) Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther 13:1758–1771
Belzacq AS, El Hamel C, Vieira HL, Cohen I, Haouzi D, Metivier D, Marchetti P, Brenner C, Kroemer G (2001) Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 20:7579–7587
Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C et al (2010) Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res 70:2465–2475
Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM (2014) Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508:108–112
Bjelakovic G, Gluud C (2007) Surviving antioxidant supplements. J Natl Cancer Inst 99:742–743
Blanchard JL, Schmidt GW (1996) Mitochondrial DNA migration events in yeast and humans: integration by a common end-joining mechanism and alternative perspectives on nucleotide substitution patterns. Mol Biol Evol 13:537–548
Boland ML, Chourasia AH, Macleod KF (2013) Mitochondrial dysfunction in cancer. Front Oncol 3:292
Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45:466–472
Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z (2007) Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A 104:11649–11654
Canter JA, Kallianpur AR, Parl FF, Millikan RC (2005) Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res 65:8028–8033
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–1657
Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Masciullo V et al (2003) Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A 100:5956–5961
Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138:628–644
Chatterjee A, Mambo E, Sidransky D (2006) Mitochondrial DNA mutations in human cancer. Oncogene 25:4663–4674
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 278:36027–36031
Chen Z, Zhang H, Lu W, Huang P (2009) Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim Biophys Acta 1787:553–560
Chinnery PF, Samuels DC, Elson J, Turnbull DM (2002) Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet 360:1323–1325
Chourasia AH, Macleod KF (2015) Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 11:1937–1938
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452:181–186
Cormio A, Milella F, Vecchiet J, Felzani G, Gadaleta MN, Cantatore P (2005) Mitochondrial DNA mutations in RRF of healthy subjects of different age. Neurobiol Aging 26:655–664
Dang CV (2010) Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res 70:859–862
Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8:870–879
Dominy JE Jr, Lee Y, Gerhart-Hines Z, Puigserver P (2010) Nutrient-dependent regulation of PGC-1alpha's acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta 1804:1676–1683
Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J, Hogg PJ (2003) A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 3:497–509
El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X (2000) Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275:223–228
Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D (2000) Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 287:2017–2019
Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T (2008) Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27:6002–6011
Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F (2011) Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7:523
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT et al (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899
Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS (2014) Targeting SOD1 reduces experimental non-small-cell lung cancer. J Clin Invest 124:117–128
Grivell LA (1983) Mitochondrial DNA. Sci Am 248:78–89
Habano W, Sugai T, Yoshida T, Nakamura S (1999) Mitochondrial gene mutation, but not large-scale deletion, is a feature of colorectal carcinomas with mitochondrial microsatellite instability. Int J Cancer 83:625–629
Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, Ames BN, Chisari FV (1994) Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci U S A 91:12808–12812
Hamanaka RB, Chandel NS (2010) Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35:505–513
Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287:19094–19104
Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, Ding Z, Ying H, Boutin AT, Zhang H et al (2012) Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148:651–663
Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ (1997) Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275:1649–1652
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW et al (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155:397–409
Jeronimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, Oliveira J, Lopes C, Fliss MS, Sidransky D (2001) Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 20:5195–5198
Jones JB, Song JJ, Hempen PM, Parmigiani G, Hruban RH, Kern SE (2001) Detection of mitochondrial DNA mutations in pancreatic cancer offers a "mass"-ive advantage over detection of nuclear DNA mutations. Cancer Res 61:1299–1304
Jung SN, Yang WK, Kim J, Kim HS, Kim EJ, Yun H, Park H, Kim SS, Choe W, Kang I et al (2008) Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 29:713–721
