
Abstract
Many progressive neurodegenerative diseases, including Alzheimer disease, Parkinson disease, Huntington disease, amyotrophic lateral sclerosis, and frontotemporal lobe dementia, are associated with the formation of insoluble intracellular proteinaceous inclusions. It is therefore imperative to understand the factors that regulate normal, as well as abnormal, protein recycling in neurons. Dysfunction of the ubiquitin-proteasome or autophagy pathways might contribute to the pathology of various neurodegenerative diseases. Induction of these pathways may offer a rational therapeutic strategy for a number of these diseases.
Similar content being viewed by others
Introduction
A common theme in neurodegeneration is the age-related accumulation of specific toxic protein species secondary to mutations or misfolding or both. Such disease-related proteins are prone to aggregation, and their oligomers cause toxicity via a gain-of-function mechanism [1]. The removal of misfolded or damaged proteins is critical for optimal cell functioning, particularly in neuronal cells, where the dynamic control of protein stability is crucial for synaptic development, function, and maintenance [2–6]. This dependence of neurons on protein recycling systems makes them particularly vulnerable to damage when these systems break down [7, 8].
Protein quality control in neurons
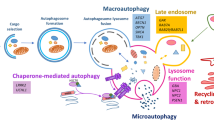
Neurons adapt to cellular stresses by maintaining the integrity of their proteome (Figure 1). This maintenance is regulated in three correlated ways by molecular chaperones: (a) refolding, (b) degrading, or (c) sequestering misfolded proteins into insoluble aggregates [1, 9]. It is not yet understood how chaperones prioritize between these fates for cellular proteins. Chaperones are also essential in assisting newly synthesized proteins to fold properly and translocate across cellular membranes. When mechanisms regulating protein recycling go awry, as happens during aging, proteins begin to lose their conformation and their state becomes energetically more favorable for the formation of insoluble aggregates [1, 10, 11]. This is a common mechanism of pathogenesis in protein conformational diseases, including neurodegenerative diseases [12–14].

Defects in protein recycling pathways in various neurodegenerative diseases. For autophagy, the defect can be at the level of autophagy induction, cargo recognition, autophagosome/lysosome fusion, or lysosomal degradation. AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; CMA, chaperone-mediated autophagy; FTD, frontotemporal lobe dementia; HD, Huntington disease; mTOR, mammalian target of rapamycin; PD, Parkinson disease; ULK-1, Unc-51 like autophagy activating kinase 1; UPS, ubiquitin-proteasome system.
Long-term cellular stress can cause high levels of protein accumulation over time and this can lead to chaperone depletion and compromise the refolding of these accumulated proteins. When refolding is not sufficient to maintain cellular homeostasis in these conditions, misfolded proteins are transferred to the degradation pathways [15, 16]. These proteins are handled by two systems: the ubiquitin-proteasome system (UPS) and autophagy. Even though the molecular machinery employed by each of these systems is unique, they essentially function in a similar manner to handle protein recycling. Each pathway has steps involved in cargo selection and tagging of the cargos, recognition and delivery of these cargos to the proteolytic machinery, degradation in the proteolytic core, and eventually recycling of the constituent amino acids [17].
The UPS is the primary intracellular proteolytic system responsible for the maintenance of rapid protein turnover in the cytosol and nuclei of cells, including the selective removal of abnormal and misfolded proteins. This degradation occurs in a two-step process: (a) the targeting of proteins through the covalent linkage of specific branched polyubiquitin chains and (b) the degradation of the tagged proteins by the downstream 26S proteasome complex. The 26S proteasome is composed of two sub-complexes: the 19S regulatory particle and the 20S core particle. In neuronal cells, dynamic control of protein stability is crucial for synaptic development and function, and the UPS plays an important role in the regulation of synaptic proteins [2]. It has been shown that depletion of the 26S proteasomes in mice leads to neurodegeneration; thus, they are essential for normal neuronal homeostasis and survival [18]. Aggregation-prone proteins may themselves impair the UPS by (a) saturating the capacity of the intracellular UPS machinery, (b) sequestering of essential components of the UPS into inclusions, or (c) clogging up the proteasome. Since the UPS is essential for cellular protein recycling, repeated attempts to degrade protein aggregates may hinder its ability to fulfill other physiologic tasks and this may ultimately lead to cellular dysfunction or death.
Autophagy represents an alternate pathway to the UPS for degradation of cytosolic proteins and organelles, including lipid structures and glycoproteins. Autophagy has a role in many biological processes, including nutrient recycling for prolonged survival during starvation and cytoprotection through degradation of insoluble inclusions in various neurodegenerative disorders. Since neurons are post-mitotic cells with polarized morphology and active protein trafficking, they are highly dependent on autophagy for their survival [3, 4]. Furthermore, suppression of basal autophagy by knocking out autophagy-related genes leads to severe neurodegeneration in mice secondary to an accumulation of ubiquitin-positive protein aggregates and neuronal loss in the cerebellar cortex, hippocampus, and cerebellum [19, 20]. Interestingly, impairment of the UPS can cause a compensatory increase in autophagy in order to maintain cellular homeostasis [21]. However, autophagy inhibition, in addition to affecting long-lived proteins, compromises the UPS function [22].
