
Abstract
Several integrated proteolytic systems contribute to the maintenance of cellular homeostasis through the continuous removal of misfolded, aggregated or oxidized proteins and damaged organelles. Among these systems, the proteasome and autophagy play the major role in protein quality control, which is a fundamental issue in non-proliferative cells such as neurons. Disturbances in the functionality of these two pathways are frequently observed in neurodegenerative diseases, like Alzheimer’s disease, and reflect the accumulation of protease-resistant, deleterious protein aggregates. In this review, we explored the sophisticated crosstalk between the ubiquitin–proteasome system and autophagy in the removal of the harmful structures that characterize Alzheimer’s disease neurons. We also dissected the role of the numerous shuttle factors and chaperones that, directly or indirectly interacting with ubiquitin and LC3, are used for cargo selection and delivery to one pathway or the other.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The maintenance of cellular homeostasis, cellular function and viability is mediated at least in part by extensive and specialized proteolytic systems that continuously process and remove intracellular misfolded proteins [1–3]. Protein degradation is critical in cell quality control, being central in numerous pathways such as cell cycle, cell growth and differentiation, apoptosis, regulation of transcriptional factors, carcinogenesis, and immune/inflammatory responses [1]. Disturbances in intracellular proteostasis trigger the accumulation of altered proteins and toxic aggregates that are widely recognized hallmarks of neurodegenerative diseases, including Alzheimer’s disease (AD) [4]. Misfolded, unfolded or oxidized proteins expose hydrophobic regions normally hidden in their native conformation, thus being highly reactive to form oligomeric complexes. The harmful effect of such modifications is a deleterious gain of toxic function. In fact, oligomers represent the effective toxic specie responsible for synaptic and neuronal dysfunctions whereas large and insoluble aggregates work as reservoirs of bioactive oligomers [5–7]. In addition, neurons are post-mitotic cells unable to dilute or remove abnormal protein aggregates via cell division and therefore are more sensitive to these toxic proteinaceous species compared to mitotic cells.
In AD, deposits of amyloid-β (Aβ) peptides and hyperphosphorylated tau protein derived from peptide/protein misfolding and aggregation that ultimately produce amyloid plaques and neurofibrillary tangles [8]. The major consequence of the accumulation of such pathogenic aggregates is the downregulation of proteolytic pathways triggering a feed-forward loop that in the end destroy essential proteolytic networks. Eukaryotic cells contain a diverse set of proteases that mediate protein degradation and protein cleavage [9]. However, two proteolytic machineries, namely the proteasomal system and autophagy, regulate the majority of protein catabolism and the interdependent regulation of these two sophisticated systems is responsible for overall protein quality control and proteostasis. Autophagy, literally “self-eating”, is a proteolytic process in charge of the recycling of both extracellular and intracellular components. Autophagic pathways are divided into macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy, depending on the mechanism by which cellular cargoes are delivered to the lysosome [10]. Proteolysis by autophagy is finally mediated by lysosomal hydrolytic enzymes, such as phosphatases, nucleases, glycosidases, proteases, peptidases, sulfatases, and lipases [11]. In details, autophagy mediates the digestion of exogenous particles and long-lived proteins, and contributes to the renewal of damaged and/or dysfunctional organelles [1, 12, 13]. Numerous papers reported on the occurrence of disturbances in autophagy in several human neurodegenerative disorders [11, 14]. For example, in AD an altered lysosomal acidification and the lysosomal proteolytic disruption are major contributors to autophagy failure and its pathological consequences [15]. The inactivation of the autophagy-related proteins 5 or 7 (Atg5 and Atg7, respectively) leads to neurodegeneration with abnormal increase of intracellular proteins in inclusions bodies [16, 17]. Moreover, AD patients with the familial amyloid precursor protein (AβPP) Swedish mutation showed the accumulation of markers of the autophagy-lysosomal pathway and their colocalization with hyperphosphorylated tau protein [18].
The proteasomal proteolytic system, in turn, mediates the degradation of short-lived, oxidatively damaged, modified and misfolded proteins (collectively accounting for more than 70–80 % of intracellular proteins) both in the cytoplasm and the nucleus [19]. Proteasome targets include also regulatory proteins such as cyclins (cyclins A, B, D and E), cyclin-dependent kinases, cyclin-dependent kinase inhibitors (p21 and p27), inhibitory proteins (Fos and Myc) and tumour suppressors (cyclin B1, p53), the degradation of which is a key event in cell cycle progression [20–28]. Proteasome activity differs from the lysosomal-dependent proteolysis because proteasome-mediated protein degradation occurs at neutral pH and does not require calcium or organelle compartmentalization. The 20S proteasome can degrade substrates either alone or in association with regulatory particles to form a complex, the 26S proteasome, which specifically recognizes ubiquitin (Ub)-tagged proteins [19, 29]. This 26S-mediated pathway of protein degradation is known as the Ub-proteasome system (UPS) and most of its substrates, as mentioned, have to be polyubiquitinated. Ubiquitination is a post-translational modification that forms an isopeptide bond between a substrate lysine residue and the C-terminus of Ub and requires a complex system of four different kinds of enzymes (known as E1–E2–E3–E4) [19, 30, 31]. The proteasome takes part in the “quality control” of newly synthesized proteins in association with the endoplasmic reticulum (ERAD, endoplasmic reticulum associated degradation). This pathway assures that improperly folded proteins in the ER are targeted for degradation by the UPS [32, 33]. Different molecular chaperones, such as heat shock proteins (Hsps), help both newly synthesized and misfolded proteins reaching/restoring their native and nontoxic conformation. If unsuccessful, irreversibly damaged proteins are driven to the UPS for final degradation [34–37]. Several studies described dysfunctions in proteasome functionality in neurodegenerative diseases, including AD [38–40]. In particular, it was observed that the accumulation of abnormal deposits of pathogenic proteins, like prion protein, α-synuclein and huntingtin protein, inhibits the functionality of some UPS components, including the proteasome [41–43]. The proteasome and autophagic pathways were long considered as distinct and independent proteolytic systems. Conversely, numerous publications recently highlighted their intimate correlation and a considerable interplay between them, with the downregulation of one clearance machinery resulting in compensatory changes in the other pathway [4, 44]. Several proteins, including sequestosome 1/p62 (SQSTM1/p62), neighbour of BRCA1 gene (NBR1), histone deacetylase 6 (HDAC6), optineurin (OPTN), nuclear dot protein 52 (NDP52), valosin containing protein (p97/VCP), autophagy-linked FYVE protein (Alfy) and Bcl-2-associated athanogene (BAG) proteins, serve as linkers between the two pathways facilitating this interplay, and possess the ability to directly or indirectly associate with both ubiquitin and components of the autophagic system. This review article focuses on the role of the proteasome and autophagy in AD, with a particular emphasis on their crosstalk and on the molecules that mediate this interplay.
The proteasomal system
The 20S proteasome
The proteasomal system regulates the intracellular degradation of oxidized proteins and transcriptional factors, and controls signal transduction, immune response, carcinogenesis, cell division, growth and differentiation, DNA repair, morphogenesis of neuronal networks, and apoptotic process [1, 19, 29]. The main particle of this machinery is the 20S proteasome that possesses the catalytic activities responsible for substrate degradation. Numerous studies suggest that its structure and biogenesis are highly conserved from yeast to mammals [31, 45]. The 20S proteasome is a large, barrel-shaped complex with a molecular weight of about 700 kDa and constitutes up to 1 % of the total cellular protein. It consists of four stacked rings, two α-rings and two β-rings, delimiting an internal cavity and arranged as follows: αββα. Each ring is made of seven distinct subunits leading to a definitive configuration α(1–7)β(1–7)β(1–7)α(1–7). The β-rings define the main internal chamber of the complex and carry the catalytic activity, whereas the outer α-rings regulate the substrate enter into the catalytic chamber and the binding of different regulatory proteins [31, 46]. Mutagenesis studies characterized subunits β1, β2 and β5 as the subunits responsible for the catalytic activities. Subunit β1 is associated with the caspase activity and possesses a limited branched chain amino acid preferring (BrAAP) activity whereas subunit β2 has the trypsin-like (T-L) activity. Subunits β5 accounts for the chymotrypsin-like (ChT-L) activity, but given its tendency to cleave after small neutral and branched side chains also the SNAAP and BrAAP activities can be assigned to this subunit [46, 47]. Substrates degradation originates small peptides with an average length of 8-12 amino acids. Studies of proteasome-mediated degradation revealed that the nucleophilic attack is mediated by the N-terminal threonine of the three catalytic β subunits [48]. This feature classifies the proteasome as a member of the N-terminal nucleophile amino-hydrolase family. The free 20S proteasome constitutes a major portion of the total amount of proteasomes in cells suggesting an independent involvement of this complex in intracellular proteolysis [49]. The 20S proteasome is usually found in its “inactive” form but regulatory proteins, unfolded proteins or proteasomal substrates can eventually activate it. Proteasome-mediated protein degradation requires the substrate unfolding and the contemporary opening of the gate formed by the N-terminal ends of the α2, α3 and α4 subunits in the outer rings. Furthermore, hydrophobic residues exposed by damaged or improperly folded proteins make the proteins susceptible to degradation and induce proteasome conformational changes favouring its proteolytic activity [50].
