
Abstract
NLRs (nucleotide-binding domain, leucine-rich repeat containing receptors) are pattern recognition receptors associated with immunity and inflammation in response to endogenous and exogenous pathogen and damage associated molecular patterns (PAMPs and DAMPs respectively). Dysregulated NLR function is associated with several diseases including cancers, metabolic diseases, autoimmune disorders and autoinflammatory syndromes. In the last decade, distinct cell and organ specific roles for NLRs have been identified however; their roles in cancer initiation, development and progression remain controversial. This review summarizes the emerging role of NLRs in cancer and their possible future as targets for cancer therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The association of chronic inflammation with cancer is well established [1–4]. Epidemiological data suggest that up to 15 % of human cancer incidence is associated with inflammation [1]. Inflammation is accepted as one of the hallmarks of cancer [5, 6]. Sustained chronic inflammation gives rise to a tumor promoting microenvironment. The tumor microenvironment comprises of stromal cells, extracellular matrix and dense vasculature as depicted in Fig. 1. Stromal cells consist of macrophages, mast cells, natural killer cells, fibroblasts, endothelial cells and pericytes. Cancer cells and stromal cells actively communicate with one other by exchange of various soluble factors including mitogenic growth factors, pro-angiogenic molecules, pro-inflammatory mediators and tumor promoting inflammation-induced factors [3, 7–13]. Cytokines produced by innate immune and stromal cells have been shown to promote cancer initiation, development and progression, as depicted in Fig. 1.

Cancer cells communicating with innate immune cells in the tumor microenvironment. Cancer cells recruit macrophages, mast cells, NK cells, fibroblast cells and endothelial cells to the tumor microenvironment. Cancer cells also induce polarization of macrophages into tumor-associated macrophages (TAMs) by cytokines and chemokines. Various pro-inflammatory mediators and mitogenic growth factors secreted from the infiltrated immune cells promote cancer cell growth, progression, extracellular matrix (ECM) remodeling and metastasis. Increased vasculature demand is supported by the recruited endothelial cells
NLRs/NOD-like receptors–the multifunctional sensors
NLRs (nucleotide-binding domain, leucine-rich repeat containing receptors) are a newly discovered family of pattern recognition receptors [14]. These germ-line encoded pattern recognition receptors are immune regulatory proteins of innate immunity as well as adaptive immunity [15]. NLRs are evolutionarily conserved from plants to animals showing high structural similarity with the R proteins [16]. NLRs recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) as well as irritants (Fig. 2).

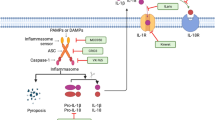
Summary of NLR inflammasome complex formation. NLRs are activated in response to specific PAMPs and DAMPs. For example, NLRP3 inflammasome activation is a two-step process. The first step includes a priming stimulus and the second stimulus leads to NLRP3 activation. NLRP3 along with ASC and pro-caspase-1 forms an inflammasome complex resulting in the maturation and release of pro-inflammatory cytokines, IL-1β and IL-18 into the extracellular environment, recruitment of other immune cells and (in some cases) pyroptosis. Similarly, other NLRs such as NLRP1 and NLRC4 also form inflammasome complexes upon activation
In humans, the NLR family consists of 22 individual NLR genes with a characteristic tripartite domain structure comprising of an N-terminal domain, a central domain and a C-terminal domain [17]. The N-terminal domain may be; a PYRIN domain (PYD), a caspase activation and recruitment domain (CARD), an acidic transactivation or a baculoviral inhibitory repeats (BIR) domain. These domains are responsible for the homotypic/heterotypic interactions with downstream molecules in the signaling pathway. The central domain consists of a nucleotide binding and oligomerization domain (NBD) which initiates self-oligomerization and is responsible for other regulatory functions. The C-terminal domain represents a series of leucine rich repeats (LRRs) which are responsible for ligand binding [18].
NLR family members are involved in multiple functions of innate immune signaling, from acting as activators of the inflammasome, nuclear factor-kB (NF-kB) and mitogen activated protein kinase (MAPK), to acting as inhibitors of inflammatory signaling, and trans-activators of MHC expression [19]. Dysregulation in NLR activation leads to various inflammatory diseases, metabolic disorders and auto-inflammatory immune disorders [20]. In the past decade there has been an exponential increase in research linking inflammasome signaling to human diseases. NLRs play dual pro- and anti-tumor roles in the initiation, progression and regression of cancer [21]. In this review we discuss emerging research in the association of NLRs with cancer and the prospective future implications in developing targeted cancer therapeutics (Table 2).
Inflammasome forming NLRs
NLRs upon activation form a multiprotein complex known as the “inflammasome” (Table 1) [22]. The inflammasome complex consists of an NLR protein, an adaptor protein: the apoptosis- associated speck -like protein (ASC) and a caspase [23]. ASC utilizes its caspase recruitment domain (CARD) to recruit the inactive pro form of caspase-1 [24]. Inflammasome activation results in auto-proteolytic cleavage of caspase-1 to form the mature active caspase-1 within the inflammasome complex (Fig. 2). Caspase-1 can cleave and activate up to 70 different substrates including pro-IL-1β and pro-IL-18 into active IL-1β and IL-18 [25]. IL-1β and IL-18 function as proinflammatory cytokines and mediate activation of various signaling pathways including NF-κB, JNK and p38 MAPK signaling pathways. Activated immune cells release IL-1β and IL-18 giving rise to an inflammatory microenvironment (Fig. 2). Inflammasome forming NLRs protect the host by modulating cytokine production, damage repair and resolution of inflammation.
NLRP3
NLRP3 is the most well studied NLR so far. The Nlrp3 gene encodes an N-terminal pyrin domain, a central NBD domain and a C-terminal LRR. NLRP3 lacks a caspase recruitment domain (CARD) and therefore, interacts with ASC to recruit procaspase-1 (Table 1). The NLRP3 inflammasome comprises of NLRP3, the adaptor protein ASC and procaspase-1. NLRP3 inflammasome activation has been observed in response to a number of PAMPs, DAMPs and irritants [22]. Mutation in human Nlrp3 is associated with cold-induced auto-inflammatory syndromes-FCAS (familial cold-induced auto-inflammatory syndrome), MWS (Muckle-Wells syndrome), and NOMID/CINCA (neonatal onset multisystem inflammatory disorder or chronic infantile neurologic cutaneous and articular syndrome) [26, 27]. Nlrp3 mutations and single-nucleotide polymorphisms (SNPs) have been associated with many inflammation-associated diseases [28]. Genetic alterations in genes encoding NLRP3 and CARD8 lead to complex inflammatory diseases such as; inflammatory bowel disease, cardiovascular disease, rheumatoid arthritis and type 1 diabetes [29, 30]. The role of NLRP3 inflammasome in metabolic diseases is well established [31]. Caspase recruitment domain-containing protein8 (CARD8; also known as TUCAN/CARDINAL) is a negative regulator of NLRP3 inflammasome activation [32]. While some studies have also shown that CARD8 knockdown did not affect IL-1β expression or IL-1β protein release, Ito et al. [33] showed that CARD8 can interact with wildtype (WT) NLRP3 and reduce IL-1β release, but not with CAPS associated mutant NLRP3. NLRP3 inflammasomes can also associate with caspase-8 to mediate IL-1β production and apoptosis in caspase-1-deficient dendritic cells and facilitate pyroptosis in wild-type dendritic cells [34]. Regulation of TH2 differentiation in CD4+ T cells by NLRP3 expression shows specific inflammasome-independent role of NLRP3 mediated via STAT5 and IL-2 signaling pathway [35]. Further in vitro analyses have confirmed specific involvement of NLRP3 in TH2 and TH17 polarization and nuclear localization of NLRP3 in TH2 cells.
NLRP3 and cancer
Very few studies have characterized the role of the NLRP3 inflammasome in cancer. Using a dextran sulfate sodium (DSS)-induced colitis model of human ulcerative colitis [36, 37], Allen et al. first demonstrated the hypersusceptibility of Nlrp3 and Casp1 deficient mice to DSS-induced colitis. This chemically induced colitis model shares pathology similar to human ulcerative colitis. Defective inflammasome activation leads to loss of epithelial integrity, massive leukocyte infiltration and increased chemokines expression in the Nlrp3 −/− and Casp1 −/− mice, leading to high mortality rates [38]. These results were supported by parallel findings that showed NLRP3 inflammasome activation as a negative regulator of tumorigenesis during colitis-associated cancer [39], with NLRP3 inflammasome dependent IL-18 production protecting against colorectal tumorigenesis. Nlrp3 deficient mice are highly susceptible to colitis-associated colorectal tumor formation and show severe inflammation, hyperplasia and tumor burden. Deregulated IL-18 expression is followed by increased chemokine expression suggestive of higher macrophage infiltration in the colon of Nlrp3 and Casp1 deficient mice. Macrophages release various tumorigenic factors in the tumor microenvironment, this is reflected in the significantly higher expression of COX-2 mRNA in colon tissue of Nlrp3 −/− and Casp1 −/− mice [40]. Hu et al. [41] showed no differences in tumor formation between the Nlrp3 −/− and WT mice. Further experiments proved that increased tumorigenesis in Casp1 −/− mice was not mediated by Nlrp3. Moreover, NLRP3 expression was primarily restricted to the hematopoietic compartment. Surprisingly, study of DSS-induced ulcerative colitis in Casp11 −/− mice indicate caspase-11-mediated non-canonical inflammasome activation, an additional pathway for production and release of pro-inflammatory cytokines, IL-1β and IL-18 [42]. Casp11 −/− mice exhibit enhanced susceptibility towards DSS and impaired IL-18 production. Casp11 −/− mice show decreased intestinal epithelial cell proliferation and disrupted epithelial cell barrier suggesting protective role of caspase-11 in maintaining intestinal epithelial cell integrity. During acute DSS-induced intestinal inflammation Casp11 −/− show a phenotype similar to that observed in the Casp1/11 −/− mice.
The NLRP3 inflammasome plays a regulatory role in the pathology of melanomas, gastric cancer and hepatocellular carcinoma; in addition NLRP3 inflammasome activation also regulates adaptive immune response to cancer vaccines (Table 2). The NLRP3 inflammasome is constitutively expressed and activated in human melanoma cells [43]. Paracrine IL-1 signaling via secreted IL-1β enhances NF-κB-dependent proIL-1β synthesis, thereby forming a positive IL-1 feedback loop in metastatic melanoma cells. In case of gastric cancer, Mycoplasma hyorhinis promotes tumor development via NLRP3 inflammasome activation, accompanied with increase in cell migration and invasion [44]. In this regard, NLRP3 activation was not cell type restricted and there was increased IL-1β and IL-18 production in M. hyorhinis infected human monocytic cells. IL-1β but not IL-18 was found to be involved in gastric cancer cell migration and invasion.
