
Abstract
Enhanced glycolysis in cancer, called the Warburg effect, is a well-known feature of cancer metabolism. Recent advances revealed that the Warburg effect is coupled to many other cancer properties, including adaptation to hypoxia and low nutrients, immortalisation, resistance to oxidative stress and apoptotic stimuli, and elevated biomass synthesis. These linkages are mediated by various oncogenic molecules and signals, such as c-Myc, p53, and the insulin/Ras pathway. Furthermore, several regulators of glycolysis have been recently identified as oncogene candidates, including the hypoxia-inducible factor pathway, sirtuins, adenosine monophosphate-activated kinase, glycolytic pyruvate kinase M2, phosphoglycerate mutase, and oncometabolites. The interplay between glycolysis and oncogenic events will be the focus of this review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As glycolysis is essential for energy production in almost all mammalian cells, impaired glycolysis was assumed to have a pathological effect in various human diseases, including diabetes mellitus and muscle atrophy [1, 2]. Among the first descriptions of enhanced glycolysis in diseased states was the Warburg effect, which was proposed by Otto Warburg [3] after he observed that cancer cells preferably covert glucose into lactate even in the presence of oxygen. Indeed, enhanced glycolysis was subsequently found to be a metabolic characteristic of many cancers [3], and the upregulation of protein levels and enzymatic activities of many glycolytic enzymes was later confirmed [4, 5]. It was initially thought that enhanced glycolysis may provide an energy boost to meet the demands of the high proliferation rate of cancer cells. However, energy generation via glycolysis is relatively inefficient, as it generates only two ATP molecules per glucose, whereas the TCA cycle in mitochondria generates 36 ATPs per glucose [6]. Thus, the reason for cancer cells to favour enhanced glycolysis cannot be simply explained by the efficiency of energy production. Recent studies have revealed causal effects of enhanced glycolysis on cancerous growth, including an increase in biomass synthesis [7, 8] and radical scavenger activities [9]. These additional aspects of the Warburg effect might partly explain the preference for enhanced glycolysis in cancer.
Inhibition of the Warburg effect has been proposed as a possible cancer therapy [6]; however, this strategy is problematic as the glycolytic pathway is also required in normal tissues. Thus, cancer therapies targeting the Warburg effect must induce cancer-specific and localised inhibition of glycolysis in order to minimise possible side effects. An important step will be to determine how glycolysis is dysregulated in cancer, while strictly regulated in healthy cells. In addition to their high proliferative capacity, cancer cells exhibit several cytological hallmarks. These include immortalisation, stress resistance mechanisms such as evasion from apoptotic stimuli, survival under nutrient-limited conditions, metastatic capacity, and anchorage-independent growth [10]. It is possible that the Warburg effect promotes these properties, which are known to be associated with genetic alterations and modulations in signalling pathways [11, 12]. Any links between the Warburg effect and oncogenic signalling pathways would be of great interest as potential targets for anticancer therapy [13, 14]. Here, we provide an overview of recent advances in our understanding of glycolysis regulation in cancer and the Warburg effect.
Cellular-context-dependent regulation of glycolysis
Glycolysis is a highly conserved metabolic process that involves sequential reactions mediated by several glycolytic enzymes. The sequences of the genes encoding these enzymes and the intermediate metabolites in glycolysis are highly conserved from bacteria to humans, implicating its fundamental importance for all living cells. It has been well established that phosphofructokinase (PFK) is the rate-limiting enzyme for the glycolytic pathway owing to its allosteric regulation, and this has been shown not only in bacteria and yeast, but also in cancerous cells and muscle cells in vitro [15, 16].
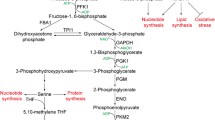
The regulation of glycolytic metabolism in mammalian cells depends on many factors, including differentiation status, growth conditions, and cellular environment (availability of oxygen, nutrients, etc.) [17, 18]. For example, normal cells might adapt to hypoxic conditions by enhancing anaerobic glycolysis and limiting energy demands. However, cancer cells continue growing even under hypoxic conditions in vivo, and this might require a maladaptive metabolic shift [19]. Thus, the fine tunings of glycolysis observed in normal cells are dysregulated in cancer cells to support their demand for excess glycolysis (Fig. 1). Indeed, recent studies have revealed that in addition to PFK, several glycolytic enzymes play key roles in establishing the Warburg effect in cancer.

Network of transcriptional and posttranscriptional regulation of glycolysis relevant to tumourigenesis. Classical oncogenic factors are indicated by green circles, while other signalling molecules are shown in blue. HIF-1 is indicated in red, and the metabolic sensor AMPK and sirtuins are shown in orange. The arrow indicates a positive effect, while the others are inhibitory effects. Pathways branching from glycolysis are described in the grey box, and some essential metabolites are in purple. See the text for additional mechanistic details and abbreviation definitions
Transport of glucose across the plasma membrane is the first rate-limiting step for glucose metabolism, which is mediated by GLUT proteins. Among them, GLUT1, GLUT3 and GLUT12 have been reported to be upregulated in some cancers [20]. Hexokinase (HK) mediates the critical first step of glycolysis; generation of glucose-6-phosphate (G-6-P) via phosphate transfer from ATP. Mammalian four isoforms of HK are designated as HK-1 to HK-4. Their intracellular localizations are variable; HK-1 and HK-2 mainly on the outer membrane of mitochondria, HK-3 in a perinuclear regions, and HK-4 in the cytosol. Their tissue distributions are also various. For example, HK-4, known as glucokinase, is mainly expressed in liver and pancreas. However, in cancer cells, HK-2 is predominantly overexpressed for following reasons. HK-1, -2, and -3 shows over 200-fold lower K m for glucose compared to that of HK-4. Moreover, HK-2 has two functionally active kinase domains, while others not. HK-2 binds to voltage-dependent anion channels (VDACs), to smoothly access to mitochondria-generated ATP. VDAC-bound HK-2 is also insensitive to feedback inhibition of G-6-P as its product. Thus, HK-2 is more efficient to restore highly glycolytic flux than others. Moreover, the interaction between HK-2 and VDACs is critical to prevent apoptosis by proapoptotic factors, Bax and Bad, in tumours [21–23]. Interestingly, Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) is also known to interact with VDACs [24].
