
Abstract
Accumulation of amyloid β (Aβ) and its aggregates in the ageing central nervous system is regarded synonymous to Alzheimer’s disease (AD) pathology. Despite unquestionable advances in mechanistic and diagnostic aspects of the disease understanding, the primary cause of Aβ accumulation as well as its in vivo roles remains elusive; nonetheless, the majority of the efforts to address pathological mechanisms for therapeutic development are focused towards moderating Aβ accumulation in the brain. More recently, Aβ deposition has been identified in the eye and is linked with distinct age-related diseases including age-related macular degeneration, glaucoma as well as AD. Awareness of the Aβ accumulation in these markedly different degenerative disorders has led to an increasing body of work exploring overlapping mechanisms, a prospective biomarker role for Aβ and the potential to use retina as a model for brain related neurodegenerative disorders. Here, we present an integrated view of current understanding of the retinal Aβ deposition discussing the accumulation mechanisms, anticipated impacts and outlining ameliorative approaches that can be extrapolated to the retina for potential therapeutic benefits. Further longitudinal investigations in humans and animal models will determine retinal Aβ association as a potential pathognomonic, diagnostic or prognostic biomarker.
Similar content being viewed by others
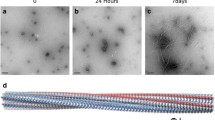

Avoid common mistakes on your manuscript.
Introduction
The retina arises as a neuro-ectodermal derivative of the forebrain during development and delineates several physiological, cellular and biochemical similarities with the brain tissue. With the advancement in technology, subtle changes in both human and animal retinas can be directly imaged and assessed in vivo and as such the retina is being increasingly used as a model to study the neurodegenerative disorders of brain particularly Alzheimer’s disease (AD) [1]. Additionally, retinal disorders such as age-related macular degeneration (AMD) and glaucoma are chronic neurodegenerative conditions that affect various retinal neurons and lead to progressive and irreversible loss of the vision. All three diseases are multifactorial with distinct pathological and clinical manifestations even though age remains the common primary risk factor and people with family history are at higher risk.
AD progressively ravages the brain and its cognitive ability and is accompanied by extreme cascade of pathological events marked by enhanced amyloid β (Aβ) levels and its deposition in the central nervous system (CNS) [2]. Several human and animal studies have reported ocular changes in AD and pathological accumulation of Aβ in the retina [1, 3]. The prominent manifestation of glaucoma is increased intraocular pressure (IOP) with defining features of irreversible retinal ganglion cell (RGC) loss and damage to the optic nerve (ON) [4]. Human post-mortem and animal studies have established aggregation of Aβ and other proteins in the retina exposed to increased IOP which then promotes RGC apoptosis [5]. AMD, a leading cause of severe vision impairment in people aged over 60 years affects the macular region of the retina which is critical for the high resolution central vision. Multiple studies have identified Aβ deposition in the retina with AMD and its progression [6]. Although AMD and glaucoma are not classified as amyloidogenic diseases, the last decade has seen many animal and human studies featuring evidence of progressive accumulation of Aβ fragments in the retina [5]. Therefore, retinal Aβ accumulation can be regarded as a common feature of these three separate disorders (Fig. 1).

Schematic representation highlighting disease specific clinical and pathophysiological observations and their overlap in AD, AMD and glaucoma
Amyloid precursor protein (APP) is expressed in various layers of the retina and plays roles in neurite outgrowth, synaptogenesis, and cell survival [7]. Sequential processing of APP generates various soluble and insoluble Aβ fragments including Aβ40 and Aβ42 [8]. Currently we cannot fully explain the mechanisms underlying Aβ accumulation in the eye and higher visual centres hindering development of possible therapeutic options. Further investigations into retinal Aβ accumulation may be potentially insightful in terms of understanding neurodegenerative mechanisms as Aβ peptides have established neurotoxic properties and play a major role in inducing pathological changes in the AD brain [9, 10]. Several studies highlight the association of AD with inner retinal and ON degenerative changes which is reminiscent of glaucoma pathology. On the contrary, AMD primarily affects the outer retina with loss of photoreceptors and reduced RPE compliance [11].
A combination of several etiological factors including; excitotoxicity, oxidative stress, neuroinflammation and chronic gliosis, proteolytic degradation, dysregulation of ocular hemodynamic parameters and ischaemia, transsynaptic degenerative changes, genetic causes and aberrant cellular signalling are thought to be involved in neurodegeneration and cell loss associated with these disorders but the exact sequence of events or molecular basis of disease remains largely unknown [12, 13]. There is currently no treatment available to prevent neuro-degeneration associated with any of these disorders notwithstanding that these diseases together comprise a major global challenge across the personal, social and economic spectrum. Based on the epidemiological studies again, we do not have a clear answer as to whether incidence of cognitive impairment or AD is more common in AMD or glaucoma subjects or otherwise if the reverse of this true. While some studies found an association between either glaucoma or AMD and cognitive impairment [14, 15]; others showed no significant association [16, 17].
A greater understanding of the nature of link between these disorders will help in the identification of overlapping molecular mechanisms as well as development of novel and effective treatment strategies. Further research should aim to explain the origins of Aβ in the retina and determine whether Aβ is causative factor in these retinal diseases or is a downstream product of a primarily cortical disease or both. This will help elucidate whether treatment for AD may protect against retinal disorders or vice versa as well as establish whether retinal Aβ can be used as a diagnostic or prognostic biomarker. It will highlight the need for clinicians to monitor the retina in AD, especially when combined with either glaucoma or AMD. In this review, we discuss the potential causes and implications of Aβ aggregation in the retina in AD, AMD and glaucoma and touch upon possible therapeutic options for reducing ocular Aβ accumulation emphasising the pathophysiological similarities and differences between these diseases.
Retinal pathology associated with amyloid β deposition
Alzheimer’s disease
Alzheimer’s disease is associated with the accumulation of soluble Aβ and its aggregated assemblies in the cerebral cortex and hippocampus leading to disrupted cellular metabolism, inflammation, neuronal cell death and cognitive decline [18]. Aβ deposition in CNS also leads to development of sensory deficits in the olfactory and visual systems [1, 19]. Mounting evidence indicates Aβ deposition in AD subjects and mice models corresponded to both retinal functional and structural deficits, reinforcing the concept of ocular pathological involvement in AD [1, 20]. We and others have shown congo red/thioflavin and antibody sensitive Aβ deposition mainly localised to the inner part and photoreceptor layer of retina and optic disc in various mice models of AD. This is accompanied by RGC loss and decline in pSTR amplitudes suggesting the retinal specific origin of these deficits [1, 7, 21, 22]. Accordingly, Perez et al. observed that ~35 % of the Aβ plaques were localised to the inner nuclear layer (INL) and recent studies highlight that there is a greater reduction in the thickness of the INL and inner plexiform layers (IPL) of the retina [1, 21]. Rescue experiments carried out using Aβ immunisation reduced Aβ plaque formation in the retina and afforded protection against loss of retinal laminar structure providing further evidence that Aβ plays a pathological role in the retina [22]. Our group and others have reported increase in TUNEL positive cells in the GCL highlighting enhanced apoptosis in the retina [1, 7]. Overall, neurotoxic effects of Aβ causing persistent synaptic transmission defects, dendritic network remodelling of cholinergic amacrine cells and insufficient acetylcholine levels are suggested to underlie these pathological changes [21, 23]. However, altered expression or localisation of mutant APP and PS1 proteins in transgenic mice as potential basis of these effects cannot be disregarded. Investigating mice models with inducible mutant APP expression in the retina or specific subset of its neurons will help corroborate retina specific roles of Aβ.
Clinically, AD subjects complain of frequent visual disturbances associated with colour discernment, stereoacuity, contrast consciousness, depth perception and comprehending structure from motion [24, 25]. A significant reduction of retinal nerve fibre layer (RNFL) thickness with loss initially occurring in the superior quadrant and reduced inferior visual field sensitivity has been reported in early AD subjects [3, 24, 26]. Moreover, functional losses characterised by changes in pattern electroretinogram (PERG) and visual evoked potential (VEP) recordings demonstrate inner retinal involvement. Reduced ganglion cell layer (GCL) thickness and ON degeneration has also been observed in post-mortem AD retinas [25, 27]. Optical coherence tomography (OCT) identified a loss in RNFL thickness at the macula and regions surrounding the optic nerve head (ONH) [3, 6]. Besides retina, there is sufficient histological evidence to support that Aβ deposits are formed in the visual cortex of AD subjects [28]. Thus apart from potential retinal Aβ neurotoxicity and associated morbidity, it is possible that retinal pathological changes may be derived through trans-synaptic degenerative activity in the higher visual centres. Interestingly, Aβ deposition has also been found in the lens leading to equatorial supra-nuclear cataract formation which bears a resemblance to the cataract formation in Down’s syndrome patients, another disease involving Aβ pathology [29]. Additionally, papillary abnormalities and altered pupil flash responses are observed in AD subjects [30]. Aβ immunoreactivity is also reported in the lens and cornea of AD mice models substantiating the human observations [10].
Another clinically relevant aspect is recognising the timeline of Aβ deposition in the retina relative to the AD brain pathology. Most animal studies show enhanced Aβ deposition in the retina at a much later point than plaque formation is observed in the brain [7, 10, 22]. However, a recent report has claimed identifying Aβ deposits in the APP/PS1 retinas well before the brain pathology [20]. While this may reflect different pathways of neuronal degeneration in APP/PS1 mice, biochemical and cellular changes in the retina may start well before Aβ deposits are observed or disease diagnosis. Accordingly, several clinical studies have identified structural and functional retinal abnormalities even at early stages of cognitive impairment [3, 24].
