
Abstract
Tissue regeneration during wound healing or cancer growth and progression depends on the establishment of a cellular microenvironment. Mesenchymal stem cells (MSC) are part of this cellular microenvironment, where they functionally modulate cell homing, angiogenesis, and immune modulation. MSC recruitment involves detachment of these cells from their niche, and finally MSC migration into their preferred niches; the wounded area, the tumor bed, and the BM, just to name a few. During this recruitment phase, focal proteolysis disrupts the extracellular matrix (ECM) architecture, breaks cell–matrix interactions with receptors, and integrins, and causes the release of bioactive fragments from ECM molecules. MSC produce a broad array of proteases, promoting remodeling of the surrounding ECM through proteolytic mechanisms. The fibrinolytic system, with its main player plasmin, plays a crucial role in cell migration, growth factor bioavailability, and the regulation of other protease systems during inflammation, tissue regeneration, and cancer. Key components of the fibrinolytic cascade, including the urokinase plasminogen activator receptor (uPAR) and plasminogen activator inhibitor-1 (PAI-1), are expressed in MSC. This review will introduce general functional properties of the fibrinolytic system, which go beyond its known function of fibrin clot dissolution (fibrinolysis). We will focus on the role of the fibrinolytic system for MSC biology, summarizing our current understanding of the role of the fibrinolytic system for MSC recruitment and the functional consequences for tissue regeneration and cancer. Aspects of MSC origin, maintenance, and the mechanisms by which these cells contribute to altered protease activity in the microenvironment under normal and pathological conditions will also be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During development, wound healing, normal organ homeostasis, or cancer progression, dynamic remodeling of the extracellular matrix (ECM) is necessary for cell migration. ECM remodeling is primarily accomplished by controlling the expression or activities of ECM enzymes like matrix metalloproteinases (MMPs), and the serine protease plasmin [1]. Although the plasminogen system is best known for its fibrinolytic activity, namely clot dissolution after thrombosis, recent evidence suggests that plasmin plays a crucial role in modulating cell migration and proliferation.
Mesenchymal stem cells (MSC) have been identified as candidates for cell-based therapies in regenerative medicine and as vehicles for delivering therapeutic agents to areas of injury and tumors. However, the signals required for homing and recruitment of stem cells to these sites are not well understood. The two major fibrinolytic factors urokinase plasminogen activator (uPA; also known as urokinase) and uPA receptor (uPAR) are up-regulated in the tumor niche of various tissue origins, where they are associated with invasive and chemo-resistant cancer phenotypes. The activation of uPA and uPAR in brain, lung, prostate, and breast cancers augments MSC tropism. This chemo-attraction of MSC to cancer cells correlates with uPAR expression levels in tumor cells, which may be important for the development of optimal stem cell-based anti-cancer therapies [2]. Therefore, MSC have been suggested as the perfect candidate for cellular drug delivery and novel cancer treatment strategies. Similarly, activation of the fibrinolytic system was observed during stress-induced hematopoiesis caused by chemotherapy or irradiation [3, 4] or by administration of growth factors [5, 6] or during inflammation.
The niche encompasses all of the elements immediately surrounding normal or malignant stem cells when they are in their naïve state, including the non-stem cells that might be in direct contact with them, as well as the ECM and soluble molecules. All of these factors act together to maintain stem cells in their undifferentiated state. MSC are one of the cellular components of the hematopoietic niche within the bone marrow (BM), but can also be found in other niches like the cancer niche, the site of a growing tumor. BM-derived MSC can also be found in peripheral locations where they interact with perivascular cells and can respond appropriately following tissue injury or during cancer progression. The hematopoietic niche, harboring hematopoietic stem cells (HSC), is one of the most studied niches. A complex interplay of cytokines, chemokines, proteases, and adhesion molecules ensures cell anchorage and the potential of cells, like stem cells, to respond to external stimuli allowing for a well-balanced cellular response within the microenvironment/niche.