The other key glycolytic enzyme is pyruvate kinase (PK), which converts phosphoenolpyruvate (PEP) into pyruvate in the final step of glycolysis. PKM1 and PKM2 are alternatively spliced isoforms of PK that differ in sequence by only 22 amino acids. PKM1 is expressed in normal adult tissues, while PKM2 is also detected in many tumours and embryonic tissues. Although there are some controversies regarding whether PKM2 is absolutely required for tumourigenesis in vivo [25, 26], PKM2 is designated as the oncogenic isoform of PK, not only because of its expression profile, but also because of its multifaceted functions in tumourigenesis [25]. The most striking evidence of PMK2 involvement in tumourigenesis is that the dimeric form of PKM2 also functions as a protein kinase that targets the tumourigenesis-associated factors STAT3 [27], β-catenin [28], histone H3 [29], BUB3 checkpoint protein [30], NF-κB p65 [31], OCT-4, CD44 (a cancer stem cell marker) [32], and HIF-1 [33]. These findings suggest that the regulation of PKM2 could be essential for cancer cell proliferation.
Interestingly, other glycolytic enzymes are involved in establishing the Warburg effect during the process of immortalisation and transformation. Normal primary cells cultured in vitro suffer irreversible cell cycle arrest, called senescence, which is induced by telomeric erosion or by stresses such as oxidative stress, DNA damage, and oncogenic insult [34–36]. The latter is designated stress-induced senescence, which is often bypassed by cell immortalising events in vitro, such as the activation of some oncogenes and the ablation of tumour suppressor genes [27]. In vivo, cellular senescence forms a protective barrier against immortalisation [37]. During the senescence process, glycolysis declines in human and mouse primary cells, while cancerous cells maintain the Warburg effect even under standard tissue culture conditions (i.e. 20 % oxygen) [19]. Recent studies have uncovered roles for glycolytic enzymes in the bypass of senescence in cancer cells.
Phosphoglycerate mutase (PGAM) was reported to be an immortalising factor in mouse fibroblasts via its radical scavenging effects [9, 38]. This finding is supported by the notion that PGAM activation suppresses mitochondrial respiration in vivo and in vitro [38, 39], followed by decreased generation of reactive oxygen species (ROS). Moreover, 2-phosphoglycerate, the metabolic product of PGAM, also activates the pentose phosphate pathway, whose product, NADPH, is essential for maintaining reducing power [8]. Hexokinase 2 (HK2) was also identified as a senescence-bypassing gene [11]. HK2-expressing cells show activation of the hexosamine biosynthetic pathway (HBP), which branches from glycolysis. The HBP affects many cellular processes through protein modification, as it further branches into N-linked glycosylation and O-linked N-acetylglucosamine (O-GlcNAc) [40]. Moreover, the ectopic expression of the glucose transporter GLUT3 renders nonmalignant breast cells susceptible to experimental transformation under 3-D culture conditions, and this occurs via HBP activation coupled with the Warburg effect [12]. These phenotypic conversions are accompanied by the activation of some oncogenic signalling factors (EGFR, AKT, MEK, and β1 integrin) [12]. Thus, the activation of different glycolytic enzymes affects various metabolic and biological pathways, whose outcome similarly promotes the proliferation of cancer cells under Warburg effect conditions. These findings indicate that investigation of the complex relationship between glycolytic regulation and cancer metabolism is essential for understanding the Warburg effect.
Adaptation to hypoxia and transcriptional regulation of glycolytic enzymes
It is quite possible that the Warburg effect is the consequence of cellular adaptation to the hypoxic environment encountered by cancer cells, particularly inside the core of solid tumours outgrowing the oxygenating capacity of neovasculatures [41]. However, the molecular mechanism of the Warburg effect was unclear until breakthrough experiments on the transcriptional regulation of glycolysis, which led to the discovery of hypoxia-inducible transcription factor 1 (HIF-1). HIF-1 was identified by DNA affinity chromatography from large-scale cultures of HeLa cells based on its ability to bind to the hypoxia response element DNA sequence [42]. Subsequently, the functional homologue HIF-2 was identified, and was found to have targets that overlapped with those of HIF-1 in addition to its own distinct target genes [43–45]. HIF-1 is required to upregulate many glycolytic enzymes under hypoxic conditions [46]. In addition, pyruvate dehydrogenase kinase 1 is also upregulated directly by HIF-1, leading to the inhibition of pyruvate entry into the TCA cycle (Fig. 1) [47, 48]. HIF-1 also regulates MCT4 (monocarboxylate transporter), which is critical to prevent the intracellular lactic acidification in tumours [49]. While intracellular lactic accumulation provokes apoptosis in cells, exported lactate might protect tumours from attack by immune systems [22].
The accumulation of HIF-1 or HIF-2 has been observed in many cancer cells, and is associated with poor prognosis of patients [50]. However, several lines of evidence suggest that the Warburg effect cannot be simply explained as an adaptation to hypoxic conditions in vivo. First, cancer cells maintain a high level of glycolysis even in tissue culture conditions under normoxia (20 % oxygen) [51]. Second, the ectopic expression of HIF-1 causes cell cycle arrest in some cell lines [52]. Third, PGAM is not upregulated by HIF-1 during hypoxia [46]. Fourth, HIF-1 knockdown hardly affects the mRNA profiles of glycolytic enzymes in some cells [12]. Fifth, recent work suggests that HIF-1 is also regulated by stimuli other than hypoxia [17]. Thus, the intriguing correlation between the Warburg effect and HIF-1 could be affected by the interplay between multiple factors in addition to hypoxia. In this context, it is noteworthy that the transcription factors STAT3 and NF-κB also regulate the transcription of glycolytic enzymes in cooperation with HIF-1 [53, 54], while ETS-1 cooperates with HIF-2 [55, 56].
Several other transcription factors are also involved in glycolytic regulation. Hepatocyte nuclear factor 1β (HNF-1β) is a homeodomain transcription factor that plays a critical role in pancreatic development, including the differentiation of pancreatic endocrine cells. HNF-1β mutations have been clinically reported in many cases of diabetes mellitus [57]. Recently, HNF-1β was reported to regulate the Warburg effect in ovarian cancer. Knockdown of HNF-1β in an ovarian clear cell carcinoma (OCCC) cell line downregulated the mRNA levels of many glycolytic enzymes, including HK, GPI, PFK, ALDO, TPI, PGK, PGAM, ENO, and LDH, leading to a reduction in glycolytic flux [58]. Interestingly, ablation of HNF-1β causes OCCC cells to proliferate more rapidly with a reduced glycolytic rate. As OCCC is known to show slow progression but a poorer prognosis than other types of ovarian cancers [59], the Warburg effect in ovarian cancer might be associated with characteristics other than its proliferative potential. AD4BP/SF-1 (NR5A1), a steroidogenic tissue-specific nuclear receptor, was also recently reported as a transcriptional regulator of glycolysis [60]. Direct regulation of many glycolytic enzymes by AD4BP/SF-1 was clearly shown using a knockdown assay and CHIP analysis; these enzymes included HK, GPI, PFK, ALDO, TPI, GAPDH, PGK, PGAM, ENO, PKM2, and LDH. It would interesting to see whether AD4BP/SF-1 is also involved in tumourigenesis in relevant tissues [60]. Furthermore, the transcription factors specificity protein 1 (SP1) and SP3 induce PKM, enolase, and aldolase [61, 62], while peroxisome proliferator-activated receptor γ (PPAR γ) activates PKM and HK2 during hepatic tumourigenesis [63]. Additionally, microRNAs have been reported to be involved in glycolytic regulation; the details of this regulation have been described in other reviews [64, 65]. Although glycolytic regulation by HIF-1 and/or other transcription factors could also be required for normal cells under hypoxia or other conditions, these factors are known to be involved in oncogenic events, and their signalling in cancer cells maintains the Warburg effect, as discussed in "Classical oncogenic signals and glycolysis".