Age-related macular degeneration
Age-related macular degeneration is a progressive retinal degenerative disease influenced by both environmental and genetic factors. The disease is characterised by accumulation of cross-linked extracellular deposits collectively called drusen that form between the retinal pigment epithelium (RPE) and Bruch’s membrane (BrM). Studies have reported Aβ deposition in drusen particularly in the retinas of advanced AMD subjects [31]. Both Aβ40 and Aβ42 species have been identified in drusen and neo-vascular lesions in AMD retinas [11]. Aβ deposits have also been identified in RPE proximity along with other histo-pathological changes and loss of scotopic electroretinogram amplitudes [11]. Both soluble and oligomerised amyloid assemblies have been detected in drusen and RPE of human donor retinas [31]. Sub-retinal administration of Aβ42 in mice produced AMD-like symptoms manifested by decline in ERG amplitudes, RPE and photoreceptor degeneration including extensive vacuolation and BrM degeneration further highlighting its pathological involvement [32]. Collectively, these findings point towards a possible association of Aβ with AMD pathogenesis.
The inheritance of the APOE4 allele is associated with elevated cholesterol levels, which is a risk factor in AMD. Transgenic animals expressing human ApoE4 exhibit a retinal phenotype that replicates many features of AMD and ERG loss associated with this model was attenuated by anti-Aβ antibody treatment, strengthening the role for Aβ toxicity in the AMD retina [11]. Interestingly, ApoE allelic variant is also a significant risk factor for sporadic type of late onset AD [33] and the commonalities between AMD and AD can be recognised in humans as well as in transgenic animals that express human ApoE4 [6, 34]. ApoE2, E3 and ApoE4 have been shown to induce synaptic and ERG impairments in the retina of mice and ApoE4 in particular is considered a susceptibility gene for AMD indicating overlapping molecular networks with AD [34]. While subjects with ApoE4 are associated with greater risk of developing sporadic early and late type of AD deficits those with ApoE2 are linked with lower risk [33, 35]. This is consistent with ApoE allele involvement in moderating Aβ proteolysis and clearance, effects on Aβ synaptic toxicity [36], reduced synaptic plasticity, increased neuroinflammation and increased cerebrovascular events in the brain in AD [33, 35].
Subjects with advanced AMD at baseline also showed an increased risk for developing AD [37]. AMD and AD share several clinical and pathological features, including underlying oxidative stress, inflammation and the formation of protein aggregates [38]. Accumulation of intracellular iron, lipofuscin deposits as well as mitochondrial and lysosomal dysfunctions seem to be common pathophysiological events in both AMD and AD [38]. Collectively, these observations suggest that the neuronal degeneration occurring in AMD and AD has at least a partial overlap of molecular mechanisms, despite these being two completely distinct diseases (Fig. 1).
Glaucoma
Glaucoma is characterised by progressive damage to the RGCs and associated excavation of the ONH region. Abnormal APP metabolism and increased Aβ expression in RGCs is associated with the development of RGC apoptosis in experimental models of glaucoma [5, 39]. Aβ co-localises with apoptotic RGCs and its administration induces significant RGC apoptosis in vivo in a quantity and time-dependent manner [39]. APP is also synthesised in RGCs and can be transported along the ON and synapses. Studies in human glaucoma tissues and rodent models of chronically elevated eye pressure indeed suggest disrupted axonal transport which may be linked to impairment of APP metabolism and contribute to upregulation of neurotoxic Aβ in the retina [5, 40].
Exogenous intra-vitreal injections of Aβ42 and Aβ25–35 fragments significantly enhanced the RGC apoptosis, peaking at 3 days post-injection. Strong APP and Aβ immunoreactivity especially close to the lamina cribrosa, GCL and ON has been detected and higher levels of Aβ40 are observed in the ON of old DBA/2 J mice [41]. Intra-vitreal administration of Aβ42 and Aβ25–35 peptides induced GC death and retinal degeneration in animal models [39]. Significant protection of the inner retina was achieved by Aβ antibody as well as β-secretase inhibitor therapy [39].
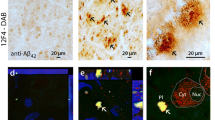
It has been shown that targeting different components of the Aβ formation and aggregation pathway can effectively reduce glaucomatous RGC apoptosis in animal models indicating that a combination of treatments may be more effective [39]. Our studies have identified enhanced levels of soluble Aβ42 in human ONH post-mortem tissues of glaucoma subjects [5] (Fig. 2a). Aβ peptide is reported to be in the detectable range in the aqueous humour in 40 % of patients with glaucoma [42]. Decreased levels of Aβ are reported in the vitreous of glaucoma patients [43] and although counterintuitive, this corresponds with the decreased CSF Aβ42 levels in AD patients. Interestingly, the impairment of brain-derived neurotrophic factor (BDNF), one of the key neurotrophic factors, has been implicated in the pathogenesis of glaucoma and RGC damage. We observed enhanced levels of Aβ42 in the ONH regions of BDNF impaired animals exposed to increased IOP [5] (Fig. 2b).

Higher levels of soluble Aβ42 are observed in the a. ONH of human post-mortem glaucoma tissues (n = 4; p < 0.01) and b mouse ONH exposed to high IOP (n = 4; p < 0.002). c Loss of inner retinal function (n = 5; p < 0.02) and d reduced GCL density is observed in mice exposed to high IOP (n = 4; p < 0.02) [5]
Glaucoma prevalence is significantly higher in AD patients than in age-matched controls with the incidence found to be 5.2-fold in a German and 2.6-fold in a Japanese population [15, 44]. Evidence of a link between glaucoma and AD has also emerged from studies showing that patients with AD have RGC loss associated with prototypal glaucomatous changes, such as optic neuropathy and visual functional impairment [3, 27]. One study reported greater visual field defects in patients with open-angle glaucoma and AD vs patients with glaucoma alone [44]. However, larger prospective studies are needed to determine the extent of epidemiological links between the two diseases.
Potential mechanisms of amyloid deposition in retina and its impact
Neurotoxic Aβ aggregates cause synaptic degeneration and interfere with long term potentiation in the brain [45]. Changes in Aβ levels can have detrimental effects on neuronal synaptic transmission of which the extent and exact mechanisms in the retina remain unknown. Similarly, whether the aggregation of Aβ starts as a protective response in the retina or is a pathological phenomenon by itself is debatable. Further, different forms of Aβ accumulation may have differential neurotoxicity. The potential mechanisms for Aβ accumulation and its biochemical, structural and functional effects on the retina with reference to AD, AMD and glaucoma are discussed here.
Metal ion and amyloid cross talk
There is much evidence to suggest that Aβ accumulation is associated with disturbances in the equilibrium of redox active transition metal ions [2]. AD brains have increased concentrations of metals including iron [46] and aluminium [2] that catalyse the production of free radicals. Increased levels of divalent metal ions such as Cu and Zn are also implicated. Both Cu and Zn bind APP; and it is believed that this can modulate the functional properties of the molecule [47]. Aβ precipitates out in the presence of these metal ions due to its high-affinity metal-binding site, and modulating this interaction can prevent the formation of Aβ from APP. Studies have demonstrated high levels of Zn and Cu in the Aβ plaques [47]. The potential significance of Zn in the aetiology of Aβ formation is demonstrated by the fact that Aβ deposition in APP transgenic mice is significantly reduced by breeding these with Zn transporter ablated animals that are unable to transport Zn into synaptic vesicles [48]. It would be interesting to determine whether retinas of these animals are protected against Aβ accumulation when this model is overlaid with Aβ associated retinal disease models. It is also worth noting that Aβ plaques are concentrated in the most Zn rich area of the brain, the hippocampus, and levels of hippocampal Zn are elevated in AD brains than age-matched controls [47]. Importantly significant high concentrations of Zn are observed in drusen deposits in retina linked with AMD [49]. In contrast, Zn administration has been reported to be protective in dry form of AMD [50] which may indicate differential effects of Zn on various stages or forms of AMD. Alternatively, it is also possible that these effects are attributed to differential localisation of the metal concentrations in retina. Cumulative oxidative damage caused by divalent metal ions may exacerbate Aβ neurotoxicity and lead to its enhanced aggregation and deposition [51].
Interestingly, ceruloplasmin- and hephaestin-deficient mice with disrupted Cu and Fe transportation and metabolism exhibit retinal degenerative changes similar to that observed in AMD which may provide further mechanistic insights into the role of metal ions in retina [52]. The effect of metal ion dysregulation has not been well characterised in glaucoma, but mitochondrial impairment in the disease points towards local disturbances in catalytic role of metal ions involved in electron transport. The role of Cu may be important in this as it is critical for the activity of many enzymes such as cytochrome-c oxidase. Accumulation of non-heme iron promotes disruption of cellular homeostasis and contributes to the mitochondrial dysfunction in AD and an analogous role of metal ion in the retinal mitochondrial impairment cannot be ruled out [53]. Further studies will unravel the causal links of chronic metal ion dysmetabolism in the retinal Aβ accumulation associated with the glaucoma, AMD and AD.
Neurotrophin impairment
Impairment of neurotrophins particularly BDNF and TrkB actions has been strongly suggested in AD and glaucoma pathologies while other neurotrophic factors like CNTF play an important role to protect the photoreceptors in AMD [5, 54]. Neurotrophins regulate Akt, Erk and Stat3 cellular signalling pathways which are critical for retinal neuroprotection in both AMD and glaucoma. Soluble forms of Aβ are implicated in mediating reduced BDNF mRNA levels and compromised BDNF signalling in the neuronal cells [55]. Aβ42 levels and Aβ42/Aβ40 ratios strongly correlate with decreased BDNF levels in the brain and retina [5, 56]. Aβ oligomerisation has also been demonstrated to impair BDNF retrograde transport along the axons [55] (Fig. 3). Briefly, Aβ upregulation is associated with decrease in BDNF levels in neurons, and in the retinal milieu this may lead to neurodegenerative changes by disrupting axonal transport and synaptic function. Brain cortical and hippocampal areas which exhibit large-scale amyloid pathology in AD demonstrate downregulation of BDNF levels [56, 57]. Similar disruption of neurotrophic factor homeostasis in ON, retina or its specific regions in advanced stages of glaucoma, AD or AMD in response to Aβ accumulation cannot be excluded. In the retina, our studies have shown an increase in soluble Aβ in the aged BDNF+/− mice indicating that BDNF perturbations may be responsible for enhanced amyloidogenesis in the retina [5]. These observations correspond with the significant inner retinal structural and functional loss in BDNF+/− animals highlighting its important role (Fig. 2c, d). Aβ levels were further exacerbated in the BDNF+/− mice exposed to experimental glaucoma and these changes commensurated with a reduced BDNF/TrkB signalling [5]. Decrease in BDNF levels and an increase in Aβ is reported in various models of experimental glaucoma as well in post-mortem tissues from glaucoma subjects [5].