The ubiquitin–proteasome system (UPS)
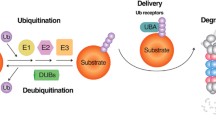
The interaction of the regulatory particle 19S with both α rings of the 20S proteasome generates a 2 MDa large complex, the so-called 26S proteasome, which is the main proteolytic component of the Ub pathway and responsible for the ATP-dependent degradation of Ub-tagged substrates [29, 51]. Ub is a 76 amino acid protein highly conserved and universally distributed among eukaryotes, with only three amino acids differing between yeast and human Ub [52]. Ub tagging controls and directs substrates for final degradation and recycling, and regulates key cellular processes including gene transcription, cell cycle progression, DNA repair, apoptosis, virus budding and receptor endocytosis [53]. Degradation of a protein via the UPS encompasses a cascade of enzymatic reactions. The first step regards the generation of a polyubiquitin chain and its covalent conjugation to the protein substrate, whereas the second step consists in the degradation of the tagged protein by the 26S proteasome (Fig. 1) [51]. Substrate ubiquitination is a very complex process that requires the presence of four different classes of enzymes: E1 (Ub activating enzymes)—E2 (Ub-carrier proteins or Ub-conjugating enzymes, Ubcs)—E3 (Ub-protein ligases)—E4 (Ub conjugation factor). E1 activates, ATP-dependently, Ub in its C-terminal glycine residue. After activation, E2 transfers Ub from E1 to a member of the Ub-protein ligase family, E3, to which the substrate protein is specifically bound [19, 51]. This enzyme catalyses the third step in the conjugation process, the covalent attachment of Ub to a lysine (K) residue of the substrate. After this, both E2 and E3 are released. The cyclic transfer of more Ub-molecules to the first Ub attached to the substrate is performed by these enzymes and by another enzyme, E4, and the so-formed chain serves as the recognition signal for the final degradation [19, 51, 54, 55]. Once the degradation is completed, deubiquitinating enzymes, such as the ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), release the single Ub-molecules from the Ub chains, in order to maintain the free pool of cellular Ub and to guarantee the formation of new chains [51]. Ub can be attached to the substrate as a single molecule at one or more amino acidic residues (respectively, monoubiquitination and multiple monoubiquitination) or as a polyubiquitin chain [56]. Monoubiquitination mainly regulate processes such as DNA repair, viral budding, and transcriptional regulation, whereas multiubiquitination is the principal signal for receptor endocytosis [57, 58]. Depending on which of the seven lysines of Ub is tagged (K6, K11, K27, K29, K33, K48, or K63) a protein will be transferred for degradation to the proteasome or to an autophagosome. Although the biological role of polyubiquitination is still under debate, the “classical” attachment of the C-terminus of one ubiquitin to the K48 of an adjacent Ub (a chain of at least four Ub moieties is necessary), is considered an established marker for proteasomal degradation, whereas substrates with single Ub-molecules or polyubiquitin chains that are attached on other lysine residues, mainly K63, are destined to the autophagosomes [53, 59]. Specific cellular proteins prevent the association of K63 Ub chains with proteasomes. Nathan et al. showed that proteins ESCRT (Endosomal Sorting Complex Required for Transport) and its components, STAM and Hrs, strongly associate with K63-ubiquitinated proteins and block their binding to proteasomes. In addition, they found that the Rad23 proteins, associate specifically with K48 conjugates and promote their binding to the 26S complex [60]. However, the possibility that also unconventional polyubiquitin chains linkages, including those on K11 and K63, are involved in proteasome-mediated proteolysis has not been excluded [53, 61, 62]. Upon K63-linked ubiquitination, adaptor molecules drive ubiquitinated substrates towards autophagy degradative pathway. The p62 protein is one of the most studied among these molecules and interacts with a higher affinity with monoubiquitinated and K63 polyubiquitinated chains through its Ub-associated (UBA) domain. Interestingly, in condition of proteasomal dysfunctions, p62 can also recognize K48-linked chains bringing them to the autophagosome [63]. Similarly, HDAC6 and NBR1, showing preference for K63-linked polyubiquitin chains, favour autophagic removal of protein aggregates [64–66]. Besides their role in directing substrates toward autophagy, K63-linked chains are involved in DNA repair, inflammation, apoptosis, internalization of plasma membrane proteins, and protein sorting to multivesicular bodies [67–71]. In a broad spectrum of human neurodegenerative disorders, the biogenesis and autophagy-mediated clearance of inclusion bodies are enhanced and facilitated by K63-linked Ub modifications [72, 73]. Components of the ubiquitination process and the nature of the Ub linkage play a critical role in AD. The overexpression of the E3 ligase parkin decreases Aβ load and gliosis in the brain of AD transgenic mice and its overexpression protects against deficits in memory, locomotion and neuropsychiatric behaviours [74]. In addition, parkin acts decreasing intracellular Aβ levels and extracellular plaque deposition, attenuating caspase activity, preventing mitochondrial dysfunction and oxidative stress and restoring neurotransmitter synthesis [75]. UCHL1 increases free Ub level and accelerates the lysosomal degradation of AβPP by promoting its ubiquitination. Furthermore, overexpression of UCHL1 reduces Aβ production and ameliorates classical AD symptoms in a transgenic mouse model suggesting that UCHL1 may be a safe and effective disease-modifying strategy to treat AD [76]. Interestingly, the final degradation of UCHL1 by the autophagy-lysosomal pathway is controlled by its parkin-mediated K63-linked polyubiquitination [77].

Schematic representation of the collaboration among UPS, autophagic pathways and molecular adaptors. a The ubiquitination process is the first event in the removal of misfolded/aggregated proteins or cellular organelles. b Attachment of the protein substrate to a K48/K11-linked Ub chain is the signal for 26S proteasome-mediated degradation. The chaperone BAG-1 selects substrates for proteasome degradation. c K63-linked Ub chains tag substrates to the autophagosome. p62, NBR1, NDP52 and OPTN directly associate with Ub and LC3 driving ubiquitinated cargo to the autophagosome for final degradation. BAG3 and Alfy vehicle substrates to the autophagic pathway. Under condition of proteasomal impairment, HDAC6 and p97 favour the autophagic removal of aggregates. d The autophagosome fuses with the lysosome and the protein cargo is finally degraded in the lysosomal lumen by the action of specific hydrolases
Autophagy
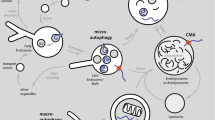
Autophagy is a highly conserved system of quality control by which cells capture intracellular components and deliver them to the lysosomal compartment where they are finally degraded [78]. The products of this degradation are recycled for the synthesis of new molecules. Nutrient deficiency or other stress conditions upregulate this catabolic process in order to either provide the cells for alternative energy metabolism pathways or remove toxic components thus maintaining cellular homeostasis [79]. Autophagy was originally considered a non-selective pathway induced in response to stressful environment but recently it emerged as a highly selective process involved in the clearance of dysfunctional organelles, protein aggregates and intracellular pathogens [80, 81]. Mounting evidences implicate autophagy flaws in numerous neurodegenerative conditions, particularly AD [15–18]. In mammalian cells, there are three distinct types of autophagy differing in the way cargo proteins are delivered to the lysosomes: macroautophagy, CMA and microautophagy [82]. Only proteins can be delivered to lysosomes via CMA, whereas macroautophagy and microautophagy concern the degradation of proteins and organelles [83].
Macroautophagy
Macroautophagy is a conserved process and the major lysosomal degradative pathway involving more than 30 autophagy-related genes (Atgs) [84]. The process starts with the formation of double-membrane vesicles (phagophore) originated from the ER/Golgi, which engulfs the cytoplasmic cargo forming the autophagosome [79, 83]. Once formed, autophagosomes move along microtubules toward the perinuclear microtubule-organizing centre (MTOC) of the neuron, where the concentration of lysosomes is higher [85]. In response to specific stimuli, such as nutrient deprivation or rapamycin treatment, the activity of autophagy-inhibitory complexes (such as the mammalian target of rapamycin or mTOR, a serine–threonine kinase) is inhibited, contributing to autophagy activation. The autophagosome formation process consists of three phases: initiation, nucleation and elongation of the membrane. The unc-51-like kinase (ULK1) complex is activated upon the dissociation from mTOR and induces the phagophore. Membrane nucleation involves the formation of a complex between beclin-1 (BECN1, the mammalian homologue of Atg6) and phosphatidylinositol 3-kinase (PI3K), whose components are BECN1, PIK3, activating molecule in BECN1-regulated autophagy protein 1 (AMBRA1), vacuolar protein sorting 34 (Vps34), vacuolar protein sorting 15 (Vps15), and Atg14. ULK1 phosphorylates AMBRA1 and, upon phosphorylation, BECN1 promotes the local production of the lipid signalling molecule phosphatidylinositol 3-phosphate (PI3P) by Vps34 that recruits other Atg proteins allowing the expansion of the autophagosomal membrane [81]. The elongation step requires Atg3, Atg4, Atg7, Atg10, and an Atg5-Atg12-Atg16L1 complex to conjugate phosphatidylethanolamine (PE) to LC3 (microtubule-associated protein light 3). LC3 is the mammalian homologue of Atg8 and exists in two forms, namely LC3-I (cytosolic) and LC3-II (membrane-bound). In this phase, LC3-I is recruited into the autophagosome where LC3-II is generated by site-specific proteolysis and covalent conjugation to PE, leading to the translocation of LC3 from cytoplasm to the membrane of the forming autophagosomes. This event contributes to the closure of the membrane and to the complete formation of the autophagosome [85, 86]. In the final step, the autophagosome merges with the lysosome that releases hydrolases, resulting in the degradation of the autolysosome content. The fusion process is regulated by several proteins such as the lysosome-associated membrane protein type 2A (LAMP2), the Rubicon-UVRAG complex, the soluble N-ethylmalemide sensitive factor attachment protein receptor (SNAREs), homotypic fusion and protein sorting (HOPS), Ras [rat sarcoma] like in rat brain (Rab), ESCRT, and LC3 [78]. Autophagy can also be induced in a mTOR-independent manner with compounds that decrease inositol (Ins) or inositol 1,4,5-trisphosphate (IP3) levels in the phosphoinositol-signalling pathway. These agents include drugs such as lithium and L-690,330 that inhibit inositol monophosphatase (IMPase), and carbamazepine and sodium valproate that inhibit inositol synthesis [87–89]. Besides the non-selective engulfment, the autophagosomal vesicle membrane can also selectively recognize its substrates, particularly in the case of damaged organelles or aggregated proteins. Selective autophagy recognizes ubiquitinated cargos through molecular adaptors including p62, NBR1, ALFY, NDP52, VCP and OPTN, which bind on one side to Ub and, on the other end, to autophagosome-specific proteins, such as LC3 (Fig. 1) [85]. These regulatory proteins together with ubiquitinated substrates and heat shock proteins, like Hsp70, are components of aggresomes. These structures are transient cytoplasmic inclusions that serve as storage sites for misfolded/damaged proteins and their aggregates that are not immediately delivered to autophagosomes or degraded by the UPS, thus minimizing their toxicity [90, 91]. In this selective form of macroautophagy, known as aggrephagy, harmful proteins are packaged into larger insoluble structures and driven to the MTOC near the nucleus with the aid of HDAC6, a key regulator of this pathway [90].