Classical oncogenic signals and glycolysis
It is known that several major oncogenic events constitute oncogenesis in vivo, including the activation of oncogenes (Ras, Myc, etc.) and inactivation of tumour suppressor genes (p53, Rb, Ink4, etc.) [66]. These classical oncogenic signals are also required for the regulation of the Warburg effect (Fig. 1).
It has been suggested that several growth factors, including insulin, IGF-1, and IGF-2, stimulate glycolysis via the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT, also known as survival kinase)/mammalian target of rapamycin (mTOR) kinase pathway or the Ras/Raf/ERK pathway in cancer cells [67, 68]. The former pathway phosphorylates 4E-BP1, resulting in enhanced translation of HIF-1 mRNA [69]. AKT kinase also promotes the translocation of the glucose transporter GLUT4 to the plasma membrane via phosphorylation of its target AS160 (AKT substrate of 160 kDa), a GTPase-activating protein of the small G protein Rab family [70]. Moreover, the ectopic expression of AKT kinase upregulates glycolysis in leukemic cells [71]. Oncogenic mutations in Ras and its downstream pathway are commonly observed in clinical and experimental tumourigenesis. Ras/Raf kinases activate the MAP kinases ERK1 and ERK2, and this is followed by the activation of MAP kinase-interacting kinases MNK1 and MNK2. Subsequently, MNK1 phosphorylates eIF-4E and promotes the translation of HIF-1. Furthermore, the Ras and insulin signalling pathways activate another small G protein, RAC1/CDC42, and its associated kinase, p21-activated protein kinase (PAK) [72, 73]. Although PAK is known to be involved in many tumourigenic processes, including cell motility, cytoskeleton reorganisation, apoptosis, and metastasis [74], its role in the Warburg effect is rather complicated. PAK directly phosphorylates and downregulates the glycolytic enzyme PGAM [75], while it facilitates insulin-stimulated GLUT4 translocation via actin remodelling [76]. These opposing roles of PAK in glycolysis are expected to be a topic of further investigation.
In early studies, Hunter et al. [77] pointed out the intriguing correlation between oncogenic kinases and glycolytic enzymes (Enolase, LDH, PGAM). More recently, PGAM was also reported to be regulated by oncogenic kinases [78], and the glycolytic enzyme PKM2 was found to be regulated by the oncogenic tyrosine kinases BCR-ABL, FGFR1, FLT3-ITD, and JAK2 [79]. Thus, phosphorylation is integral to the regulation of the Warburg effect. The counteracting activity of phosphatases might also be involved, as might other posttranscriptional modifications.
The function of HIF-1 is largely affected by two major cancer-related transcriptional regulators (c-MYC and p53) [80]. The Warburg effect is also induced by c-MYC activation or p53 inactivation, and this is associated with the senescence-bypassing ability of cancer cells [81, 82]. Several cancers frequently harbour oncogenic mutations or amplification of c-Myc, which directly affects the expression of several glycolytic enzymes including HK, PFK, TPI, GAPDH, ENO, and LDH [83, 84]. Moreover, c-MYC enhances the alternative splicing of PKM2 rather than PKM1 via upregulation of the RNA-binding proteins hnRNPA1, hnRNPA2, and hnRNPI [85, 86]. The tumour suppressor p53 also has several effects on glycolysis-related factors. For example, the inactivation of tumour suppressor p53 upregulates GLUT3 via NF-κB activation, and activates HK [82, 87]. Moreover, TP53-induced glycolysis and apoptosis regulator (TIGAR) is another glycolytic target of p53 [88]. The TIGAR protein shows a weak similarity to the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2), but lacks the kinase domain. While fructose-2,6-bisphosphate, generated by PFK-2, is known as the most potent allosteric effector of PFK, the accumulation of fructose-6-phosphate generated by FBPase-2 or TIGAR inhibits PFK. Thus, the ectopic expression of TIGAR inhibits glycolysis but enhances the prosurvival ability of some cancer cells (U2OS and H1299), as TIGAR increases PPP activity, leading to increased reducing power and decreased ROS in cells. In this setting, p53 attenuates the Warburg effect to protect cancer cells from ROS-induced apoptosis.
While the knockdown of p53 mainly upregulates transcription of glycolytic enzymes or glycolytic flux in cancer cells [89], there is one exception; the glycolytic enzyme PGAM is positively regulated by p53 in muscle cells [90]. It is noteworthy that PGAM is also exempt from the regulation of glycolytic enzymes by other transcription factors, including HIF-1 and c-MYC. Thus, it is still not clear how the transcriptional regulation of PGAM is linked to the Warburg effect, although recent works have suggested that PGAM is subject to a high degree of posttranscriptional regulation. It was recently discovered that PGAM is posttranscriptionally regulated by the ubiquitin/proteasome pathway in primary cells under senescence-inducing stress, DNA damage, or oncogenic stress [91]. Proteolysis is an irreversible reaction that constitutes a regulatory mechanism for many cellular processes. Ubiquitination requires a substrate-specific E3 ubiquitin ligase and a substrate-nonspecific E1 ubiquitin-activating enzyme and E2 ubiquitin-conjugating enzyme [92]. Ubiquitinated proteins are degraded by proteasome pathway, unless ubiquitination is reversed by a deubiquitinase. Generally, ubiquitination requires an advance modification of the substrate (e.g. phosphorylation, acetylation). The RING finger protein MDM2, a transcriptional target of p53, is the ubiquitin ligase for PGAM, while PAK1 works as a priming kinase by facilitating the interaction between PGAM and MDM2 under stress [91]. MDM2 has been perceived as an oncogene, because MDM2 also ubiquitinates the tumour suppressor p53 [93, 94]. Indeed, in certain cancers, gene amplification of MDM2 is observed [95, 96]; however, in contrast, MDM2 has also been reported to be a tumour suppressor [97, 98]. Thus, MDM2 may have opposing effects on the two different substrates, p53 and PGAM, in a cellular-context dependent manner. Under senescence-inducing stress, PGAM is degraded by the p53/MDM2 axis, whereas in the presence of some oncogenic signals, such as Ras-G12V and MDM2-M459I, PGAM is stabilised while p53 is impaired. In conclusion, p53 may regulate glycolysis directly by its transcriptional role or posttranscriptionally via its target MDM2 [91].