Flow chart illustrating the impact of Aβ on the visual structure and function in AD, AMD and glaucoma
Increased Aβ-related cognitive impairment has also been reported in individuals having BDNF Val66Met polymorphism along with the apolipoprotein ApoEε4 allele in AD [58]. Although BDNF/TrkB signalling plays important roles in the retina and its impairment is associated with retinal Aβ perturbations, association of BDNF SNPs in retinal disorders or in mediating Aβ changes in the retina in AD and other retinal neurodegenerative disorders so far is speculative. Amyloid β aggregation may also lead to down-regulation of BDNF levels through sequestration, axonal transport obstruction and synaptic impairment [56]. This corresponds with the observations that Aβ oligomerisation strongly inhibits the BDNF-induced increase of activity-regulated cytoskeleton-associated protein (Arc) which plays a role in synaptic plasticity [59]. Further, it has also been shown that Aβ exposure results in up-regulation of full-length TrkB expression and a down-regulation in the levels of truncated TrkB in the neuronal cells indicating differential effects on various receptor subtypes and consequently different effects on downstream signalling have been observed [60].
Similar to BDNF involvement, a causal link has been suggested between NGF imbalance and activation of the amyloidogenic pathway in AD [61]. Prolonged treatment with NGF is suggested to be beneficial in improving visual acuity and electrophysiological responses in AMD [62]. CNTF administration provided significant protection against high IOP induced RGC degeneration in animal model. It is already being tested in clinical trials for non-exudative AMD and reported to be effective in stabilising visual acuity [54, 63]. Given that CNTF treatment was effective in shielding against synaptic impairment and mitigating memory loss in mouse model of AD [64], it may have potential beneficial effects for the retina in AD. TrkA and TrkB activation imparts neuroprotection to the RGCs in experimental models of glaucoma [5, 65, 66] and Aβ binding to TrkB-FL receptor negatively affects the synaptic plasticity [67]. Aβ binding to the p75NTR has similarly been demonstrated to induce apoptosis and p75NTR suppression protected RGCs following ON injury indicating potential pathological roles of Aβ in the retina [65, 68].
Association with inflammatory pathways
Aβ is associated with microglia, astrocyte and dendritic cell activation and is also an activator of the complement factors [69]. AD mice models depict significant upregulation of inflammatory markers in their retinas. Elevated GFAP and IBA1 levels were observed in various layers of Tg2576 mice retinas indicating astrocyte and microglial upregulation [22]. The APP/PS1 mouse model of AD also depicted increased levels of F4/80 macrophage marker in the retina indicating activation of microglial cells [21]. Astrocyte activation has been suggested to regulate ONH excavation and RNFL loss through the stimulation of matrix metalloproteases in glaucoma [70]. In contrast, AMD is a chronic inflammatory disease involving defective regulation of alternative complement pathway which generates inflammatory mediators. Drusen deposits are the major site of local chronic inflammation in AMD and Aβ co-localises with activated complement components in drusen deposits [71, 72]. It is still not precisely known what triggers the complement cascade; however, Aβ in drusen may be a candidate peptide, as it has a known role in promoting the complement pathways in AD [73]. Also, low levels of complementary factor H were observed in the retinas of various AD models suggesting a link of complement pathway dysregulation with amyloid deposition [74]. C1q which is the first element in the complement pathway exhibits increased neuronal expression both in AD as well as in animal models of glaucoma [75, 76].
Moreover, increased monocyte chemotactic protein (MCP1) staining in the GCL coinciding with sites of Aβ deposition was observed in APP/PS1 animals along with TUNEL positive cells [7]. Inflammatory cytokines, TNFα and its receptors TNFR1α/β have also been noted to be upregulated in the retinas of glaucoma patients [77]. β amyloid stimulates secretion of TNFα which stimulates iNOS production through sphingomyelinase pathway [78]. Interestingly, TNFR1β ablation affords robust neuroprotection to RGCs in mouse model of elevated IOP. Gene-profiling studies have also indicated upregulation of serum amyloid A which is an inflammatory marker in the trabecular meshwork and retina in human glaucoma [79]. Reduction in Aβ-derived neuro-inflammation through minocycline treatment which leads to microglial inhibition has protective effects in AD as well as on the RGCs in glaucoma [6, 80]. Overall, increased glial number and inflammatory markers may contribute to degenerative changes in the retina by releasing cytokines and ROS, although these may also be playing a role in the elimination of Aβ assemblies [81].
Imbalance of proteolytic system in the retina
Physiological regulation of proteolytic processes is fundamental to both the generation and clearance of toxic Aβ species. α, β and γ-secretase activity mediates generation of various Aβ fragments and these enzymes are, thus, imperative focus to modulate Aβ accumulation [82]. β-secretase 1 (BACE1) in particular initiates the formation of β-amyloid, and the fact that BACE1 levels are elevated in the AD brain provide compelling reasons to develop therapies directed at BACE1 inhibition to reduce β-amyloid generation. BACE1 changes in the retina under pathological conditions are not known although treatment with β-secretase inhibitors exhibits a protective effect on the RGCs against damage caused by excitotoxicity. There is evidence of neuroprotective effects of BACE1 inhibition in experimental models of ON damage [83]. More recent studies, however, highlighted that BACE−/− mice illustrate retinal thinning, apoptosis, loss of retinal blood architecture and lipofuscin accumulation. Pharmacological inhibition of BACE1 also resulted in increased choroidal neo-vascularization in animals [84]. These along with observations made by Devi and Ohno that partial BACE1 suppression did not reduce cerebral Aβ deposition in AD mice [85] suggest that targeting BACE1 might not be a valid strategy mainly due to associated side effects in the retina.
γ-secretase, on the other hand, regulates VEGFR signalling in RPE as well as vascular permeability and angiogenesis in retinal endothelial cells [86]. It also affects endothelial function through its modulation of notch signalling [87]. Targeting γ-secretase–presenilin complex may therefore find applications in pathology associated with abnormal vascularisation in retina such as in wet AMD. Furthermore, Aβ once generated is subject to proteolytic degradation by Aβ-degrading proteases such as endothelin-converting enzyme (ECE) and insulin-degrading enzyme although not much is known about their roles in the retina. Extracellular Aβ breakdown is accomplished mainly through metalloproteases like neprilysin and angiotensin converting enzyme (ACE). Interestingly, mice that lack neprilysin develop RPE degeneration, and exhibit sub-RPE Aβ deposits similar to that observed in AMD in humans [88]. ACE is also expressed in retina and vitreous humour in human and animal tissues [89]. Activation of endogenous ACE2 has been suggested as a potential therapeutic strategy for glaucoma treatment [89]. Plasmin and its activators have also been shown to be both activated by and involved in the degradation of Aβ aggregates [90]. Activation of plasminogen system mediates excitotoxicity induced injury to the RGCs and is associated with glaucomatous damage [91]. We have shown that Aβ aggregates form complexes with the protease inhibitor neuroserpin in the retina [92] and this can potentially limit proteolytic actions of plasminogen system on the Aβ assemblies and obstruct the clearance mechanisms.
Lysosomal and autophagic dysregulation
There is reasonable evidence to support disturbances of the lysosomal vacuolar system and its hydrolytic enzymes associated with Aβ generation and neurotoxicity in AD [93]. Aβ has been identified in various components of the lysosomal system, such as rab5 endosomes; autophagic vacuoles and multivesicular bodies. Several studies have recognised the endosomal/lysosomal pathway as an important regulator of APP processing. Lysosomal involvement in Aβ generation is further discernible as these organelles contain both APP as substrate and β- and γ-secretases as its processing enzymes [94]. Endosomes are involved in Aβ production from APP and mediate the uptake of Aβ and soluble APP. Aβ accumulates within neuronal lysosomes in AD mice brains and may undergo deposition in the retina by similar mechanisms [95]. Among a wide spectrum of lysosomal enzymes, cathepsins are associated with both production and degradation of Aβ directly as well as through activation of other proteases [96]. Cathepsins are well expressed in the retina and are upregulated in AMD and glaucoma conditions [97]. These changes reflect biochemical facilitation to increase the clearance of toxic and degradation products and can play crucial role in modulating Aβ levels in the retina. With the Aβ build-up in vacuolar structures, lysosomal dysfunction ensues and eventually the cellular degeneration contributes to discharge of Aβ and its oligomeric forms into the extracellular space.
Interestingly, inheritance of the ApoE4 allele is reported to exacerbate endocytosis upregulation and potentiate Aβ induced lysosomal impairment [98]. More recent studies also support impaired lysosomal autophagy in RPE dysfunction with implications in AMD [99]. Lipofuscin and drusen accumulate within lysosomes in RPE along with Aβ and negatively affect phagocytosis of casted photoreceptor outer segments [38]. Autophagosomes were also observed to be increased in the GCL, 1–4 weeks after exposure to elevated IOP in a chronic model of experimental glaucoma. Upregulation of autophagy indicators LC3-II and p62 subsequent to IOP elevation suggests potential autophagic flux disturbances [100]. Autophagy activation is suggested to exert a protective after ON injury and protect RGCs under stress conditions [101].