Autophagy can be subdivided into the macroautophagy-lysosomal system (MALS), chaperone-mediated autophagy (CMA), and mitophagy. MALS is responsible for bulk lysosomal degradation of cytosolic proteins, including protein aggregates and organelles. Mitophagy is the selective degradation of mitochondria. Unlike other types of autophagy, which rely on vesicle-mediated delivery of substrates to the lysosome, CMA is a selective process that involves direct translocation of substrates across the lysosomal membrane. When MALS is induced, a double-membrane compartment called an autophagosome sequesters an area of the cytoplasm and eventually fuses with the lysosome, where lysosomal enzymes degrade its contents. The MALS pathway has been implicated in several neurodegenerative diseases. As neurological disease progresses, there is an accumulation of autophagosomes or autophagic vacuoles in the diseased brains. In general, neurodegeneration is associated with defects at various steps in the autophagic process: (a) the disruption of autophagosome formation, (b) the disruption of autophagosome maturation, and (c) the disruption of autophagosome clearance.
Several adaptor proteins or shuttling factors are involved in the transport of protein substrates. Such proteins usually possess ubiquitin-associated (UBA) and ubiquitin-like (UBL) domains and include the ubiquilins (UBQLN) and SQSTM1/p62 [23]. They bind the substrates with their UBA domain and transport them to the proteasome, where they subsequently bind to the 19S regulatory particle with their UBL domain [23]. Interestingly, the UBQLNs and SQSTM1/p62 have also been proposed to form the link between UPS and autophagy [23].
We will now discuss the involvement of protein recycling pathways in the most common age-related neurodegenerative diseases, focusing on the UPS, MALS, and mitophagy pathways (Figure 1). CMA has been reviewed in detail by others and will not be the focus of this review [24].
Alzheimer disease
Alzheimer disease (AD) is the most common cause of senile dementia and is characterized by progressive dementia accompanied by personality changes, psychosis, and language problems. Neuropathology is characterized mainly by extracellular senile plaques, consisting primarily of β-amyloid (Aβ), and intracellular neurofibrillary tangles, with hyperphosphorylated microtubule associated protein tau as a main constituent. Dystrophic neurites surrounding the plaques are ubiquitinated and consist of several ubiquitin-binding proteins, such as UBQLNs and SQSTM1/p62. There is not much evidence that the UPS is involved in degradation of Aβ or its precursor, the β-amyloid precursor protein (βAPP). On the other hand, tau is inefficiently degraded by the UPS, although the non-canonical 20S proteasomal degradation rather than the ubiquitin-dependent 26S proteasomal degradation seems to have a predominant role [25]. Despite the limited role for the UPS in the recycling of Aβ and tau, there is evidence that both of these proteins can impair UPS function [26, 27] and such impairments have been observed in several brain regions of patients with AD [28]. Interestingly, a frame-shift mutant of ubiquitin B (UBB+1) has been found to accumulate in the dystrophic neurites and neurofibrillary tangles of AD [29] as well as other tauopathies and in polyglutamine diseases but not in synucleinopathies [30]. Although low levels of UBB+1 are degraded by the UPS, high levels of UBB+1 are incompletely degraded by the UPS, resulting in inhibition [31]. It has been reported that ubiquitination of UBB+1 is mediated by E2-25 K, which is essential for Aβ toxicity in animal AD models [32]. There is also emerging evidence that the ubiquitin-like protein, UBQLN1, is important in the regulation of βAPP and the AD-related protein, presenilin 1 [33].
Abundant autophagic vacuoles were first identified by electron microscopy studies on AD brain [34] and later confirmed by immunolabeling studies to be a major reservoir of intracellular Aβ [35]. This accumulation of autophagic vacuoles seems to be secondary to a combination of increased autophagy induction and their defective clearance [36, 37]. Hyperactivation of mammalian target of rapamycin (mTOR) by Aβ results in decreased autophagy and contributes to tau hyperphosphorylation [38–40]. Furthermore, genetic inhibition of autophagy enhances Aβ-induced toxicity in cultured neurons [41]. Lee and colleagues [42] have previously reported that loss of presenilin 1 activity led to impairments in autophagosomal function as a consequence of lysosomal alkalinization, caused by failed maturation of the proton translocating V0a1 subunit of the vacuolar (H+)-ATPase and targeting to the lysosome. Several groups have supported these findings under presenilin 1 loss-of-function conditions [43–46]. However, these findings were not supported by two recent independent studies [47, 48]. Reliable lysosomal pH measurement requires an appropriate probe and optimal experimental conditions and controls, and the differing observations by these groups may be due to differences in experimental approaches employed [44]. Interestingly, genetic enhancement of lysosomal activity has been shown to reduce Aβ accumulation and prevented the development of cognitive deficits in a mouse model of AD [49].
There is some evidence supporting a potential beneficial effect of inducing autophagy with rapamycin in AD [50]. For instance, it has been shown that rapamycin-mediated mTOR inhibition reduces Aβ and tau accumulation in an autophagy-dependent manner and reduces cognitive deficits in two different mouse models of AD [38, 51]. Furthermore, induction of autophagy by viral vector-mediated expression of beclin1, which is involved in autophagic vesicle nucleation, reduces Aβ pathology in a mouse model of AD [52]. However, it must be noted that a possible dysregulation of mTOR-protein synthesis by rapamycin may have detrimental effects on learning and memory [53, 54]. Also, if autophagosome clearance is impaired, as has been suggested by a lysosomal acidification defect in AD, autophagy induction may cause further accumulation of autophagosomes and autophagy substrates. For instance, it has been shown that rapamycin treatment decreases lifespan of flies overexpressing Aβ42 [55]. Collectively, these observations suggest that UPS and autophagy play a critical role in the pathogenesis of AD and that mTOR or beclin 1 signaling or the activation (or a combination of these) may be important therapeutic targets in AD.