Chaperone-mediated autophagy (CMA)
Chaperone-mediated autophagy is a selective form of autophagy that differs from the above-described autophagic pathway in that vesicles are not involved in the transport of the cytosolic component to the lysosomes. Indeed, it regards the degradation of cytosolic proteins harbouring in their primary sequence the consensus pentapeptide motif KFERQ, which is recognized by the cytosolic chaperone heat shock cognate protein of 70 kDa (Hsc70). This binding is necessary for the lysosomal degradation of the substrate protein [92]. CMA is a multi-step process including cargo recognition and lysosomal targeting, substrate binding and unfolding, substrate translocation and lysosomal degradation. Once identified, the substrate is unfolded and targeted by Hsc70 in the presence of co-chaperones (BAG1, Hip, Hop, and Hsp40/DNAJB1, Hsp90) that modulate the interaction [93]. At this point, the complex moves to the lysosomal membrane where it interacts with the cytosolic region of the monomeric form of the protein LAMP-2A. The multimerization of this single-span membrane protein is then necessary for substrate translocation into the lysosomal lumen where the rapid degradation finally occurs [94, 95]. Interestingly, dynamic organization of LAMP-2A at the lysosomal membrane is specifically regulated by the functional interaction between Hsc70 and Hsp90. In details, Hsc70 was suggested to promote the organization of LAMP-2A into monomers or smaller complexes whereas the stability of LAMP-2A during the formation of the multimeric complex is maintained through its interaction with a form of Hsp90 located at the lysosomal membrane [96]. Clearly, the coordinated activity of all the chaperones participating in this process is essential for the correct unfolding of the substrate and for its translocation and final degradation in the lysosome. Interestingly, an internal direct crosstalk exists between CMA and macroautophagy, with the blockage of macroautophagy leading to up-regulation of CMA [97].
Microautophagy
Microautophagy is the third form of autophagy, well characterized in yeast but not completely in eukaryotic cells [98]. It is a constitutive process but both rapamycin and starvation can induce it. Major functions of microautophagy are the maintenance of organelles size, membrane composition and cell survival under condition of nitrogen restriction [99]. In microautophagy, the transfer of protein substrates into the lysosomes occurs through a direct invagination of the lysosomal membrane resulting in vesicles budding into the lumen of the lysosome. These vesicles then pinch off into the lysosomal lumen and are degraded by lysosomal proteases [79, 100]. For small particles and some proteins (non-selective microautophagy), the vacuolar membrane forms tubular invaginations from which small microautophagic vesicles pinch off. Conversely, when the degradation regards larger structures, including organelles, localized interactions with the vacuolar membrane and/or finger-like protrusions of the vacuole surround the targeted cellular component destined for degradation [100, 101]. Selective forms of microautophagy were mainly described in yeasts and regard the degradation of peroxisomes (micropexophagy), of non-essential portions of the nucleus, and of mitochondria (micromitophagy) [99]. A novel form of microautophagy, named endosomal microautophagy, was shown to share molecular components, such as the Hsc70 protein, with both the other autophagic pathways and to contribute to the degradation of cytosolic elements upon internalization in late endosomes [102, 103].
UPS and autophagy in AD
UPS and autophagy play a crucial role in the processing of proteins involved in the onset of AD and their activities are heavily downregulated by protein aggregates in AD neurons. The proteasome participates in Aβ degradation and, at the same time, is affected by Aβ. In a 2003 research, Lopez Salon et al. demonstrated that the inhibition of the 26S proteasome with lactacystin promoted a marked decrease in Aβ42 in primary cultures of cortical neurons and astrocytes, suggesting that the peptide could be a possible substrate of this enzymatic complex [104]. Treatment of neuronal cells with different Aβ peptides and with aggregated forms of the amyloid protein induced a strong inhibition of the proteasomal complex [104–106]. Aβ oligomers accumulation is responsible for neuronal cells death in transgenic mice through the induction of ER stress, endosomal/lysosomal leakage, and mitochondrial dysfunction [107]. Furthermore, through the degradation of the γ-secretase activating protein (GSAP) the proteasome system regulates AβPP metabolism and Aβ formation [108]. In addition, several research groups established that full-length AβPP undergo degradation by the UPS upon ubiquitination and that, when proteasome activity is inhibited, AβPP co-localizes and interacts with aggresome markers [109–112]. There are evidences that indicate the UPS as responsible for tau protein degradation, albeit other studies suggest that tau is not an effective substrate of the proteasome [113, 114]. In AD neurons, tau becomes hyperphosphorylated and forms filamentous inclusions called paired helical filaments (PHFs), which are the main constituents of neurofibrillary tangles (NFT). In vivo and in vitro experiments using lactacystin showed the accumulation, insolubility and ubiquitination of tau proteins and of phosphorylated tau, respectively [115, 116]. Numerous evidences indicate the presence of tau aggregates in immunoprecipitates of proteasome subunits and the localization and accumulation of Ub in both PHFs and NFTs [39, 117]. These findings identify bound PHF-tau as the reason for impaired proteasome function in AD brain [39]. In addition, it was shown that tau removal might be accomplished by the 20S and 26S proteasomes, thus both in Ub-independent and Ub-dependent (and ATP-dependent) manner [118, 119]. The development of proteasome inhibition in AD can be also due to Aβ- and tau-independent mechanisms, including mutations in genes encoding for components of the UPS and excessive oxidative stress. A mutant form of Ub B, the UBB+1 protein, is known to accumulate in disease-specific aggregates, to inhibit the proteasome and therefore to contribute to the disease progression [120, 121]. Recently, Bilguvar et al. identified a homozygous missense mutation within the Ub-binding domain of UCHL1 that almost completely abolish the hydrolase activity leading to a childhood-onset multisystem neurodegenerative syndrome and definitely linking the loss of UCHL1 function with a broad range of neurodegenerations [122]. Abnormal levels of oxidative and nitrosative stress favour both structural modifications of the proteasome, such as protein carbonyls, 4-hydroxynonenal-conjugation, neuroprostane-conjugation, and increased levels of oxidized substrates, this latter associating with the loss of the 20S proteasome activity [38, 123, 124]. UCHL1 is an extremely susceptible target of oxidative damage and numerous findings, based also on redox proteomics analysis, evidenced its specific oxidative modifications in AD brains, including carbonyl formation, methionine and cysteine oxidation providing an additional direct link between oxidative damage to the proteasomal machinery and the pathogenesis of AD [125–127].
Autophagy is considerably involved in amyloid degradation [128]. Autophagy and the BECN1–PIK3C3 complex regulate AβPP processing in AD [129]. Autophagic vacuoles contain AβPP and are highly enriched in active enzymes needed to generate Aβ (γ-secretase components, β-secretase activity and, to a lesser extent, BACE and γ-secretase activity) [130]. The autophagic-lysosomal system plays a role in the clearance of tau and the use of autophagy inhibitors delays tau degradation and favours the formation of high molecular weight species of tau including oligomers and insoluble aggregates [131]. Additionally, autophagy stimulation successfully reduces the number of tau inclusions and improves nerve cell survival in a mouse model of human tauopathy [132]. The lysosomal hydrolase cathepsin D was shown to degrade tau proteins in cultured hippocampal slices [133]. The involvement of autophagy in the pathology of AD is extensively documented. The observation in AD human brain tissues of accumulated autophagic and lysosomal markers indicated a defect of the autophagosome–lysosome pathway that contribute to the development of tau pathology [18]. The analysis of neocortical biopsies from AD brains revealed the striking accumulation of immature autophagic vacuoles in dystrophic neurites, suggesting that their transport and maturation to lysosomes may be impaired in such neurodegenerative condition [134]. Again, AD-associated disruption of lysosomal proteolysis slowed the axonal transport of autolysosomes, late endosomes, and lysosomes and caused their selective accumulation within dystrophic axons [135]. Presenili-1 was shown to be fundamental for correct lysosomal acidification and its mutations that are responsible for an early-onset form of AD determine a defective lysosomal proteolysis [136]. In the brain at early stages of sporadic AD and in the PS1/AβPP transgenic mouse model of AD pathology, macroautophagy is both induced and impaired, leading to the accumulation of Aβ-containing autophagic vacuoles within affected neurons [137]. Defective autophagic recycling of mitochondria and consequent mitochondria accumulation were observed in hippocampal brain samples of sporadic AD patients [138]. CMA is involved in normal tau degradation upon Hsc70 recognition of one of the two targeting motifs in the tau sequence. Differently, mutant tau variants, once bound to LAMP-2A, are only partially internalized and they remained associated with the lysosomal surface where they form oligomeric structures. This process alters the lysosomal membrane integrity and blocks the normal lysosomal functionality finally contributing to AD pathogenesis [139]. Besides tau, the regulator of calcineurin 1 (RCAN1) represents another link between CMA and AD. The degradation of RCAN1 is mediated by the proteasome and CMA and, as observed in AD and Down syndrome patients, their inhibition contributes to RCAN1 overexpression in the brain, which may eventually lead to disrupted neural function and neurodegeneration [140]. It has been suggested that the effects of mutant tau and the abnormal levels of RCAN1 are interconnected and both contribute to the severity of the AD pathology [95].
The AD neuropathology, with the associated alterations in both UPS and autophagy, frequently characterizes Down Syndrome (DS)-affected subjects, with an early onset usually after the age of 40. Since numerous similarities exist between the two pathological conditions, DS is considered an optimal model for the study of the pathophysiological events that occur early in AD. This disorder is a genetic condition due to the partial or complete triplication of chromosome 21, which harbours the gene encoding for the AβPP resulting in premature and excessive amyloid production and deposition. Recent findings suggest that DS brains are extremely vulnerable to oxidative stress and, prior to significant AD pathology, show early disturbances of the proteostasis network possibly linked to this increased oxidative condition. In details, Di Domenico et al. found general oxidative damage in lipids and proteins, such as glucose-regulated protein 78, UCH-L1, cathepsin D, V0-type proton ATPase subunit B and glial fibrillary acidic protein, which coupled with decreased activity of the proteasome and impaired autophagy [141, 142]. These data indicate that oxidative damage accumulation is a central event in DS and that, dramatically altering proteostasis network, it ultimately contributes to the development of AD pathogenesis.