Mitochondrial dysfunction
Mitochondrial dysfunction is a prominent feature of Aβ induced neuronal toxicity in AD. APP and Aβ can cross the mitochondrial membrane, where they can interact with mitochondrial components, obstruct the transport of proteins to mitochondria, disrupt the electron transport chain, increase ROS production and actuate mitochondrial damage leading to neuronal dysfunction [102]. Aβ has been shown to promote formation of permeability transition pore in mitochondria and promote apoptosis [103]. Mitochondrial impairment makes less energy available to carry out the repair processes predisposing retinal cells to apoptosis [104]. Aβ is also associated with modulation of mitochondrial Ca2+ levels leading to alterations in organelle physiology and enhancing self fragmentation [105]. Being metabolically highly active, aberrant mitochondrial function in photoreceptors and RPE can have a significant impact in AMD. Mitochondrial dysfunction and disruption of mitochondrial axonal transport is strongly suggested as a pathophysiological feature in glaucoma, which may disturb the local energy balance and increase susceptibility of RGC to degeneration. Inhibition of histone deacetylation can rescue the hippocampal neurons from Aβ-induced impairment of mitochondrial axonal transport. This inhibition also provides significant protection to RGCs in ON injury models [106]. Failure to meet the energy demands in axonal and dendritic terminals may eventually cause synaptic dysfunction in the retinal neurons. Elucidating the impact of Aβ accumulation on retinal mitochondria in AD, AMD and glaucoma may uncover novel therapeutic targets to help protect the retina.
Oxidative stress
There is evidence supporting widespread oxidative stress triggered by increased ROS production and a dysfunctional antioxidant system in these three disorders [6] (Fig. 1). Prolonged exposure to increased oxidative stress is characterised by biochemical changes including lipid peroxidation, nitration, accumulation of advanced glycation end products (AGEs) and protein/nucleic acid damage [107]. In brain, oxidative stress markers may appear several years prior to significant Aβ deposition [107] and it is likely that similar changes occur in the retina in AD and other retinal disorders linked with Aβ accumulation. Oxidative stress is suggestively involved in the amyloidogenic processing of APP, thereby contributing to elevated intracellular Aβ levels [108]. Aβ also tends to undergo increased aggregation in the presence of free radicals, which enhances pro-oxidative environment by generating more ROS advancing cellular injury. Aβ deposition may in turn further amplify the neurotoxic effects by increasing neuronal sensitivity to oxidative stress [108]. Epidemiologic, genetic, and biochemical studies support a role for oxidative stress in AMD pathogenesis and RGC and ON damage in glaucoma. The macular region is particularly exposed to the high levels of oxidative stress that is increased with ageing [109]. Lipofuscin deposition in AMD is believed to further augment this by inducing an increased level of free radical formation. Similar to brain associated AD pathology, APP processing is likely to be influenced in the retina by exposure to an altered pro-oxidative environment and Aβ can both undergo increased aggregation under such conditions and augment the cellular sensitivity to its neurotoxic effects [108]. AGE receptors (RAGE) bind to Aβ and promote its influx into the CNS across the blood–brain barrier [110]. RAGE are also well expressed in the retina and functionally may play a role analogous to that of the brain by modulating Aβ endocytosis and its flux across the blood retinal barrier. Enhanced RAGE expression has accordingly been observed in the optic disc region of AD subjects [111].
Involvement of vascular changes
Retinal vascular changes may be early indicators of neurovascular uncoupling in AD [24, 112]. Decrease in retinal blood flow mimicking a situation similar to that associated with Aβ microvascular deposition in brain leading to cerebral hypoperfusion has been suggested [24]. Micro-vascular Aβ deposition is also reported in the retinas of Tg2576 mouse model of AD [7, 22]. Such amyloid deposition could contribute to local retinal vascular endothelial dysfunction, thereby encompassing dysregulation of vascular parameters in the ambit of Aβ associated retinal disorders [113]. Visual disturbances observed in AD patients may be linked with local narrowing of retinal and choroidal vessels or endothelial dysfunction mediated by Aβ toxicity [3, 24]. Interestingly, a correlation has been observed between retinal microvasculature changes and cognitive performance [114].
Aβ is shown to impair eNOS activity in endothelial cells caused by alterations in intracellular Ca2+ levels and posttranslational modifications [115]. Transport of Aβ through the blood–brain barrier is affected by eNOS and its inhibition may result in vascular dysfunction, impairment of microcirculation and promote Aβ tissue deposition [116]. Conversely, increased expression of APP and BACE1 as well as increased production of Aβ peptides is detected in the cerebral microvasculature of eNOS-deficient mice [117]. The relevance of these findings in the retina are not yet known although NO is expressed in all retinal cells that plays crucial role to synthesise cGMP and regulate phototransduction [118].
Aβ antibody mediated treatments in AD mice resulted in significant increases in vascular aggregation signifying the involvement of blood vessels in mediating Aβ clearance [119]. This along with the observation that Aβ deposits are observed in choroidal vessels raises concerns that Aβ deposition may also affect the endothelial function, blood supply and vascular elasticity or permeability in AMD. Interestingly, Aβ induces a marked increase in VEGF production by RPE cells [86] and thus it is likely to have significant implications in retinal neovascularisation in wet AMD. Glaucoma, particularly normal tension glaucoma, is also thought to have a strong vascular component, with reported changes in vascular regulation, retinal vascular pulsation, optic disc haemorrhages and upregulation of hypoxia inducible factor in the retina [6, 120]. High levels of both Aβ40 and Aβ42 are reported in the optic disc and region surrounding lamina cribrosa in experimental glaucoma models and human glaucoma ONH [5]. Such Aβ accumulation may cause local disturbances in hemodynamic parameters and also lead to the gradual failure of the elimination of APP cleavage products which can further exacerbate cytotoxic Aβ deposition. Advances in ocular OCT angiography will elucidate the extent of retinal vascular association with each of these diseases and determine potential relationship with Aβ deposition.
Targeting amyloid β in the retina
Deposition of Aβ in retina signifies increased Aβ formation, its aggregation, blunted clearance mechanisms or a combined effect of these deficits. The major question is whether Aβ itself represents one of the pathological causes or is an outcome of the associated retinal pathology. Nevertheless, success in protecting the retina against Aβ accumulation and associated neurotoxicity will stem from greater familiarity with the underlying pathological mechanisms. In this context, efforts to develop drugs for amelioration of Aβ-induced retinal toxicity will benefit from our prior experience in developing brain related therapies. Intervention at early stages before the onset of irreversible neurodegenerative changes in the retina is expected to have better prognosis. In addition to the disease stage, high heterogeneity amongst human populations and varied cohort, sex and age associated responses can lead to differential treatment outcomes. AD management includes using acetylcholinesterase inhibitors for early stages of AD and memantine as the disease progresses [121]. Other drugs such as valproic acid, monoclonal antibodies to Aβ (bapineuzumab, solanezumab, GSK-933776, MABT-5102A), γ-secretase inhibitors (semagacestat, begacestat, etc.) and statins (simvastatin, pitavastatin, atorvastatin, etc.) have been extensively investigated, but the design of clinical trials has not been able to clearly establish statistical significant primary outcomes in AD and no effective drug has been approved by FDA in the last decade [122]. Some of these therapies may find applications in protecting the retina against Aβ toxicity and can potentially be administered locally.
Can amyloid β generation in the retina be scaled down?
Pre-clinical studies indicate that treatments with β and γ secretase inhibitors and with anti-β secretase antibodies results in decreased levels of Aβ40 and Aβ42 species in the brain [123] (Table 1). In the retina, Guo et al. demonstrated that inhibiting Aβ generation minimises ganglion cell apoptosis in a rodent model of elevated IOP indicating that therapies targeting Aβ generation may have a protective effect in Aβ mediated retinal pathologies [39]. Intravitreal administration of enzyme modulators or neutralising antibodies can lead to decreased Aβ generation in the retina, with minimal systemic side effects. Thiazolidinedione derivative diabetes drugs rosiglitazone and pioglitazone stimulate the nuclear peroxisome proliferator-activated receptor γ (PPARγ) and are potent β secretase inhibitors and may find applications in reducing retinal Aβ build-up. PPARγ receptor activation suppresses APP expression and promotes its ubiquitination and subsequent degradation [124]. γ secretase inhibitors such as semagacestat ameliorate Aβ accumulation and additionally cause alterations in the notch signalling pathway [87]. Notch-signalling has been relatively well characterised in the Muller glial cells in mature retina and blocking notch activity prior to damage is suggested to be protective in various retinal neurons [125]. Non-steroidal anti-inflammatory drugs like ibuprofen and indomethacin selectively affect γ secretase cleavage site and block APP proteolysis leading to reduced Aβ production; however, comprehensive assessment of the safety and efficacy of these drugs in the retinal context needs to be carried out [126]. Similarly, statins in addition to their established cholesterol lowering properties, reduce Aβ production thereby ameliorating Aβ neurotoxicity and might potentially play a role in managing Aβ associated retinal disorders [127].
Experimental and epidemiological data suggest that decline in sex steroid hormone levels is associated with increased AD risk [128]. Interpretation of results from several clinical trials however indicated that hormone replacement therapy (HRT) in women does not improve cognition and one study even showed HRT to have negative effects on cognition [129]. AD subjects depict significant elevation in serum luteinizing hormone (LH) concentrations [130]. Treatments targeting LH can modulate cognitive behaviour in aged APP transgenic mice, and also reduce Aβ deposition. Gonadotropin-releasing hormone (GRH) agonist leuprolide, suppresses LH by down-regulating pituitary GRH receptors and is suggested to be an effective treatment for AD [131]. LH receptors are expressed in the neural retina and their pharmacological modulation may help prevent Aβ accumulation.