Parkinson disease
Parkinson disease (PD) is a progressive neurodegenerative disorder caused by a selective death of dopaminergic neurons in the substantia nigra and results in a resting tremor, rigidity, slowness of movement, and postural instability. The characteristic pathology of PD includes cytoplasmic inclusions called Lewy bodies, whose major constituent is α-synuclein. It has been reported that the UPS is the main degradation pathway for α-synuclein under normal conditions in vivo while, with increased α-synuclein burden, MALS is recruited [56]. It has been shown that expression of the A53T mutant of α-synuclein induces alterations of the UPS [57]. Providing further evidence for a role of the UPS in PD pathogenesis, Bedford and colleagues [18] reported that depletion of the 26S proteasome leads to an accumulation of α-synuclein and the development of Lewy-like inclusions. There is also limited evidence for a direct genetic role for UPS involvement in PD pathogenesis. A putative causal mutation in ubiquitin C-terminal hydrolase L1 has been identified in two German siblings with PD [58]. Mutations in the E3 ligase parkin cause juvenile PD [59, 60], and the mechanism was linked to diminished affinity for the 19S particle for at least some of the mutations [61]. Parkin is reportedly involved in proteasomal degradation of several substrates, including α-synuclein, and in mitophagy.
Autophagy has a vital role to play in the pathogenesis of PD. Conditional deletion of the autophagy-related protein, Atg7, in the substantia nigra dopaminergic neurons recapitulates many of the pathologic features of PD [62]. Moreover, it has been shown that α-synuclein inclusions are preferred targets for SQSTM1/p62-dependent autophagy [63]. The familial PD-linked proteins, such as α-synuclein, DJ-1, parkin, PINK1, and LRRK2, are also known to be involved in the autophagic pathway. For instance, it is known that overexpression of wild-type α-synuclein impairs macroautophagy by causing mislocalization of the autophagy protein, Atg9, and decreased formation of autophagosome precursors called omegasomes [64]. In addition, overexpression of the A53T mutant of human α-synuclein induces an accumulation of autophagic vacuoles [57, 65]. Functional deficiency of DJ-1 also leads to altered autophagy via an increase in autophagic flux in murine and human cell lines [66]. Moreover, cells transfected with mutant LRRK2 show marked accumulation of autophagic vacuoles [67]. The full-length PINK1 interacts with beclin 1, and it has been shown that the overexpression of PINK1 significantly enhances both basal and starvation-induced autophagy. Of note, the W437X mutant of PINK1 shows an impaired interaction with beclin 1 and lacks the ability to induce autophagy [68]. Interestingly, it has been shown that beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in an α-synuclein model of PD [69].
It is known that damaged mitochondria accumulate with normal aging. Mitochondrial dysfunction has also been implicated in the pathogenesis of PD. It has been shown that parkin is selectively recruited to damaged mitochondria and is important in their selective elimination [70]. Moreover, damage to mitochondria facilitates the rapid accumulation of PINK1, a mitochondrial serine/threonine kinase, in the mitochondria which recruits parkin to induce mitophagy in a process which involves the recruitment of SQSTM1/p62 and VDAC1 [71, 72]. This PINK1/parkin-mediated mitophagy is compromised by PD-linked mutations [73]. Consistent with this, it has been shown that there is an accumulation of damaged mitochondria in knockout models of PINK1 and parkin [74–76].
Mutations in genes encoding lysosomal proteins, such as glucocerebrosidase (GBA) and lysosomal type 5 P-type ATPase (ATP13A2), have also been linked to PD [77]. GBA mutations impair lysosomal function, leading to α-synuclein accumulation, which further decreases lysosomal GBA activity by impairing the trafficking of GBA from the endoplasmic reticulum-Golgi to lysosomes, leading to neurodegeneration [77]. Recent work has shown that deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity [78, 79]. Collectively, these observations suggest that impairments of UPS, autophagy, and mitophagy may underlie PD pathogenesis and that these pathways may serve as relevant targets for the design of rational therapies in PD.
Amyotrophic lateral sclerosis and frontotemporal lobe dementia
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease and is caused by selective degeneration of motor neurons in the brain and spinal cord. Progressive weakness leads to paralysis that is ultimately fatal, in most cases, within 5 years of symptom onset. About 5 % of patients with ALS develop features of overt and disabling dementia, usually of the frontotemporal lobar type (FTD) [80]. The presence of ubiquitin-positive proteinaceous inclusions in motor neurons is the signature pathological feature of ALS, and it has been proposed that dysfunction of the UPS might play a role in this phenomenon. Indeed, motor neuron-specific disruption of proteasomes results in an ALS-like phenotype in mice [81]. Functional alterations of the UPS occur in motor neurons of mutant SOD1-linked ALS mice and may play a role in disease progression [82]. It is also known that mutant SOD1 is degraded by the proteasome and that partial inhibition of proteasome activity leads to the formation of large SOD1-containing aggregates [83–87].