UPS and autophagy crosstalk in AD
The UPS and autophagy were extensively considered as two essentially independent cellular catabolic pathways with difference in substrates, mechanisms and speed of degradation (autophagy is a slower process). Nevertheless, recent advances strongly suggest that their activities are carefully orchestrated and some crosstalk mechanisms have been suggested. Interestingly, the two pathways can both degrade ubiquitinated substrates and share common substrates, such as α-synuclein and regulatory proteins [143]. In addition, under specific conditions, autophagy can selectively degrade short-lived proteins, whereas the UPS can degrade long-lived proteins [144, 145]. In neurodegenerative conditions, where the accumulation of toxic species becomes prominent, cells can reorganize proteolysis regulating the communication between the two proteolytic pathways. Furthermore, the accumulation of amyloid-β plaques, a major hallmark of the pathology, is influenced by both the UPS system and autophagy [146, 147]. When one proteolytic system is damaged and shows a reduced functionality, the enhanced activity of the other pathway may become a compensatory mechanism necessary to protect neuronal cells against the accumulation of toxic species. An example of the inter-regulation between the UPS and autophagy is the observation that impairment in the UPS-mediated degradation leads to an increased autophagic function. The activation of this compensatory mechanism allows cells to reduce the number of aggregates formed in response to proteasomal inhibition [148, 149]. Pandey et al. found that HDAC6-dependent compensatory autophagy was induced in Drosophila melanogaster in response to mutations affecting the proteasome, suggesting that damages to the autophagic pathway might predispose to neurodegenerative processes [149, 150]. Similarly, Iwata et al. demonstrated the induction of the autophagosome formation in response to impaired UPS activity. They described an HDAC6-dependent retrograde transport on microtubules responsible for the recruitment of autophagy-related proteins and lysosomes to pericentriolar cytoplasmic inclusion bodies. This mechanism efficiently and selectively enhanced the autophagic degradation of aggregated huntingtin [151]. We previously dissected the regulation of the two proteolytic systems in SH-SY5Y cells overexpressing either the wild-type AβPP gene or the 717 valine-to-glycine AβPP-mutated gene. The overexpression of the AβPP, besides increasing oxidative stress, correlated with a reorganization of the cellular proteolytic machineries with marked inhibition of proteasome activities, impairment in the autophagic flux and increased HDAC6 expression as an attempt to activate compensatory autophagy [152]. Interestingly, we observed that the induction of pharmacological inhibition of one system promoted a compensatory reaction of the other, with a bidirectional effect [153]. Furthermore, the wild-type or mutated AβPP sequence influenced proteasome or autophagy activities in response to treatment with specific inhibitors and, upon MG132 administration, significantly enhanced the induction in cathepsin B [153]. Using the same AD model, we recently demonstrated that ghrelin, an orexigenic hormone involved in the onset and progression of neurodegenerative disorders, affects the crosstalk between UPS and autophagy successfully promoting the proteasome functionality in response to a compromised autophagy [154]. The induction of proteasomal inhibition in embryonic rat cortical neurons activated macroautophagy and the lysosomal pathway with the resulting dissolution of ubiquitinated inclusions into small aggregates, without a direct influence on neuronal cell death [155].
Molecules mediators of the crosstalk
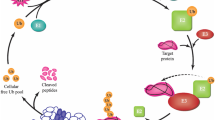
Numerous proteins mediators with crucial regulatory roles in the UPS-autophagy crosstalk in AD have been identified and classified based on their ability to bind or not the ubiquitin protein and the autophagy-related protein LC3. Proteins and their domains are summerized in Fig. 2 and their role and function are reviewed below.

Proteins involved in the mediation of the crosstalk between the UPS and autophagy in Alzheimer’s disease. They are classified based on their ability to interact with Ub and LC3. BUZ, Ub-binding zinc-finger domain; PB1, Phox and Bem1p domain; ZZ, Zinc-finger domain; TB, TRAF6 binding domain; LIR, LC3-interacting domain; KIR, Keap1-interacting region; UBA, Ub-associated domain; CC, coiled-coil domain; SKICH, SKIP–carboxyl homology domain; Gal-8, Galectin-8 binding domain; UBZ, Ub-binding zinc-finger domain; NLS, nuclear localization signal; BAG, Bcl-2-associated athanogene 1 domain; BEACH, BEACH domain; WD40, WD40 repeats; FYVE, Fab1,YOTB/ZK632.12, Vac1, and EEA1 domain; WW, WW domain
p62
p62, also called SQSTM1, is a highly conserved cytosolic protein that functions as an adaptor molecule driving ubiquitinated proteins to the autophagic cascade [156]. p62 was detected in inclusion bodies in many protein conformational diseases, including Lewy bodies-containing α-synuclein in Parkinson’s disease, neurofibrillary tangles of tau protein in AD and huntingtin aggregates in Huntington’s disease [157, 158]. p62 itself is then removed by autophagy and accumulates in the cell when this pathway is altered. p62 contains numerous interacting motifs through which it associates with several proteins to regulate their homeostasis, trafficking, aggregation and degradation [159]. The ability of p62 to act as shuttle for autophagic cargo depends on its specific interaction with Atg8/LC3/GABARAP proteins. Specifically, p62 shows a short LIR (LC3-interacting region) sequence of 22-amino acids responsible for the interaction with LC3-II, the active form of LC3, an autophagosomal marker (Fig. 2) [160]. As previously mentioned, p62 harbours in its C-terminal region an Ub-associated (UBA) domain through which the protein interacts non-covalently with ubiquitinated proteins [161]. In addition, at its N-terminus p62 has a Phox-BEM1 (PB1) domain, a protein–protein interaction domain that can assume an Ub-like folding and can directly bind to the proteasomal subunit S5a and other proteins containing the same domain, including p62. p62 supports tau polyubiquitination by the ligase TRAF6, shuttles polyubiquitinated tau to the proteasome and favours the autophagy-mediated clearance of aggregated tau [162]. In addition, Seibenhener et al. demonstrated that p62 favours tau degradation binding the proteasome through its N-terminal PB1 domain and interacting with polyubiquitinated tau through the UBA domain [161]. p62 plays a key role in the regulation of aggregation and in the formation of inclusion bodies. A decline in p62 expression or a decrease in proteasome activity contributed to accumulation of insoluble/aggregated K63-polyubiquitinated tau [161]. Studies on p62 knockout mice clearly demonstrated that the deficiency of the p62 protein leads to accumulation of hyperphosphorylated tau and neurofibrillary tangles accompanied by evident other symptoms of neurodegeneration [163]. Other studies in tauopathies and synucleinopathies demonstrated that Ub-containing protein inclusions show positive staining for p62 [157]. The same authors evidenced that p62 immunoreactivity appears early during neurofibrillary pathogenesis and is invariably and stably present in neurofibrillary tangles [164]. In a p62-null mouse model, the lack of p62 inhibited the formation of Ub-positive protein aggregates in neurons with impaired autophagy, indicating that p62 plays an important role in inclusion body formation [165].
NBR1
NBR1 was originally cloned as a candidate gene for the ovarian cancer antigen CA125 [166] and then proposed as another autophagic receptor for ubiquitinated substrates [64]. NBR1 directly interacts with p62. The two proteins show a poorly conserved primary sequence with NBR1 more than twice as large as p62 but with similar interacting motifs [64]. In fact, NBR1 contains both a LIR domain through which it interacts with LC3-II and a C-terminal UBA domain interacting with Ub, with a preference toward the K63-linked polyUb chain (Fig. 2). The interaction with p62 is mediated by the PB1 domain and its homodimerization occurs via the N-terminal two coiled-coil domains. NBR1 is an autophagic substrate, it is degraded in a LIR-dependent manner and, as shown in p62-deficient cells, in the absence of p62 [64]. Proteasome inhibition did not affect NBR1 levels, which in turn were dramatically increased upon blocking lysosomal acidification [64]. NBR1, together with p62, plays a role in the sequestration of misfolded and ubiquitinated proteins in p62 bodies and both proteins are necessary for the final autophagic-mediated degradation of such substrates [167]. Lamark et al. described a model for the degradation of ubiquitinated cargo by selective autophagy where oligomers of NBR1 and polymeric forms of p62 act as adaptors or cargo receptors linking the ubiquitinated substrate to the nascent autophagosome [167].
OPTN
OPTN is a 577 amino acid protein encoded by the OPTN gene, whose mutations are associated with normal tension glaucoma and amyotrophic lateral sclerosis. It is a cytoplasmic protein ubiquitously expressed in the heart, brain, skeletal muscle, liver, and the eye [168]. This protein contains several domains including an Ub-binding domain (UBD), that allows the binding to both K63 and linear chains, and a LC3-interacting region (LIR), through which it binds and brings polyubiquitinated cargoes to autophagosomes (Fig. 2) [169]. OPTN contributes to a wide range of cellular functions such as vesicle trafficking, maintenance of the Golgi apparatus, NF-κB pathway, antibacterial and antiviral signalling, cell division control, and autophagy. As p62, OPTN can polymerize and become a substrate for autophagic degradation [169]. Its role as an autophagy receptor can be both Ub-dependent and independent [169, 170]. The processing of endogenous OPTN is mainly mediated by the UPS. Overexpression of wild-type or mutant E50 K OPTN in RGC-5 cells downregulated the level of proteasome β5 subunit and enhanced LC3-II indicating that the UPS function was compromised whereas autophagy was induced [171]. Cho et al. demonstrated that OPTN favours the autophagic clearance of extracellular Aβ by microglia mediating the interaction of the amyloid protein with LC3-II [172]. OPTN immunoreactivity was widely detected not only in inclusions in amyotrophic lateral sclerosis but also in senile plaques and neurofibrillary tangles in AD and other neurodegenerations [173]. Interestingly, these aggregates show also positive staining for Ub and p62. The same authors suggested that the expression of OPTN could be upregulated in these pathological conditions [173].
NDP52
NDP52 is expressed in neurons, microglia and astrocytes [174]. NDP52 was principally characterized for its role in the autophagic removal of cytosolic bacteria. On this regard, several mechanisms of action were described and one of them is based on the property of the protein to detect galectin-8-positive bacteria. Galectin-8 controls endosomal and lysosomal integrity and individuate bacteria presence by binding host glycans exposed on damaged Salmonella-containing vacuoles. Upon recognition, galectin 8 recruits NDP52 and activates antibacterial autophagy [175]. In addition, other studies showed how human cells utilize the Ub system and NDP52 to activate autophagy against bacteria [176, 177]. The Ub-binding preference of NDP52 is not established. In AD, NDP52 co-localizes with both phosphorylated tau and intracellular Aβ indicating a role in the autophagic clearance of both proteins [174]. On this regard, NDP52 is strongly regulated by Nrf2 and plays a role in the amelioration of AD symptoms through the clearance of phosphorylated tau [178].