New regulators for glycolysis and their oncogenic involvement
Besides hypoxia, low nutrient or low glucose conditions constitute critical metabolic stresses against rapidly growing solid tumours in vivo [19]. Recent advances in aging research have uncovered how adaption to low glucose modulates organismal longevity. Calorie restriction (CR) is a popular aging model proposed by McCay and Crowell in 1934 [99]. It has been well established that CR activates two crucial posttranscriptional regulators: adenosine monophosphate-activated kinase (AMPK) and sirtuins. AMPK is activated by an increase in the AMP/ATP ratio, while sirtuin is an NAD+-dependent deacetylase that is activated by the accumulation of nicotinamide adenine dinucleotide (NAD), a by-product of activated respiration during CR. Both molecules form an essential physiological energy sensor to regulate energy balance in vivo and in vitro [100]. Moreover, activation of mTOR signalling is also tightly linked to metabolic stress (starvation of amino acid or glucose) or hypoxia [101].
The core of mTOR signalling is mediated by mTORC (mTOR complex) kinase, which is activated by GTP-bound Rheb small G protein. TSC1/TSC2, the tuberous sclerosis complex (TSC) tumour suppressors, are GTPase-activating protein (GAP) for Rheb. TSC1/TSC2 is targeted by several kinases, AMPK, Akt kinase, ERK, and so on, as mTORC activation is essentially required for protein synthesis, autophagy, lipid synthesis and others. Interestingly, mTORC1 also upregulates glycolysis via enhanced translation of HIF-1 mRNA. It is noteworthy that mTORC1 is aberrantly activated in 40–90 % of ten most frequently occurring cancers [102].
It is difficult to conclude whether AMPK behaves as an oncogene by supporting cancer survival under metabolic stress, or functions as a tumour suppressor by inhibiting anabolic metabolism. Several lines of evidence support the former model. AMPK is frequently amplified in human cancers [18], and is activated by oncogenic Ras-G12V [103]. AMPK directly activates the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, PFKFB3, leading to an increase in fructose-2,6-bisphosphate, which is an allosteric effector of PFK. Moreover, AMPK-dependent degradation of thioredoxin-interacting protein (TXNIP) enhances glucose uptake by activating its binding partner GLUT1 [104]. In support of the tumour suppressor model, AMPK belongs to the LKB1/mTOR tumour suppressor pathway; mutations to components of this pathway are known to cause predisposition to Peutz–Jeghers syndrome. Furthermore, AMPK directly activates p53 under low glucose conditions [105], and in a MYC-overexpressing state, AMPK ablation increases HIF-1-coupled glycolysis [106]. Thus, AMPK might augment or attenuate the Warburg effect in a cellular-context-dependent manner.
Sirtuins are the mammalian homologues of the S. cerevisiae silent information regulator 2 (SIR2) gene, which was initially identified as a pro-longevity gene under CR conditions. The sirtuin protein family has seven members, SIRT1-SIRT7, which share a central catalytic deacetylase domain and have distinct structures in the N- and C-termini. Initially, histones were proposed as the target for deacetylation by sirtuins [107]; however, recent studies revealed that sirtuins deacetylate not only histones, but also other metabolic regulators, including PGC-1, HIF-1, and MYC [99, 108]. As cancer cells adapt to different forms of metabolic stress, there has been keen interest as to whether sirtuins also function as metabolic modulators in cancer. Interestingly, many sirtuin knockout mice (SIRT2, SIRT3, SIRT4 and SIRT6) display a cancer-prone phenotype [109–112], while overexpression of the brain-specific SIRT1 and SIRT6 extended organismal lifespan in mice [113, 114].
Although elevated expression of SIRT1 has been observed in several cancers [115–117], opposing effects of SIRT1 on HIF-1 and MYC have been reported [118, 119], and it is not clear whether SIRT1 regulates the Warburg effect positively or negatively. The link between SIRT6 and glycolysis is more clear, as enhanced glycolysis in SIRT6 knockout conditions was observed both in vivo and in vitro, consistent with its tumourigenic phenotype [112, 120]. Interestingly, several sirtuins (SIRT3, SIRT6, and SIRT7) inactivate HIF-1 and suppress the Warburg effect [120–122]. However, the inhibition of MYC by sirtuins (SIRT4, SIRT6, and SIRT7) has little effect on its glycolytic regulation [111, 112, 123], suggesting that unknown accessory regulation is operating for the MYC-induced Warburg effect. In addition, the deacetylase HDAC4 was also found to regulate and promote HIF-1 stability in a renal cancer cell line [124].
Glycolytic enzymes are also regulated by acetylation/deacetylation. The acetylation of LDH-A is downregulated in pancreatic cancer by SIRT2-mediated deacetylation, leading to increased LDH-A enzymatic activity due to inhibition of protein degradation [125]. SIRT2 also regulates PGAM, although both negative and positive regulation has been reported [126, 127], and PGAM is also downregulated by SIRT1 [128]. Acetylation of GAPDH by PCAF increases its enzymatic activity and promotes cell proliferation after glucose stimuli, while GAPDH deacetylation by HDAC5 downregulates its enzymatic activity [129]. The acetylation of PKM2 is differently regulated by several different stimuli: glucose facilitates Lys305 acetylation of PKM2, leading to autophagic degradation [130], while oncogenic stimuli induce Lys433 acetylation by p300, which activates PKM2 kinase activity [131].
Regulation by ubiquitination and metabolites
Glycolysis is also controlled and greatly affected by the ubiquitin/proteasome system. While PGAM is degraded by MDM2 under stress, the HIF-1 protein is very unstable under normoxic conditions [42]. The E3 ubiquitin ligase for HIF-1 is the von Hippel–Lindau (VHL) protein, whose loss-of-function mutations are responsible for a renal cancer predisposition, termed VHL syndrome [132, 133], which involves the accumulation of HIF protein [134]. The competence of HIF-1 for ubiquitination is dependent upon hydroxylation of its proline-402 and -564 residues, which is induced under high oxygen conditions by the proyly-4-hydroxylase domain (PHD) proteins PHD1, PHD2, and PHD3 [135]. Hydroxylated HIF-1 binds more tightly to VHL and is therefore ubiquitinated more readily [134]. As PHD proteins are a subtype of dioxygenase, O2 and α-ketoglutarate are utilised as substrates [17], and thus the dioxygenase activity of PHD proteins is impaired by ROS generated from dysfunctional mitochondria or from oncogenic signalling [136]. However, the activation of MnSOD by SIRT3-dependent deacetylation protects PHDs from ROS-dependent inactivation and facilitates HIF-1 activity [121, 137]. Together, these findings indicate that ubiquitin-mediated proteolysis is a key regulator of the Warburg effect.