Additionally, several plant extracts and natural compounds have been demonstrated to promote non-amyloidogenic APP processing and reduce toxic Aβ generation and prevent its aggregation by multiple and complex mechanisms. Epigallocatechin-3-gallate for example, is present in green tea, which reduced amyloidogenesis in animal models of AD [132]. Antioxidants such as lipoic acid, ginkgo biloba extract, n-acetyl cysteine, flavonoid derivatives, vitamin E, curcumin, ubiquinone are also demonstrated to have protective effects in AD and reduce amyloidogenesis [133]. Interestingly some of these supplements have been suggested to exhibit protective effects in retina in AMD and glaucomatous conditions [134]. Dietary antioxidative supplements containing vitamins C, E, Zn and β carotene significantly reduced the AMD progression. Similarly, over-expression of antioxidant enzymes thioredoxins 1/2 showed a protective effect on RGCs in glaucoma while SOD1 deficient mice exhibited increased RGC loss [135, 136].
Can neurotoxic effects of Aβ be silenced by preventing its aggregation?
Amyloid β monomers tend to self-aggregate into various assemblies such as monomers, oligomers, protofibrils, fibrils and amyloid plaques. It is suggested that Aβ monomers can be neuroprotective, though conversely the aggregation of Aβ may contribute to AD pathology (Fig. 3). Soluble Aβ oligomers are also reported as being more harmful than monomeric or fibrillar forms and promote synaptic loss [137]. Several natural and synthetic molecules have been shown to prevent Aβ aggregation [132, 133]. Treatment with curcumin and nitrophenol for example, disrupted existing amyloid plaques and in some studies even partially restored the distorted neurite network [133]. Curcumin is shown to bind Aβ aggregates in the retina and protect the retinal neurons against excitotoxicity and oxidative damage [20, 138].
Clioquinol is a specific Cu2+, Zn2+ chelator and markedly inhibited cerebral Aβ aggregation in animal models of AD as well as in the patients in phase 2 trials. It readily penetrates the BBB due to its hydrophobic properties and may find therapeutically effective role in preventing Aβ deposition in the retina; however, careful investigations need to be carried out in the light of subacute myelo-optico neuropathy reports associated with its administration [139]. PBT2 also binds Cu2+ and Zn2+ ions and has even higher BBB permeability than clioquinol and its treatment reduced levels of soluble and insoluble Aβ. In the phase 2 trials, PBT2 was effective in reducing CSF Aβ42 levels in AD subjects [140]. PBT2 may, thus, have more bioavailability and effectively reduce Aβ load in vitreous and retina. Similarly, peptidic and non-peptidic compounds (RS-0406, SEN1269) that bind to Aβ and block its self-aggregation seem to work well in culture conditions [141], and given the ease of delivery and relatively lower Aβ load, retina will be a good model system to investigate their efficacy in vivo. Further, several antibodies targeting Aβ conformation have been developed to target its aggregation and tested positively in animal models of AD [142, 143] and some of these are currently undergoing clinical trials (Gammagard-Baxter, Gantenerumab-Roche, AAB-002-Elan) [144] (Table 1).
Will enhanced amyloid β clearance from retina be helpful?
Promoting Aβ clearance from retina has the potential to slow down or rescue the structural and functional defects associated with Aβ linked retinal disorders. Several pharmacological and immunotherapies have been tested while others are under development and have illustrated varying degrees of success in modulating Aβ levels in preclinical and clinical trials in AD [39, 143, 145, 146]. Aβ42 vaccination in young Tg2576 mice protected against Aβ deposition in brain while in aged animals it was not as efficacious in eroding already formed Aβ assemblies. Aβ42 immunisation also resulted in cognitive benefits and reduction in soluble Aβ in aged 3xTg mice suggesting that treatment may be more effective before the Aβ deposits are formed [143, 146]. However, Aβ42 immunisation approach (AN-1792) met with serious side effects such as meningoencephalitis possibly attributed to increased influx of T lymphocytes in brain and this needs to be considered when developing analogous therapies for the retina [147]. Various vaccines such as Lu AF20513, CAD-106, ACC-001, V-950, ACI-24, UB-311 have been developed with the aim to minimise the cellular and maximise the humoral immune responses [145, 148]. Phase 2 and 3 longitudinal trials in AD subjects are being conducted using next generation vaccines (CAD106) comprising much smaller Aβ sequences to minimise the immune response [149]. Nucleic acid-Aβ immunotherapy is another approach which is being evaluated in mice models of AD for non-inflammatory immune responses to target Aβ [150].
Aβ antibody therapies which target amino or carboxy terminal, middle part, different conformational states and aggregated forms of Aβ are under investigation. Antibody studies indicate reduction in amyloid load, plaque formation and improvement of cognitive function in mouse models of AD [151]. Humanised monoclonal antibodies (bapineuzumab, ponezumab and solanezumab) directed against different regions of the Aβ peptide have been tested in the clinic (Table 1). Although initial trials showed various cognitive benefits especially in ApoE/E4 non-carriers, overall results from larger phase 3 trials failed to achieve expected end-points and were also associated with vasogenic edema and associated MRI signal abnormalities and microhemorrhages [152, 153]. Modifications in antibody design may help ameliorate some of these side effects and phase 3 trials on such Aβ antibodies including aducanumab, gantenerumab, crenezumab, BAN2401, etc. are currently in progress in both MCI and AD subjects [121, 144, 149]. The deglycosylated form of an anti-Aβ40 (2H6-D) treatment in Tg2576 mice quite interestingly exhibited a significantly lower incidence of haemorrhagic and inflammatory brain lesions [142]. Setbacks associated with immunotherapies do not necessarily indicate that Aβ is the wrong target as these shortcomings could also arise from low passage of antibodies across the BBB and consequent inability to achieve required tissue levels. Besides, immunotherapeutic approaches could be most effective in the initial stages of the disease and effects may not be evident if the intervention is too late. Anti-Aβ40/42 mAb therapy targeting c-terminal of Aβ peptides resulted in decreased AMD-like pathology, improved visual acuity and protected against RPE structural changes and retinal functional deficits in a dose-dependent manner [11]. Antibody therapy also resulted in reduced RGC apoptosis in animal model of glaucoma [39]. Reduction in Aβ deposition and functional improvements indicated that approaches to enhance Aβ clearance represent a viable therapeutic option that needs to be investigated in the retinal context and refined to enhance efficacy and minimise side effects [20]. Efficacy of Aβ antibody therapies has not been evaluated for the retina in animal models of AD but represents a valid option to target soluble and aggregated Aβ through intravitreal access, which will also help minimise the systemic side effects.
Aβ levels can also potentially be modulated by stimulating increased uptake by glial cells. In this regard, activation of retinoid x-receptors (RXRs) in brain using RXR agonist bexarotene was explored to promote glial uptake of soluble Aβ to enhance clearance [154]. Independent follow-up studies, however, failed to confirm the protective effects of the drug or establish any beneficial effects on memory and cognition in animal models, with only few groups being able to confirm lowering of soluble Aβ levels [155, 156]. Given that RXRs are well expressed in the retina, alternative strategies to target them can have differential tissue specific effects on Aβ levels in the retina and help unravel the pathophysiological cross talk between RXR and Aβ. The soluble form of RAGE (sRAGE) has also been shown to compete for Aβ binding with the membrane-linked RAGE, thus promoting removal of circulating Aβ and its antagonists are being tested in clinical trials [157]. Monitoring in vivo retinal changes will provide significant inputs about potential applications of these treatments in Aβ linked ocular pathologies.
Do we have efficient neuroprotective therapies to buffer detrimental effects of Aβ in retina?
No medications or treatment strategies are yet credibly established in the clinic to specifically increase cerebral or retinal tolerance to neurotoxic effects of Aβ although several preclinical and clinical studies have suggested neuroprotective effects of various therapies in AD. Memantine, for example, reduces neurotransmission rates and is approved for dementia management and promotes reduction in Aβ toxicity in AD and vascular dementia; prevents tau hyperphosphorylation; decreases microglial activation and promotes release of neurotrophic factors from astroglia [158]. This drug also protected RGCs in mice models of inner retinal damage [159], but phase 3 glaucoma trial did not achieve primary endpoints [160]. Nevertheless, its role in decreasing Aβ or imparting protection against its toxicity in the retina was not considered in these studies. With the advent of newer in vivo Aβ and apoptotic cell imaging technologies, potential protective effects of memantine on Aβ toxicity in retina could be evaluated in further detail in AMD, AD and glaucoma. Latrepirdine, which is an antihistamine drug, inhibited the formation of mitochondrial permeability transition pore and protected neuronal mitochondria from Aβ mediated toxicity [161]. Latrepirdine may, therefore, find applications in protecting retinal mitochondrial dysregulation in AMD and glaucoma against Aβ effects. Acetylcholinesterase inhibitors such as donepezil, rivastigmine, galantamine are also widely used in AD management [162]. These are also suggested to boost retinal vasoprotection and enhance ocular blood flow in experimental models of glaucoma [163]. Detailed investigations on whether acetylcholinesterase inhibitor mediated retinal protection is facilitated by modulation of Aβ neurotoxic effects will provide mechanistic insights and unravel new lines of disease management. Another potential approach could be pharmacologically or genetically targeting group IV phospholipase A2 which is well expressed in the retinal tissues to reduce toxic effects of Aβ deposition as downregulation of this enzyme has previously been shown to exhibit protective effects in animal model of AD [164] (Table 1). Similarly, T cell mediated microglial activation was associated with reduced Aβ deposition and enhanced neurogenesis. Administration of T-cell modulating antigens exerted protective effects in AMD by reducing the drusen deposition [6, 165].