Direct etiological evidence for the involvement of UPS and MALS pathways was provided by the identification of mutations in UBQLN2 and SQSTM1/p62 in ALS and FTD [23, 88, 89]. Pathologic inclusions, containing ubiquitin, SQSTM1/p62, and UBQLN2, are a common feature in a wide spectrum of ALS and ALS-FTD, implying a functional convergence at the level of abnormal turnover of ubiquitinated proteins [23, 80, 88]. Indeed, it has been shown that mutant UBQLN2 impairs UPS function in vitro[88], implying that induction of UPS function can be a viable therapeutic option in ALS and ALS-FTD.
Although a defective UPS has been suggested to produce ALS-associated protein aggregates, recent studies have revealed a prominent role for autophagy [90]. Surviving motor neurons from mutant SOD1-linked ALS mice at the end stage of disease show accumulation of autophagic vacuoles within the cytosol in a progressive and protein aggregation-related manner [91]. Mutant SOD1 can be recognized by SQSTM1/p62 in a ubiquitin-independent fashion and targeted for autophagy [92]. Autophagy induction clears aggregated proteins and rescues motor neuron degeneration in ALS mice [90, 93]. However, it has also been shown that rapamycin treatment in mutant SOD1-linked ALS mice resulted in exacerbation of ALS pathology, earlier disease onset, and faster disease progression [94]. Moreover, although lithium-mediated autophagy induction was originally reported to be beneficial in both mice and patients with ALS [91], these findings could not be reproduced by subsequent studies [95–97]. Interestingly, a recent study has suggested a potential use of trehalose and enhancers of mTOR-independent autophagy for the treatment of ALS [98]. Therefore, further studies are required to clarify the potential role of autophagy induction in ALS.
An autophagy defect has also been suggested by genetic studies of ALS and FTD. For instance, mutations in UBQLN2 and SQSTM1/p62 have been reported in ALS and FTD [88, 89, 99]. UBQLNs were previously thought to be involved in protein degradation via the UPS. Recently, several studies have provided convincing evidence for their role in autophagy [100–102]. UBQLNs are present in autophagosomes and bind LC3 in a complex [101, 103]. Overexpression of UBQLNs protects cells from starvation-induced death (via autophagy and UBA-domain dependent mechanisms), whereas depletion renders cells more susceptible [100]. Depletion of UBQLNs also regulates formation and maturation of autophagic vacuoles [100, 101]. It has been reported that SQSTM1/p62 binds ubiquitinated proteins and LC3 [104]. Under conditions of impaired autophagy, SQSTM1/p62 mediates the aggregation of ubiquitinated proteins and sequesters them from the UPS [22, 105]. Similarly, mutations in VCP and OPTN cause ALS [106, 107]. VCP mutations also cause the syndrome of inclusion body myopathy with Paget disease of bone and FTD [108]. Interestingly, both VCP and OPTN are also involved in protein degradation via autophagy [109–111]. Recently, the ALS-linked protein FIG 4 was also shown to be involved in autophagy [112]. Similarly, mutations in the endosomal ESCRTIII-complex subunit CHMP2B cause FTD [113]. VCP and ESCRT family members, including CHMP2B, are known to participate in autophagosome-lysosome fusion.
The ALS-FTD-associated protein TDP43 is degraded by both the UPS and autophagy pathways, and it has been shown that overexpression of SQSTM1/p62 reduces TDP43 aggregation in an autophagy- and proteasome-dependent manner [114–116]. Deficiency of VCP and ESCRT family members has been shown to lead to the accumulation of the TDP43 [109, 117, 118]. Interestingly, overexpression of UBQLN1 has been shown to potentiate the aggregation of TDP43 and modify toxicity in a drosophila model of ALS [119, 120]. Future studies to address the precise molecular mechanism of UPS and autophagy dysfunction in ALS and FTD are essential to identify appropriate therapeutic targets.
Huntington disease
Huntington disease (HD) is caused by a polyglutamine repeat expansion in the huntingtin (htt) gene, resulting in protein aggregation and causing a syndrome of involuntary movements and dementia. Although it is clear that the proteasome is involved in the degradation of mutant htt, the role of proteasomes remains contradictory [121]. Interestingly, a recent unbiased screen has identified both UBQLN1 and UBQLN2 to be highly associated with htt inclusions [122]. Altered autophagy has been observed in post-mortem specimens from HD patients and animal models [123, 124]. It has been suggested that wild-type htt may function physiologically as an autophagy regulator, and it has been shown that cells expressing mutant htt have an increase in autophagic vacuoles [125]. A cargo recognition defect has been suggested as the mechanism underlying autophagic dysfunction in HD [126]. Another mechanism may be related to recruitment of beclin 1 by mutant htt, which impairs beclin 1-mediated long-lived protein turnover [127]. The involvement of autophagy in HD has been further demonstrated by the sequestration of mTOR in polyglutamine aggregates in cell models, transgenic mice, and human brains. This sequestration of mTOR impairs its kinase activity and induces autophagy. This mechanism protects against polyglutamine toxicity, as autophagy induction attenuates htt accumulation and cell death in cell models of HD, whereas the inhibition of autophagy has opposite effects [128–130]. Thus, the therapeutic induction and recovery of autophagy may be useful to enhance the clearance of mutant htt and reduce its neurotoxicity.
Conclusions
Because of their post-mitotic nature, polarized morphology and complex arborization, neurons are particularly sensitive to alterations in protein homeostasis. A plethora of evidence has firmly established the essential role for the UPS and autophagy pathways in both normal physiological functioning as well as pathophysiological conditions of the nervous system.