Hepatocellular carcinoma (HCC) is the third leading cause of cancer deaths worldwide. The expression of NLRP3 inflammasome components is down-regulated in hepatic parenchymal cells in HCC. Complete loss of NLRP3 inflammasome activation positively correlates with a higher HCC pathological grade suggesting protective role of NLRP3 against HCC cancer development and progression [45]. NLRP3 inflammasome is activated during anticancer chemotherapy (oxaliplatin). Autophagic tumor (EL4 or EG7 thymoma) cells release ATP sensed by the P2X7 receptors of dendritic cells leading to NLRP3 inflammasome activation [46]. Antigen presenting dendritic cells prime CD8+ T lymphocytes generating IFN-γ-dependent antitumor responses. Functional P2X7 receptor dependent activation of NLRP3 inflammasome and IL-1β secretion, are necessary for T cell priming. Notably, this study conflicts with recent results that NLRP3 dampens anti-tumor responses elicited by dendritic cells vaccination. NLRP3 expression was found to be upregulated in tumor-associated myeloid derived suppressor cells (MDSCs). Nlrp3 −/− mice had fewer MDSCs accumulating at the tumor site and increased survival rate upon DC vaccination [47]. Increased survival curves were obtained for both metastatic melanoma (B16-F10) and lymphoma (EG7-OVA) tumor cells. In vitro analysis showed that Nlrp3 −/− MDSCs are morphologically and functionally equivalent to the wild type (WT) MDSCs. However, the WT dendritic cells generated significant amount of IL-12. In summary, NLRP3 expression in the presence of IL-12, dampens antitumor immunity and reduces the efficacy of dendritic cell vaccine by excessive infiltration of MDSCs to the tumor site. Both groups focused on different immune cells associated with adaptive immunity and therefore differences in findings may be attributed to difference in tumor cell types, stimuli for NLRP3 inflammasome activation and experimental protocols.
Several studies have implicated increased IL-1β secretion in the tumor microenvironment promoting inflammation, early angiogenic response, tumor induction and progression [48, 49]. In a study utilizing U87, U251 and patient derived-glioblastoma multiforme cell lines, IL-1 acted as a primary signal stimulating IL-1β expression and NLRP3 inflammasome activation. IL-1 induced glioma cells showed activated STAT3 expression, increased angiogenesis and release of neurotoxic substances leading to tumor progression [50]. IL-1β microenvironment significantly increased migration and invasion in the human glioma U87MG and U251MG cell lines [51]. These findings need validation in mouse models and patient data; however, they suggest possible novel strategies to increase the anti-tumor activity of cancer drugs and vaccines by strategic modulation of either inflammasome components or their products, such as IL-1β and IL-18.
In summary, the contrasting role of NLRP3 in cancer, hints at the complexity of function of the NLRP3 inflammasome, its multi-faceted functions and regulation of inflammatory pathways across multiple stages of carcinogenesis.
NLRC4
The Nlrc4 gene encodes an N-terminal CARD domain, a central NACHT domain, and C-terminal leucine-rich repeats containing protein (Table 1). Nlrc4 is predominantly expressed in hematopoietic tissues and cells [52]. Recently reported crystal structure of mouse NLRC4 revealed an autoinhibitory mechanism of NLRC4 wherein the LRR sequesters mNLRC4 in a monomeric state. Disruption in the ADP-mediated NBD-WHD (winged-helix domain) or NBD-HD2/NBD-LRR interactions resulted in constitutive activation of NLRC4 [53].The LRR domain of NLRC4 interacts directly with the chaperone heat-shock protein HSP90, and an ubiquitin ligase-associated protein SGT1 to form an inhibited but receptive oligomeric state. The interaction leads to LRR deletion, resulting in NLRC4 activation [54]. Phosphorylation of NLRC4 at a single, evolutionarily conserved residue, Ser 533, by PKCδ kinase is required for NLRC4 inflammasome assembly formation [55]. NLRC4 inflammasome is activated in response to gram-negative bacteria such as Salmonella typhimurium, Shigella flexneri, Legionella pneumophila and Pseudomonas aeruginosa [56]. Activation in response to intracellular bacteria is critical for caspase-1 mediated pyroptosis as well as maturation and extracellular release of IL-1β and IL-18 [57]. NLRC4 senses cytoplasmic flagellin and bacterial type III secretion apparatus [58]. NLRC4 inflammasome activation in response to S.flexneri requires bacterial type III secretion system and is independent of flagellin [59]. Both NLRP3 and NLRC4 inflammasome activation occur in response to S.typhimurium infection [60]. Inflammasome activation in macrophages infected with S.typhimurium causes recruitment of both NLRC4 and NLRP3 to the same inflammasome complex. ASC is present in the outer ring structure while NLRs and caspases remain internally localized. NLRP3, NLRC4, caspase-1 and caspase-8 are present in the same ASC speck for robust IL-1β production [61]. NLRC4 dependent IL-1β production helps in discriminating pathogenic bacteria from commensal bacteria. Therefore, NLRC4 provides protection against enteric pathogens in intestinal epithelial cells [62].
NLRC4 and cancer
Azoxymethane (AOM)-dextran sodium sulfate (DSS) is a model for inflammation induced colorectal cancer (CAC) [36]. This model has been used for studying function of inflammasomes in colitis and CAC. AOM (a potent carcinogen) and DSS (chronic colitis inducing agent) are systemically administered to induce inflammation-associated colon tumorigenesis in mice. Allen et al. found that Nlrc4 −/− mice showed no significant difference in tumorigenesis as compared to similarly treated wild type (WT) mice. NLRP3 expression and function in hematopoietic cells but not in intestinal epithelial or stromal cells is responsible for protection against increased tumorigenesis [38]. In contrast, Hu et al. showed that the regulation of inflammation-induced tumorigenesis is mediated by NLRC4 and caspase-1 [63]. No difference in inflammation between WT and Casp1 −/− mice was observed during acute DSS-colitis. In the AOM-DSS inflammation-induced colorectal cancer model, Casp1 −/− and Nlrc4 −/− mice exhibited increased tumor load and tumor number per mice. In addition, enhanced colon epithelial and tumor cell proliferation was observed in the Casp1 −/− mice. Caspase-1 and NLRC4 were relatively highly expressed in both colonic epithelial cells and CD45+ hematopoietic cells in the colon. These results conclude that an intrinsic epithelial cell effect exacerbates tumorigenesis in the absence of Caspase-1 or NLRC4 activity. While results from both the groups are conflicting and present contradictory findings, the differences may have arisen from the experiment models, colon tissue and protocols resulting in differences in susceptibility to infection and epithelial integrity. The most significant conclusion from these studies is the protective role of NLRs against increased tumorigenesis in colitis and colitis-associated cancer.
NAIP/BIRC1/NLRB1
NAIPs are a family of NLRs with seven paralogs in mice (NAIP1–7) and one member in humans (hNAIP). The human Naip gene encodes the Neuronal apoptosis and inhibitor protein (NAIP) also termed as BIRC1 baculoviral IAP repeat-containing protein. NAIP consists of 3 domains; an N-terminal BIR domain, a central NACHT domain and a C-terminal LRR domain (Table 1). NAIP lacks the RING zinc finger domain present in other baculovirus and human IAPs. In 1995, Roy et al. first reported partial deletion of Naip gene in patients with spinal muscular atrophy (SMA) [64]. Years later, deletion in Naip gene was positively correlated with the clinical severity of spinal muscular atrophy patients [65]. NAIP protein is expressed in brain, placenta, kidney, spleen, and heart tissues. NAIP expression is highest in the placenta and lowest in the heart. NAIP expression is also found in peripheral blood mononuclear cells while differentiating into macrophages [66]. NAIP mediates suppression of apoptosis and increased cell survival in the Chinese hamster ovary (CHO) cell line after serum deprivation or menadione induced oxidative injury [67]. Naip gene expression inhibits caspase-3 and caspase-7 mediated apoptotic pathways [68]. NAIP directly interacts via its BIR3 domain with procaspase-9. Association of Naip with procaspase-9 prevents the autoproteolysis of caspase-9 in the apoptosome complex, putting a brake to caspase-9-mediated cell death pathways [69]. Integrity of both NOD (nucleotide-binding oligomerization domain) and BIR domains of Naip gene are necessary for effective inhibition of procaspase-9 auto-proteolysis. NAIP also forms NOD–NOD domain interactions with the APAF-1 and BIR3 mediated interaction with procaspase-9 [70]. hNaip (human NAIP) gene is involved in bacterial sensing and inducing pyroptosis in human macrophages and epithelial cells [71]. NLRC4 recognizes the T3SS needle protein with the help of hNAIP and forms hNAIP-NLRC4 inflammasome complex [72].
In the brain, NAIP interaction with hippocalcin mediates protection to neurons from calcium induced cell death via caspase-3-dependent and -independent pathways [73]. Upregulation of Naip expression at 6 h post traumatic brain injury inhibits apoptosis of neurons and oligodendrocytes. While 24 h post-traumatic brain injury, NAIP and procaspase-3 expression is decreased and PARP1 (poly(ADP-ribose) polymerase) cleavage increases, leading to increased apoptosis [74]. NAIP plays critical role in generating innate immune inflammatory response via caspase-1, 4, 5 activation [75].
NAIP and cancer
Cancer triggers molecular events that result in inhibition of apoptotic pathways [76]. Many anti-apoptotic proteins are overexpressed during cancer [77]. Upregulation of these proteins results in increased tumor cell survival and tumor progression. NAIP expression is significantly elevated in malignantly transformed oral squamous cell carcinomas as compared to the corresponding oral potentially malignant disorders. In support of this data, Naip allele was found to be methylated in normal oral mucosa tissues suggesting, Naip expression could be one of the early events in carcinogenesis [78]. Naip is not expressed in normal breast tissue but is expressed at high levels in breast cancer with other unfavorable clinical features such as tumor stage and size [79]. These results offer strong possibility of using NAIP as a biomarker for early diagnosis of squamous cell carcinomas, breast cancer and other malignancies. These studies need to be replicated with large data sets. Mazrouei et al. observed no significant change in NAIP expression in lymphoma tissues and controls. NAIP expression is significantly different in reactive lymphoid hyperplasia (RLH) lymph nodes and normal lymph nodes indicating that Naip function is not restricted to apoptosis regulation but is actively involved in inflammatory responses as well [80]. Overexpression of IAP family members excluding NAIP has been detected in prostate cancer [81]. Later, Chiu et al. [82] observed increased NAIP expression in response to androgen deprivation in prostate cancer. A link between increased NF-kB DNA-binding activity and Naip expression was observed but the mechanism behind NF-kB activation and increased Naip transcription remains unclear. In well differentiated adenocarcinomas younger patients exhibit higher NAIP expression in comparison to elderly patients [83]. Expression was similar for both age-groups in case of normal colonic mucosa. NAIP expression was reported to be significantly more in normal tissues than in well differentiated adenocarcinoma, however, the small sample size and relatively broad age group demand further exploration in this direction. Stronger NAIP expression has been seen in esophageal cancer, though the results have not been statistically significant [84]. This may be related to the number of tissue samples available for the study.