Gene therapy approaches to modulate neurotrophins such as BDNF have been demonstrated as beneficial in animal model of AD [166]. BDNF plays a critical role in the protection of RGCs in glaucoma and its impairment is associated with increased retinal Aβ [5]. BDNF gene therapy can rescue the retina, particularly RGCs, but the protective effects are transient and repeated administration or higher doses do not generally prolong the effects. Therefore, upstream regulators or downstream effectors of BDNF/TrkB signalling may serve as better therapeutic targets to improve BDNF/TrkB signalling [5, 66]. Both APP and Aβ bind to the Nogo receptor and its downregulation promoted increased levels of Aβ in the brain and plaque formation in animal models of AD. Treatment with soluble Nogo receptor was shown to reduce Aβ aggregation [167] and interestingly, soluble Nogo-66 receptor also prevented synaptic impairment and rescued RGC loss in chronic glaucoma, while Nogo upregulation induced RGC death [168]. Nogo signalling thus can be another potential therapeutic target for gene therapy or pharmacological intervention directed towards modulating Aβ effects.
Limitations and future directions
This review has highlighted the involvement of Aβ accumulation in retina in AD, AMD and glaucoma and its complex pathophysiological effects on the eye. Various strategies to manage Aβ accumulation and ameliorate its effects are discussed; however, predominantly non-mendelian genetic basis of these diseases designate that a single cure may not be easily achievable in the near future. Accumulating evidence from human and animal studies suggests that these three are distinct disease conditions but with overlapping Aβ related neurodegenerative processes. Analysing Aβ characteristics including fragment size, oligomerisation and anatomical distribution within the retina holds “reasonable promise” to provide unique retinal signatures and differentiate between various Aβ associated disease conditions. For instance, AMD-associated Aβ deposition is more restricted to RPE and likely to be identified as complex with drusen [71]. In glaucoma, expression of Aβ could be found in the inner retina [5, 39] along with ON excavation. Retinal Aβ deposition in AD is geographically more wide spread and again associated with degenerative changes in the inner retina and ON [1]. Successful identification of differences and similarities will drive drug development and mechanism based pathophysiological research. Imaging patterns of Aβ deposition in the eye could be of diagnostic and prognostic value when considered with other ocular or biochemical markers and play imperative role in disease monitoring. AD is mainly a brain disease and clinical evaluation of retinal involvement promises to help in AD diagnosis and prognosis. Development of newer Aβ imaging technologies will greatly accelerate research into mechanism based therapies for these neurodegenerative disorders. Longitudinal studies in larger cohorts and rescue experiments will support the hypothesis that Aβ deposition does exert neurotoxic effects on the retina. Concluding, the significance of Aβ deposition in mechanistic understanding of retinal pathology, disease diagnosis, prognosis or as a treatment target is highly relevant considering its known neurotoxic effects and the high incidence of these diseases in ageing populations.
Abbreviations
- AD:
-
Alzheimer’s disease
- AMD:
-
Age-related macular degeneration
- Aβ:
-
Amyloid beta
- RGC:
-
Retinal ganglion cell
- APP:
-
Amyloid precursor protein
- RNFL:
-
Retinal nerve fibre layer
- PERG:
-
Pattern electroretinogram
- VEP:
-
Visual evoked potential
- OCT:
-
Optical coherence tomography
- RPE:
-
Retinal pigment epithelium
- BrM:
-
Bruch’s membrane
- IOP:
-
Intra ocular pressure
- BDNF:
-
Brain derived neurotrophic factor
- CNTF:
-
Ciliary neurotrophic factor
- GDNF:
-
Glial derived neurotrophic factor
- Arc:
-
Activity regulated cytoskeleton-associated protein
- BACE:
-
β Secretase
- ACE:
-
Angiotensin converting enzyme
- AGE:
-
Advanced glycation end product
- VEGF:
-
Vascular endothelial growth factor
- INL:
-
Inner nuclear layer
- ROS:
-
Reactive oxygen species
- ONH:
-
Optic nerve head
- NSAID:
-
Non-steroidal anti-inflammatory drugs
- HRT:
-
Hormone replacement therapy
- LH:
-
Luteinizing hormone
- GRH:
-
Gonadotrophin releasing hormone
- SOD:
-
Superoxide dismutase
References
Gupta VK et al (2016) Amyloid beta accumulation and Inner retinal degenerative changes in Alzheimer’s disease transgenic mouse. Neurosci Lett 623:52–56
Gupta VB et al (2005) Aluminium in Alzheimer’s disease: are we still at a crossroad? Cell Mol Life Sci 62:143–158
Iseri PK, Altinas O, Tokay T, Yuksel N (2006) Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J Neuroophthalmol 26:18–24
Casson RJ, Chidlow G, Wood JP, Crowston JG, Goldberg I (2012) Definition of glaucoma: clinical and experimental concepts. Clin Exp Ophthalmol 40:341–349
Gupta V, You Y, Li J, Gupta V, Golzan M, Klistorner A, van den Buuse M, Graham S (2014) BDNF impairment is associated with age-related changes in the inner retina and exacerbates experimental glaucoma. Biochim Biophys Acta 1842:1567–1578
Sivak JM (2013) The aging eye: common degenerative mechanisms between the Alzheimer’s brain and retinal disease. Invest Ophthalmol Vis Sci 54:871–880
Ning A, Cui J, To E, Ashe KH, Matsubara J (2008) Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci 49:5136–5143
Lista S, Garaci FG, Ewers M, Teipel S, Zetterberg H, Blennow K, Hampel H (2014) CSF Abeta1-42 combined with neuroimaging biomarkers in the early detection, diagnosis and prediction of Alzheimer’s disease. Alzheimers Dement 10:381–392
Zhao Y, Bhattacharjee S, Jones BM, Hill JM, Clement C, Sambamurti K, Dua P, Lukiw WJ (2015) Beta-amyloid precursor protein (betaAPP) processing in Alzheimer’s disease (AD) and age-related macular degeneration (AMD). Mol Neurobiol 52:533–544
Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG (2009) Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefes Arch Clin Exp Ophthalmol 247:1213–1221
Ding JD et al (2011) Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci USA 108:E279–E287
Almasieh M, Wilson AM, Morquette B, Vargas JLC, Di Polo A (2012) The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res 31:152–181
Tham Y-C, Li X, Wong TY, Quigley HA, Aung T, Cheng C-Y (2014) Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 121:2081–2090
Woo SJ, Park KH, Ahn J, Choe JY, Jeong H, Han JW, Kim TH, Kim KW (2012) Cognitive impairment in age-related macular degeneration and geographic atrophy. Ophthalmology 119:2094–2101
Tamura H et al (2006) High frequency of open-angle glaucoma in Japanese patients with Alzheimer’s disease. J Neurol Sci 246:79–83
Keenan TD, Goldacre R, Goldacre MJ (2014) Associations between age-related macular degeneration, Alzheimer disease, and dementia: record linkage study of hospital admissions. JAMA Ophthalmol 132:63–68
Kessing LV, Lopez AG, Andersen PK, Kessing SV (2007) No increased risk of developing Alzheimer disease in patients with glaucoma. J Glaucoma 16:47–51
Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2:a006346
Wu N, Rao X, Gao Y, Wang J, Xu F (2013) Amyloid-beta deposition and olfactory dysfunction in an Alzheimer’s disease model. J Alzheimers Dis 37:699–712
Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, Schwartz M, Farkas DL (2011) Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54(Suppl 1):S204–S217
Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S (2009) Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. Invest Ophthalmol Vis Sci 50:793–800
Liu B, Rasool S, Yang Z, Glabe CG, Schreiber SS, Ge J, Tan Z (2009) Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer’s transgenic mice. Am J Pathol 175:2099–2110
Bentley P, Driver J, Dolan RJ (2008) Cholinesterase inhibition modulates visual and attentional brain responses in Alzheimer’s disease and health. Brain 131:409–424
Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL (2007) Retinal abnormalities in early Alzheimer’s disease. Invest Ophthalmol Vis Sci 48:2285–2289
Jackson GR, Owsley C (2003) Visual dysfunction, neurodegenerative diseases, and aging. Neurol Clin 21:709–728
Paquet C, Boissonnot M, Roger F, Dighiero P, Gil R, Hugon J (2007) Abnormal retinal thickness in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett 420:97–99
Parisi V, Restuccia R, Fattapposta F, Mina C, Bucci MG, Pierelli F (2001) Morphological and functional retinal impairment in Alzheimer’s disease patients. Clin Neurophysiol 112:1860–1867
McKee AC, Au R, Cabral HJ, Kowall NW, Seshadri S, Kubilus CA, Drake J, Wolf PA (2006) Visual association pathology in preclinical Alzheimer disease. J Neuropathol Exp Neurol 65:621–630
Moncaster JA et al (2010) Alzheimer’s disease amyloid-β links lens and brain pathology in Down syndrome. PLoS One 5:e10659
Fotiou DF, Brozou CG, Haidich AB, Tsiptsios D, Nakou M, Kabitsi A, Giantselidis C, Fotiou F (2007) Pupil reaction to light in Alzheimer’s disease: evaluation of pupil size changes and mobility. Aging Clin Exp Res 19:364–371
Isas JM, Luibl V, Johnson LV, Kayed R, Wetzel R, Glabe CG, Langen R, Chen J (2010) Soluble and mature amyloid fibrils in drusen deposits. Invest Ophthalmol Vis Sci 51:1304–1310
Liu C, Cao L, Yang S, Xu L, Liu P, Wang F, Xu D (2015) Subretinal injection of amyloid-beta peptide accelerates RPE cell senescence and retinal degeneration. Int J Mol Med 35:169–176
Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118
Malek G et al (2005) Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci U S A 102:11900–11905
Zlokovic BV (2013) Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol 70:440–444
Verghese PB et al (2013) ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci 110:E1807–E1816
Klaver CC, Ott A, Hofman A, Assink JJ, Breteler MM, de Jong PT (1999) Is age-related maculopathy associated with Alzheimer’s Disease? The Rotterdam Study. Am J Epidemiol 150:963–968
Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M (2011) Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J Alzheimers Dis 24:615–631
Guo L et al (2007) Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci USA 104:13444–13449
Kipfer-Kauer A, McKinnon SJ, Frueh BE, Goldblum D (2010) Distribution of amyloid precursor protein and amyloid-beta in ocular hypertensive C57BL/6 mouse eyes. Curr Eye Res 35:828–834
Goldblum D, Kipfer-Kauer A, Sarra G-M, Wolf S, Frueh BE (2007) Distribution of amyloid precursor protein and amyloid-β immunoreactivity in DBA/2J glaucomatous mouse retinas. Invest Ophthalmol Vis Sci 48:5085–5090
Janciauskiene S, Krakau T (2001) Alzheimer’s peptide: a possible link between glaucoma, exfoliation syndrome and Alzheimer’s disease. Acta Ophthalmol Scand 79:328–329
Yoneda S, Hara H, Hirata A, Fukushima M, Inomata Y, Tanihara H (2005) Vitreous fluid levels of β-amyloid(1–42) and tau in patients with retinal diseases. Jpn J Ophthalmol 49:106–108
Bayer AU, Ferrari F (2002) Severe progression of glaucomatous optic neuropathy in patients with Alzheimer’s disease. Eye (Lond) 16:209–212
Trillaud-Doppia E, Paradis-Isler N, Boehm J (2016) A single amino acid difference between the intracellular domains of amyloid precursor protein and amyloid-like precursor protein 2 enables induction of synaptic depression and block of long-term potentiation. Neurobiol Dis 91:94–104
Belaidi AA, Bush AI (2015) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem. doi:10.1111/jnc.13425
Avan A, Hoogenraad TU (2015) Zinc and copper in Alzheimer’s disease. J Alzheimers Dis 46:89–92
Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY (2002) Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc Natl Acad Sci USA 99:7705–7710
Lengyel I et al (2007) High concentration of zinc in sub-retinal pigment epithelial deposits. Exp Eye Res 84:772–780
Age-Related Eye Disease Study 2 Research Group (2013) Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 309:2005–2015
Greenough MA, Camakaris J, Bush AI (2013) Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 62:540–555
Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL, Dunaief JL (2004) Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci USA 101:13850–13855
Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1802:2–10
Zhang K et al (2011) Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci USA 108:6241–6245
Garzon DJ, Fahnestock M (2007) Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci 27:2628–2635
Peng S et al (2009) Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci 29:9321–9329
Garzon D, Yu G, Fahnestock M (2002) A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J Neurochem 82:1058–1064
Lim YY et al (2015) APOE and BDNF polymorphisms moderate amyloid beta-related cognitive decline in preclinical Alzheimer’s disease. Mol Psychiatry 20:1322–1328
Echeverria V, Berman DE, Arancio O (2007) Oligomers of beta-amyloid peptide inhibit BDNF-induced arc expression in cultured cortical Neurons. Curr Alzheimer Res 4:518–521
Wong J, Higgins M, Halliday G, Garner B (2012) Amyloid beta selectively modulates neuronal TrkB alternative transcript expression with implications for Alzheimer’s disease. Neuroscience 210:363–374
Cattaneo A, Capsoni S, Paoletti F (2008) Towards non invasive nerve growth factor therapies for Alzheimer’s disease. J Alzheimers Dis 15:255–283
Lambiase A, Coassin M, Tirassa P, Mantelli F, Aloe L (2009) Nerve growth factor eye drops improve visual acuity and electrofunctional activity in age-related macular degeneration: a case report. Ann Ist Super Sanita 45:439–442
Pease ME et al (2009) Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci 50:2194–2200
Garcia P et al (2010) Ciliary neurotrophic factor cell-based delivery prevents synaptic impairment and improves memory in mouse models of Alzheimer’s disease. J Neurosci 30:7516–7527
Lebrun-Julien F, Morquette B, Douillette A, Saragovi HU, Di Polo A (2009) Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol Cell Neurosci 40:410–420
Gupta VK, You Y, Klistorner A, Graham SL (2012) Shp-2 regulates the TrkB receptor activity in the retinal ganglion cells under glaucomatous stress. Biochim Biophys Acta 1822:1643–1649
Jeronimo-Santos A et al (2015) Dysregulation of TrkB receptors and BDNF function by amyloid-beta peptide is mediated by calpain. Cereb Cortex 25:3107–3121
Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE (2009) Selective vulnerability in Alzheimer’s disease: amyloid precursor protein and p75(NTR) interaction. Ann Neurol 65:294–303
Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009) Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87:181–194
Rogers RS, Dharsee M, Ackloo S, Sivak JM, Flanagan JG (2012) Proteomics analyses of human optic nerve head astrocytes following biomechanical strain. Mol Cell Proteomics 11(M111):012302
Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV (2004) Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res 78:243–256
Buschini E, Piras A, Nuzzi R, Vercelli A (2011) Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol 95:14–25
Zhao T et al (2015) Age-related increases in amyloid beta and membrane attack complex: evidence of inflammasome activation in the rodent eye. J Neuroinflamm 12:121
Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ (2011) Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. Neuroreport 22:623–627
Fonseca MI, Chu SH, Berci AM, Benoit ME, Peters DG, Kimura Y, Tenner AJ (2011) Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J Neuroinflamm 8:4
Stasi K, Nagel D, Yang X, Wang RF, Ren L, Podos SM, Mittag T, Danias J (2006) Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci 47:1024–1029
Tezel G (2008) TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res 173:409–421
Zeng C, Lee JT, Chen H, Chen S, Hsu CY, Xu J (2005) Amyloid-beta peptide enhances tumor necrosis factor-alpha-induced iNOS through neutral sphingomyelinase/ceramide pathway in oligodendrocytes. J Neurochem 94:703–712
Wang WH, McNatt LG, Pang IH, Hellberg PE, Fingert JH, McCartney MD, Clark AF (2008) Increased expression of serum amyloid A in glaucoma and its effect on intraocular pressure. Invest Ophthalmol Vis Sci 49:1916–1923
Bosco A et al (2008) Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2 J mouse model of glaucoma. Invest Ophthalmol Vis Sci 49:1437–1446
Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE (2009) Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci 29:4252–4262
Zhang H, Ma Q, Zhang YW, Xu H (2012) Proteolytic processing of Alzheimer’s beta-amyloid precursor protein. J Neurochem 120(Suppl 1):9–21
Yamamoto R, Yoneda S, Hara H (2004) Neuroprotective effects of beta-secretase inhibitors against rat retinal ganglion cell death. Neurosci Lett 370:61–64
Cai J et al (2012) beta-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med 4:980–991
Devi L, Ohno M (2013) Mechanisms that lessen benefits of beta-secretase reduction in a mouse model of Alzheimer’s disease. Transl Psychiatry 3:e284
Boulton ME, Cai J, Grant MB (2008) gamma-Secretase: a multifaceted regulator of angiogenesis. J Cell Mol Med 12:781–795
Gupta VB, Gupta VK, Martins R (2013) Semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369:1660–1661
Ohno-Matsui K (2011) Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog Retin Eye Res 30:217–238
Foureaux G et al (2013) Antiglaucomatous effects of the activation of intrinsic Angiotensin-converting enzyme 2. Invest Ophthalmol Vis Sci 54:4296–4306
Barker R, Love S, Kehoe PG (2010) Plasminogen and plasmin in Alzheimer’s disease. Brain Res 1355:7–15
Chintala SK (2016) Tissue and urokinase plasminogen activators instigate the degeneration of retinal ganglion cells in a mouse model of glaucoma. Exp Eye Res 143:17–27
Gupta V, Wall RV, Gupta V, Graham S (2016) Interaction with neuroserpin may be involved in the impairment of protease mediated amyloid β clearance from the brain and retina. Alzheimers Dement 11:864–865
De Kimpe L, van Haastert ES, Kaminari A, Zwart R, Rutjes H, Hoozemans JJ, Scheper W (2013) Intracellular accumulation of aggregated pyroglutamate amyloid beta: convergence of aging and Abeta pathology at the lysosome. Age (Dordr) 35:673–687
Zheng L, Cedazo-Minguez A, Hallbeck M, Jerhammar F, Marcusson J, Terman A (2012) Intracellular distribution of amyloid beta peptide and its relationship to the lysosomal system. Transl Neurodegener 1:19
Van Broeck B et al (2008) Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer’s disease. Neurobiol Aging 29:241–252
Wang C, Sun B, Zhou Y, Grubb A, Gan L (2012) Cathepsin B degrades amyloid-beta in mice expressing wild-type human amyloid precursor protein. J Biol Chem 287:39834–39841
Im E, Kazlauskas A (2007) The role of cathepsins in ocular physiology and pathology. Exp Eye Res 84:383–388
Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, Bu G (2012) Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem 287:44593–44601
Viiri J et al (2013) Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One 8:e69563
Kitaoka Y, Munemasa Y, Kojima K, Hirano A, Ueno S, Takagi H (2013) Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis 4:e860
Rodriguez-Muela N, Germain F, Marino G, Fitze PS, Boya P (2012) Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ 19:162–169
Hansson Petersen CA et al (2008) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA 105:13145–13150
Ren R, Zhang Y, Li B, Wu Y, Li B (2011) Effect of beta-amyloid (25-35) on mitochondrial function and expression of mitochondrial permeability transition pore proteins in rat hippocampal neurons. J Cell Biochem 112:1450–1457
Schrier SA, Falk MJ (2011) Mitochondrial disorders and the eye. Curr Opin Ophthalmol 22:325–331
Hung CH, Ho YS, Chang RC (2010) Modulation of mitochondrial calcium as a pharmacological target for Alzheimer’s disease. Ageing Res Rev 9:447–456