Impairments in the quality control of proteins and organelles in the neuronal soma, axon, and synapses led to an aggregation of toxic proteins and, thereby, cause neuronal dysfunction and neurodegeneration in diseases such as AD, PD, ALS, FTD, and HD.
As reviewed above, several independent studies have shown that autophagy induction can be beneficial as a disease-modifying treatment in experimental models of various neurodegenerative diseases. Of note, rapamycin has been in clinical practice for several years. But as a disease-modifying therapy in neurodegeneration, long-term use of rapamycin may have adverse effects on cognition by dysregulating physiological mTOR signaling, which is important for axonal growth, dendritic arborization, and synaptic plasticity [50, 131]. However, such adverse effects have not been reported so far in patients undergoing long-term rapamycin therapy. Autophagy induction therapy would have to be initiated either before disease onset or in the very early stages before pathological protein aggregates have become too large to overwhelm the cellular protein recycling pathways. Moreover, understanding the step(s) affected in the autophagic process is essential to developing targeted therapeutic approaches based on the modulation of autophagy. For instance, if autophagosome-lysosome fusion or lysosomal acidification is impaired, autophagy induction would lead to an accumulation of non-degradable autophagosomes and this could be detrimental. It should also be noted that long-term induction of autophagy may reduce cell viability by reducing the cells’ responsiveness to stress [50].
Mechanistic correlates for abnormal UPS and autophagy in these diseases have yet to be fully understood. Since the cargo destined for recycling may need to be transported over great distances in a neuron, it is likely that complex processes involving abnormal protein-protein interactions or impaired trafficking (or both) may be involved. A better understanding of these mechanisms will aid in the design of rational therapies for neurodegenerative diseases.
Abbreviations
- AD:
-
Alzheimer disease
- ALS:
-
amyotrophic lateral sclerosis
- ATP13A2:
-
lysosomal type 5 P-type ATPase
- Aβ:
-
β-amyloid
- CMA:
-
chaperone-mediated autophagy
- FTD:
-
frontotemporal lobar dementia
- GBA:
-
glucocerebrosidase
- HD:
-
Huntington disease
- htt:
-
huntingtin
- MALS:
-
macroautophagy-lysosomal system
- mTOR:
-
mammalian target of rapamycin
- PD:
-
Parkinson disease
- UBA:
-
ubiquitin-associated
- UBB+1:
-
frame-shift mutant of ubiquitin B
- UBL:
-
ubiquitin-like
- UBQLN:
-
ubiquilins
- UPS:
-
ubiquitin-proteasome system
- βAPP:
-
β-amyloid precursor protein.
References
Gestwicki JE, Garza D: Protein quality control in neurodegenerative disease. Prog Mol Biol Transl Sci. 2012, 107: 327-353.
Ding M, Shen K: The role of the ubiquitin proteasome system in synapse remodeling and neurodegenerative diseases. Bioessays. 2008, 30: 1075-1083. 10.1002/bies.20843.
Mizushima N, Kuma A: Autophagosomes in GFP-LC3 transgenic mice. Methods Mol Biol. 2008, 445: 119-124. 10.1007/978-1-59745-157-4_7.
Tsvetkov AS, Mitra S, Finkbeiner S: Protein turnover differences between neurons and other cells. Autophagy. 2009, 5: 1037-1038. 10.4161/auto.5.7.9291.
Hegde AN: The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010, 17: 314-327. 10.1101/lm.1504010.
Tai HC, Schuman EM: Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008, 9: 826-838. 10.1038/nrn2499.
Dennissen FJ, Kholod N, van Leeuwen FW: The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim?. Prog Neurobiol. 2012, 96: 190-207. 10.1016/j.pneurobio.2012.01.003.
Son JH, Shim JH, Kim KH, Ha JY, Han JY: Neuronal autophagy and neurodegenerative diseases. Exp Mol Med. 2012, 44: 89-98. 10.3858/emm.2012.44.2.031.
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE: Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009, 78: 959-991. 10.1146/annurev.biochem.052308.114844.
Baldwin AJ, Knowles TP, Tartaglia GG, Fitzpatrick AW, Devlin GL, Shammas SL, Waudby CA, Mossuto MF, Meehan S, Gras SL, Christodoulou J, Anthony-Cahill SJ, Barker PD, Vendruscolo M, Dobson CM: Metastability of native proteins and the phenomenon of amyloid formation. J Am Chem Soc. 2011, 133: 14160-14163. 10.1021/ja2017703.
Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A: Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005, 1: 131-140. 10.4161/auto.1.3.2017.
Walker LC, LeVine H: The cerebral proteopathies: neurodegenerative disorders of protein conformation and assembly. Mol Neurobiol. 2000, 21: 83-95. 10.1385/MN:21:1-2:083.
Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI: Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006, 311: 1471-1474. 10.1126/science.1124514.
Douglas PM, Dillin A: Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010, 190: 719-729. 10.1083/jcb.201005144.
Morimoto RI: Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22: 1427-1438. 10.1101/gad.1657108.
Douglas PM, Summers DW, Cyr DM: Molecular chaperones antagonize proteotoxicity by differentially modulating protein aggregation pathways. Prion. 2009, 3: 51-58. 10.4161/pri.3.2.8587.
Wong E, Cuervo AM: Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010, 2: a006734-
Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ: Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci. 2008, 28: 8189-8198. 10.1523/JNEUROSCI.2218-08.2008.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N: Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006, 441: 885-889. 10.1038/nature04724.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K: Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006, 441: 880-884. 10.1038/nature04723.