MSC display robust reparative properties through their ability to limit apoptosis, enhance angiogenesis, and direct positive tissue remodeling. In this review, we will discuss various aspects of the fibrinolytic system for cell migration of both myeloid cells and MSC and the implications for normal cell homeostasis and diseased tissues.
MSC origin and functional properties
MSC are adult stem cells and were first described in human tissues by Friedenstein nearly 50 years ago [7–9]. Stromal cells fulfilling MSC characteristics can be isolated from almost every type of connective tissue [10]. Human MSC have been isolated from BM [11], adipose tissues [12], cord blood [13], amniotic fluid [14], and umbilical cord tissues [15]. BM-MSC are thought to be derived from the BM stromal compartment [16]. MSC have mainly been characterized after isolation from the BM, where they make up about 0.001–0.01 % of the BM cells. To date, the colony-forming unit—fibroblast assay has been considered as one of the gold standards for determining the incidence of clonogenic BM-MSC and quantifying functional MSC in vitro (Fig. 1).

Characterization of MSC. MSC are multipotent cells that are characterized by their fibroblastic shape (see in the center cultured murine MSC), their ability to differentiate into mesenchymal lineages, i.e., osteoblasts, chondrocytes and adipocytes, their expression of species-specific surface antigen marker sets, their ability to secrete chemokines or growth factors (e.g., CXCR5, MCP-1, TGF-β, hepatic growth factors, and interleukin (IL)-6/-10), and their involvement in diseases like inflammatory diseases, ischemia, tissue lesions, and cancer
The International Society for Cellular Therapy defines the minimal criteria for all human MSC populations as follows. Human MSC (a) remain plastic adherent under standard culture conditions; (b) express CD105, CD73, and CD90, and lack expression of CD45, CD34, CD14 or CD11b, CD79a or CD19, and HLA-DR; and (c) differentiate into osteoblasts, adipocytes, and chondrocytes in vitro [17, 18] (Fig. 1). Mouse MSC lack the expression of the hematopoietic leukocyte marker CD45 and the erythroid lineage marker Ter119, but show positivity for platelet-derived growth factor receptor α and stem cell antigen 1 [19].
BM-MSC secrete the following cytokines at sufficient high levels (Fig. 1): interleukin (IL)-6, interleukin-8, tissue inhibitor of metalloproteinase 2, monocyte chemoattractant protein-1 (MCP-1), vascular endothelial growth factor-A (VEGF-A) and osteoprotegerin [20]. Other studies have demonstrated that MSC secrete the following cytokines: macrophage-, granulocyte–macrophage-, granulocyte-colony-stimulating factor, IL-11, IL-7, thrombopoietin, kit ligand (also known as stem cell actor), FMS-like tyrosine kinase 3 ligand, hepatocyte growth factor (HGF), stromal-derived factor-1 (also known as C-X-C motif chemokine 12), transforming growth factor-β (TGF-β, insulin-like growth factor-1, certain platelet-derived growth factors, and fibroblast growth factor-2 (also known as FGF-2) [20].
MSC are currently widely used in clinical trials for cell therapy and regenerative medicine applications, with around 300 registered clinical trails of MSC-based therapy on ClinicalTrails.gov. Whether MSC are used in basic research or in translational studies, MSC should be expanded to meet the required cell amounts for clinical use. The in vitro expansion of MSC can be achieved using the following growth factors: platelet-derived growth factor-A, platelet-derived growth factor-BB, FGF-2, epidermal growth factor, TGF-β3, VEGF-A, and bone morphogenic protein-3. MSC possess self-renewal ability and show multilineage differentiation into not only mesoderm lineages, such as chondrocytes, osteocytes and adipocytes, but also ectodermic cells and endodermic cells [21].