Kang N, Zhang JH, Qiu F, Tashiro S, Onodera S, Ikejima T (2010) Inhibition of EGFR signaling augments oridonin-induced apoptosis in human laryngeal cancer cells via enhancing oxidative stress coincident with activation of both the intrinsic and extrinsic apoptotic pathways. Cancer Lett 294:147–158
Karbowski M, Youle RJ (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 10:870–880
Kelly PN, Strasser A (2011) The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ 18:1414–1424
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325–337
Larman TC, DePalma SR, Hadjipanayis AG, Cancer Genome Atlas Research Network, Protopopov A, Zhang J, Gabriel SB, Chin L, Seidman CE, Kucherlapati R et al (2012) Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci U S A 109:14087–14091
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H et al (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15:110–121
Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277:20336–20342
Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP (2003) Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J 22:5501–5510
Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25:6225–6234
Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B et al (2008) Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A 105:2169–2174
Lightowlers RN, Chinnery PF, Turnbull DM, Howell N (1997) Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet 13:450–455
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44:479–496
Liu Z, Li D, Zheng X, Wang E, Wang J (2013) Selective induction of apoptosis: promising therapy in pancreatic cancer. Curr Pharm Des 19:2259–2268
Luciakova K, Kuzela S (1992) Increased steady-state levels of several mitochondrial and nuclear gene transcripts in rat hepatoma with a low content of mitochondria. Eur J Biochem 205:1187–1193
Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA (2002) Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion 1:425–435
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25:4777–4786
Maximo V, Soares P, Lima J, Cameselle-Teijeiro J, Sobrinho-Simoes M (2002) Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hurthle cell tumors. Am J Pathol 160:1857–1865
Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J (2009) A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A 106:11960–11965
Mitsuishi Y, Motohashi H, Yamamoto M (2012) The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol 2:200
Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P et al (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142:889–901
Morrish F, Hockenbery D (2014) MYC and mitochondrial biogenesis. Cold Spring Harb Perspect Med 4
Nagaraj R, Gururaja-Rao S, Jones KT, Slattery M, Negre N, Braas D, Christofk H, White KP, Mann R, Banerjee U (2012) Control of mitochondrial structure and function by the Yorkie/YAP oncogenic pathway. Genes Dev 26:2027–2037
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106
Neuzil J, Dyason JC, Freeman R, Dong LF, Prochazka L, Wang XF, Scheffler I, Ralph SJ (2007) Mitocans as anti-cancer agents targeting mitochondria: lessons from studies with vitamin E analogues, inhibitors of complex II. J Bioenerg Biomembr 39:65–72
Okami J, Simeone DM, Logsdon CD (2004) Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res 64:5338–5346
Ostman A, Frijhoff J, Sandin A, Bohmer FD (2011) Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem 150:345–356
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3):607–614
Owusu-Ansah E, Banerjee U (2009) Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461:537–541
Parker DJ, Iyer A, Shah S, Moran A, Hjelmeland AB, Basu MK, Liu R, Mitra K (2015) A new mitochondrial pool of cyclin E, regulated by Drp1, is linked to cell-density-dependent cell proliferation. J Cell Sci 128:4171–4182
Parrella P, Xiao Y, Fliss M, Sanchez-Cespedes M, Mazzarelli P, Rinaldi M, Nicol T, Gabrielson E, Cuomo C, Cohen D et al (2001) Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res 61:7623–7626
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S et al (2009) The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8:3984–4001
Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH et al (2005) mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A 102:719–724
Pollak M (2014) Overcoming drug development bottlenecks with repurposing: repurposing biguanides to target energy metabolism for cancer treatment. Nat Med 20:591–593
Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B (1998) Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet 20:291–293
Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B (2012) Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci 125:5745–5757
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF et al (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475:231–234
Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 108:10190–10195
Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL (2012) Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 26:2175–2186
Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA (2004) Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64:985–993