NLRP6
NLRP6 consists of an N-terminal PYRIN domain, a central nucleotide-binding domain and C-terminal leucine-rich repeat as shown in Table 1 [85]. Nlrp6 is highly expressed in the intestine and helps in maintaining intestinal homeostasis by regulating gut microflora [86]. Nlrp6 deficient mice are highly resistant to infection with the bacterial pathogens Listeria monocytogenes, S.typhimurium and Escherichia coli. NLRP6 acts as negative regulator of canonical NF-κB and MAPK-dependent inflammatory signaling. Increased release of canonical NF-κB and MAPK-dependent cytokines and chemokines was found in Nlrp6 −/− mice. Both haematopoietic and non-haematopoietic cells contribute to NLRP6-mediated inhibition of bacterial clearance [87]. NLRP6 inflammasome deficiency leads to altered colonic microbiota and increased risk for colitis occurrence. NLRP6 is recruited to the “specks” formed by ASC oligomerization present in the cytoplasm, leading to procaspase-1 activation and release of active IL-1β and IL-18. Decreased IL-18 levels were present in colonic epithelial cells of Nlrp6 deficient mice, along with characteristics of spontaneous intestinal hyperplasia, inflammatory cell recruitment and exacerbation of dextran sodium sulfate-induced colitis via induction of CCL5 chemokine release. Bacterial components were found to be responsible for transferrable colitis phenotype in the Nlrp6 deficient mice [88] .
NLRP6 and cancer
The association of NLRP6 with colon cancer was established in 2011, when Chen et al. observed that Nlrp6 deficient mice were more susceptible to DSS-induced colitis and colitis associated colon tumorigenesis as compared to wild type controls. Nlrp6 deficient mice were unable to efficiently resolve inflammation and repair damaged tissue, resulting in excessive epithelial tissue proliferation and tumor induction. Nlrp6 function in hematopoietic-derived cells is significantly important in conferring protection against colitis-induced tumorigenesis. IL-18 serum levels are decreased in Nlrp6 deficient mice both before and after DSS-treatment. Although, there is an increase in other pro-inflammatory mediators (MIP2, TNF-α, IL-6, IL-1β, IFN-ϒ) in Nlrp6 deficient mice [89]. NLRP6 controls epithelial self-renewal and colorectal carcinogenesis upon injury (DSS-induced colitis). There is enhanced colitis-associated tumor growth in the Nlrp6 deficient mice. Lack of NLRP6 inflammasome renders injury-induced mice prone to developing relapsing colitis. Taken together, all these results provide evidence for the protective role of NLRP6 in regulation and maintenance of intestinal inflammation [90]. Recently, role of aberrant inflammasome induced microbiota in colorectal cancer development was investigated. In Asc −/− mice, there was enhanced tendency for inflammation induced tumorigenesis, driven by their altered microflora. As expected, microflora transferred from the Nlrp6 −/− mice played critical role in the enhanced susceptibility to inflammation-induced tumorigenesis. Further experimental analysis revealed enhanced tumorigenesis in co-housed WT mice was driven by the microbiota, mediated by IL-18 and CCL5. Colitogenic microflora-induced NLRP6 inflammasome promotes excessive epithelial cell proliferation via IL-6 signaling in transmissible cancer [91]. These studies reveal the critically important role of NLRP6 in maintaining the gut microflora homeostasis (Table 2). Notably, NLRP6 regulates specific host immune response towards different microflora components.
NLRP1
NLRP1 was the first member of the NLR family to be characterized with inflammasome assembly and caspase-1 activation [92]. NLRP1 is a sensor for muramyl dipeptide, Toxoplasma gondii and Bacillus anthracis lethal toxin [93–96]. NLRP1 consists of both Pyrin and CARD domains along with the function to find domain (FIIND domain). Unlike other NLRs, NLRP1 directly recruits procaspase-1 through its CARD domain. The auto-proteolytic processing within the FIIND/ZU5-UPA domains is essential for NLRP1 inflammasome activation [97]. The anti-apoptotic proteins Bcl-2 or Bcl-XL and HHV8 are cellular and viral regulators of NLRP1 that bind and suppress NLRP1 activation [98, 99]. NLRP1 functions include forming inflammasome complex with ASC and caspase-1, binding to the patched receptor via DRAL adaptor protein and regulating caspase-9 mediated apoptosis by binding to Apaf-1 [100, 101]. NLRP1 polymorphisms have been associated with various pathologies like vitiligo, rheumatoid arthritis, Crohn’s disease, Alzheimer’s and melanoma [43, 102, 103]. In vitro analyses show increased NLRP1-mediated caspase-1-dependent pyroptosis in cortical neurons in response to amyloid-β in models of Alzheimer’s disease [104]. Nlrp1 and caspase-1 -deficient mice exhibit significantly reduced neuronal pyroptosis and reversed cognitive impairments. The neuronal NLRP1 inflammasome consists of caspase-1, caspase-11, NLRP1, ASC, the inhibitor of apoptosis protein XIAP, and pannexin1 [105]. Therapeutic neutralization of ASC interferes with NLRP1 inflammasome signaling and significantly reduces caspase-1 activation. Therefore, NLRP1 is an important part of innate immunity in the brain reducing damage caused by post-traumatic brain inflammation. NLRP1 also acts as a cellular sentinel to activate caspase-1 in response to haematopoietic and infectious stress [106]. NLRP1a generates caspase-1-containing inflammasome in vivo, independently of caspase-11 and ASC. The NLRP1 inflammasome formation drives IL-1β-dependent inflammatory disease, which is negatively regulated by IL-18. NLRP1a-induced pyroptosis was observed in the haematopoietic progenitor cells, leading to leucopenia and anemia. Kovarova et al. have shown that NLRP1 inflammasome-induced pyroptosis, mediated by caspase-1 causes acute lung injury and morbidity in mice [107].
NLRP1 and cancer
NLR polymorphisms have been investigated for association with certain cancers. Nlrp1 allele was found to be significantly more frequent in malignant mesothelioma suggesting NLRP1 as a novel factor possibly involved in the development of mesothelioma [108]. Williams et al. have shown significant dysregulation of NLRP1, inflammasome forming NLR in inflammatory bowel disease and colitis-associated cancer [109]. Nlrp1b −/− mice models of experimental colitis were studied using DSS and AOM-DSS-mediated inflammation induced tumorigenesis. NLRP1 inflammasome attenuated gastrointestinal inflammation and progression of colitis-induced tumorigenesis. Enhanced tumorigenesis observed in the Nlrp1b −/− mice is correlated with the attenuated levels of IL-1β and IL-18 in the colon. The Nlrp1b −/− mice exhibited increased cell death, epithelial barrier disruption and disease severity. Bone marrow reconstitution experiments demonstrated association of NLRP1 inflammasome with the non-hematopoietic cells to generate host immune response in case of inflammatory bowel disease and cancer. The regulatory role of NLRP1 in colon cancer is consistent with the view that multiple NLR inflammasomes are involved in maintaining disease pathology and intestinal homeostasis.
AIM2
AIM2 (absent in melanoma 2) was originally identified while screening for tumor suppressor genes associated with melanoma [110]. Aim2-an interferon-inducible gene also known as PYHIN4, is an NLR-like gene encoding cytosolic protein. The AIM2 protein consists of a N-terminal pyrin domain, mediating homotypic interactions with ASC and a C-terminal HIN-200 domain for DNA binding [111]. AIM2 recognizes cytoplasmic DNA and forms specific AIM2/ASC specks, suggesting a novel inflammasome platform inducing activation of ASC-mediated apoptotic and pyroptotic cell death pathways [112–114]. The AIM2 pyrin domain drives filament formation and dsDNA binding, the key signal transduction mechanism responsible for the AIM2 inflammasome formation [115]. Upon activation, AIM2 triggers innate immunity and provides host defense against both bacterial and viral pathogens, such as Francisella tularensis, Listeria monocytogenes and Mycobacterium tuberculosis infection [116–120]. Mitochondrial ROS production, the transcription factor IRF1 and guanylate-binding proteins (GBPs) target AIM2 inflammasome activation by Francisella tularensis infection [121, 122]. Innate immune signaling in response to Porphyromonas gingivalis-induced periodontitis, hemazoin and DNA during malaria involves activation of both NLRP3 and AIM2 inflammasomes and release of IL-1β and IL-18 cytokines and pyroptotic cell death [123, 124]. IFN-inducible IFI16 along with AIM2 acts as innate immune sensor for cytosolic double-stranded DNA in human fibroblasts [125]. Nuclear factor E2-related Factor-2 (Nrf2), a key transcription factor for cellular redox homeostasis regulation was found to be an essential mediator for NLRP3 and AIM2 inflammasome signaling in Myd88-dependent manner [126]. The crystal structure of AIM2 pyrin domain reveals a death domain fold with a short α3 helix that is buttressed by a highly conserved lysine residue at the α2 helix, stabilizing the α3 helix for potential interaction with partner domains. The structural characterization of AIM2 has provided novel insights into the PYD-HIN and PYD–PYD interactions important for AIM2 autoinhibition and inflammasome assembly [127]. The microtubule-associated protein EB1 links AIM2 inflammasome activation and non-classical secretion of IL-1β induced by LC3-dependent autophagy [128].
Recently, the role of AIM2 inflammasome was explored in the brain. AIM2 inflammasome mediated regulation was observed during acute Staphylococcus aureus-induced central nervous system (CNS) inflammation [129]. Distinct survival patterns and susceptibility to infection observed in the Aim2 −/− mice, explains critical role of AIM2 in immune regulation of CNS during bacterial infection. Significantly reduced expression of IL-1β and other key inflammatory mediators, including IL-6, CXCL1, CXCL10, and CCL2 in the CNS of Aim2 −/− and Asc −/− mice, implicates autocrine/paracrine actions of IL-1β in CNS inflammation. Both AIM2 and NLRC4 inflammasomes contribute in acute brain injury independently of NLRP [130]. In fact, the Aim2 −/− and Nlrc4 −/− mice show reduced injury and improved behavioral outcomes. The study suggests novel role of inflammasomes in brain injury, especially AIM2 and NLRC4 as key drivers for sterile inflammation responses in the brain.