HDAC6
HDAC6 is a multidomain microtubule-associated deacetylase not only dedicated to genomic functions, but also involved in cytoplasmic pathways. HDAC6 has a number of cytoplasmic substrates including α-tubulin, cortactin, Hsp90 and peroxiredoxin. Differently from p62 and NBR1, HDAC6 possesses no LIR. It binds to Ub via a highly conserved Zn-finger Ub-binding domain and it shows preference for K63-linked Ub chains. The binding to polyubiquitinated proteins and dynein molecular motors provides HDAC6 with the ability to act as a physical link between ubiquitinated cargo and transport machinery. In addition, HDAC6 is involved in the formation and clearance of aggresomes, structures required for the autophagic degradation of abnormal aggregated proteins [151, 179]. Cells deficient in HDAC6 fail to clear misfolded protein aggregates from the cytoplasm, cannot form aggresomes properly, and are hypersensitive to the accumulation of misfolded proteins [179]. HDAC6 also facilitates the fusion of autophagosomes with lysosomes leading to autophagic clearance of substrates [180]. Pharmacological inhibition of HDAC6 in oligodendrocytes alters the assembly of protein aggregates formed in response to proteasomal inhibition and leads to the accumulation of autophagosomal vacuoles and increase in LC3-II immunoreactivity as a consequent of impairment of the autophagic flux [181]. Yan et al. suggested a crucial role for p62 in regulating HDAC6 activity [182]. They identified a specific binding domain of p62 which interacts with a catalytic domain of HDAC6 resulting in the modulation of the deacetylase activity. Lack of p62 favours HDAC6 hyper-activation with elevated de-acetylation of the HDAC6 specific substrates α-tubulin and cortactin [182]. HDAC6 activity can be also modulated by tau protein that, upon binding, decreases the functionality of the deacetylase with a consequent increase in tubulin acetylation, as observed in AD [183]. An excess of tau protein, as a HDAC6 inhibitor, prevents the induction of autophagy upon inhibition of proteasome function [183]. Recent evidences suggested a role for HDCA6 in the mediation of the crosstalk between the UPS and autophagy. Using Drosophila models of neurodegenerative diseases, Pandey et al. revealed that HDAC6 was able to suppress degeneration associated with proteasome mutations and impairment through the activation of autophagy as a compensatory degradation system [149, 150]. The authors suggested that increasing HDAC6 levels could be a strategy to enhance autophagy in neurodegeneration thus favouring the elimination of toxic species [149]. In addition, in an AD cellular model with evident impairment in proteasome functionality we described an increased expression of HDAC6 as an attempt to activate compensatory autophagy [152].
p97 (VCP)
p97/VCP is a AAA+ protein with an ATPase activity involved in various cellular processes such as membrane fusion, apoptosis, cell cycle regulation, DNA damage repair, regulation of transcription, metabolic modulation, and protein degradation [184–187]. Phosphorylation and acetylation at specific amino-acidic residues and interaction with a wide number of cofactors regulates the activity of the enzyme [184, 188, 189]. p97 acts as a Ub-selective chaperone using the energy generated from the hydrolysis of ATP to induce conformational changes of target proteins. These ubiquitinated substrates are separated from their protein complexes and then released for proteasomal degradation or recycling [186]. p97/VCP has also the ability to interact with HDAC6 and to modulate its functionality, originating a system able to determine the fate of ubiquitinated cellular proteins [190]. Interestingly, in condition of proteasomal impairment, the two proteins encourage the accumulation of misfolded proteins in aggresomes. p97 contributes also to the autophagosome formation. Mutations in p97 were associated with the IBMPFD (inclusion body myopathy with early-onset Paget disease and frontotemporal dementia) disease. Cells expressing the mutant form of p97 that correlates with this pathology display increased levels of the autophagosome markers p62 and LC3 II [191]. In addition, autophagic vesicles that accumulate in response to p97 mutations are extremely rich in Ub suggesting that p97 may be selectively required for autophagic degradation of ubiquitinated substrates [192]. Halawani et al. demonstrated that p97 is a substrate of Caspase-6 and that the cleavage generates a fragment able to impair UPS-mediated protein degradation in AD [193]. A role for p97 was described also in selective autophagy, specifically in the removal of mitochondria (mitophagy), peroxisomes (pexophagy) and 60S ribosomal subunit (ribophagy) [189].
Alfy
Alfy is another molecular link between autophagy and the proteasome system, but differently from the previously described molecules, it does not bind Ub. Alfy is a large protein member of the FYVE-domain family of proteins and is implicated in membrane trafficking. The FYVE domain is a zinc-finger domain shown to interact specifically with PI3P, which plays an important role in endosomal and autophagosomal membrane traffic. Alfy co-localizes with autophagic but not endocytic markers [194]. Simonsen et al. proposed that Alfy might recognize protein aggregates and then act as a scaffold for the autophagic machinery [195]. In normal condition, Alfy localizes in the nucleus but upon induction of autophagy, accumulation of Alfy-positive structures was detected in the cytoplasm, with interactions with autophagic markers. In addition, the inhibition of proteasome-mediated degradation caused a strong increase in the number of cytoplasmic Alfy-positive structures [195]. Alfy and p62 were shown to interact forming protein bodies that contain misfolded and ubiquitinated substrates then degraded by autophagy [196]. The importance of Alfy in removing toxic aggregates was confirmed by a study on a fruit flies mutant for the Alfy homologue, blue cheese (Bchs), which showed a reduced life span due to accumulation of ubiquitinated protein aggregates in the brain [197].
BAG-1 and BAG-3
Other co-chaperones known to be involved in the regulation of the interplay between the UPS and autophagy are members of the BAG protein family. BAG1 and BAG3 regulate the trafficking of polyubiquitinated substrates: BAG1 directs them to the proteasomal system, whereas BAG3, facilitates the degradation of substrates via the autophagic process interaction with p62 [198]. Although BAG3 is poorly expressed in young cells, an increase in the BAG3/BAG1 ratio was observed in ageing, indicating that autophagy is predominant in aged cells, because of accumulated protein aggregates that cannot be degraded by the proteasome [198]. These proteins can bind through their BAG domain to chaperones of the Hsc/Hsp70 family, thus modulating chaperone function [198]. Both BAG1 and BAG3 are involved in tau degradation in AD. Elliott et al. demonstrated the Hsc70-dependent interaction between BAG-1 and tau protein. They found that BAG-1 favours accumulation of tau protein by inhibiting its proteasomal degradation and that it co-localizes with aggregated tau in an AD mouse model suggesting an involvement of BAG-1 in the AD pathogenesis [199]. The same authors evidenced a significant increase of the BAG-1M isoform in the hippocampus of AD patients. In addition, BAG-1 was also found to co-localize and physically associate with intracellular tau and amyloid [200]. BAG3 regulates the clearance of tau in neurons through selective autophagy. In fact, the activation of autophagy consequent to proteasome inhibition resulted in upregulation of BAG3 and in a significant decrease in tau and phospho-tau levels [201].
Conclusion
The proteasome and autophagy are two proteolytic systems with a fundamental role in protein quality control and in the maintenance of cellular homeostasis. Alterations that reduce their functionality favour accumulation of toxic protein aggregates that alter neuronal trafficking and trigger neurons death as reported in numerous protein conformational disorders, including AD. In AD, amyloid peptides and tau protein tend to aggregate and form oligomers with a high β-sheet content that favours the formation of extracellular plaques and neurofibrillary tangles. These inclusions can impair the proteasome as well as autophagy interacting with various strategic components of the two proteolytic pathways. In this review, we described the sophisticated crosstalk between UPS and autophagy in the removal of such deleterious structures. The fine collaboration between these two pathways, with the inhibition of one system favouring the activation of the other, is essential to protein quality control in neurons. This interplay is rigorously coordinated by numerous shuttle factors and chaperones that are used for cargo selection and delivery to one system or the other. Specific protein domains allow these components to interact with Ub and LC3 thus determining the fate of the substrate. Besides their role in the control of protein degradation, they are also extremely useful to the cell to sequester toxic species in inclusion bodies, especially if proteolytic pathways are for some reasons defective. Thanks to this ability, which prevents harmful molecules from interfering and altering other fundamental intracellular processes, these shuttle molecules gain a role also in cell survival and in the delay of the progression of the neurodegeneration [179, 202]. The thorough control and manipulation of all the actors playing in cellular proteolysis could be therefore a promising strategy in the view of developing pharmacological interventions for therapeutic goals in AD and other neuropathies characterized by detrimental inclusions. Undoubtedly, additional data are needed to gain a better understanding of the connections between UPS and autophagy.