Glycolytic regulation by metabolites has been well studied, but remains an intense focus of investigation. It has been well established that PFK1 is allosterically inhibited by the metabolites, citrate, and ATP, and allosterically activated by AMP and fructose 2,6-bisphosphate [16]. Thus, PFK is the rate-limiting step for glycolysis in cells. Surprisingly, recent developments in metabolomic analysis led to the identification of additional metabolites involved in glycolysis regulation. For example, lactate, fumarate, and succinate have been discovered to inhibit PHD activity under normoxic conditions, leading to an increase in HIF-1 stability [138–140]. It is noteworthy that α-ketoglutarate-dependent dioxygenases, which are PHD proteins, are competitively inhibited by another metabolite, 2-hydroxyglutarate (2-HG), which has been designated as an oncometabolite [141]. 2-HG is generated by oncogenic mutants of IDH1 and IDH2, which are observed frequently in gliomas and acute myeloid leukaemia, while their normal counterparts generate α-ketoglutarate (α-KG). Thus, in cancer cells bearing IDH mutations, the accumulation of 2-HG would disrupt the connection between environmental stress (oxygen or ROS condition) and the stabilisation of HIF-1, thereby causing constitutive activation of HIF-1.
PKM2 is also subject to metabolite-dependent regulation, including allosteric activation by fructose-1,6-bisphosphate, serine, and succinyl-5-aminoimidazole-4-carboxamide-1-ribose-50-phosphate (SAICAR), which is generated during de novo purine nucleotide biosynthesis. Curiously, oncogenic PKM2 shows much less pyruvate kinase activity than PKM1 [142], and PEP consequently accumulates in PKM2-expressing cancer cells. In this setting, phosphate from PEP is transferred to the catalytic histidine His11 on another glycolytic enzyme, PGAM, leading to a significant enhancement of PGAM activity [143]. Subsequently, pyruvate is generated from PEP by PGAM as an alternative glycolytic pathway in cancer cells [143]. This connection between PKM2 and PGAM via metabolites forms another positive feedback loop that maintains the Warburg effect. These findings suggest the possibility that as-yet-unknown metabolites could modulate the Warburg effect and potentially serve as anticancer therapies in the future. Indeed, the plant metabolite AICAR, which activates AMPK, has successfully been developed as a drug for the treatment of diabetes [144]. Human aetiology disclosed the positive statistical link between diabetes and several cancers (liver, pancreas, colon, etc.) [145], while recent data suggest that AICAR inhibits the proliferation of cancer in vitro [146]. Thus AICAR could potentially be a candidate for anticancer drug especially in diabetic cases.
In conclusion, the Warburg effect is not simply an energy boost mechanism in cancer cells. Rather, glycolysis in cancer is affected by several key factors, including hypoxia, ROS, metabolic stress, senescence-inducing stress, and growth factors. These factors are also coupled with other properties of cancer through the modulation of oncogenic signalling pathways. Furthermore, it is possible that oncogenic mutations or oncometabolites may disrupt the tight connection between glycolytic enzymes and their regulators, thereby maintaining a constitutively high flux of glycolysis. Thus, the Warburg effect connects many aspects of cancer to a metabolic shift that results from genetic reprogramming and oncogenic signalling.
References
Tarui S (1995) Glycolytic defects in muscle: aspects of collaboration between basic science and clinical medicine. Muscle Nerve Suppl 3:S2–S9. doi:10.1002/mus.880181404
Bouche C, Serdy S, Kahn CR, Goldfine AB (2004) The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev 25(5):807–830. doi:10.1210/er.2003-0026
Warburg O (1956) On respiratory impairment in cancer cells. Science 124(3215):269–270
Durany N, Joseph J, Campo E, Molina R, Carreras J (1997) Phosphoglycerate mutase, 2,3-bisphosphoglycerate phosphatase and enolase activity and isoenzymes in lung, colon and liver carcinomas. Br J Cancer 75(7):969–977
Altenberg B, Greulich KO (2004) Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 84(6):1014–1020. doi:10.1016/j.ygeno.2004.08.010
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033. doi:10.1126/science.1160809
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149(3):656–670. doi:10.1016/j.cell.2012.01.058
Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, Gu TL, Jin P, Aleckovic M, Leroy G, Kang Y, Sudderth JA, Deberardinis RJ, Luan CH, Chen GZ, Muller S, Shin DM, Owonikoko TK, Lonial S, Arellano ML, Khoury HJ, Khuri FR, Lee BH, Ye K, Boggon TJ, Kang S, He C, Chen J (2012) Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22(5):585–600. doi:10.1016/j.ccr.2012.09.020
Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D (2005) Glycolytic enzymes can modulate cellular life span. Cancer Res 65(1):177–185
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Gitenay D, Wiel C, Lallet-Daher H, Vindrieux D, Aubert S, Payen L, Simonnet H, Bernard D (2014) Glucose metabolism and hexosamine pathway regulate oncogene-induced senescence. Cell Death Dis 5:e1089. doi:10.1038/cddis.2014.63
Onodera Y, Nam JM, Bissell MJ (2014) Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J Clin Investig 124(1):367–384. doi:10.1172/JCI63146
Granchi C, Minutolo F (2012) Anticancer agents that counteract tumor glycolysis. ChemMedChem 7(8):1318–1350. doi:10.1002/cmdc.201200176
Jang M, Kim SS, Lee J (2013) Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med 45:e45. doi:10.1038/emm.2013.85
Banaszak K, Mechin I, Obmolova G, Oldham M, Chang SH, Ruiz T, Radermacher M, Kopperschlager G, Rypniewski W (2011) The crystal structures of eukaryotic phosphofructokinases from baker’s yeast and rabbit skeletal muscle. J Mol Biol 407(2):284–297. doi:10.1016/j.jmb.2011.01.019
Hasawi NA, Khandari MA, Luqmani YA (2014) Phosphofructokinase: a mediator of glycolytic flux in cancer progression. Crit Rev Oncol Hematol. doi:10.1016/j.critrevonc.2014.05.007
Prabhakar NR, Semenza GL (2012) Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev 92(3):967–1003. doi:10.1152/physrev.00030.2011
Liang J, Mills GB (2013) AMPK: a contextual oncogene or tumor suppressor? Cancer Res 73(10):2929–2935. doi:10.1158/0008-5472.CAN-12-3876
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4(11):891–899. doi:10.1038/nrc1478
Macheda ML, Rogers S, Best JD (2005) Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 202(3):654–662. doi:10.1002/jcp.20166
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25(34):4777–4786. doi:10.1038/sj.onc.1209603
Pedersen PL (2007) Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr 39(3):211–222. doi:10.1007/s10863-007-9094-x
Mathupala SP, Ko YH, Pedersen PL (2009) Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol 19(1):17–24. doi:10.1016/j.semcancer.2008.11.006