Rumsey WL, Schlosser C, Nuutinen EM, Robiolio M, Wilson DF (1990) Cellular energetics and the oxygen dependence of respiration in cardiac myocytes isolated from adult rat. J Biol Chem 265:15392–15402
Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C et al (2011) Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470:359–365
Salani B, Marini C, Rio AD, Ravera S, Massollo M, Orengo AM, Amaro A, Passalacqua M, Maffioli S, Pfeffer U et al (2013) Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci Rep 3:2070
Sanchez-Cespedes M, Parrella P, Nomoto S, Cohen D, Xiao Y, Esteller M, Jeronimo C, Jordan RC, Nicol T, Koch WM et al (2001) Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res 61:7015–7019
Sasaki R, Suzuki Y, Yonezawa Y, Ota Y, Okamoto Y, Demizu Y, Huang P, Yoshida H, Sugimura K, Mizushina Y (2008) DNA polymerase gamma inhibition by vitamin K3 induces mitochondria-mediated cytotoxicity in human cancer cells. Cancer Sci 99:1040–1048
Scarpulla RC, Vega RB, Kelly DP (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23:459–466
Schumacker PT (2006) Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 10:175–176
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48:158–167
Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N, Jacks T (2011) Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc Natl Acad Sci U S A 108:8773–8778
Sherr CJ, McCormick F (2002) The RB and p53 pathways in cancer. Cancer Cell 2:103–112
Shimoda R, Nagashima M, Sakamoto M, Yamaguchi N, Hirohashi S, Yokota J, Kasai H (1994) Increased formation of oxidative DNA damage, 8-hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res 54:3171–3172
Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N et al (2011) Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20:674–688
Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118:3930–3942
Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C, Harris AL (2003) Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol 201:573–580
Tan EY, Campo L, Han C, Turley H, Pezzella F, Gatter KC, Harris AL, Fox SB (2007) BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin Cancer Res 13:467–474
Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C et al (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J 28:1589–1600
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8:579–591
Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF (2007) BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 27:6229–6242
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Umeda S, Muta T, Ohsato T, Takamatsu C, Hamasaki N, Kang D (2000) The D-loop structure of human mtDNA is destabilized directly by 1-methyl-4-phenylpyridinium ion (MPP+), a parkinsonism-causing toxin. Eur J Biochem 267:200–206
Van den Bogert C, Muus P, Haanen C, Pennings A, Melis TE, Kroon AM (1988) Mitochondrial biogenesis and mitochondrial activity during the progression of the cell cycle of human leukemic cells. Exp Cell Res 178:143–153
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM et al (2013) PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 23:287–301
Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137:413–431
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698
Warburg O (1956) On respiratory impairment in cancer cells. Science 124:269–270
Wenz T (2013) Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion 13:134–142
Wong LJ, Lueth M, Li XN, Lau CC, Vogel H (2003) Detection of mitochondrial DNA mutations in the tumor and cerebrospinal fluid of medulloblastoma patients. Cancer Res 63:3866–3871
Yarosh W, Monserrate J, Tong JJ, Tse S, Le PK, Nguyen K, Brachmann CB, Wallace DC, Huang T (2008) The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet 4, e6
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B (2013) Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc Natl Acad Sci U S A 110:18226–18231
Zhang J, Ney PA (2009) Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 16:939–946
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL (2007) HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11:407–420
Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z (2011) Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A 108:16259–16264
Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA et al (2014) Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun 5:3295
Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313:459–465
Acknowledgments
I thank Drs. Harpreet Singh and James Barrett (Drexel University College of Medicine) for helpful discussions, and Dr. Frances Munoz and Kajol Shah for proof reading of the book chapter. The work was supported by a CURE grant.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gururaja Rao, S. (2016). Mitochondrial Changes in Cancer. In: Singh, H., Sheu, SS. (eds) Pharmacology of Mitochondria. Handbook of Experimental Pharmacology, vol 240. Springer, Cham. https://doi.org/10.1007/164_2016_40
Download citation
DOI: https://doi.org/10.1007/164_2016_40
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57311-3
Online ISBN: 978-3-319-57313-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)