The route of administration is an important factor determining the fate of MSC for treatment. The favorite route of administration in human is intravenous, as it allows the administration of large amounts of MSC. MSC are short-lived and do not migrate beyond the lungs after intravenous infusion [22–25], most likely due to their size (between 15 and 19 μm in diameter) [26]. Administration of MSC via alternative routes leads to detainment of MSC in other filtering organs. For instance, MSC administered via the portal vein are found in the liver [27], while MSC administered in tissues like muscle, spine, and fat pads remain present locally up to several weeks [28].
The fibrinolytic system: from ECM protein degradation to MMP activation
Activators and their receptors of the fibrinolytic system
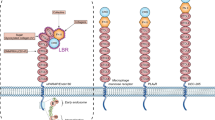
The fibrinolytic system has been implicated in embryogenesis, wound healing and angiogenesis, and in a variety of pathological conditions such as infection, tumor growth, and metastasis [29–31]. The molecule at the center of the fibrinolytic cascade is the serine protease plasmin (Fig. 2). Plasminogen, the inactive precursor of plasmin, is produced by the liver and circulates at relative concentrations of around 2 μM, making it an abundant zymogen in plasma and interstitial fluids, where its concentration can be increased upon inflammation or injury through exudation from the vascular system [32]. Plasmin is generated through the activation of plasminogen by tissue-type plasminogen activator (tPA), uPA, or kallikrein (Fig. 2). Although tPA and uPA catalyze the same reaction, tPA and uPA have traditionally been regarded as conveying different signals: tPA is thought to activate plasminogen during fibrinolysis, whereas uPA activates cell-associated plasminogen during cell migration or in tissues experiencing persistent tissue remodeling, such as wounds or the tumor microenvironment [33].

MSC generate a local proteolytic niche that alters the cellular and chemical composition of the niche. MSC are part of the stromal microenvironment. Urokinase-type plasminogen activator receptor (uPAR)-expressing MSC can bind the inactive zymogen pro-uPA (pro-urokinase-type plasminogen activator) and convert it in its active form uPA, which in turn causes pericellular proteolysis at the cell surface by converting plasminogen to plasmin. Tissue-type plasminogen activator (tPA) or kallikrein can convert plasminogen into plasmin in circulation. uPA can be inhibited by, e.g., plasminogen activator inhibitor type 1 (PAI-1) and α2-antiplasmin. Generated plasmin can activate proteases like MMPs, modulate growth factors or bioactive fragments of ECM molecules, thereby regulating the recruitment of inflammatory cells or MSC. Plasmin can break down cell–matrix interactions with receptors, such as integrins, and control cell adhesion through a delicate balance between uPA/PAI-1/fibrin/uPAR. Plasmin and PAI-1 promote cellular motility by regulating the interaction between uPAR and vitronectin: PAI-1, which has a higher affinity to vitronectin can compete for uPAR binding to vitronectin. Complexes of full-length uPAR and its ligands interact with α and β integrin co-receptors for intracellular signal transduction with signaling pathways that are involved in cell proliferation/maintenance/survival (RAS-ERK 1/2, Akt-PI3 K), cell differentiation (RhoA-ROCK), and cell apoptosis (FAK-JNK/p38)
Since the major components of the uPA/plasmin systems are secreted proteins, activation of plasminogen by uPA can occur extracellularly, albeit in close proximity to the uPA-expressing endothelial or cancer cell. uPAR is associated with the external surface of the plasma membrane by a glycosyl phosphatidylinositol anchor [34] and localizes uPA and pro-uPA to the cell surface. uPA is synthesized as a single-chain protein with little intrinsic activity. Single-chain uPA (pro-uPA) can bind to uPAR (CD87) and will be converted into the two-chain active uPA by plasmin. Activated uPA cleaves the zymogen plasminogen, generating plasmin, which reciprocally cleaves and activates pro-uPA [35–39]. Alternatively, kallikreins 2, 4 and 12 can activate single-chain uPA. uPAR is expressed by monocytes, macrophages, neutrophils, vascular endothelial, smooth muscle, epithelial cells, and MSC [40].