Abbreviations
- AD:
-
Alzheimer’s disease
- CMA:
-
Chaperone-mediated autophagy
- UPS:
-
Ubiquitin–proteasome system
- mTOR:
-
Mammalian target of rapamycin
- LC3:
-
Microtubule-associated protein light 3
- NBR1:
-
Neighbour of BRCA1 gene
- Alfy:
-
Autophagy-linked FYVE protein
- HDAC6:
-
Histone deacetylase 6
- NDP52:
-
Nuclear dot protein 52
- VCP:
-
Valosin containing protein
- OPTN:
-
Optineurin
- BAG:
-
Bcl-2-associated athanogene
- AβPP:
-
Amyloid precursor protein
References
Ciechanover A (2005) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ 12(9):1178–1190. doi:10.1038/sj.cdd.4401692
Fecto F, Esengul YT, Siddique T (2014) Protein recycling pathways in neurodegenerative diseases. Alzheimers Res Ther 6(2):13. doi:10.1186/alzrt243
Tanaka K (1843) Matsuda N (2014) Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim Biophys Acta 1:197–204. doi:10.1016/j.bbamcr.2013.03.012
Nijholt DAT, De Kimpe L, Elfrink HL, Hoozemans JJM, Scheper W (2011) Removing protein aggregates: the role of proteolysis in neurodegeneration. Curr Med Chem 18(16):2459–2476
Stadtman ER (2006) Protein oxidation and aging. Free Radical Res 40(12):1250–1258. doi:10.1080/10715760600918142
Stadtman ER (2001) Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci 928:22–38
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8(2):101–112. doi:10.1038/nrm2101
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harbor perspectives in medicine 1(1):a006189. doi:10.1101/cshperspect.a006189
Varshavsky A (2005) Regulated protein degradation. Trends Biochem Sci 30(6):283–286. doi:10.1016/j.tibs.2005.04.005
Wang DW, Peng ZJ, Ren GF, Wang GX (2015) The different roles of selective autophagic protein degradation in mammalian cells. Oncotarget 6(35):37098–37116. doi:10.18632/oncotarget.5776
Kirkegaard T, Jaattela M (2009) Lysosomal involvement in cell death and cancer. Biochim Biophys Acta 1793(4):746–754. doi:10.1016/j.bbamcr.2008.09.008
Cuervo AM (2004) Autophagy: in sickness and in health. Trends Cell Biol 14(2):70–77. doi:10.1016/j.tcb.2003.12.002
Massey AC, Zhang C, Cuervo AM (2006) Chaperone-mediated autophagy in aging and disease. Curr Top Dev Biol 73:205–235. doi:10.1016/S0070-2153(05)73007-6
Martinez-Vicente M, Cuervo AM (2007) Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 6(4):352–361. doi:10.1016/S1474-4422(07)70076-5
Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA (2013) Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Euro J Neurosci 37(12):1949–1961. doi:10.1111/ejn.12169
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889. doi:10.1038/nature04724
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095):880–884. doi:10.1038/nature04723
Piras A, Collin L, Gruninger F, Graff C, Ronnback A (2016) Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4(1):22. doi:10.1186/s40478-016-0292-9
Jung T, Catalgol B, Grune T (2009) The proteasomal system. Mol Aspects Med 30(4):191–296. doi:10.1016/j.mam.2009.04.001
Tu Y, Chen C, Pan J, Xu J, Zhou ZG, Wang CY (2012) The Ubiquitin Proteasome Pathway (UPP) in the regulation of cell cycle control and DNA damage repair and its implication in tumorigenesis. Int J Clin Experim Pathol 5(8):726–738
Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M (1997) Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 3(2):231–234
Spataro V, Norbury C, Harris AL (1998) The ubiquitin-proteasome pathway in cancer. Br J Cancer 77(3):448–455
Hu X, Bryington M, Fisher AB, Liang X, Zhang X, Cui D, Datta I, Zuckerman KS (2002) Ubiquitin/proteasome-dependent degradation of D-type cyclins is linked to tumor necrosis factor-induced cell cycle arrest. J Biol Chem 277(19):16528–16537. doi:10.1074/jbc.M109929200
Yam CH, Siu WY, Lau A, Poon RY (2000) Degradation of cyclin A does not require its phosphorylation by CDC2 and cyclin-dependent kinase 2. J Biol Chem 275(5):3158–3167
Dimova NV, Hathaway NA, Lee BH, Kirkpatrick DS, Berkowitz ML, Gygi SP, Finley D, King RW (2012) APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat Cell Biol 14(2):168–176. doi:10.1038/ncb2425
Ikeuchi K, Marusawa H, Fujiwara M, Matsumoto Y, Endo Y, Watanabe T, Iwai A, Sakai Y, Takahashi R, Chiba T (2009) Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced Parkin in human colorectal cancers. Int J Cancer J Int du Cancer 125(9):2029–2035. doi:10.1002/ijc.24565
Gregory MA, Hann SR (2000) c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 20(7):2423–2435
He H, Qi XM, Grossmann J, Distelhorst CW (1998) c-Fos degradation by the proteasome. An early, Bcl-2-regulated step in apoptosis. J Biol Chem 273(39):25015–25019
Ciechanover A (1998) The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J 17(24):7151–7160. doi:10.1093/emboj/17.24.7151
Tramutola A, Di Domenico F, Barone E, Perluigi M, Butterfield DA (2016) It Is All about (U)biquitin: Role of Altered Ubiquitin-Proteasome System and UCHL1 in Alzheimer Disease. Oxidat Med Cell Long 2016:2756068. doi:10.1155/2016/2756068
Baumeister W, Walz J, Zuhl F, Seemuller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92(3):367–380
Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4(3):181–191. doi:10.1038/nrm1052
Ruggiano A, Foresti O, Carvalho P (2014) Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol 204(6):869–879. doi:10.1083/jcb.201312042
Luo GR, Le WD (2010) Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Curr Pharm Biotechnol 11(2):180–187
Sulistio YA, Heese K (2016) The ubiquitin-proteasome system and molecular chaperone deregulation in Alzheimer’s disease. Mol Neurobiol 53(2):905–931. doi:10.1007/s12035-014-9063-4
Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, Cashman NR, Wilson MR, Ecroyd H (2016) Walking the tightrope: Proteostasis and neurodegenerative disease. J Neurochem. doi:10.1111/jnc.13575
Goldbaum O, Oppermann M, Handschuh M, Dabir D, Zhang B, Forman MS, Trojanowski JQ, Lee VM, Richter-Landsberg C (2003) Proteasome inhibition stabilizes tau inclusions in oligodendroglial cells that occur after treatment with okadaic acid. J Neurosci 23(26):8872–8880
Cecarini V, Ding Q, Keller JN (2007) Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radical Res 41(6):673–680. doi:10.1080/10715760701286159
Keck S, Nitsch R, Grune T, Ullrich O (2003) Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem 85(1):115–122
Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce-Keller AJ, Keller JN (2004) Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem 279(20):20699–20707. doi:10.1074/jbc.M313579200
McKinnon C, Goold R, Andre R, Devoy A, Ortega Z, Moonga J, Linehan JM, Brandner S, Lucas JJ, Collinge J, Tabrizi SJ (2016) Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol 131(3):411–425. doi:10.1007/s00401-015-1508-y
Dasgupta S, Fishman MA, Mahallati H, Castro LM, Tashima AK, Ferro ES, Fricker LD (2015) Reduced Levels of Proteasome Products in a Mouse Striatal Cell Model of Huntington’s Disease. PLoS ONE 10(12):e0145333. doi:10.1371/journal.pone.0145333
Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, Jensen PH (2004) Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 279(13):12924–12934. doi:10.1074/jbc.M306390200
Zheng Q, Li J, Wang X (2009) Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int J Physiol Pathophysiol Pharmacol 1(2):127–142
Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386(6624):463–471. doi:10.1038/386463a0
Jung T, Grune T (2013) The proteasome and the degradation of oxidized proteins: Part I-structure of proteasomes. Redox Biol 1:178–182. doi:10.1016/j.redox.2013.01.004
Groll M, Bochtler M, Brandstetter H, Clausen T, Huber R (2005) Molecular machines for protein degradation. Chem Bio Chem 6(2):222–256. doi:10.1002/cbic.200400313
Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W (1995) Proteasome from Thermoplasma acidophilum: a threonine protease. Science 268(5210):579–582
Orlowski M, Wilk S (2003) Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys 415(1):1–5
Shringarpure R, Grune T, Davies KJ (2001) Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci 58(10):1442–1450
Ciechanover A, Orian A, Schwartz AL (2000) The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Suppl 34:40–51
Wilkinson KD (2000) Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Semin Cell Dev Biol 11(3):141–148. doi:10.1006/scdb.2000.0164
Shaid S, Brandts CH, Serve H, Dikic I (2013) Ubiquitination and selective autophagy. Cell Death Differ 20(1):21–30. doi:10.1038/cdd.2012.72
Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96(5):635–644
Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19(1):94–102. doi:10.1093/emboj/19.1.94
Ciechanover A (1843) Stanhill A (2014) The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim Biophys Acta 1:86–96. doi:10.1016/j.bbamcr.2013.07.007
Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5(5):461–466. doi:10.1038/ncb983
Hicke L (2001) Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2(3):195–201. doi:10.1038/35056583
Tenno T, Fujiwara K, Tochio H, Iwai K, Morita EH, Hayashi H, Murata S, Hiroaki H, Sato M, Tanaka K (2004) Shirakawa M (2004) Structural basis for distinct roles of Lys63- and Lys48-linked polyubiquitin chains. Genes Cells 9(11):1137. doi:10.1111/j.1365-2443.2004.00810.x
Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL (2013) Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J 32(4):552–565. doi:10.1038/emboj.2012.354
Saeki Y, Kudo T, Sone T, Kikuchi Y, Yokosawa H, Toh-e A, Tanaka K (2009) Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J 28(4):359–371. doi:10.1038/emboj.2008.305
Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137(1):133–145. doi:10.1016/j.cell.2009.01.041
Benbrook DM, Long A (2012) Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Experimental oncology 34(3):286–297
Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33(4):505–516. doi:10.1016/j.molcel.2009.01.020
Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS (2007) Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol 178(6):1025–1038. doi:10.1083/jcb.200611128
Li W, Ye Y (2008) Polyubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci 65(15):2397–2406. doi:10.1007/s00018-008-8090-6
Spence J, Sadis S, Haas AL, Finley D (1995) A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol 15(3):1265–1273
Lauwers E, Jacob C, Andre B (2009) K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J Cell Biol 185(3):493–502. doi:10.1083/jcb.200810114
Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, Gygi SP, Yates NA, Sorkin A (2013) Lysine 63-linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci USA 110(39):15722–15727. doi:10.1073/pnas.1308014110
Sato M, Konuma R, Sato K, Tomura K, Sato K (2014) Fertilization- induced K63-linked ubiquitylation mediates clearance of maternal membrane proteins. Development 141(6):U1268–U1324. doi:10.1242/dev.103044
Fritsch J, Stephan M, Tchikov V, Winoto-Morbach S, Gubkina S, Kabelitz D, Schutze S (2014) Cell fate decisions regulated by K63 ubiquitination of tumor necrosis factor receptor 1. Mol Cell Biol 34(17):3214–3228. doi:10.1128/MCB.00048-14
Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17(3):431–439. doi:10.1093/hmg/ddm320
Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci 25(8):2002–2009. doi:10.1523/JNEUROSCI.4474-04.2005
Hong X, Liu J, Zhu G, Zhuang Y, Suo H, Wang P, Huang D, Xu J, Huang Y, Yu M, Bian M, Sheng Z, Fei J, Song H, Behnisch T, Huang F (2014) Parkin overexpression ameliorates hippocampal long-term potentiation and beta-amyloid load in an Alzheimer’s disease mouse model. Hum Mol Genet 23(4):1056–1072. doi:10.1093/hmg/ddt501
Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE (2011) Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet 20(11):2091–2102. doi:10.1093/hmg/ddr091
Zhang M, Cai F, Zhang S, Zhang S, Song W (2014) Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Scient Rep 4:7298. doi:10.1038/srep07298
McKeon JE, Sha D, Li L, Chin LS (2015) Parkin-mediated K63-polyubiquitination targets ubiquitin C-terminal hydrolase L1 for degradation by the autophagy-lysosome system. Cell Mol Life Sci 72(9):1811–1824. doi:10.1007/s00018-014-1781-2
Lee KM, Hwang SK, Lee JA (2013) Neuronal autophagy and neurodevelopmental disorders. Exp Neurobiol 22(3):133–142. doi:10.5607/en.2013.22.3.133
Park C, Cuervo AM (2013) Selective autophagy: talking with the UPS. Cell Biochem Biophys 67(1):3–13. doi:10.1007/s12013-013-9623-7
Reggiori F, Komatsu M, Finley K, Simonsen A (2012) Autophagy: more than a nonselective pathway. Int J Cell Biol 2012:219625. doi:10.1155/2012/219625
Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16(6):495–501. doi:10.1038/ncb2979
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451(7182):1069–1075. doi:10.1038/nature06639
Wong E, Cuervo AM (2010) Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2(12):a006734. doi:10.1101/cshperspect.a006734
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y (2003) A unified nomenclature for yeast autophagy-related genes. Dev Cell 5(4):539–545
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997. doi:10.1038/nm.3232
Wong AS, Cheung ZH (1812) Ip NY (2011) Molecular machinery of macroautophagy and its deregulation in diseases. Biochim Biophys Acta 11:1490–1497. doi:10.1016/j.bbadis.2011.07.005
Williams RS, Cheng L, Mudge AW, Harwood AJ (2002) A common mechanism of action for three mood-stabilizing drugs. Nature 417(6886):292–295. doi:10.1038/417292a
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170(7):1101–1111. doi:10.1083/jcb.200504035
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16(1):46–56. doi:10.1038/cdd.2008.110
Hyttinen JM, Amadio M, Viiri J, Pascale A, Salminen A, Kaarniranta K (2014) Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev 18:16–28. doi:10.1016/j.arr.2014.07.002
Lamark T, Johansen T (2012) Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012:736905. doi:10.1155/2012/736905
Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246(4928):382–385
Agarraberes FA, Dice JF (2001) A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 114(Pt 13):2491–2499
Kon M, Cuervo AM (2010) Chaperone-mediated autophagy in health and disease. FEBS Lett 584(7):1399–1404. doi:10.1016/j.febslet.2009.12.025
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24(1):92–104. doi:10.1038/cr.2013.153
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28(18):5747–5763. doi:10.1128/MCB.02070-07
Kaushik S, Massey AC, Mizushima N, Cuervo AM (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19(5):2179–2192. doi:10.1091/mbc.E07-11-1155
Marzella L, Ahlberg J, Glaumann H (1981) Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Archiv B, Cell pathology including molecular pathology 36(2–3):219–234
Li WW, Li J, Bao JK (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69(7):1125–1136. doi:10.1007/s00018-011-0865-5
Mijaljica D, Prescott M, Devenish RJ (2011) Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7(7):673–682
Todde V, Veenhuis M, van der Klei IJ (2009) Autophagy: principles and significance in health and disease. Biochim Biophys Acta 1792(1):3–13. doi:10.1016/j.bbadis.2008.10.016
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20(1):131–139. doi:10.1016/j.devcel.2010.12.003
Santambrogio L, Nosotti M, Palleschi A, Gazzano G, De Simone M, Cioffi U (2011) Primary pulmonary glomangioma: a coin lesion negative on PET study. Case report and literature review. Thorac Cardiovasc Surg 59(6):380–382. doi:10.1055/s-0030-1250577
Lopez Salon M, Pasquini L, Besio Moreno M, Pasquini JM, Soto E (2003) Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol 180(2):131–143
Cecarini V, Bonfili L, Amici M, Angeletti M, Keller JN, Eleuteri AM (2008) Amyloid peptides in different assembly states and related effects on isolated and cellular proteasomes. Brain Res 1209:8–18. doi:10.1016/j.brainres.2008.03.003
Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM (2008) Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 29(11):1607–1618. doi:10.1016/j.neurobiolaging.2007.04.014
Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H (2011) Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res 89(7):1031–1042. doi:10.1002/jnr.22640
Chu J, Li JG, Hoffman NE, Madesh M, Pratico D (2015) Degradation of gamma secretase activating protein by the ubiquitin-proteasome pathway. J Neurochem 133(3):432–439. doi:10.1111/jnc.13011
Dehvari N, Mahmud T, Persson J, Bengtsson T, Graff C, Winblad B, Ronnback A, Behbahani H (2012) Amyloid precursor protein accumulates in aggresomes in response to proteasome inhibitor. Neurochem Int 60(5):533–542. doi:10.1016/j.neuint.2012.02.012
Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall’Armi C, Simoes S, Point Du, Jour KS, McCabe BD, Small SA, Di Paolo G (2013) Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 4:2250. doi:10.1038/ncomms3250
El Ayadi A, Stieren ES, Barral JM, Boehning D (2012) Ubiquilin-1 regulates amyloid precursor protein maturation and degradation by stimulating K63-linked polyubiquitination of lysine 688. Proc Natl Acad Sci USA 109(33):13416–13421. doi:10.1073/pnas.1206786109
Wang H, Saunders AJ (2014) The role of ubiquitin-proteasome in the metabolism of amyloid precursor protein (APP): implications for novel therapeutic strategies for Alzheimer’s disease. Dis Med 18(97):41–50
Brown MR, Bondada V, Keller JN, Thorpe J, Geddes JW (2005) Proteasome or calpain inhibition does not alter cellular tau levels in neuroblastoma cells or primary neurons. J Alzheimer’s Dis JAD 7(1):15–24
Delobel P, Leroy O, Hamdane M, Sambo AV, Delacourte A, Buee L (2005) Proteasome inhibition and Tau proteolysis: an unexpected regulation. FEBS Lett 579(1):1–5. doi:10.1016/j.febslet.2004.11.018
Ren QG, Liao XM, Wang ZF, Qu ZS, Wang JZ (2006) The involvement of glycogen synthase kinase-3 and protein phosphatase-2A in lactacystin-induced tau accumulation. FEBS Lett 580(10):2503–2511. doi:10.1016/j.febslet.2006.03.073
Liu YH, Wei W, Yin J, Liu GP, Wang Q, Cao FY, Wang JZ (2009) Proteasome inhibition increases tau accumulation independent of phosphorylation. Neurobiol Aging 30(12):1949–1961. doi:10.1016/j.neurobiolaging.2008.02.012
Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y (1993) Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 10(6):1151–1160
Zhang JY, Liu SJ, Li HL, Wang JZ (2005) Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J Neural Trans 112(4):547–555. doi:10.1007/s00702-004-0196-x
David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG (2002) Proteasomal degradation of tau protein. J Neurochem 83(1):176–185
van Tijn P, de Vrij FM, Schuurman KG, Dantuma NP, Fischer DF, van Leeuwen FW, Hol EM (2007) Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J Cell Sci 120(Pt 9):1615–1623. doi:10.1242/jcs.03438
Irmler M, Gentier RJ, Dennissen FJ, Schulz H, Bolle I, Holter SM, Kallnik M, Cheng JJ, Klingenspor M, Rozman J, Ehrhardt N, Hermes DJ, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Meyer HE, Hopkins DA, Van Leeuwen FW, Beckers J (2012) Long-term proteasomal inhibition in transgenic mice by UBB(+1) expression results in dysfunction of central respiration control reminiscent of brainstem neuropathology in Alzheimer patients. Acta Neuropathol 124(2):187–197. doi:10.1007/s00401-012-1003-7
Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, Delil S, Caglayan AO, Baranoski JF, Erturk O, Yalcinkaya C, Karacorlu M, Dincer A, Johnson MH, Mane S, Chandra SS, Louvi A, Boggon TJ, Lifton RP, Horwich AL, Gunel M (2013) Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci USA 110(9):3489–3494. doi:10.1073/pnas.1222732110
Dasuri K, Ebenezer P, Zhang L, Fernandez-Kim SO, Bruce-Keller AJ, Markesbery WR, Keller JN (2010) Increased protein hydrophobicity in response to aging and Alzheimer disease. Free Radic Biol Med 48(10):1330–1337. doi:10.1016/j.freeradbiomed.2010.02.012
Petropoulos I, Conconi M, Wang X, Hoenel B, Bregegere F, Milner Y, Friguet B (2000) Increase of oxidatively modified protein is associated with a decrease of proteasome activity and content in aging epidermal cells. J Gerontol Series A Biol Sci Med Sci 55(5):B220–B227
Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L (2004) Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem 279(13):13256–13264. doi:10.1074/jbc.M314124200
Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA (2002) Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med 33(4):562–571
Swomley AM, Butterfield DA (2015) Oxidative stress in Alzheimer disease and mild cognitive impairment: evidence from human data provided by redox proteomics. Arch Toxicol 89(10):1669–1680. doi:10.1007/s00204-015-1556-z
Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA (2011) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain J Neurol 134(Pt 1):258–277. doi:10.1093/brain/awq341
Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T (2010) Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One 5(6):e11102. doi:10.1371/journal.pone.0011102
Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, Nixon RA (2004) Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int J Biochem Cell Biol 36(12):2531–2540. doi:10.1016/j.biocel.2004.05.010
Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, Deture M, Ko LW (2008) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Euro J Neurosci 27(5):1119–1130. doi:10.1111/j.1460-9568.2008.06084.x
Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M (2012) Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain J Neurol 135(Pt 7):2169–2177. doi:10.1093/brain/aws143
Bednarski E, Lynch G (1996) Cytosolic proteolysis of tau by cathepsin D in hippocampus following suppression of cathepsins B and L. J Neurochem 67(5):1846–1855
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64(2):113–122
Lee S, Sato Y, Nixon RA (2011) Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 31(21):7817–7830. doi:10.1523/JNEUROSCI.6412-10.2011
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141(7):1146–1158. doi:10.1016/j.cell.2010.05.008
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA (2005) Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171(1):87–98. doi:10.1083/jcb.200505082
Martin-Maestro P, Gargini R, Perry G, Avila J, Garcia-Escudero V (2016) PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet 25(4):792–806. doi:10.1093/hmg/ddv616