AIM2 and cancer
AIM2 plays a protective role against colorectal cancer progression. The lack of AIM2 or its reduced expression was positively correlated with poor survival in colorectal cancer patients [131]. AIM2 decreases the likelihood of colorectal cancer development by tight regulation of tumorigenic susceptibility and intestinal stem cell proliferation independently of inflammasomes [132]. AIM2 interacts with and limits the activation of DNA-dependent protein kinase (DNA-PK), a PI3 K-family member, to reduce Akt phosphorylation for suppression of colon tumorigenesis [133]. Therefore, AIM2 limits tumor burden and cancer progression in an inflammasome-independent fashion. The protective role of AIM2 has been identified in breast and colon cancer. AIM2 suppresses breast cancer cell growth in vitro (MCF-7 cell line) and tumor formation in vivo. Subsequently, AIM2 also suppresses the viability and tumorigenicity of breast cancer cells. In vivo experiments show AIM2-mediated inhibition of breast cancer cell proliferation by suppression of NF-κB transcriptional activity and desensitizing TNF-α-mediated NF-κB activation [134]. Further, in vitro analysis observed restoration of AIM2 induces G2/M arrest causing reduction in cell proliferation and confers invasive phenotype in colon cancer cells [135]. The constitutive levels of interferon-inducible AIM2 mRNA and protein were significantly lowered in prostate cancer cells as compared to the benign prostate hyperplasia cells [136]. The microarray based gene expression profiling of AIM2-responsive target genes suggests novel role of interferon/AIM2/Interferon-stimulated genes cascade in colorectal tumors. Several interferon-stimulated genes including, TLR3 and CIITA (MHC Class II transactivator) were found to be significantly upregulated. AIM2 activation resulted in activation of both IFN-γ independent and dependent interferon stimulated genes. The authors conclude that the recently identified role of AIM2 in inflammasome-mediated cell death was not observed here in colorectal cancer cells [137]. Gene expression profiling by Liu et al. [138] presents both NLRP3 and AIM2 as potential biomarkers for colorectal cancer and cancer progression. Extensive analysis of multiple cancer datasets revealed significantly decreased expression levels of NLRs—NLRP1, NLRP3, NLRC3, NLRC4 and AIM2 in human colorectal cancer. Together, these results link AIM2 with tumor suppression activity, and keeping in mind the already recognized roles of NLRs in tumorigenesis, AIM2 could also function as a double-edged sword.
ASC/TMS1/PYCARD
ASC (apoptosis-associated speck-like protein containing CARD) also known as TMS1 (target of methylation-induced silencing1) is an adaptor molecule that mediates inflammatory and apoptotic signaling. ASC contains the Pyrin/PAAD death domain in addition to the CARD protein–protein interaction domain (Table 1) [139]. Asc gene is predominantly expressed in monocytes and mucosal epithelial cells. The functions of ASC include; regulation of procaspase-1 activation, maturation of the cytokines-IL-1β and IL-18 and activation of NF-kB [140]. Members of NLR family including NLRP1, NLRP3 and NLRC4, and the adaptor protein, ASC are critical components of the inflammasome that results in procaspase-1 activation [23]. Later, inflammasome mediated cell death activity was linked with caspase-8 [57].Co-expression of NLRC4 with ASC induces NF-kB activation and apoptosis mediated by IkB kinase (IKK) and caspase-8, respectively [141]. ASC is central to several death pathways including p53–Bax mitochondrial apoptosis pathway, NF-kB and caspase-8-dependent apoptosis [142]. ASC directs NF-kB activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions [143]. AIM2 and NLRP3 inflammasomes recruit and activate caspase-8 and -1 via ASC leading to both apoptotic and pyroptotic cell death [114]. Though, in vitro studies suggest an axis between ASC/NLRC4/NF-κB activation, these findings require confirmation in vivo.
ASC and cancer
Asc gene is overexpressed in several tumors, triggering apoptosis and formation of ASC specks from its intra-nuclear localization to punctate cytosolic structures. Recent studies have proved methylation-associated silencing of Asc across many cancer types [144]. However, the epigenetic mechanisms underlying regulation of Asc silencing or overexpression remains unknown. The aberrant methylation of Asc and gene silencing was first reported in human breast cancer cells. Asc is inactivated in almost 40 % of the breast cancers [145]. Supporting evidence came from a study reporting Asc methylation in colorectal cancer tissues and cell lines lacking ASC protein expression [146]. Later on, aberrant methylation was also well correlated with loss of ASC expression in ovarian cancer. Methylation-mediated Asc silencing in tumor cells provides an extended survival and escape to apoptosis. In addition to methylation, histone deacetylation is also responsible for Asc gene silencing in ovarian cancer [147]. Machida et al. found hypermethylation of Asc as a marker for late lung cancer cells and in sputum could predict prognosis in patients resected for early-stage disease [148]. Though the ASC protein levels were reduced in all lung cancer types, the hypermethylation particularly correlated with late tumor stages being present in 60 % of late-stage tumors. Methylation of Asc gene promoter is both a frequent and early event in prostate cancer carcinogenesis and is associated with the aggressive prostate cancer [149]. Recent study of Asc methylation in glioblastoma shows reduced or absent expression of ASC in human glioblastoma cell lines and loss in expression was associated with aberrant methylation of CpG Island in the promoter of the Asc gene. Further analysis showed inverse correlation between the degree of methylation and level of ASC expression [150]. Another research group found hypermethylation of Asc gene promoter in 57.1 % of long term survival glioblastoma, distinguishing it from the classic GBM having 16 % of the cases with hypermethylation [151]. Zhang et al. observed epigenetic inactivation of Asc in hepatocellular carcinoma cells. Here, Asc inactivation is regulated by promoter hypermethylation associated gene silencing, accompanied with histone H3 Lysine 9 (H3K9) hypoacetylation and trimethylation modifications in a coordinated way [152]. The dual role of ASC in human melanoma tumorigenesis was identified by Liu et al. ASC expression in metastatic melanoma was found to be down-regulated as compared to primary melanoma [153]. As expected, silencing of Asc in metastatic melanoma resulted in reduced cell viability and suppressed tumorigenesis. Asc knockdown resulted in inhibition of caspase-1 activation and IL-1β secretion in both primary and metastatic melanoma. It also resulted in activation/suppression of phosphorylated IkB kinase (IKK) α/β expression and NF-kB activity in metastatic/primary melanoma. Asc was also found to be highly expressed in mouse models of medulloblastoma. Asc deficiency profoundly reduced proliferation and extended survival rate. ASC plays the role of tumor promoter in medulloblastoma [154]. Interestingly, ASC has always been identified as a tumor suppressor in specific cancer models. These results reveal complex role played by ASC in regulating cell proliferation. Based on these findings, ASC represents a potential modulator of inflammatory responses that may help in coordinating the activity of NLRs and cytokine activating caspases in mammalian cells. DNA hypermethylated gene promoter sequences have always been extremely promising cancer markers. Certainly, they can be used for early diagnosis or prognosis depending on the change in gene expression during tumor induction and progression.
Non-inflammasome forming NLRs
Non-inflammasome forming NLRs, such as NLRX1, NLRP12, NLRC3, NOD1 and NOD2 form a significantly important subgroup of the NLR family that can both positively and negatively regulate inflammation. These NLRs do not form an inflammatory complex upon activation but regulate inflammation associated pathways by other mechanisms [155]. Non-inflammasome forming NLRs modulate NF-kB and other major inflammation regulatory pathways, which are crucial in chronic inflammation and inflammation-induced tumorigenesis [156]. NLRs modulate these pathways through interaction with a specific upstream/downstream molecule belonging to that pathway. Their known functions reveal the fact that dysregulation in NF-kB and other inflammation associated signaling pathways by these NLRs are very important and play critical role in tumor induction and progression (Table 2).
NLRP12
NLRP12 also known as Monarch-I and PYPAF7, belongs to the non-inflammasome forming subgroup of NLRs [157]. It is one of the first NLR proteins to be studied. It has a tripartite domain structure, with an N-terminal PYRIN domain, a central nucleotide binding site domain, and a C-terminal domain composed of at least 12 leucine-rich repeat motifs as shown in Table 1 [158]. In humans, Nlrp12 is expressed in peripheral blood leukocytes, including granulocytes, eosinophils, monocytes, and dendritic cells (DCs) [159]. Recent in vitro research studies have shown NLRP12 negatively regulates inflammation by attenuating both canonical (via interaction with IRAK1) and non-canonical (via interaction with TRAF3 and NF-kB inducing kinase (NIK)) NF-kB pathway [160–162]. NLRP12 also down-regulates MAPK pathway by inhibiting ERK signaling [163]. S. typhimurium induces NLRP12-mediated dampening of host immune defenses to persist and survive inside the host. During salmonellosis, NLRP12 inhibits NF-κB and ERK activation by suppressing phosphorylation of IκBα and ERK, preventing efficient clearance of bacterial burden [164].
NLRP12 and cancer
Zaki et al. first observed that Nlrp12 deficient mice are hyper-susceptible to DSS-induced colitis and have increased colitis-associated colorectal tumorigenesis. NLRP12 plays a critical role in dampening the inflammatory response in myeloid cells and during DSS-induced colitis. The expression of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α, IL-17, IL-15 and chemokines (G-CSF, eotaxin, KC, IP-10, MIP-1α, MIP-1β, MIP2) were significantly higher in the colon of Nlrp12 −/− mice, leading to hyperplasia and increased tumorigenesis. Therefore, Nlrp12 deficiency leads to increased and prolonged inflammatory responses in colon tissue. Significantly higher activation levels of MAPK, NF-κB, STAT and AKT were observed, accompanied with massive infiltration of macrophages and increased proliferation of epithelial cells particularly in the hyperplastic colon regions of the AOM/DSS treated Nlrp12 −/− colon tissue [165]. Collectively, these results suggest NLRP12 signaling in the hematopoietic cells is critical for protection against colitis and CAC. Allen et al. confirmed increased susceptibility of Nlrp12 −/− mice towards inflammation-driven colon tumorigenesis. The ex vivo contribution of NLRP12 is reflected in terms of enhanced non-canonical NF-kB signaling and MAPK activation, observed in the myeloid dendritic cells isolated from the Nlrp12 −/− mice. NLRP12 functionally interacts with TRAF3, which in turn directly interacts with NIK, inducing NIK degradation. The pathogenesis of Nlrp12 −/− mice was found to be derived from both the hematopoietic and non-hematopoietic compartments. Further experimental analysis, suggested an early role for NLRP12 in the AOM + DSS model through both hematopoietic and non-hematopoietic compartments. Coincidental with the polyp formation, the effect of NLRP12 was derived primarily from the non-hematopoietic compartment [163]. In summary, the study indicates how NLRP12 attenuates the development of experimental colitis and suppresses colitis-associated cancer. These studies strongly suggest that functions of NLRP12 might be cell/tissue-specific for specific stage of inflammation/tumorigenesis. Both ex vivo and in vivo studies have proved a critical role played by NLRP12 in regulation of major signaling pathways associated with inflammation and inflammation-associated tumorigenesis.