Inhibitors of the fibrinolytic system
The activity of the fibrinolytic system is regulated by plasmin inhibitors such as α2-antiplasmin and α2-macroglobulin, and PA inhibitors (PAIs) such as PA inhibitor type 1 (PAI-1, also known as serpin E1) and PAI-2. The principal PAIs are PAI-1 [41], PAI type 2 (PAI-2), also known as placental-type PAI [42], and PAI type 3, which is identical to protein C inhibitor [43, 44]. PAI-1 is produced by endothelial cells, megakaryocytes, smooth muscle cells, fibroblasts, monocytes/macrophages, adipocytes, endometrium, peritoneum, liver cells, mesothelial cells, cardiac myocytes, and—as discussed later in more detail—in MSC [45, 46]. PAI-1 is mainly stored in platelets, but it also can be deposited on the subendothelial matrix. PAI-1 in the blood stream is present in an active form or, more frequently, complexed with either tPA or vitronectin (a relatively thermo-stable glycoprotein, which is able to stabilize and convert PAI-1 into an active form) [47]. uPAR-bound uPA is active and susceptible to inhibition by PAI-1 and PAI-2.
Plasmin as an upstream regulator of MMPs
Plasmin can degrade additional components of the ECM through its ability to convert pro-MMPs to active MMPs, including MMP-1, MMP-2, MMP-3, and MMP-9 [3, 48, 49] (Fig. 2). Therefore, plasmin has been suggested as an important upstream regulator of extracellular proteolysis [29]. Thus, once activated, MMP-2, -3, -7, -9, and -12 can initiate a negative feedback signal by degrading plasminogen [50, 51]. Degradation of plasmin-suppressing serpin proteinase inhibitors (e.g., α2-antiplasmin [52]) by, e.g., MMP-3, which promotes the conversion of pro-MMPs, is an example of how both proteolytic systems control each other.
Fibrinolytic factors involvement during hematopoietic cell regeneration
Stem cell fate is regulated by a combination of intrinsic and extrinsic mechanisms. Intrinsic mechanisms include specific transcription factors expressed by cells. Extrinsic mechanisms are signals provided by the local microenvironment (niche), including growth factors, the ECM [53], and protease activation in the niche (“proteolytic niche”). One of the best-studied niches within the body is the HSC niche within the BM, where HSC can differentiate and proliferate in response to hematopoietic stress (e.g., myelosuppression or ionizing irradiation), thereby ensuring a well-regulated supply of mature and immature hematopoietic cells within the circulation and prompt adjustment of blood cell levels within normal ranges [4]. The HSC niche includes perivascular MSC, macrophages, sinusoidal endothelial cells, sympathetic nerve fibers, and osteoblasts [54]. These niche cells harbor dormant and self-renewing HSC closely associated with nestin-expressing MSC [55]. Nestin+ MSC are present in the vicinity of the endosteum and tightly associated with adrenergic nerve fibers of the sympathetic nervous system that regulate the circadian oscillations in circulating HSC [56, 57].
The recovery of tissues and organs from irradiation therapy or chemotherapy is dependent on stromal cells and resident HSC, which repopulate the BM cavity and give rise to differentiated, functional blood cells. HSC, and their progeny, BM stromal cells, and the related vasculature/stromal cells can be damaged by myelosuppressive stress [58, 59]. Therefore, it is necessary that both the hematopoietic and the stromal compartment recover after myelosuppression. The presence of fibrinolytic factors within the BM after myelosuppression in vivo suggests that localized proteolysis occurs during hematopoietic reconstitution. BM cell recovery occurred after treatment with the myelosuppressive drug 5-fluorouracil in plasminogen wild type, but not plasminogen deficient mice [3], suggesting a necessity for fibrinolytic factor activation during hematopoietic regeneration. We delineated a mechanism whereby myelosuppression activates the proteolytic cascade involving the successive activation of plasminogen and MMP-9 that ultimately leads to the release of the hematopoietic cytokine kit ligand that accelerates hematopoietic recovery [3]. The importance of the activation of the fibrinolytic system was also shown in mice after total body irradiation [60]. Genetic disruption of the PAI-1 gene or pharmacological inhibition of PAI-1 activity improved myeloablation-related mortality and promoted rapid hematopoietic recovery after HSC transplantation. These studies set forth the idea that activation of the fibrinolytic system can regulate cytokine and growth factor bioavailability necessary for tissue regeneration.