Ganapathy-Kanniappan S, Kunjithapatham R, Geschwind JF (2012) Glyceraldehyde-3-phosphate dehydrogenase: a promising target for molecular therapy in hepatocellular carcinoma. Oncotarget 3(9):940–953
Li Z, Yang P, Li Z (2014) The multifaceted regulation and functions of PKM2 in tumor progression. Biochim Biophys Acta 1846(2):285–296. doi:10.1016/j.bbcan.2014.07.008
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, Burga LN, Xie J, Jurczak MJ, DePinho RA, Clish CB, Jacks T, Kibbey RG, Wulf GM, Di Vizio D, Mills GB, Cantley LC, Vander Heiden MG (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155(2):397–409. doi:10.1016/j.cell.2013.09.025
Gao X, Wang H, Yang JJ, Liu X, Liu ZR (2012) Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 45(5):598–609. doi:10.1016/j.molcel.2012.01.001
Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z (2011) Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480(7375):118–122. doi:10.1038/nature10598
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z (2012) PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150(4):685–696. doi:10.1016/j.cell.2012.07.018
Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, Aldape K, Wei C, Guo F, Chen Y, Lu Z (2014) PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell 53(1):75–87. doi:10.1016/j.molcel.2013.11.001
Kwon OH, Kang TW, Kim JH, Kim M, Noh SM, Song KS, Yoo HS, Kim WH, Xie Z, Pocalyko D, Kim SY, Kim YS (2012) Pyruvate kinase M2 promotes the growth of gastric cancer cells via regulation of Bcl-xL expression at transcriptional level. Biochem Biophys Res Commun 423(1):38–44. doi:10.1016/j.bbrc.2012.05.063
Lee J, Kim HK, Han YM, Kim J (2008) Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol 40(5):1043–1054. doi:10.1016/j.biocel.2007.11.009
Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL (2011) Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145(5):732–744. doi:10.1016/j.cell.2011.03.054
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88(5):593–602. doi:10.1016/S0092-8674(00)81902-9
Chen Q, Ames BN (1994) Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci USA 91(10):4130–4134
Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5(8):741–747. doi:10.1038/ncb1024
Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, Beach D, Serrano M (2005) Tumour biology: senescence in premalignant tumours. Nature 436(7051):642. doi:10.1038/436642a
Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, Gil J, Beach D (2007) A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal 9(3):293–299. doi:10.1089/ars.2007.9.ft-14
Okuda J, Niizuma S, Shioi T, Kato T, Inuzuka Y, Kawashima T, Tamaki Y, Kawamoto A, Tanada Y, Iwanaga Y, Narazaki M, Matsuda T, Adachi S, Soga T, Takemura G, Kondoh H, Kita T, Kimura T (2013) Persistent overexpression of phosphoglycerate mutase, a glycolytic enzyme, modifies energy metabolism and reduces stress resistance of heart in mice. PLoS One 8(8):e72173. doi:10.1371/journal.pone.0072173
Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O (2011) Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80:825–858. doi:10.1146/annurev-biochem-060608-102511
Helmlinger G, Yuan F, Dellian M, Jain RK (1997) Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med 3(2):177–182. doi:10.1038/nm0297-177
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92(12):5510–5514
Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B (2006) HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev 20(5):557–570. doi:10.1101/gad.1399906
Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM (2006) Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J Biol Chem 281(22):15215–15226. doi:10.1074/jbc.M511408200
Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL (2003) Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res 63(19):6130–6134
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12(2):149–162. doi:10.1101/gad.12.2.149
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3(3):177–185. doi:10.1016/j.cmet.2006.02.002
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3(3):187–197. doi:10.1016/j.cmet.2006.01.012
Kennedy KM, Dewhirst MW (2010) Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol 6(1):127–148. doi:10.2217/fon.09.145
Maynard MA, Ohh M (2007) The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci CMLS 64(16):2170–2180. doi:10.1007/s00018-007-7082-2
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11(5):325–337. doi:10.1038/nrc3038
Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE (2004) HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J 23(9):1949–1956. doi:10.1038/sj.emboj.7600196
Gariboldi MB, Ravizza R, Monti E (2010) The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem Pharmacol 80(4):455–462. doi:10.1016/j.bcp.2010.05.011
Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A (2007) Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell 18(12):4691–4697. doi:10.1091/mbc.E07-04-0391
Elvert G, Kappel A, Heidenreich R, Englmeier U, Lanz S, Acker T, Rauter M, Plate K, Sieweke M, Breier G, Flamme I (2003) Cooperative interaction of hypoxia-inducible factor-2alpha (HIF-2alpha) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J Biol Chem 278(9):7520–7530. doi:10.1074/jbc.M211298200
Le Bras A, Lionneton F, Mattot V, Lelievre E, Caetano B, Spruyt N, Soncin F (2007) HIF-2alpha specifically activates the VE-cadherin promoter independently of hypoxia and in synergy with Ets-1 through two essential ETS-binding sites. Oncogene 26(53):7480–7489. doi:10.1038/sj.onc.1210566
Bingham C, Hattersley AT (2004) Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dial Transplant Off Publication Eur Dial Transpl Assoc Eur Renal Assoc 19(11):2703–2708. doi:10.1093/ndt/gfh348
Okamoto T, Mandai M, Matsumura N, Yamaguchi K, Kondoh H, Amano Y, Baba T, Hamanishi J, Abiko K, Kosaka K, Murphy SK, Mori S, Konishi I (2013) Hepatocyte nuclear factor-1beta (HNF-1beta) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol Carcinog. doi:10.1002/mc.22072
Itamochi H, Kigawa J, Terakawa N (2008) Mechanisms of chemoresistance and poor prognosis in ovarian clear cell carcinoma. Cancer Sci 99(4):653–658. doi:10.1111/j.1349-7006.2008.00747.x
Baba T, Otake H, Sato T, Miyabayashi K, Shishido Y, Wang CY, Shima Y, Kimura H, Yagi M, Ishihara Y, Hino S, Ogawa H, Nakao M, Yamazaki T, Kang D, Ohkawa Y, Suyama M, Chung BC, Morohashi K (2014) Glycolytic genes are targets of the nuclear receptor Ad4BP/SF-1. Nat Commun 5:3634. doi:10.1038/ncomms4634
Discher DJ, Bishopric NH, Wu X, Peterson CA, Webster KA (1998) Hypoxia regulates beta-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a conserved GC element. J Biol Chem 273(40):26087–26093. doi:10.1074/jbc.273.40.26087
Schafer D, Hamm-Kunzelmann B, Brand K (1997) Glucose regulates the promoter activity of aldolase A and pyruvate kinase M2 via dephosphorylation of Sp1. FEBS Lett 417(3):325–328. doi:10.1016/S0014-5793(97)01314-8