NLRX1
NLRX1 is the first non-cytoplasmic NLR protein discovered. NLRX1 is localized in mitochondria however; the exact localization within mitochondria remains to be characterized [166, 167]. Its structure constitutes a highly conserved nucleotide-binding domain (NBD) and leucine-rich repeats (LRR) as seen in Table 1. Nlrx1 expression is highest in mitochondria-rich tissues such as muscle and heart. Major functions of Nlrx1 includes negative regulation of anti-viral inflammatory response via MAVS-RIG1 signaling pathway or TLR-induced NF-kB signaling by targeting TRAF6 and IKK signaling pathway [168, 169]. NLRX1 inhibits NF-kB activation by inhibition of IKKα and IKKβ phosphorylation. These results show specific knockdown of Nlrx1 resulted in increased gene expression of cytokines; TNF-α and IL-6 and chemokines; CCL2 and CXCL10 in response to LPS treatment [169]. NLRX1 also exerts positive control of NF-kB and JNK signaling pathway for active production of reactive oxygen species (ROS) in response to TNF-α, poly (I:C) and pathogens [170]. NLRX1 binds to the ssRNA, dsRNA, poly (I:C) but not with DNA [171]. A study by Soares et al. [172] discovered that NLRX1 is not involved in inhibition of MAVS-dependent anti-viral signaling. NLRX1 interacts with a mitochondrial matrix protein, UQCRC2 to induce ROS production but exact association between the two is yet to be shown. NLRX1 also forms complex with mitochondrial Tu translation elongation factor protein, TUFM, that dually regulates IFN-ϒ production and promotes autophagy during viral infection [173]. Recently, crystal structure of NLRX1 C-terminal was elucidated, describing association of LRR and neighboring helical domains to form a hexameric platform which promotes the interaction/binding of NLRX1 with its target proteins. It’s only recently that researchers have started exploring NLRX1, therefore, its major regulatory functions, RNA recognition by NLRX1 and NLRX1 –RNA interaction mechanism are still under investigation.
NLRX1 and cancer
NLRX1 plays an important role in regulating the balance between intrinsic and extrinsic apoptosis in cancer cells. NLRX1 positively regulates apoptosis in response to intrinsic apoptosis signals and that may be this is why Nlrx1 expression is down regulated in cancer cells. Nlrx1 −/− mice develop fewer tumors than wild type mice in the AOM-induced colorectal cancer murine model. Nlrx1 deficiency reduced cancer progression in this cancer model. In contrast, in a AOM/DSS treated colitis-associated cancer model, Nlrx1 −/− mice developed a more severe pathology, showing increased sensitivity to DSS colitis. NLRX1 protects against DSS-induced damage and AOM/DSS-triggered colorectal cancer. This differential role of NLRX1 might be consequence of an exacerbated rate of apoptotic cell death in response to DSS treatment followed by increased epithelial proliferation as part of repair of damaged tissue [174]. Recent data indicates NLRX1 attenuates tumorigenesis through the negative regulation of AKT and NF-κB signaling [168]. NLRX1 sensitizes cells to TNF-α induced cell death by activating caspase-8. In vitro studies suggest that NLRX1 expression suppresses clonogenic ability, anchorage-independent growth and migration of cancer cells. Interestingly, NLRX1 may also contribute to the metabolic switch toward glycolysis in these tumor cells. Study extended to in vivo models has shown suppression of tumorigenicity in nude mice by NLRX1 [175]. Given the current understanding of NLRX1 and its role in innate immune responses, NLRX1 might serve as a promising target for manipulating immune response in inflammation-associated diseases and cancer pathology.
NOD1 and NOD2
NOD1 and NOD2 (nucleotide-binding oligomerization domain-containing protein 1 and 2, also known as NLRC1 and NLRC2, respectively) were the first NLRs to be characterized. The NOD-proteins are amongst the most prominent members of the NLR (NOD-LRR) family of proteins that contain nucleotide-binding NACHT domains, receptor-like LRR domains and CARD domains. NOD1 contains a single amino-terminal caspase recruitment domain (CARD), and NOD2 contains tandem N-terminal CARD domains that mediate interactions with the CARD domains of receptor-interacting protein 2 (RIP2) [176]. While Nod1 is ubiquitously expressed, Nod2 is expressed more in hematopoietic cells [177]. Several E3 ubiquitin ligases have been suggested to be involved in the activation of NOD1 and NOD2 signaling, including cellular inhibitor of apoptosis protein 1 (CIAP1), CIAP2, and TRAF6 [178]. Interestingly, both NOD1 and NOD2 can be recruited to the plasma membrane during infection and ligand recognition. NOD2 promotes the membrane recruitment of RICK, a serine-threonine kinase involved in NF-κB activation downstream of NOD2 [179]. Ubiquitin is also able to bind to the caspase recruitment domains of NOD1 and NOD2, competing with RIP2 for binding and reducing activation. Furthermore, polyubiquitylated RIP2 is a substrate for the deubiquitylating enzyme, A20, which negatively regulates NOD2 signaling.
Nod1 and Nod2 recognize muropeptides derived from cell walls of Gram-positive and Gram-negative bacteria, such as Listeria monocytogenes and Shigella flexneri. Nod1 and Nod2 are important for microbial recognition and host defense after TLR stimulation [180]. NOD1 mediates selective recognition of bacteria through detection of iE-DAP-containing peptidoglycan [181]. NOD1-dependent responses play a critical role in host resistance against Trypanosoma cruzi, Pseudomonas aeruginosa, and Helicobacter pylori infections [182–184]. NOD1 also participates in the induction of a noninfectious pancreatitis via its response to commensal organisms [185]. NOD1 and NOD2 proteins contribute to the maintenance of mucosal homeostasis and the induction of mucosal inflammation [186]. Nod2 acts as an intracellular receptor for sensing of muramyl dipeptide [187]. Card8 negatively regulates Nod2-mediated signaling, displaying a novel molecular switch involved in the endogenous regulation of Nod2-dependent inflammatory processes [188]. The molecular chaperone protein, HSP70 binds to and stabilizes NOD2 mutants, providing an effective therapy for Crohn’s disease [189].
NOD1 and NOD2 in cancer
The activation of NOD1 and NOD2 triggers recruitment of the adaptor proteins RICK and CARD9, resulting in K63-linked ubiquitylation and activation of NF-κB and the MAP kinase signaling cascades [190]. Critical role of NOD1 signaling in maintaining intestinal epithelial barrier permeability and balancing inflammatory responses has been shown using AOM/DSS-induced colon-associated tumorigenesis [191]. NOD1, RIP2 and Caspase12 have been described as potentially novel biomarkers for oral squamous cell carcinoma development and progression [192].
The role of NOD2 is being studied extensively in colitis and colorectal tumorigenesis because of its well-recognized genetic association with inflammatory bowel disease. Nod2 mutations and polymorphisms have been linked with increased to colorectal cancer [193]. In addition to its well-characterized role in NF-κB and MAPK activation, NOD2 is also involved in the regulation of autophagy, which plays a crucial role in intestinal homeostasis and tumorigenesis. Nod2 deficiency-induced dysbiosis, gives rise to a reversible transmissible colitis and colitis-associated carcinogenesis in mice. Results show that Nod2/Rip2 deficiency confers a maternally transmissible colitis risk on immunocompetent host [194]. Additionally, fecal dysbiosis in Nod2-deficient mice also sensitized the colonic mucosa to DSS-induced chemical injury. Researchers have shown that the NOD2 3020insC mutation may be a genetic predisposing factor for breast, lung cancer and colorectal cancer [195]. Nod1/Nlrc1 and Nod2/Nlrc2 gene polymorphisms have been identified in cancer etiology. Several Nod1/Nod2 polymorphisms may be associated with altered risk of gastric, colorectal, breast, ovarian, prostate, testicular, lung, skin cancer and various other cancers [196].
Current cancer therapeutics
Cancer cells overcome the host innate immune surveillance system to promote tumorigenesis and constitute a tumor-promoting microenvironment. The inflammatory tumor microenvironment plays major role in cancer development and progression as depicted in Fig. 1. The microenvironment consists of increased expression of pro-inflammatory cytokines, chemokines and other major inflammatory molecules facilitating interaction between normal and cancer cells. Given the major role played by cytokines during chronic inflammation, tumor induction and tumor development, cytokines became a common target of therapeutics for auto-inflammatory diseases. IL-1 dysregulation has been reported in several auto-inflammatory diseases [197]. Therefore, IL-1 signaling pathway inhibitors are being used for effective treatment of cold induced periodic syndromes including NOMID, MWS, and FCAS syndromes associated with various Nlrp3 mutations. The first such IL-1 inhibitor was anakinra followed by rilonacept and canakinumab [198]. The direct blockade of important immunoregulators like IL-1β compromises the host immune response to infectious agents or damage-associated molecules and limits the anticancer immune response elicited by immunogenic cell death inducers. Apart from IL-1β, IL-18 signaling can also be blocked by neutralizing antibodies or recombinant IL-18BP (Interleukin-18 binding protein). The inhibition of inflammasomes or therapeutic neutralization of their products would exert profound effects on carcinogenesis and tumor progression [199].
Over the years, multiple therapeutic strategies have been utilized for targeting cancer cells. Recently, liposomes packaged siRNA targeting the Pkn3 gene (protein kinase N3) for lung metastasis inhibition has been approved for evaluation in a human clinical trial [200]. siRNAs are potent activators of innate immunity but do have some major off-target effects on gene expression, antibody response against delivery vehicle and immune-toxicities due to excessive cytokine release. Re-expressing tumor-suppressive miRNAs holds great promise for cancer therapy. It is well accepted that aberrant miRNA expression is linked to cancer, hence delivering tumor–suppressive miRNAs and silencing oncogenic miRNAs has been successful in various mouse models [11]. Reprogrammed viruses have also been utilized as oncolytic vectors for developing virus based therapeutics for cancer. Currently, DNA viruses in clinical trials include adenovirus (AD), herpes simplex virus 1 (HSV1) and vaccinia virus. The selection of HSV to treat glioblastoma utilizing this approach gives great hope [201]. The only engineered oncolytic RNA virus undergoing clinical trials is the measles virus. Oncolytic viruses hold great potential as self-amplifying cancer therapeutics and can be applied in combination with radiation and chemotherapy. Desirable protease targets (for example-matrix metalloproteinases) for oncolytic viruses are either expressed preferentially or at high levels by cancer cells. In fact, Ad H101 is the first reprogrammed virus to be approved as a cancer drug and has been administered to hundreds of patients with head and neck carcinoma in China [202]. Today, peptide-vaccination targeting the epidermal growth factor receptor (EGFR) mutation III (vIII) is one of the most prominent examples of immunotherapy for glioblastoma [203]. Treatment of glioblastoma patients with cediranib, pan-VEGF receptor tyrosine kinase inhibitor resulted in increased tumor perfusion, and improved survival rate because of the vascular normalization [204].