Role of the fibrinolytic system for cell migration
Cell migration of inflammatory cells requires the coordinated activation of integrins, coagulation/fibrinolysis, and endocytosis. uPA–uPAR-mediated plasminogen activation facilitates cell migration by enhancing pericellular proteolysis [36]. uPAR lacks transmembrane and intracellular domains, so it requires co-receptors like integrins or vitronectin to control migration. The binding of uPA to uPAR promotes cell adhesion by increasing the affinity of uPAR for both vitronectin and integrins [61–63] (Fig. 2). PAI-1 can detach cells by disrupting uPAR–vitronectin and integrin–vitronectin interactions and it does so by binding to the uPA present in uPA–uPAR–integrin complexes on the cell surface [64, 65]. It has been proposed that the endocytic clearance of the complexes of integrins, uPA, uPAR, and PAI-1 can lead to the disengagement of integrins from the ECM and cell detachment [66], a process necessary for cancer metastasis. The idea that PAI-1 is a deadhesive factor toward a variety of cells growing on different ECM could explain why high PAI-1 levels can be detected in patients with human metastatic disease and are often associated with a poor prognosis.
Colocalization of uPAR with integrins, like α4β1, at the leading edge of migrating cells focuses uPA activity in the direction of movement [67]. Similarly, the Mac-1 integrin (also called M2 or CD11b/18) and plasmin activation have been shown to augment macrophage accumulation in the peritoneal cavity [68–70].
uPAR consists of three disulfide-bonded domains (D1, D2, and D3). Domains D1 and D3 represent a composite binding site for uPA. Cleavage in the D1–D2 linker by uPA [71], plasmin, and MMPs [72] creates a soluble D1 fragment and a D2–D3 fragment that can be membrane associated or shed [72]. Soluble uPAR (suPAR) is released from cell membrane-bound uPAR [71]. It can be found in blood, urine, and pleural fluid. suPAR has been implicated as a biomarker for inflammatory and immune diseases. The peptide sequence Ser-Arg-Ser-Arg-Tyr, near the N terminus of the D2–D3 fragment, interacts with the G protein-coupled receptor formyl peptide receptor-like 1, inducing cell migration [73]. The cleaved form of suPAR binds and activates fMet-Leu-Phe receptors and regulates the activity of MCP-1, Chemokine (C–C motif) ligand 5 and C-X-C chemokine receptor type 4. Soluble D2–D3 is a chemoattractant for formyl peptide receptor-like 1-expressing monocytes and basophils [74]. Plasmin cleaves uPAR on HSC, causing HSC release into circulation [75]. suPAR is up-regulated during granulocyte colony-stimulating factor-induced HSC mobilization in humans in vitro and in vivo [76, 77].
Whereas HSC mobilization is a well-established fact due to their appearance in the circulation, the notion of MSC mobilization is still debated. MSC could not be detected in the blood of healthy individuals [78]. In contrast, under conditions of severe organ injury (e.g., a trauma patient with multiple bone fractures or hip fractures) MSC could be found in circulation. No circulating MSC were detected in the blood of patients with end-stage kidney failure, end-stage liver failure, or during rejection episode after heart transplantation [79]. MSC as BM niche cells express uPAR [80]. A role for uPAR in MSC mobilization, similar to HSC mobilization, was proposed. MSC mobilization after granulocyte colony-stimulating factor administration did not occur in uPAR−/− mice [80]. These studies suggest an important role of the fibrinolytic system in cell migration.