Nedelsky NB, Todd PK, Taylor JP: Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008, 1782: 691-699. 10.1016/j.bbadis.2008.10.002.
Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC: Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009, 33: 517-527. 10.1016/j.molcel.2009.01.021.
Fecto F, Siddique T: UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve. 2012, 45: 157-162. 10.1002/mus.23278.
Koga H, Cuervo AM: Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis. 2011, 43: 29-37. 10.1016/j.nbd.2010.07.006.
Grune T, Botzen D, Engels M, Voss P, Kaiser B, Jung T, Grimm S, Ermak G, Davies KJ: Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions. Arch Biochem Biophys. 2010, 500: 181-188. 10.1016/j.abb.2010.05.008.
Keck S, Nitsch R, Grune T, Ullrich O: Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem. 2003, 85: 115-122. 10.1046/j.1471-4159.2003.01642.x.
Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM: Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008, 29: 1607-1618. 10.1016/j.neurobiolaging.2007.04.014.
Keller JN, Hanni KB, Markesbery WR: Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000, 75: 436-439.
van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Köycü S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM: Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science. 1998, 279: 242-247. 10.1126/science.279.5348.242.
Dennissen FJ, Kholod N, Steinbusch HW, Van Leeuwen FW: Misframed proteins and neurodegeneration: a novel view on Alzheimer’s and Parkinson’s diseases. Neurodegener Dis. 2010, 7: 76-79. 10.1159/000285510.
van Tijn P, de Vrij FM, Schuurman KG, Dantuma NP, Fischer DF, van Leeuwen FW, Hol EM: Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J Cell Sci. 2007, 120: 1615-1623. 10.1242/jcs.03438.
Ko S, Kang GB, Song SM, Lee JG, Shin DY, Yun JH, Sheng Y, Cheong C, Jeon YH, Jung YK, Arrowsmith CH, Avvakumov GV, Dhe-Paganon S, Yoo YJ, Eom SH, Lee W: Structural basis of E2-25 K/UBB + 1 interaction leading to proteasome inhibition and neurotoxicity. J Biol Chem. 2010, 285: 36070-36080. 10.1074/jbc.M110.145219.
Haapasalo A, Viswanathan J, Bertram L, Soininen H, Tanzi RE, Hiltunen M: Emerging role of Alzheimer’s disease-associated ubiquilin-1 in protein aggregation. Biochem Soc Trans. 2010, 38: 150-155. 10.1042/BST0380150.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM: Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005, 64: 113-122.
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA: Macroautophagy - a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005, 171: 87-98. 10.1083/jcb.200505082.
Nixon RA: Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007, 120: 4081-4091. 10.1242/jcs.019265.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA: Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008, 28: 6926-6937. 10.1523/JNEUROSCI.0800-08.2008.
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S: Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010, 285: 13107-13120. 10.1074/jbc.M110.100420.
Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magri A, Oddo S: Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem. 2011, 286: 8924-8932. 10.1074/jbc.M110.180638.
Meske V, Albert F, Ohm TG: Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3 -dependent phosphorylation of Tau. J Biol Chem. 2008, 283: 100-109. 10.1074/jbc.M704292200.
Hung SY, Huang WP, Liou HC, Fu WM: Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009, 5: 502-510. 10.4161/auto.5.4.8096.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA: Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010, 141: 1146-1158. 10.1016/j.cell.2010.05.008.
Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H: Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013, 288: 1295-1306. 10.1074/jbc.M112.409250.
Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA: Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013, 37: 1949-1961. 10.1111/ejn.12169.
Torres M, Jimenez S, Sanchez-Varo R, Navarro V, Trujillo-Estrada L, Sanchez-Mejias E, Carmona I, Davila JC, Vizuete M, Gutierrez A, Vitorica J: Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Mol Neurodegener. 2012, 7: 59-10.1186/1750-1326-7-59.
Dobrowolski R, Vick P, Ploper D, Gumper I, Snitkin H, Sabatini DD, De Robertis EM: Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2012, 2: 1316-1328. 10.1016/j.celrep.2012.09.026.
Zhang X, Garbett K, Veeraraghavalu K, Wilburn B, Gilmore R, Mirnics K, Sisodia SS: A role for presenilins in autophagy revisited: normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci. 2012, 32: 8633-8648. 10.1523/JNEUROSCI.0556-12.2012.
Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, Wuytack F, Hiesinger PR, Grinstein S, Annaert W: Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012, 198: 23-35. 10.1083/jcb.201201076.
Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA: Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011, 134: 258-277. 10.1093/brain/awq341.
Bove J, Martinez-Vicente M, Vila M: Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011, 12: 437-452. 10.1038/nrn3068.
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V: Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010, 5: e9979-10.1371/journal.pone.0009979.
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T: The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008, 118: 2190-2199.
Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey CH, Kandel ER: A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999, 99: 221-237. 10.1016/S0092-8674(00)81653-0.
Tischmeyer W, Schicknick H, Kraus M, Seidenbecher CI, Staak S, Scheich H, Gundelfinger ED: Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur J Neurosci. 2003, 18: 942-950. 10.1046/j.1460-9568.2003.02820.x.
Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM: Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One. 2009, 4: e4201-10.1371/journal.pone.0004201.
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK: Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci. 2011, 31: 14508-14520. 10.1523/JNEUROSCI.1560-11.2011.
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA: Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001, 21: 9549-9560.