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18(21):4153–4170. doi:10.1093/hmg/ddp367
Liu H, Wang P, Song W, Sun X (2009) Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J Off Publ Feder Am Soc Exp Biol 23(10):3383–3392. doi:10.1096/fj.09-134296
Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C (1832) Perluigi M (2013) Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta 8:1249–1259. doi:10.1016/j.bbadis.2013.04.013
Di Domenico F, Pupo G, Tramutola A, Giorgi A, Schinina ME, Coccia R, Head E, Butterfield DA, Perluigi M (2014) Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: clues for understanding the development of Alzheimer disease. Free Radic Biol Med 71:270–280. doi:10.1016/j.freeradbiomed.2014.03.027
Kraft C, Peter M, Hofmann K (2010) Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12(9):836–841. doi:10.1038/ncb0910-836
Fuertes G, Villarroya A, Knecht E (2003) Role of proteasomes in the degradation of short-lived proteins in human fibroblasts under various growth conditions. Int J Biochem Cell Biol 35(5):651–664
Fuertes G, Martin De Llano JJ, Villarroya A, Rivett AJ, Knecht E (2003) Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J 375(Pt 1):75–86. doi:10.1042/BJ20030282
van Tijn P, Dennissen FJ, Gentier RJ, Hobo B, Hermes D, Steinbusch HW, Van Leeuwen FW, Fischer DF (2012) Mutant ubiquitin decreases amyloid beta plaque formation in a transgenic mouse model of Alzheimer’s disease. Neurochem Int 61(5):739–748. doi:10.1016/j.neuint.2012.07.007
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC (2013) Abeta secretion and plaque formation depend on autophagy. Cell Reports 5(1):61–69. doi:10.1016/j.celrep.2013.08.042
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171(2):513–524. doi:10.2353/ajpath.2007.070188
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447(7146):859–863. doi:10.1038/nature05853
Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP (2007) HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 3(6):643–645
Iwata A, Riley BE, Johnston JA, Kopito RR (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280(48):40282–40292. doi:10.1074/jbc.M508786200
Cecarini V, Bonfili L, Cuccioloni M, Mozzicafreddo M, Rossi G, Buizza L, Uberti D, Angeletti M (1822) Eleuteri AM (2012) Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer’s disease. Biochim Biophys Acta 11:1741–1751. doi:10.1016/j.bbadis.2012.07.015
Cecarini V, Bonfili L, Cuccioloni M, Mozzicafreddo M, Rossi G, Keller JN, Angeletti M (1842) Eleuteri AM (2014) Wild type and mutant amyloid precursor proteins influence downstream effects of proteasome and autophagy inhibition. Biochim Biophys Acta 2:127–134. doi:10.1016/j.bbadis.2013.11.002
Cecarini V, Bonfili L, Cuccioloni M, Keller JN, Bruce-Keller AJ, Eleuteri AM (2015) Effects of Ghrelin on the Proteolytic Pathways of Alzheimer’s Disease Neuronal Cells. Mol Neurobiol. doi:10.1007/s12035-015-9227-x
Rideout HJ, Lang-Rollin I, Stefanis L (2004) Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol 36(12):2551–2562. doi:10.1016/j.biocel.2004.05.008
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7(3):279–296
Kuusisto E, Salminen A, Alafuzoff I (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuro Report 12(10):2085–2090
Nagaoka U, Kim K, Jana NR (2004) Doi H, Maruyama M, Mitsui K, Oyama F, Nukina N Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J Neurochem 91(1):57–68. doi:10.1111/j.1471-4159.2004.02692.x
Salminen A, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H, Alafuzoff I (2012) Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog Neurobiol 96(1):87–95. doi:10.1016/j.pneurobio.2011.11.005
Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34(3):259–269. doi:10.1016/j.molcel.2009.04.026
Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24(18):8055–8068. doi:10.1128/MCB.24.18.8055-8068.2004
Babu JR, Geetha T, Wooten MW (2005) Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem 94(1):192–203. doi:10.1111/j.1471-4159.2005.03181.x
Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106(1):107–120. doi:10.1111/j.1471-4159.2008.05340.x
Kuusisto E, Salminen A, Alafuzoff I (2002) Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol 28(3):228–237
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131(6):1149–1163. doi:10.1016/j.cell.2007.10.035
Campbell IG, Nicolai HM, Foulkes WD, Senger G, Stamp GW, Allan G, Boyer C, Jones K, Bast RC Jr, Solomon E (1994) A novel gene encoding a B-box protein within the BRCA1 region at 17q21.1. Hum Mol Genet 3(4):589–594
Lamark T, Kirkin V, Dikic I, Johansen T (2009) NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8(13):1986–1990
Liu YH, Tian T (2011) Hypothesis of optineurin as a new common risk factor in normal-tension glaucoma and Alzheimer’s disease. Med Hypotheses 77(4):591–592. doi:10.1016/j.mehy.2011.06.040
Ying H, Yue BY (2015) Optineurin: The autophagy connection. Exp Eye Res. doi:10.1016/j.exer.2015.06.029
Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I (2013) Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126(Pt 2):580–592. doi:10.1242/jcs.114926
Shen X, Ying H, Qiu Y, Park JS, Shyam R, Chi ZL, Iwata T, Yue BY (2011) Processing of optineurin in neuronal cells. J Biol Chem 286(5):3618–3629. doi:10.1074/jbc.M110.175810
Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, Kim HM, Kim DH, Yoon SY (2014) Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 10(10):1761–1775. doi:10.4161/auto.29647
Osawa T, Mizuno Y, Fujita Y, Takatama M, Nakazato Y, Okamoto K (2011) Optineurin in neurodegenerative diseases. Neuropathol Off J Japn Soc Neuropathol 31(6):569–574. doi:10.1111/j.1440-1789.2011.01199.x
Kim S, Lee D, Song JC, Cho SJ, Yun SM, Koh YH, Song J, Johnson GV, Jo C (2014) NDP52 associates with phosphorylated tau in brains of an Alzheimer disease mouse model. Biochem Biophys Res Commun 454(1):196–201. doi:10.1016/j.bbrc.2014.10.066
Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F (2012) Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482(7385):414–418. doi:10.1038/nature10744
Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10(11):1215–1221. doi:10.1038/ni.1800
Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P (2011) p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem 286(30):26987–26995. doi:10.1074/jbc.M111.223610
Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV (2014) Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun 5:3496. doi:10.1038/ncomms4496
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115(6):727–738
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29(5):969–980. doi:10.1038/emboj.2009.405
Leyk J, Goldbaum O, Noack M, Richter-Landsberg C (2015) Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J Molecul Neurosci MN 55(4):1031–1046. doi:10.1007/s12031-014-0460-y
Yan J, Seibenhener ML, Calderilla-Barbosa L, Diaz-Meco MT, Moscat J, Jiang J, Wooten MW, Wooten MC (2013) SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS One 8(9):e76016. doi:10.1371/journal.pone.0076016
Perez M, Santa-Maria I, Gomez de Barreda E, Zhu X, Cuadros R, Cabrero JR, Sanchez-Madrid F, Dawson HN, Vitek MP, Perry G, Smith MA, Avila J (2009) Tau–an inhibitor of deacetylase HDAC6 function. J Neurochem 109(6):1756–1766. doi:10.1111/j.1471-4159.2009.06102.x
Yang FC, Lin YH, Chen WH, Huang JY, Chang HY, Su SH, Wang HT, Chiang CY, Hsu PH, Tsai MD, Tan BC, Lee SC (2013) Interaction between salt-inducible kinase 2 (SIK2) and p97/valosin-containing protein (VCP) regulates endoplasmic reticulum (ER)-associated protein degradation in mammalian cells. J Biol Chem 288(47):33861–33872. doi:10.1074/jbc.M113.492199
Uchiyama K, Kondo H (2005) p97/p47-Mediated biogenesis of Golgi and ER. J Biochem 137(2):115–119. doi:10.1093/jb/mvi028
Yamanaka K, Sasagawa Y (1823) Ogura T (2012) Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta 1:130–137. doi:10.1016/j.bbamcr.2011.07.001
Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22(2):626–634
Mori-Konya C, Kato N, Maeda R, Yasuda K, Higashimae N, Noguchi M, Koike M, Kimura Y, Ohizumi H, Hori S, Kakizuka A (2009) p97/valosin-containing protein (VCP) is highly modulated by phosphorylation and acetylation. Genes Cells Devoted Molecul Cell Mechan 14(4):483–497. doi:10.1111/j.1365-2443.2009.01286.x
Dargemont C, Ossareh-Nazari B (2012) Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim Biophys Acta 1823(1):138–144. doi:10.1016/j.bbamcr.2011.07.011
Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Muller CW, Khochbin S (2006) HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J 25(14):3357–3366. doi:10.1038/sj.emboj.7601210
Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187(6):875–888. doi:10.1083/jcb.200908115
Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6(2):217–227
Halawani D, Tessier S, Anzellotti D, Bennett DA, Latterich M, LeBlanc AC (2010) Identification of Caspase-6-mediated processing of the valosin containing protein (p97) in Alzheimer’s disease: a novel link to dysfunction in ubiquitin proteasome system-mediated protein degradation. J Neurosci 30(17):6132–6142. doi:10.1523/JNEUROSCI.5874-09.2010
Isakson P, Holland P, Simonsen A (2013) The role of ALFY in selective autophagy. Cell Death Differ 20(1):12–20. doi:10.1038/cdd.2012.66
Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, Yoshimori T, Slagsvold T, Brech A, Stenmark H (2004) Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci 117(Pt 18):4239–4251. doi:10.1242/jcs.01287
Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A, Johansen T (2010) p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6(3):330–344
Finley KD, Edeen PT, Cumming RC, Mardahl-Dumesnil MD, Taylor BJ, Rodriguez MH, Hwang CE, Benedetti M, McKeown M (2003) Blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J Neurosci 23(4):1254–1264
Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C (2009) Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J 28(7):889–901. doi:10.1038/emboj.2009.29
Elliott E, Tsvetkov P, Ginzburg I (2007) BAG-1 associates with Hsc70.Tau complex and regulates the proteasomal degradation of Tau protein. J Biol Chem 282(51):37276–37284. doi:10.1074/jbc.M706379200
Elliott E, Laufer O, Ginzburg I (2009) BAG-1M is up-regulated in hippocampus of Alzheimer’s disease patients and associates with tau and APP proteins. J Neurochem 109(4):1168–1178. doi:10.1111/j.1471-4159.2009.06047.x
Lei ZN, Brizzee C, Johnson GVW (2015) BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol Aging 36(1):241–248. doi:10.1016/j.neurobiolaging.2014.08.012
Paine MG, Babu JR, Seibenhener ML, Wooten MW (2005) Evidence for p62 aggregate formation: role in cell survival. FEBS Lett 579(22):5029–5034. doi:10.1016/j.febslet.2005.08.010
Acknowledgments
This work was in part funded by the University of Camerino.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cecarini, V., Bonfili, L., Cuccioloni, M. et al. The fine-tuning of proteolytic pathways in Alzheimer’s disease. Cell. Mol. Life Sci. 73, 3433–3451 (2016). https://doi.org/10.1007/s00018-016-2238-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2238-6