Panasyuk G, Espeillac C, Chauvin C, Pradelli LA, Horie Y, Suzuki A, Annicotte JS, Fajas L, Foretz M, Verdeguer F, Pontoglio M, Ferre P, Scoazec JY, Birnbaum MJ, Ricci JE, Pende M (2012) PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat Commun 3:672. doi:10.1038/ncomms1667
Singh PK, Mehla K, Hollingsworth MA, Johnson KR (2011) Regulation of Aerobic Glycolysis by microRNAs in Cancer. Mol Cell Pharmacol 3(3):125–134
Yang F, Zhang H, Mei Y, Wu M (2014) Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol Cell 53(1):88–100. doi:10.1016/j.molcel.2013.11.004
Sherr CJ, McCormick F (2002) The RB and p53 pathways in cancer. Cancer Cell 2(2):103–112. doi:10.1016/S1535-6108(02)00102-2
Feng Z, Levine AJ (2010) The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol 20(7):427–434. doi:10.1016/j.tcb.2010.03.004
Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL (2002) Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem 277(41):38205–38211. doi:10.1074/jbc.M203781200
Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39(2):171–183. doi:10.1016/j.molcel.2010.06.022
Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278(17):14599–14602. doi:10.1074/jbc.C300063200
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64(11):3892–3899. doi:10.1158/0008-5472.CAN-03-2904
Ro TB, Holien T, Fagerli UM, Hov H, Misund K, Waage A, Sundan A, Holt RU, Borset M (2013) HGF and IGF-1 synergize with SDF-1alpha in promoting migration of myeloma cells by cooperative activation of p21-activated kinase. Exp Hematol 41(7):646–655. doi:10.1016/j.exphem.2013.03.002
Sun J, Khalid S, Rozakis-Adcock M, Fantus IG, Jin T (2009) P-21-activated protein kinase-1 functions as a linker between insulin and Wnt signaling pathways in the intestine. Oncogene 28(35):3132–3144. doi:10.1038/onc.2009.167
Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R (2009) PAK signaling in oncogenesis. Oncogene 28(28):2545–2555. doi:10.1038/onc.2009.119
Shalom-Barak T, Knaus UG (2002) A p21-activated kinase-controlled metabolic switch up-regulates phagocyte NADPH oxidase. J Biol Chem 277(43):40659–40665. doi:10.1074/jbc.M206650200
Tunduguru R, Chiu TT, Ramalingam L, Elmendorf JS, Klip A, Thurmond DC (2014) Signaling of the p21-activated kinase (PAK1) coordinates insulin-stimulated actin remodeling and glucose uptake in skeletal muscle cells. Biochem Pharmacol. doi:10.1016/j.bcp.2014.08.033
Cooper JA, Reiss NA, Schwartz RJ, Hunter T (1983) Three glycolytic enzymes are phosphorylated at tyrosine in cells transformed by Rous sarcoma virus. Nature 302(5905):218–223. doi:10.1038/302218a0
Hitosugi T, Zhou L, Fan J, Elf S, Zhang L, Xie J, Wang Y, Gu TL, Aleckovic M, LeRoy G, Kang Y, Kang HB, Seo JH, Shan C, Jin P, Gong W, Lonial S, Arellano ML, Khoury HJ, Chen GZ, Shin DM, Khuri FR, Boggon TJ, Kang S, He C, Chen J (2013) Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat Commun 4:1790. doi:10.1038/ncomms2759
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Science Signal 2(97):ra73. doi:10.1126/scisignal.2000431
Yeung SJ, Pan J, Lee MH (2008) Roles of p53, MYC and HIF-1 in regulating glycolysis—the seventh hallmark of cancer. Cell Mol Life Sci CMLS 65(24):3981–3999. doi:10.1007/s00018-008-8224-x
Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV (1997) c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci USA 94(13):6658–6663
Kawauchi K, Araki K, Tobiume K, Tanaka N (2008) p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol 10(5):611–618. doi:10.1038/ncb1724
Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275(29):21797–21800. doi:10.1074/jbc.C000023200
Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 24(13):5923–5936. doi:10.1128/MCB.24.13.5923-5936.2004
David CJ, Chen M, Assanah M, Canoll P, Manley JL (2010) HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463(7279):364–368. doi:10.1038/nature08697
Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR (2010) The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA 107(5):1894–1899. doi:10.1073/pnas.0914845107
Mathupala SP, Heese C, Pedersen PL (1997) Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem 272(36):22776–22780. doi:10.1074/jbc.272.36.22776
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126(1):107–120. doi:10.1016/j.cell.2006.05.036
Burns DM, Richter JD (2008) CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev 22(24):3449–3460. doi:10.1101/gad.1697808
Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS Jr, Chien KR, Gualberto A (1999) p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ 10(5):295–306
Mikawa T, Maruyama T, Okamoto K, Nakagama H, Lleonart ME, Tsusaka T, Hori K, Murakami I, Izumi T, Takaori-Kondo A, Yokode M, Peters G, Beach D, Kondoh H (2014) Senescence-inducing stress promotes proteolysis of phosphoglycerate mutase via ubiquitin ligase Mdm2. J Cell Biol 204(5):729–745. doi:10.1083/jcb.201306149
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479. doi:10.1146/annurev.biochem.67.1.425
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387(6630):296–299. doi:10.1038/387296a0
Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387(6630):299–303. doi:10.1038/387299a0
Leach FS, Tokino T, Meltzer P, Burrell M, Oliner JD, Smith S, Hill DE, Sidransky D, Kinzler KW, Vogelstein B (1993) p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res 53(10 Suppl):2231–2234
Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP (1993) Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 53(12):2736–2739
Brown DR, Thomas CA, Deb SP (1998) The human oncoprotein MDM2 arrests the cell cycle: elimination of its cell-cycle-inhibitory function induces tumorigenesis. EMBO J 17(9):2513–2525. doi:10.1093/emboj/17.9.2513
Manfredi JJ (2010) The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 24(15):1580–1589. doi:10.1101/gad.1941710
McCay CM, Crowell MF (1934) Prolonging the life span. Sci Mon 39(5):405–414
Canto C, Auwerx J (2009) PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20(2):98–105. doi:10.1097/MOL.0b013e328328d0a4
Yecies JL, Manning BD (2011) mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med 89(3):221–228. doi:10.1007/s00109-011-0726-6
Menon S, Manning BD (2008) Common corruption of the mTOR signaling network in human tumors. Oncogene 27(Suppl 2):S43–S51. doi:10.1038/onc.2009.352
Rios M, Foretz M, Viollet B, Prieto A, Fraga M, Costoya JA, Senaris R (2013) AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res 73(8):2628–2638. doi:10.1158/0008-5472.CAN-12-0861
Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC (2013) AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49(6):1167–1175. doi:10.1016/j.molcel.2013.01.035
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18(3):283–293. doi:10.1016/j.molcel.2005.03.027
Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM, Jones RG (2013) AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17(1):113–124. doi:10.1016/j.cmet.2012.12.001