The glycosylphosphatidylinositol-anchored uPAR regulates cell migration, adhesion, proliferation, and differentiation through activation of an intracellular signaling network (Fig. 2): the prosurvival phosphatidylinositol-4,5-bisphosphate 3-kinase/Akt and ERK1/2 signaling, and focal adhesion kinase (FAK) pathways [81–83]. uPAR also associates with endocytic receptor 180, a constitutively recycling collagen receptor of the mannose receptor family [84]. This interaction leads to activation of Rho GTPases Rac and Cdc42, which regulate filamentous actin assembly and directional migration, driving chemotaxis up a gradient of catalytically inactive uPA [85].
Plasmin controls chemokine/cytokine expression
The ECM anchors soluble growth factors, increasing the local concentration of agonists to which target populations in the niche are exposed [4, 86–88]. MMPs and plasmin regulate the repertoire of available extracellular growth factors by enzymatic activation, inactivation, or degradation e.g., the conversion of the latent form of TGF-β or platelet-derived growth factor C [3, 89–91].
Plasmin has been called an inflammatory molecule [92]. We found that plasmin is activated during acute graft versus host disease, a disease occurring after allogeneic cell transplantation, and that inhibition of plasmin prevented the disease by reducing the chemokine–cytokine-driven myeloid cell influx and cytokine storm. Plasmin induced macrophage infiltration by enhancing MCP-1 signaling through the release of a MCP-1 fragment with improved chemoattractive ability for macrophages [93, 94]. CD11b+F4/80+ macrophage recruitment into the growing tumor was dependent on plasmin and MMP activation in a murine model of lymphoma [95].
Besides macrophage infiltration, various reports showed the importance of plasmin for neutrophil recruitment. Plasmin enhanced Gr-1+ neutrophil-driven neoangiogenesis during recovery from hindlimb ischemia [46], whereby these cells provided the ischemic tissue with FGF-2 and VEGF-A. Enterocyte-derived CXC receptor type 5 can attract CXCR2+ neutrophils into the gut tissues [96]. Plasmin, by altering MMP2 and MMP9 activity, can process CXCL5 and can promote neutrophil recruitment in models of peritonitis [97] and inflammatory bowel disease [88]. Even hormones can be controlled by plasmin, e.g., decreased serum testosterone levels in plasminogen deficient mice were found as a consequence of impaired secretion of the pituitary luteinizing hormone under steady-state conditions [98].
Fibrinolytic factors modify MSC adhesion and survival during wound healing
It was shown that kallikrein-mediated plasmin enhanced wound healing in mice [99]. The importance of plasmin for wound healing was further demonstrated in Plg−/− mice, which showed severely retarded wound healing [100]. In humans, loss of plasmin similarly leads to abnormal wound healing. Patients with advanced diabetes mellitus show impaired wound healing, which is accompanied by elevated circulating PAI-1 levels [101]. Mimicking the situation in human disease, diabetic mice showed high circulating PAI-1 levels. Plasminogen treatment improved the healing of acute burn wounds and chronic diabetic wounds in these mice [102, 103].