Chindasub P, Lindsey JD, Duong-Polk K, Leung CK, Weinreb RN (2013) Inhibition of histone deacetylases 1 and 3 protects injured retinal ganglion cells. Invest Ophthalmol Vis Sci 54:96–102
Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, Smith MA (2010) Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology 59:290–294
Tamagno E, Guglielmotto M, Monteleone D, Tabaton M (2012) Amyloid-beta production: major link between oxidative stress and BACE1. Neurotox Res 22:208–219
Chiras D, Kitsos G, Petersen MB, Skalidakis I, Kroupis C (2015) Oxidative stress in dry age-related macular degeneration and exfoliation syndrome. Crit Rev Clin Lab Sci 52:12–27
Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, Martins RN (2009) Clearance mechanisms of Alzheimer’s amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatry 14:469–486
Wang MY, Ross-Cisneros FN, Aggarwal D, Liang CY, Sadun AA (2009) Receptor for advanced glycation end products is upregulated in optic neuropathy of Alzheimer’s disease. Acta Neuropathol 118:381–389
Nicolakakis N, Hamel E (2011) Neurovascular function in Alzheimer’s disease patients and experimental models. J Cereb Blood Flow Metab 31:1354–1370
Hoh Kam J, Lenassi E, Jeffery G (2010) Viewing ageing eyes: diverse sites of amyloid beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS One 5:e13127
Wong TY et al (2002) Retinal microvascular abnormalities and cognitive impairment in middle-aged persons: the Atherosclerosis Risk in Communities Study. Stroke 33:1487–1492
Gentile MT et al (2004) Mechanisms of soluble beta-amyloid impairment of endothelial function. J Biol Chem 279:48135–48142
Provias J, Jeynes B (2014) The role of the blood–brain barrier in the pathogenesis of senile plaques in Alzheimer’s disease. Int J Alzheimers Dis 2014:191863
Austin SA, Santhanam AV, Hinton DJ, Choi DS, Katusic ZS (2013) Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J Neurochem 127:691–700
Eldred WD, Blute TA (2005) Imaging of nitric oxide in the retina. Vision Res 45:3469–3486
Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN (2007) Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience 144:950–960
Graham SL, Butlin M, Lee M, Avolio AP (2013) Central blood pressure, arterial waveform analysis, and vascular risk factors in glaucoma. J Glaucoma 22:98–103
Galimberti D, Scarpini E (2016) Emerging amyloid disease-modifying drugs for Alzheimer’s disease. Expert Opin Emerg Drugs 21:5–7
Schneider LS et al (2014) Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med 275:251–283
Rakover I, Arbel M, Solomon B (2007) Immunotherapy against APP beta-secretase cleavage site improves cognitive function and reduces neuroinflammation in Tg2576 mice without a significant effect on brain abeta levels. Neurodegener Dis 4:392–402
Landreth G, Jiang Q, Mandrekar S, Heneka M (2008) PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 5:481–489
Ghai K, Zelinka C, Fischer AJ (2010) Notch signaling influences neuroprotective and proliferative properties of mature Muller glia. J Neurosci 30:3101–3112
Kukar TL et al (2008) Substrate-targeting gamma-secretase modulators. Nature 453:925–929
Solomon A, Kivipelto M (2009) Cholesterol-modifying strategies for Alzheimer’s disease. Expert Rev Neurother 9:695–709
Pike CJ, Carroll JC, Rosario ER, Barron AM (2009) Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol 30:239–258
Henderson VW (2014) Alzheimer’s disease: review of hormone therapy trials and implications for treatment and prevention after menopause. J Steroid Biochem Mol Biol 142:99–106
Butchart J, Birch B, Bassily R, Wolfe L, Holmes C (2013) Male sex hormones and systemic inflammation in Alzheimer disease. Alzheimer Dis Assoc Disord 27:153–156
Blair JA, Palm R, Chang J, McGee H, Zhu X, Wang X, Casadesus G (2016) Luteinizing hormone downregulation but not estrogen replacement improves ovariectomy-associated cognition and spine density loss independently of treatment onset timing. Horm Behav 78:60–66
Mandel SA, Amit T, Kalfon L, Reznichenko L, Weinreb O, Youdim MB (2008) Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: special reference to epigallocatechin gallate (EGCG). J Alzheimers Dis 15:211–222
Craggs L, Kalaria RN (2011) Revisiting dietary antioxidants, neurodegeneration and dementia. Neuroreport 22:1–3
Rhone M, Basu A (2008) Phytochemicals and age-related eye diseases. Nutr Rev 66:465–472
Chew EY, Clemons TE, Agron E, Sperduto RD, Sangiovanni JP, Kurinij N, Davis MD, Age-Related Eye Disease Study Research Group (2013) Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no 35. Ophthalmology 120:1604–1611 e4
Caprioli J, Munemasa Y, Kwong JM, Piri N (2009) Overexpression of thioredoxins 1 and 2 increases retinal ganglion cell survival after pharmacologically induced oxidative stress, optic nerve transection, and in experimental glaucoma. Trans Am Ophthalmol Soc 107:161–165
Giuffrida ML, Caraci F, De Bona P, Pappalardo G, Nicoletti F, Rizzarelli E, Copani A (2010) The monomer state of beta-amyloid: where the Alzheimer’s disease protein meets physiology. Rev Neurosci 21:83–93
Wang LL, Sun Y, Huang K, Zheng L (2013) Curcumin, a potential therapeutic candidate for retinal diseases. Mol Nutr Food Res 57:1557–1568
Takeda A, Takada S, Ando M, Itagaki K, Tamano H, Suzuki M, Iwaki H, Oku N (2010) Impairment of recognition memory and hippocampal long-term potentiation after acute exposure to clioquinol. Neuroscience 171:443–450
Lannfelt L et al (2008) Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol 7:779–786
Amijee H, Scopes DI (2009) The quest for small molecules as amyloid inhibiting therapies for Alzheimer’s disease. J Alzheimers Dis 17:33–47
Wilcock DM et al (2006) Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci 26:5340–5346
Wang A, Das P, Switzer RC 3rd, Golde TE, Jankowsky JL (2011) Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J Neurosci 31:4124–4136
Lannfelt L, Moller C, Basun H, Osswald G, Sehlin D, Satlin A, Logovinsky V, Gellerfors P (2014) Perspectives on future Alzheimer therapies: amyloid-beta protofibrils—a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6:16
Vellas B et al (2013) Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement 9:438–444
Lemere CA, Masliah E (2010) Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol 6:108–119
Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F (2004) Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol 14:11–20
Davtyan H et al (2013) Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J Neurosci 33:4923–4934
Wisniewski T, Goni F (2015) Immunotherapeutic approaches for Alzheimer’s disease. Neuron 85:1162–1176
Lambracht-Washington D, Rosenberg RN (2015) A noninflammatory immune response in aged DNA Abeta42-immunized mice supports its safety for possible use as immunotherapy in AD patients. Neurobiol Aging 36:1274–1281
Demattos RB et al (2012) A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 76:908–920
Salloway S et al (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370:322–333
Doody RS et al (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370:311–321
Cramer PE et al (2012) ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335:1503–1506
LaClair KD, Manaye KF, Lee DL, Allard JS, Savonenko AV, Troncoso JC, Wong PC (2013) Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener 8:18
Ulrich JD et al (2013) In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegener 8:13
Walker D, Lue LF, Paul G, Patel A, Sabbagh MN (2015) Receptor for advanced glycation endproduct modulators: a new therapeutic target in Alzheimer’s disease. Expert Opin Investig Drugs 24:393–399
Wu HM et al (2009) Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34:2344–2357
Kim TW, Kim DM, Park KH, Kim H (2002) Neuroprotective effect of memantine in a rabbit model of optic nerve ischemia. Korean J Ophthalmol 16:1–7
Osborne NN (2009) Recent clinical findings with memantine should not mean that the idea of neuroprotection in glaucoma is abandoned. Acta Ophthalmol 87:450–454
Porter T, Bharadwaj P, Groth D, Paxman A, Laws SM, Martins RN, Verdile G (2016) The effects of latrepirdine on amyloid-beta aggregation and toxicity. J Alzheimers Dis 50:895–905
Birks J (2006) Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev 25:CD005593
Almasieh M, MacIntyre JN, Pouliot M, Casanova C, Vaucher E, Kelly ME, Di Polo A (2013) Acetylcholinesterase inhibition promotes retinal vasoprotection and increases ocular blood flow in experimental glaucoma. Invest Ophthalmol Vis Sci 54:3171–3183
Ong WY, Farooqui T, Kokotos G, Farooqui AA (2015) Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci 6:814–831
Landa G, Butovsky O, Shoshani J, Schwartz M, Pollack A (2008) Weekly vaccination with Copaxone (glatiramer acetate) as a potential therapy for dry age-related macular degeneration. Curr Eye Res 33:1011–1013
Nagahara AH et al (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15:331–337
Park JH, Strittmatter SM (2007) Nogo receptor interacts with brain APP and Abeta to reduce pathologic changes in Alzheimer’s transgenic mice. Curr Alzheimer Res 4:568–570
Liu X, Zuo Z, Liu W, Wang Z, Hou Y, Fu Y, Han Y (2014) Upregulation of Nogo receptor expression induces apoptosis of retinal ganglion cells in diabetic rats. Neural Regen Res 9:815–820
Ghosh AK, Osswald HL (2014) BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem Soc Rev 43:6765–6813
Mecocci P, Polidori MC (2012) Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim Biophys Acta 1822:631–638
Acknowledgments
We acknowledge research funding from the NHMRC, ORIA, MQRDG and Hillcrest foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gupta, V., Gupta, V.B., Chitranshi, N. et al. One protein, multiple pathologies: multifaceted involvement of amyloid β in neurodegenerative disorders of the brain and retina. Cell. Mol. Life Sci. 73, 4279–4297 (2016). https://doi.org/10.1007/s00018-016-2295-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2295-x