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403(6771):795–800. doi:10.1038/35001622
Zwaans BM, Lombard DB (2014) Interplay between sirtuins, MYC and hypoxia-inducible factor in cancer-associated metabolic reprogramming. Dis Models Mech 7(9):1023–1032. doi:10.1242/dmm.016287
Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, Li C, Veenstra TD, Li B, Yu H, Ji J, Wang XW, Park SH, Cha YI, Gius D, Deng CX (2011) SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20(4):487–499. doi:10.1016/j.ccr.2011.09.004
Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17(1):41–52. doi:10.1016/j.ccr.2009.11.023
Jeong SM, Lee A, Lee J, Haigis MC (2014) SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J Biol Chem 289(7):4135–4144. doi:10.1074/jbc.M113.525949
Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, Greenson JK, MacDonald AI, McGlynn L, Maxwell F, Edwards J, Giacosa S, Guccione E, Weissleder R, Bernstein BE, Regev A, Shiels PG, Lombard DB, Mostoslavsky R (2012) The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151(6):1185–1199. doi:10.1016/j.cell.2012.10.047
Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18(3):416–430. doi:10.1016/j.cmet.2013.07.013
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483(7388):218–221. doi:10.1038/nature10815
Chen HC, Jeng YM, Yuan RH, Hsu HC, Chen YL (2012) SIRT1 promotes tumorigenesis and resistance to chemotherapy in hepatocellular carcinoma and its expression predicts poor prognosis. Ann Surg Oncol 19(6):2011–2019. doi:10.1245/s10434-011-2159-4
Cha EJ, Noh SJ, Kwon KS, Kim CY, Park BH, Park HS, Lee H, Chung MJ, Kang MJ, Lee DG, Moon WS, Jang KY (2009) Expression of DBC1 and SIRT1 is associated with poor prognosis of gastric carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res 15(13):4453–4459. doi:10.1158/1078-0432.CCR-08-3329
Noguchi A, Kikuchi K, Zheng H, Takahashi H, Miyagi Y, Aoki I, Takano Y (2014) SIRT1 expression is associated with a poor prognosis, whereas DBC1 is associated with favorable outcomes in gastric cancer. Cancer Med. doi:10.1002/cam4.310
Yuan J, Minter-Dykhouse K, Lou Z (2009) A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 185(2):203–211. doi:10.1083/jcb.200809167
Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38(6):864–878. doi:10.1016/j.molcel.2010.05.023
Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140(2):280–293. doi:10.1016/j.cell.2009.12.041
Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC (2011) SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 19(3):416–428. doi:10.1016/j.ccr.2011.02.014
Hubbi ME, Hu H, Kshitiz, Gilkes DM, Semenza GL (2013) Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J Biol Chem 288(29):20768–20775. doi:10.1074/jbc.M113.476903
Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K, Li P, Cheng HL, Murphy AJ, Valenzuela DM, Luo H, Kapahi P, Krauss R, Mostoslavsky R, Yancopoulos GD, Alt FW, Chua KF, Chen D (2013) SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 5(3):654–665. doi:10.1016/j.celrep.2013.10.007
Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, Qian DZ (2011) HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem 286(44):38095–38102. doi:10.1074/jbc.M111.257055
Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, Guan KL (2013) Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 23(4):464–476. doi:10.1016/j.ccr.2013.02.005
Tsusaka T, Guo T, Yagura T, Inoue T, Yokode M, Inagaki N, Kondoh H (2014) Deacetylation of phosphoglycerate mutase in its distinct central region by SIRT2 down-regulates its enzymatic activity. Genes Cells Devoted Mol Cell Mech 19(10):766–777. doi:10.1111/gtc.12176
Xu Y, Li F, Lv L, Li T, Zhou X, Deng CX, Guan KL, Lei QY, Xiong Y (2014) Oxidative stress activates SIRT2 to deacetylate and stimulate phosphoglycerate mutase. Cancer Res 74(13):3630–3642. doi:10.1158/0008-5472.CAN-13-3615
Hallows WC, Yu W, Denu JM (2012) Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem 287(6):3850–3858. doi:10.1074/jbc.M111.317404
Li T, Liu M, Feng X, Wang Z, Das I, Xu Y, Zhou X, Sun Y, Guan KL, Xiong Y, Lei QY (2014) Glyceraldehyde-3-phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J Biol Chem 289(6):3775–3785. doi:10.1074/jbc.M113.531640
Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42(6):719–730. doi:10.1016/j.molcel.2011.04.025
Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, Jiang Y, Zhou X, Li TT, Guan KL, Lei QY, Xiong Y (2013) Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell 52(3):340–352. doi:10.1016/j.molcel.2013.09.004
Kaelin WG Jr, Maher ER (1998) The VHL tumour-suppressor gene paradigm. Trends Genet TIG 14(10):423–426. doi:10.1016/S0168-9525(98)01558-3
Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW (2000) Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA 97(19):10430–10435. doi:10.1073/pnas.190332597
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275. doi:10.1038/20459
Chan DA, Sutphin PD, Yen SE, Giaccia AJ (2005) Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol 25(15):6415–6426. doi:10.1128/MCB.25.15.6415-6426.2005
Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F (2004) JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 118(6):781–794. doi:10.1016/j.cell.2004.08.025
Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40(6):893–904. doi:10.1016/j.molcel.2010.12.013
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8(2):143–153. doi:10.1016/j.ccr.2005.06.017
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7(1):77–85. doi:10.1016/j.ccr.2004.11.022
Lu H, Forbes RA, Verma A (2002) Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 277(26):23111–23115. doi:10.1074/jbc.M202487200
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19(1):17–30. doi:10.1016/j.ccr.2010.12.014
Keller KE, Tan IS, Lee YS (2012) SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 338(6110):1069–1072. doi:10.1126/science.1224409
Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC (2010) Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329(5998):1492–1499. doi:10.1126/science.1188015
Musi N, Goodyear LJ (2003) AMP-activated protein kinase and muscle glucose uptake. Acta Physiol Scand 178(4):337–345. doi:10.1046/j.1365-201X.2003.01168.x
Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D (2010) Diabetes and cancer: a consensus report. Diabet Care 33(7):1674–1685. doi:10.2337/dc10-0666
Jung JW, Park SB, Lee SJ, Seo MS, Trosko JE, Kang KS (2011) Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS One 6(11):e28068. doi:10.1371/journal.pone.0028068
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mikawa, T., LLeonart, M.E., Takaori-Kondo, A. et al. Dysregulated glycolysis as an oncogenic event. Cell. Mol. Life Sci. 72, 1881–1892 (2015). https://doi.org/10.1007/s00018-015-1840-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1840-3