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH: The ubiquitin pathway in Parkinson’s disease. Nature. 1998, 395: 451-452. 10.1038/26652.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998, 392: 605-608. 10.1038/33416.
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ: Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001, 293: 263-269. 10.1126/science.1060627.
Safadi SS, Barber KR, Shaw GS: Impact of autosomal recessive juvenile Parkinson’s disease mutations on the structure and interactions of the parkin ubiquitin-like domain. Biochemistry. 2011, 50: 2603-2610. 10.1021/bi200065g.
Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM: Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J Neurosci. 2012, 32: 16503-16509. 10.1523/JNEUROSCI.0209-12.2012.
Watanabe Y, Tatebe H, Taguchi K, Endo Y, Tokuda T, Mizuno T, Nakagawa M, Tanaka M: p62/SQSTM1-dependent autophagy of Lewy body-like alpha-synuclein inclusions. PLoS One. 2012, 7: e52868-10.1371/journal.pone.0052868.
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O’Kane CJ, Rubinsztein DC: alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010, 190: 1023-1037. 10.1083/jcb.201003122.
Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A: Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002, 22: 2780-2791.
Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M, Lutz AK, Rousseaux MW, Bevilacqua L, Jahani-Asl A, Callaghan S, MacLaurin JG, Winklhofer KF, Rizzu P, Rippstein P, Kim RH, Chen CX, Fon EA, Slack RS, Harper ME, McBride HM, Mak TW, Park DS: Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010, 19: 3734-3746. 10.1093/hmg/ddq288.
Plowey ED, Cherra SJ, Liu YJ, Chu CT: Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008, 105: 1048-1056. 10.1111/j.1471-4159.2008.05217.x.
Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, Torosantucci L, Cassina L, Russo MA, Dallapiccola B, Valente EM, Casari G: The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010, 17: 962-974. 10.1038/cdd.2009.200.
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E: Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009, 29: 13578-13588. 10.1523/JNEUROSCI.4390-09.2009.
Narendra D, Tanaka A, Suen DF, Youle RJ: Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008, 183: 795-803. 10.1083/jcb.200809125.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ: PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8: e1000298-10.1371/journal.pbio.1000298.
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W: PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010, 12: 119-131. 10.1038/ncb2012.
Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W: The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010, 6: 871-878. 10.4161/auto.6.7.13286.
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ: Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003, 100: 4078-4083. 10.1073/pnas.0737556100.
Gautier CA, Kitada T, Shen J: Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008, 105: 11364-11369. 10.1073/pnas.0802076105.
Shim JH, Yoon SH, Kim KH, Han JY, Ha JY, Hyun DH, Paek SH, Kang UJ, Zhuang X, Son JH: The antioxidant Trolox helps recovery from the familial Parkinson’s disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion. 2011, 11: 707-715. 10.1016/j.mito.2011.05.013.
Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E: Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013, 28: 725-732. 10.1002/mds.25462.
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D: Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012, 32: 4240-4246. 10.1523/JNEUROSCI.5575-11.2012.
Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, Sidransky E, Krainc D, Shull GE: Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013, 22: 2067-2082. 10.1093/hmg/ddt057.
Fecto F, Siddique T: Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia. J Mol Neurosci. 2011, 45: 663-675. 10.1007/s12031-011-9637-9.
Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H, Sakimura K, Ito H, Takahashi R: Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012, 287: 42984-42994. 10.1074/jbc.M112.417600.
Cheroni C, Marino M, Tortarolo M, Veglianese P, De Biasi S, Fontana E, Zuccarello LV, Maynard CJ, Dantuma NP, Bendotti C: Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet. 2009, 18: 82-96.
Puttaparthi K, Wojcik C, Rajendran B, DeMartino GN, Elliott JL: Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes. J Neurochem. 2003, 87: 851-860.
Di Noto L, Whitson LJ, Cao X, Hart PJ, Levine RL: Proteasomal degradation of mutant superoxide dismutases linked to amyotrophic lateral sclerosis. J Biol Chem. 2005, 280: 39907-39913. 10.1074/jbc.M506247200.
Hoffman EK, Wilcox HM, Scott RW, Siman R: Proteasome inhibition enhances the stability of mouse Cu/Zn superoxide dismutase with mutations linked to familial amyotrophic lateral sclerosis. J Neurol Sci. 1996, 139: 15-20. 10.1016/0022-510X(96)00031-7.
Hyun DH, Lee M, Halliwell B, Jenner P: Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J Neurochem. 2003, 86: 363-373.
Johnston JA, Dalton MJ, Gurney ME, Kopito RR: Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000, 97: 12571-12576. 10.1073/pnas.220417997.
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T: Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011, 477: 211-215. 10.1038/nature10353.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T: SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011, 68: 1440-1446. 10.1001/archneurol.2011.250.
Ferrucci M, Fulceri F, Toti L, Soldani P, Siciliano G, Paparelli A, Fornai F: Protein clearing pathways in ALS. Arch Ital Biol. 2011, 149: 121-149.
Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A: Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008, 105: 2052-2057. 10.1073/pnas.0708022105.
Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H: Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009, 111: 1062-1073. 10.1111/j.1471-4159.2009.06388.x.
Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH: XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23: 2294-2306. 10.1101/gad.1830709.
Zhang X, Li L, Chen S, Yang D, Wang Y, Wang Z, Le W: Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011, 7: 412-425. 10.4161/auto.7.4.14541.
Pizzasegola C, Caron I, Daleno C, Ronchi A, Minoia C, Carri MT, Bendotti C: Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler. 2009, 10: 221-228. 10.1080/17482960902803440.