The pivotal role of IKKβ/NF-κB signaling pathway in inhibition of programmed cell death and constitutive expression in various malignancies, strongly suggest the use of IKKβ and/or NF-κB inhibitors for cancer treatment. Currently available drugs are non-specific Ikkβ or NF-kB inhibitors inclusive of anti-inflammatory agents, NSAIDs, cyclopentenone prostaglandins, proteasome inhibitors, and glucocorticoids. IKKβ/NF-κB inhibitors block NF-κB activation in infiltrating inflammatory cells, which are an important source of tumor growth and survival factors [205]. Targeting specific NF-kB-regulated ligands for regulating particular gene expression makes this a promising approach for cancer therapy.
The critical role played by NLRs in major inflammatory pathways associated with cancer has provided a new direction to cancer therapeutics [21]. Recently, the crystal structures for NLR domains such as pyrin, CARD, NBD have been elucidated. The focus is on developing drugs that directly interact with the NLR domains for selective activation or inhibition of a NLR. Distinct NLR ligands may be used as vaccine adjuvants for enhancement of innate immune response. Depending on the particular cancer and its stage, inflammasome activation or inhibition can be utilized as cancer therapeutics. The emerging role of inflammasomes in host innate immune responses suggests inflammasome components as direct drug targets for cancer. Opsona Therapeutics recently published that the cytokine release inhibitory drug 3 (CRID3, also known as CP-456,773) targets ASC oligomerization during NLRP3 and AIM2 inflammasome activation (Coll and O’Neill, 2011) [206]. Anticancer chemotherapeutics are particularly efficient when they succeed in killing tumor cells through immunogenic cell death, and thereby converting dying cells into therapeutic vaccines [207]. Earlier, immunomodulators such as thalidomide (anti-inflammatory and anti-angiogenic) were used that may exert inhibitory effects on the NLPR3 inflammasome but also have strong teratogenic activity. The anti-inflammatory activity of thalidomide is mediated via caspase-1 in mice. Thalidomide has been approved for the treatment of inflammatory skin diseases and cancer [206]. NLRs present promising option for novel therapeutics targets for transformation of the tumor-promoting microenvironment into an anti-tumor microenvironment (Fig. 3).

Possible therapeutic targets. NLR dependent pathways may present opportunities as novel therapeutics targets for transformation of the tumor-promoting microenvironment into an anti-tumor microenvironment
Future directions
Inflammasomes display contrasting roles across multiple stages of tumorigenesis. Even though NLRs play important roles in innate immunity and inflammation, information related to NLR-ligand interactions, inflammasome activation and NLR-associated intracellular and extracellular signaling pathways remain largely unknown. Further insights into the upstream and downstream signaling molecules participating in NLR-associated signaling are needed. Hence, it is important to unravel the molecular mechanisms responsible for activation of anti-tumor or tumor-promoting inflammasome-associated pathways. Keeping in view, the future prospects of NLRs, inflammasomes and their products as cancer biomarkers some questions remain unanswered.
-
While the association of NLRs and major inflammatory pathways with cancer has opened up a new field of cancer therapeutics, the regulatory mechanisms of NLR dependent signaling pathways remain unclear. These regulatory molecules might serve as valuable biomarkers as well as highly specific drug targets to modulate an NLR associated pathway.
-
The molecular mechanism behind NLRs affecting the activation/inhibition of other NLRs remains largely unknown.
-
The regulatory molecules anchoring ligands towards the cytoplasmic NLRs and the feedback loops responsible for balanced activation of the NLR inflammatory pathways need further investigation.
References
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444
Philip M, Rowley DA, Schreiber H (2004) Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol 14:433–439
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30:1073–1081
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H (2012) The brain tumor microenvironment. Glia 60:502–514
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA (2013) Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13:759–771
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21:309–322
Kasinski AL, Slack FJ (2011) MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer 11:849–864
Lin W-W, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Investig 117:1175
Porta C, Larghi P, Rimoldi M, Totaro MG, Allavena P, Mantovani A, Sica A (2009) Cellular and molecular pathways linking inflammation and cancer. Immunobiology 214:761–777
Ting JPY, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot J-P, Inohara N, MacKenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V, Ward PA (2008) The NLR gene family: a standard nomenclature. Immunity 28:285–287
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
DeYoung BJ, Innes RW (2006) Plant NBS-LRR proteins in pathogen sensing and host defense. Nat Immunol 7:1243–1249
Ting JPY, Davis BK (2005) CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol 23:387–414
Ye Z, Ting JP-Y (2008) NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr Opin Immunol 20:3–9
Kufer TA, Sansonetti PJ (2011) NLR functions beyond pathogen recognition. Nat Immunol 12:121–128
Zhong Y, Kinio A, Saleh M (2013) Functions of NOD-like receptors in human diseases. Front Immunol 4:333
Kent A, Blander JM (2014) Nod-like receptors: key molecular switches in the conundrum of cancer. Front Immunol 5:185
Schroder K, Tschopp J (2010) The Inflammasomes. Cell 140:821–832
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241–247
Srinivasula SM (2002) The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 277:21119–21122
Keller M, Ruegg A, Werner S, Beer HD (2008) Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818–831
Feldmann J, Prieur A-M, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, Basile GDS (2002) Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 71:198–203
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29:301–305
Conforti-Andreoni C, Ricciardi-Castagnoli P, Mortellaro A (2011) The inflammasomes in health and disease: from genetics to molecular mechanisms of autoinflammation and beyond. Cell Mol Immunol 8:135–145
Verma D, Lerm M, Blomgran-Julinder R, Eriksson P, Soderkvist P, Sarndahl E (2008) Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases? Arthritis Rheum 58:888–894
Paramel GV, Sirsjö A, Fransén K (2015) Role of genetic alterations in theNLRP3andCARD8Genes in health and disease. Mediators Inflamm 2015:1–10
De Nardo D, Latz E (2011) NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 32:373–379
Ito S, Hara Y, Kubota T (2014) CARD8 is a negative regulator for NLRP3 inflammasome, but mutant NLRP3 in cryopyrin-associated periodic syndromes escapes the restriction. Arthritis Res Ther 16:R52
Fransén K (2012) Role of NLRP3 and CARD8 in the regulation of TNF-α induced IL-1β release in vascular smooth muscle cells. Int J Mol Med 30:697–702
Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, Mocarski ES, Dubyak GR (2015) Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J Biol Chem 290:20167–20184
Bruchard M, Rebé C, Derangère V, Togbé D, Ryffel B, Boidot R, Humblin E, Hamman A, Chalmin F, Berger H, Chevriaux A, Limagne E, Apetoh L, Végran F, Ghiringhelli F (2015) The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol 16:859–870
Wirtz S, Neufert C, Weigmann B, Neurath MF (2007) Chemically induced mouse models of intestinal inflammation. Nat Protoc 2:541–546
Chassaing B, Aitken JD, Malleshappa M, Vijay‐Kumar M (2014) Dextran sulfate sodium (DSS)‐induced colitis in mice. Curr Protoc Immunol 15.25. 11–15.25. 14
Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207:1045–1056
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32:379–391
Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD (2010) IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 185:4912–4920
Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA (2010) Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA 107:21635–21640
Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, Corr SC, Kay EW, O’Neill LA, Mills KH (2015) Protective role for caspase-11 during acute experimental murine colitis. J Immunol 194:1252–1260
Verma D, Bivik C, Farahani E, Synnerstad I, Fredrikson M, Enerbäck C, Rosdahl I, Söderkvist P (2012) Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res 25:506–513
Xu Y, Li H, Chen W, Yao X, Xing Y, Wang X, Zhong J, Meng G (2013) Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLoS ONE 8:e77955
Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, Zhao W, Huai W, Guo P, Han L (2013) Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest 94:52–62
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini J-L, Schlemmer F, Tasdemir E, Uhl M, Génin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, André F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat Med 15:1170–1178
van Deventer HW, Burgents JE, Wu QP, Woodford RMT, Brickey WJ, Allen IC, McElvania-Tekippe E, Serody JS, Ting JPY (2010) The inflammasome component Nlrp3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res 70:10161–10169
Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, Abutbul S, Huszar M, Dinarello CA, Apte RN, Voronov E (2013) The role of IL-1 in the early tumor cell-induced angiogenic response. J Immunol 190:3500–3509
Tarassishin L, Lim J, Weatherly DB, Angeletti RH, Lee SC (2014) Interleukin-1-induced changes in the glioblastoma secretome suggest its role in tumor progression. J Proteom 99:152–168
Tarassishin L, Casper D, Lee SC (2014) Aberrant expression of interleukin-1β and inflammasome activation in human malignant gliomas. PLoS ONE 9:e103432
Fathima K, Hurmath P, Ramaswamy DN (2014) Nandakumar, IL-1β microenvironment promotes proliferation, migration, and invasion of human glioma cells. Cell Biol Int 38:1415–1422
Poyet JL (2001) Identification of Ipaf, a human caspase-1-activating protein related to apaf-1. J Biol Chem 276:28309–28313
Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, Wang R, Zhang Y, Martinon F, Miao D (2013) Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341:172–175
Mayor A, Martinon F, De Smedt T, Pétrilli V, Tschopp J (2007) A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol 8:497–503
Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Kömüves L, Cupp JE, Arnott D, Monack D, Dixit VM (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490:539–542
Lage SL, Longo C, Branco LM, da Costa TsB, Buzzo CdL, Bortoluci KR (2014) Emerging concepts about NAIP/NLRC4 inflammasomes. Front Immunol 5:309
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11:1136–1142
Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, Liu L, Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600
Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nuñez G (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in shigella-infected macrophages. PLoS Pathog 3:e111
Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207:1745–1755
Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, Bryant CE (2014) Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci 111:7403–7408
Nordlander S, Pott J, Maloy KJ (2013) NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol 7(4):775–785
Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA (2010) Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci 107:21635–21640
Roy N, Mahadevan MS, McLean M, Shutter G, Yaraghi Z, Farahani R, Baird S, Besner-Johnston A, Lefebvre C, Kang X, Salih M, Aubry H, Tamai K, Guan X, Ioannou P, Crawford TO, de Jong PJ, Surh L, Ikeda J-E, Korneluk RG, MacKenzie A (1995) The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 80:167–178
Watihayati MS, Fatemeh H, Marini M, Atif AB, Zahiruddin WM, Sasongko TH, Tang TH, Zabidi-Hussin Z, Nishio H, Zilfalil BA (2009) Combination of SMN2 copy number and NAIP deletion predicts disease severity in spinal muscular atrophy. Brain Dev 31:42–45
Maier JKX, Balabanian S, Coffill CR, Stewart A, Pelletier L, Franks DJ, Gendron NH, MacKenzie AE (2007) Distribution of neuronal apoptosis inhibitory protein in human tissues. J Histochem Cytochem 55:911–923
Listen P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda J-E, Mackenzie A, Korneluk RG (1996) Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379:349–353
Maier JK, Lahoua Z, Gendron NH, Fetni R, Johnston A, Davoodi J, Rasper D, Roy S, Slack RS, Nicholson DW (2002) The neuronal apoptosis inhibitory protein is a direct inhibitor of caspases 3 and 7. J Neurosci 22:2035–2043
Davoodi J, Ghahremani M-H, Es-haghi A, Mohammad-gholi A, MacKenzie A (2010) Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int J Biochem Cell Biol 42:958–964
Karimpour S, Davoodi J, Ghahremani M-H (2011) Integrity of ATP binding site is essential for effective inhibition of the intrinsic apoptosis pathway by NAIP. Biochem Biophys Res Commun 407:158–162
Vinzing M, Eitel J, Lippmann J, Hocke AC, Zahlten J, Slevogt H, N’Guessan PD, Gunther S, Schmeck B, Hippenstiel S, Flieger A, Suttorp N, Opitz B (2008) NAIP and Ipaf control Legionella pneumophila replication in human cells. J Immunol 180:6808–6815
Yang J, Zhao Y, Shi J, Shao F (2013) Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci 110:14408–14413
Mercer EA, Korhonen L, Skoglösa Y, Olsson P-A, Kukkonen JP, Lindholm D (2000) NAIP interacts with hippocalcin and protects neurons against calcium-induced cell death through caspase-3-dependent and -independent pathways. EMBO J 19:3597–3607
Hutchison JS, Derrane RE, Johnston DL, Gendron N, Barnes D, Fliss H, King WJ, Rasquinha I, MacManus J, Robertson GS, MacKenzie AE (2001) Neuronal apoptosis inhibitory protein expression after traumatic brain injury in the mouse. J Neurotrauma 18:1333–1347
Martinon F, Tschopp J (2006) Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ 14:10–22
Bai L, Wang S (2014) Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med 65:139–155
Evan GI, Vousden KH (2001) Proliferation, cell cycle and apoptosis in cancer. Nature 411:342–348
Chen Y-K, Huse S-S, Lin L-M (2010) Expression of inhibitor of apoptosis family proteins in human oral squamous cell carcinogenesis. Head Neck 33:985–998
Choi J, Hwang YK, Choi YJ, Yoo KE, Kim JH, Nam SJ, Yang JH, Lee SJ, Yoo KH, Sung KW, Koo HH, Im Y-H (2007) Neuronal apoptosis inhibitory protein is overexpressed in patients with unfavorable prognostic factors in breast cancer. J Korean Med Sci 22:S17
Mazrouei S, Ziaei A, Tanhaee A, Keyhanian K, Esmaeili M, Baradaran A, Salehi M (2012) Apoptosis inhibition or inflammation: the role of NAIP protein expression in Hodgkin and non-Hodgkin lymphomas compared to non-neoplastic lymph node. J Inflamm 9:4
Krajewska M, Krajewski S, Banares S, Huang X, Turner B, Bubendorf L, Kallioniemi O-P, Shabaik A, Vitiello A, Peehl D (2003) Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin Cancer Res 9:4914–4925
Chiu HHL, Yong TMK, Wang J, Wang Y, Vessella RL, Ueda T, Wang Y-Z, Sadar MD (2010) Induction of neuronal apoptosis inhibitory protein expression in response to androgen deprivation in prostate cancer. Cancer Lett 292:176–185
Endo T, Abe S, Seidlar HBK, Nagaoka S, Takemura T, Utsuyama M, Kitagawa M, Hirokawa K (2004) Expression of IAP family proteins in colon cancers from patients with different age groups. Cancer Immunol Immunother 53:770–776
Nemoto T, Kitagawa M, Hasegawa M, Ikeda S, Akashi T, Takizawa T, Hirokawa K, Koike M (2004) Expression of IAP family proteins in esophageal cancer. Exp Mol Pathol 76:253–259
Grenier JM, Wang L, Manji GA, Huang W-J, Al-Garawi A, Kelly R, Carlson A, Merriam S, Lora JM, Briskin M, DiStefano PS, Bertin J (2002) Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-κB and caspase-1. FEBS Lett 530:73–78
Chen GY (2014) Role of Nlrp6 and Nlrp12 in the maintenance of intestinal homeostasis. Eur J Immunol 44:321–327
Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti T-D (2012) NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488:389–393
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145(2011):745–757
Chen GY, Liu M, Wang F, Bertin J, Nunez G (2011) A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186:7187–7194
Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M (2011) Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci 108:9601–9606
Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, Jin C, Wunderlich C, Wunderlich T, Eisenbarth SC, Flavell RA (2013) Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci 110:9862–9867
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10:417–426
Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, Karin M (2008) A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci USA 105:7803–7808
Ewald SE, Chavarria-Smith J, Boothroyd JC (2014) NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun 82:460–468
Chavarría-Smith J, Vance RE (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9(6):e1003452
Chavarría-Smith J, Vance RE (2015) The NLRP1 inflammasomes. Immunol Rev 265:22–34
Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin JJ, Gough PJ (2012) Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem 287:25030–25037
Bruey J-M, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A (2007) Bcl-2 and Bcl-X L regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129:45–56
Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S-I, Reed JC, Ting JP, Damania B (2011) Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331:330–334
Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25:713–724
Mille F, Thibert C, Fombonne J, Rama N, Guix C, Hayashi H, Corset V, Reed JC, Mehlen P (2009) The Patched dependence receptor triggers apoptosis through a DRAL–caspase-9 complex. Nat Cell Biol 11:739–746
Pontillo A, Catamo E, Arosio B, Mari D, Crovella S (2012) NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord 26:277–281
Sui J, Li H, Fang Y, Liu Y, Li M, Zhong B, Yang F, Zou Q, Wu Y (2012) NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in Han Chinese. Arthritis Rheum 64:647–654
Tan M, Tan L, Jiang T, Zhu X, Wang H, Jia C, Yu J (2014) Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis 5:e1382
de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29(7):1251–1261
Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ (2012) NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37:1009–1023
Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP, Koller BH (2012) NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 189:2006–2016
Girardelli M, Maestri I, Rinaldi RR, Tognon M, Boldorini R, Bovenzi M, Crovella S, Comar M (2012) NLRP1 polymorphisms in patients with asbestos-associated mesothelioma. Infect Agent Cancer 7:25
Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, Heid B, Cecere TE, Allen IC (2015) The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol 194:3369–3380
DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA, Meltzer PS, Trent JM (1997) Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 15:453–457
Davis BK, Wen H, Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518
Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, Miao EA, Aderem A (2010) Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol 185:818–821
Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ (2013) AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20:1149–1160
Morrone SR, Matyszewski M, Yu X, Delannoy M, Egelman EH, Sohn J (2015) Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat Commun 6:7827
Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11:395–402
Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, Takeda K (2012) Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol 24:637–644
Sauer J-D, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA (2010) Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7:412–419
Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11:385–393
Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, Henry T (2012) AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ 19:1709–1721
Crane DD, Bauler TJ, Wehrly TD, Bosio CM (2014) Mitochondrial ROS potentiates indirect activation of the AIM2 inflammasome. Front Microbiol 5:438
Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, Yamamoto M, Lamkanfi M, Kanneganti T-D (2015) The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16:467–475
Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, Fitzgerald KA (2014) Dual Engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during Malaria. Cell Rep 6:196–210
Park E et al (2014) Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect Immun 82:112–123
Duan X, Ponomareva L, Veeranki S, Panchanathan R, Dickerson E, Choubey D (2011) Differential Roles for the interferon-inducible IFI16 and AIM2 innate immune sensors for cytosolic DNA in cellular senescence of human fibroblasts. Mol Cancer Res 9(5):589–602
Zhao C, Gillette DD, Li X, Zhang Z, Wen H (2014) Nuclear Factor E2-related Factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J Biol Chem 289:17020–17029
Jin T, Perry A, Smith P, Jiang J, Xiao TS (2013) Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem 288:13225–13235
Wang L-J, Huang H-Y, Huang M-P, Liou W, Chang Y-T, Wu C-C, Ojcius DM, Chang Y-S (2014) The microtubule-associated protein EB1 links AIM2 inflammasomes with autophagy-dependent secretion. J Biol Chem 289:29322–29333
Hanamsagar R, Aldrich A, Kielian T (2014) Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J Neurochem 129:704–711
Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J, Rothwell N, Allan SM, Brough D (2015) AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc Natl Acad Sci 112:4050–4055
Dihlmann S, Tao S, Echterdiek F, Herpel E, Jansen L, Chang-Claude J, Brenner H, Hoffmeister M, Kloor M (2014) Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int J Cancer 135:2387–2396
Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, Sharma D, Li L, Malireddi RKS, Gurung P, Neale G, Olsen SR, Carter RA, McGoldrick DJ, Wu G, Finkelstein D, Vogel P, Gilbertson RJ, Kanneganti T-D (2015) Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 162:45–58
Wilson JE, Petrucelli AS, Chen L, Koblansky AA, Truax AD, Oyama Y, Rogers AB, Brickey WJ, Wang Y, Schneider M, Mühlbauer M, Chou W-C, Barker BR, Jobin C, Allbritton NL, Ramsden DA, Davis BK, Ting JPY (2015) Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med 21:906–913
Chen IF (2006) AIM2 suppresses human breast cancer cell proliferation in vitro and mammary tumor growth in a mouse model. Mol Cancer Ther 5:1–7
Patsos G, Germann A, Gebert J, Dihlmann S (2009) Restoration of absent in melanoma 2 (AIM2) induces G2/M cell cycle arrest and promotes invasion of colorectal cancer cells. Int J Cancer
Ponomareva L, Liu H, Duan X, Dickerson E, Shen H, Panchanathan R, Choubey D (2013) AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol Cancer Res 11:1193–1202
Lee J, Li L, Gretz N, Gebert J, Dihlmann S (2012) Absent in Melanoma 2 (AIM2) is an important mediator of interferon-dependent and-independent HLA-DRA and HLA-DRB gene expression in colorectal cancers. Oncogene 31:1242–1253
Liu R, Truax A, Chen L, Hu P, Li Z, Chen J, Song C, Ting J (2015) Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget 6(32):33456–33469
Hasegawa M, Imamura R, Motani K, Nishiuchi T, Matsumoto N, Kinoshita T, Suda T (2009) Mechanism and repertoire of ASC-mediated gene expression. J Immunol 182:7655–7662
Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC (2003) Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol 171:6154–6163
Masumoto J, Dowds TA, Schaner P, Chen FF, Ogura Y, Li M, Zhu L, Katsuyama T, Sagara J, Taniguchi SI, Gumucio DL, Núñez G, Inohara N (2003) ASC is an activating adaptor for NF-B and caspase-8-dependent apoptosis. Biochem Biophys Res Commun 303:69–73
Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA, Lee SW (2004) ASC is a Bax adaptor and regulates the p53–Bax mitochondrial apoptosis pathway. Nat Cell Biol 6:121–128
Sarkar A, Duncan M, Hart J, Hertlein E, Guttridge DC, Wewers MD (2006) ASC Directs NF-B activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions. J Immunol 176:4979–4986
McConnell BB, Vertino PM (2004) TMS1/ASC: the cancer connection. Apoptosis 9:5–18
Levine JJ, Stimson-Crider KM, Vertino PM (2003) Effects of methylation on expression of TMS1/ASC in human breast cancer cells. Oncogene 22:3475–3488
Yokoyama T, Sagara J, Guan X, Masumoto J, Takeoka M, Komiyama Y, Miyata K, Higuchi K (2003) S.i. Taniguchi, Methylation of ASC/TMS1, a proapoptotic gene responsible for activating procaspase-1, in human colorectal cancer. Cancer Lett 202:101–108
Terasawa K (2004) Epigenetic Inactivation of TMS1/ASC in Ovarian Cancer. Clin Cancer Res 10:2000–2006
Machida EO (2006) Hypermethylation of ASC/TMS1 is a sputum marker for late-stage lung cancer. Cancer Res 66:6210–6218
Das PM, Ramachandran K, Vanwert J, Ferdinand L, Gopisetty G, Reis IM, Singal R (2006) Methylation mediated silencing of TMS1/ASC gene in prostate cancer. Mol Cancer 5:28
Stone AR, Bobo W, Brat DJ, Devi NS, Van Meir EG, Vertino PM (2004) Aberrant methylation and down-regulation of TMS1/ASC in human glioblastoma. Am J Pathol 165:1151–1161
Martinez R, Schackert G, Esteller M (2006) Hypermethylation of the proapoptotic gene TMS1/ASC: prognostic importance in glioblastoma multiforme. J Neurooncol 82:133–139
Zhang C, Li H, Zhou G, Zhang Q, Zhang T, Li J, Zhang J, Hou J, Liew CT, Yin D (2007) Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J Pathol 212:134–142
Liu W, Luo Y, Dunn JH, Norris DA, Dinarello CA, Fujita M (2012) Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J Investig Dermatol 133:518–527
Knight ERW, Patel EY, Flowers CA, Crowther AJ, Ting JP, Miller CR, Gershon TR, Deshmukh M (2014) ASC deficiency suppresses proliferation and prevents medulloblastoma incidence. Oncogene 34:394–402
Ting JPY, Duncan JA, Lei Y (2010) How the noninflammasome NLRs function in the innate immune system. Science 327:286–290
Allen IC (2014) Non-inflammasome forming NLRs in inflammation and tumorigenesis. Front Immunol 5:169
Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, Geddes BJ, Briskin M, DiStefano PS, Bertin J (2002) PYPAF7, a Novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem 277:29874–29880
Pinheiro AS, Eibl C, Ekman-Vural Z, Schwarzenbacher R, Peti W (2011) The NLRP12 pyrin domain: structure, dynamics, and functional insights. J Mol Biol 413:790–803
Williams KL, Taxman DJ, Linhoff MW, Reed W, Ting JPY (2003) Cutting Edge: monarch-1: a pyrin/nucleotide-binding domain/leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J Immunol 170:5354–5358
Williams KL (2005) The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor-, and mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem 280:39914–39924
Ye Z, Lich JD, Moore CB, Duncan JA, Williams KL, Ting JPY (2007) ATP binding by monarch-1/NLRP12 is critical for its inhibitory function. Mol Cell Biol 28:1841–1850
Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, Ting JPY (2007) Cutting edge: monarch-1 suppresses non-canonical NF-B activation and p52-dependent chemokine expression in monocytes. J Immunol 178:1256–1260
Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RMT, Davis BK, Uronis JM, Herfarth HH, Jobin C, Rogers AB, Ting JPY (2012) NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36:742–754
Zaki MH, Man SM, Vogel P, Lamkanfi M, Kanneganti TD (2013) Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci 111:385–390
Zaki MH, Vogel P, Malireddi RKS, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, Kanneganti T-D (2011) The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20:649–660
Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JPY (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451:573–577
Xiao TS, Ting JPY (2012) NLRX1 has a tail to tell. Immunity 36:311–312
Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, Ting JPY (2011) NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 34:854–865
Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, Wang R-F (2011) NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 34:843–853
Tattoli I, Carneiro LA, Jéhanno M, Magalhaes JG, Shu Y, Philpott DJ, Arnoult D, Girardin SE (2008) NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep 9:293–300
Hong M, Yoon S-I, Wilson IA (2012) Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 36:337–347
Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE (2012) NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun 19:438–448
Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, Swanson KV, Wen K-W, Damania B, Moore CB, Giguère PM, Siderovski DP, Hiscott J, Razani B, Semenkovich CF, Chen X, Ting JPY (2012) The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36:933–946
Soares F, Tattoli I, Rahman MA, Robertson SJ, Belcheva A, Liu D, Streutker C, Winer S, Winer DA, Martin A, Philpott DJ, Arnoult D, Girardin SE (2014) The mitochondrial protein NLRX1 controls the balance between extrinsic and intrinsic apoptosis. J Biol Chem 289:19317–19330
Singh K, Poteryakhina A, Zheltukhin A, Bhatelia K, Prajapati P, Sripada L, Tomar D, Singh R, Singh AK, Chumakov PM, Singh R (1853) NLRX1 acts as tumor suppressor by regulating TNF-α induced apoptosis and metabolism in cancer cells. Biochim Biophys Acta 2015:1073–1086
Correa RG, Milutinovic S, Reed JC (2012) Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep 32:597–608
Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE (2013) NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14:9–23
Bertrand MJM, Doiron K, Labbé K, Korneluk RG, Barker PA, Saleh M (2009) Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30:789–801
Lecine P, Esmiol S, Metais JY, Nicoletti C, Nourry C, McDonald C, Nunez G, Hugot JP, Borg JP, Ollendorff V (2007) The NOD2-RICK complex signals from the plasma membrane. J Biol Chem 282:15197–15207
Kim Y-G, Park J-H, Shaw MH, Franchi L, Inohara N, Núñez G (2008) The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to toll-like receptor ligands. Immunity 28:246–257
Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 4:702–707
Travassos LH (2005) Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J Biol Chem 280:36714–36718
Grubman A, Kaparakis M, Viala J, Allison C, Badea L, Karrar A, Boneca IG, Le Bourhis L, Reeve S, Smith IA, Hartland EL, Philpott DJ, Ferrero RL (2010) The innate immune molecule, NOD1, regulates direct killing ofHelicobacter pyloriby antimicrobial peptides. Cell Microbiol 12:626–639
Silva GK, Gutierrez FRS, Guedes PMM, Horta CV, Cunha LD, Mineo TWP, Santiago-Silva J, Kobayashi KS, Flavell RA, Silva JS, Zamboni DS (2009) Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against trypanosoma cruzi infection. J Immunol 184:1148–1152
Tsuji Y, Watanabe T, Kudo M, Arai H, Strober W, Chiba T (2012) Sensing of commensal organisms by the intracellular sensor NOD1 mediates experimental pancreatitis. Immunity 37:326–338
Strober W, Murray PJ, Kitani A, Watanabe T (2005) Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 6:9–20
Mo J, Boyle JP, Howard CB, Monie TP, Davis BK, Duncan JA (2012) Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem 287:23057–23067
von Kampen O, Lipinski S, Till A, Martin SJ, Nietfeld W, Lehrach H, Schreiber S, Rosenstiel P (2010) Caspase recruitment domain-containing protein 8 (CARD8) negatively regulates NOD2-mediated signaling. J Biol Chem 285:19921–19926
Mohanan V, Grimes CL (2014) The molecular chaperone HSP70 binds to and stabilizes NOD2, an important protein involved in crohn disease. J Biol Chem 289:18987–18998
Kanneganti T-D, Lamkanfi M, Núñez G (2007) Intracellular NOD-like receptors in host defense and disease. Immunity 27:549–559
Chen GY, Shaw MH, Redondo G, Núñez G (2008) The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res 68:10060–10067
Wang X, Jiang W, Duan N, Qian Y, Zhou Q, Ye P, Jiang H, Bai Y, Zhang W, Wang W (2014) NOD1, RIP2 and Caspase12 are potentially novel biomarkers for oral squamous cell carcinoma development and progression. Int J Clin Exp Pathol 7:1677
Zaki MH (2013) NOD-like receptors in colon tumorigenesis. Immunogastroenterology 2:90
Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A (2013) NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Investig 123:700
Kurzawski G, Suchy J, Kładny J, Grabowska E, Mierzejewski M, Jakubowska A, Dȩbniak T, Cybulski C, Kowalska E, Szych Z (2004) The NOD2 3020insC mutation and the risk of colorectal cancer. Cancer Res 64:1604–1606
Kutikhin AG (2011) Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum Immunol 72:955–968
Gabay C, Lamacchia C, Palmer G (2010) IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 6:232–241
Dhimolea E (2011) Interleukin-1beta; inhibitors for the treatment of cryopyrin-associated periodic syndrome. Appl Clin Genet 4:21–27
Zitvogel L, Kepp O, Galluzzi L, Kroemer G (2012) Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol 13:343–351
Shen H, Sun T, Ferrari M (2012) Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther 19:367–373
Friedman A (2006) Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity. Cancer Res 66:2314–2319
Cattaneo R, Miest T, Shashkova EV, Barry MA (2008) Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Micro 6:529–540
Hegi ME, Rajakannu P, Weller M (2012) Epidermal growth factor receptor. Curr Opin Neurol 25:774–779
Sorensen AG, Emblem KE, Polaskova P, Jennings D, Kim H, Ancukiewicz M, Wang M, Wen PY, Ivy P, Batchelor TT, Jain RK (2012) Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res 72:402–407
Luo JL (2005) IKK/NF-B signaling: balancing life and death: a new approach to cancer therapy. J Clin Investig 115:2625–2632
López-Castejón G, Pelegrín P (2012) Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs 21:995–1007
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P (2012) Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12:860–875
Acknowledgments
SJ’s laboratory is funded by grants from the Department of Science and Technology (Young scientist scheme, SB/YS/LS-282/2013) and Board of Research in Nuclear Sciences (2013/36/72-BRNS/2415), Government of India. The software application Science Slides (VisiScience) was used to generate parts of Figs. 1, 2 and 3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sharma, N., Jha, S. NLR-regulated pathways in cancer: opportunities and obstacles for therapeutic interventions. Cell. Mol. Life Sci. 73, 1741–1764 (2016). https://doi.org/10.1007/s00018-015-2123-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2123-8