Gill A, Kidd J, Vieira F, Thompson K, Perrin S: No benefit from chronic lithium dosing in a sibling-matched, gender balanced, investigator-blinded trial using a standard mouse model of familial ALS. PLoS One. 2009, 4: e6489-10.1371/journal.pone.0006489.
Chiò A, Borghero G, Calvo A, Capasso M, Caponnetto C, Corbo M, Giannini F, Logroscino G, Mandrioli J, Marcello N, Mazzini L, Moglia C, Monsurrò MR, Mora G, Patti F, Perini M, Pietrini V, Pisano F, Pupillo E, Sabatelli M, Salvi F, Silani V, Simone IL, Sorarù G, Tola MR, Volanti P, Beghi E, LITALS Study Group: Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose-finding trial. Neurology. 2010, 75: 619-625. 10.1212/WNL.0b013e3181ed9e7c.
Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C: Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013, 9: 1308-1320. 10.4161/auto.25188.
Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, Bruni AC, St George-Hyslop PH, Scarpini E, Gallone S, Pinessi L, TODEM Study Group: SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012, 79: 1556-1562. 10.1212/WNL.0b013e31826e25df.
N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ: PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10: 173-179. 10.1038/embor.2008.238.
Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ: Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010, 19: 3219-3232. 10.1093/hmg/ddq231.
Rothenberg C, Monteiro MJ: Ubiquilin at a crossroads in protein degradation pathways. Autophagy. 2010, 6: 979-980. 10.4161/auto.6.7.13118.
Komatsu M, Ichimura Y: Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584: 1374-1378. 10.1016/j.febslet.2010.02.017.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T: p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007, 282: 24131-24145. 10.1074/jbc.M702824200.
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K: Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007, 131: 1149-1163. 10.1016/j.cell.2007.10.035.
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H: Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010, 465: 223-226. 10.1038/nature08971.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G, Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ, ITALSGEN Consortium: Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010, 68: 857-864. 10.1016/j.neuron.2010.11.036.
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE: Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004, 36: 377-381. 10.1038/ng1332.
Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC: Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009, 187: 875-888. 10.1083/jcb.200908115.
Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP: VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010, 6: 217-227. 10.4161/auto.6.2.11014.
Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I: Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011, 333: 228-233. 10.1126/science.1205405.
Ferguson CJ, Lenk GM, Meisler MH: Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet. 2009, 18: 4868-4878. 10.1093/hmg/ddp460.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sørensen SA, Gydesen S, Fisher EM, Collinge J: Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005, 37: 806-808. 10.1038/ng1609.
Wang X, Fan H, Ying Z, Li B, Wang H, Wang G: Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010, 469: 112-116. 10.1016/j.neulet.2009.11.055.
Urushitani M, Sato T, Bamba H, Hisa Y, Tooyama I: Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J Neurosci Res. 2010, 88: 784-797.
Brady OA, Meng P, Zheng Y, Mao Y, Hu F: Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011, 116: 248-259. 10.1111/j.1471-4159.2010.07098.x.
Gitcho MA, Strider J, Carter D, Taylor-Reinwald L, Forman MS, Goate AM, Cairns NJ: VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J Biol Chem. 2009, 284: 12384-12398. 10.1074/jbc.M900992200.
Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A: Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007, 179: 485-500. 10.1083/jcb.200702115.
Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ, Tibbetts RS: Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J Biol Chem. 2009, 284: 8083-8092. 10.1074/jbc.M808064200.
Hanson KA, Kim SH, Wassarman DA, Tibbetts RS: Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J Biol Chem. 2010, 285: 11068-11072. 10.1074/jbc.C109.078527.
Schipper-Krom S, Juenemann K, Reits EA: The ubiquitin-proteasome system in Huntington’s disease: are proteasomes impaired, initiators of disease, or coming to the rescue?. Biochem Res Int. 2012, 2012: 837015-
Rutherford NJ, Lewis J, Clippinger AK, Thomas MA, Adamson J, Cruz PE, Cannon A, Xu G, Golde TE, Shaw G, Borchelt DR, Giasson BI: Unbiased screen reveals ubiquilin-1 and -2 highly associated with huntingtin inclusions. Brain Res. 2013, 1524: 62-73.
Tellez-Nagel I, Johnson AB, Terry RD: Studies on brain biopsies of patients with Huntington’s chorea. J Neuropathol Exp Neurol. 1974, 33: 308-332. 10.1097/00005072-197404000-00008.
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M: Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000, 20: 7268-7278.
Atwal RS, Truant R: A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy. 2008, 4: 91-93.
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM: Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010, 13: 567-576. 10.1038/nn.2528.
Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J: Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem. 2006, 281: 14474-14485. 10.1074/jbc.M600364200.
Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC: Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum Mol Genet. 2003, 12: 985-994. 10.1093/hmg/ddg109.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC: Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004, 36: 585-595. 10.1038/ng1362.
Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Kane CJ, Schreiber SL, Rubinsztein DC: Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007, 3: 331-338. 10.1038/nchembio883.
Garelick MG, Kennedy BK: TOR on the brain. Exp Gerontol. 2011, 46: 155-163. 10.1016/j.exger.2010.08.030.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Fecto, F., Esengul, Y.T. & Siddique, T. Protein recycling pathways in neurodegenerative diseases. Alz Res Therapy 6, 13 (2014). https://doi.org/10.1186/alzrt243
Published:
DOI: https://doi.org/10.1186/alzrt243