A disrupted blood flow can bring tissue regeneration and wound healing processes to a deadlock [104]. MSC are recruited into wounds [105, 106] where they can accelerate wound healing [107]. Mouse and human MSC under steady-state conditions express uPA, uPAR, and PAI-1 [108–110]. Interestingly, uPAR and PAI-1 are hypoxia inducible factor α (HIF-1α) targets [111]. In MSC, HIF-1α activation in MSC mediates the upregulation of FGF-2 and HGF, whereas HIF-2α upregulates VEGF-A [112]. All these factors can promote wound healing. In addition, HGF enhances the recruitment of MSC into wounded tissue [113]. Ischemia and hypoxia are major causes of wound repair dysregulation. Even though the natural habitats of MSC are tissues with low oxygen level, e.g., the BM or adipose tissues, in vivo survival of transplanted MSC limits their overall effectiveness and affects their clinical usage [114]. Transplanted cells often die under unfavorable niche conditions as found in wound, scar, or hypoxic tissue [115–117]. During the early phase of the wound healing process, plasminogen bound to inflammatory cells is transported to the wound area, where high plasminogen levels can be detected [118, 119]. Neuss et al. reported that cultured human uPAR-expressing MSC show fibrinolytic activity [120]. During wound healing, migrating cells increase the expression of PAs. The balance between uPA (proadhesive) and PAI-1 (nonadhesive) regulates MSC adhesion to vitrogen and matrigel matrices [109]. PAI-1, which has a higher affinity to vitronectin can compete for uPAR binding with vitronectin [121] thereby determining an adhesive or nonadhesive effect [109, 122, 123]. It was reported that complex formation between PAI-1 and PAs results in loss of PAI-1 affinity for vitronectin and restores cell migration. These studies indicate that fibrinolytic factors secreted by MSC affect MSC survival and adhesion under hypoxic conditions (Fig. 2).
Fibrinolytic factors accelerate MSC recruitment into the tumor microenvironment
Promigratory conditioning of the tumor-associated tissue increases the invasion and dissemination of tumor cells, and the motility and activity of stromal cells (fibroblasts, macrophages and MSC). The promigratory tumor niche/microenvironment is dominated by proteases. Proteases support pericellular proteolysis and process extracellular chemokines and growth factors released by tumor or activated stromal cells that induce the transition from tissue-anchored to mobile cells.
Studies have shown that differential gene expression of chemokine receptors and adhesion molecules in MSC [124] induces MSC to migrate toward a specific tumor microenvironment. uPAR, which is consistently expressed by MSC, is a poor prognostic factor under various pathological conditions, including inflammation or cancer [36]. The activation of uPA and uPAR in malignant solid tumors (brain, lung, prostate, and breast) promotes the recruitment of MSC to the tumor site [125–127]. uPA and uPAR are involved in chemotaxis and cell guidance. Improved tumor tropic properties of MSC were reported after overexpression of uPA in cord blood-MSC [128]. uPAR down-regulation inhibits human MSC migration [80]. In addition, uPAR down- or upregulation results in inhibition or stimulation of MSC differentiation into vascular smooth muscle cells, correspondingly.
uPA and uPAR are up-regulated in tumors of various origins, where they play critical roles in the development of invasive and chemo-resistant cancer phenotypes. Colon cancer cell/MSC interactions seem to regulate colon cancer progression [129]. BM-MSC were shown to stimulate invasion, survival, and tumorigenesis of colorectal cancer (CRC) cells through the release of soluble neuregulin 1, thereby activating the human epidermal growth factor receptor 2/3 (HER2/3)-dependent PI3K/AKT signaling cascade in CRC cells. Similarly, tumor-associated mesenchymal cells in CRC demonstrated high transmembrane neuregulin 1 expression. High neuregulin 1 expression was associated with advanced cancer stage and invasion depth in human cancer tissues and decreased 5-year progression-free patient survival [129]. A strong correlation has been found between HER2, uPAR expression, and breast cancer progression [130]. Further studies will be necessary to understand the role of MSC in this context.
Sier et al. demonstrated a tenfold increase in PAI-1 and a two- to threefold increase in PAI-2 between normal colonic epithelium and colonic adenocarcinoma [131]. It was reported that MSC-secreted PAI-1 enhances the migration of colon cancer cells [126]. This might be due to the fact that uPAR and PAI-1 have vitronectin binding sites and thus PAI-1, which has a higher affinity to vitronectin, could compete for uPAR binding to vitronectin [121] enhancing cell detachment, and migration. uPA–uPAR knockdown in PC3 prostate cancer cells inhibited tumor-specific migration of uPA-overexpressing MSC [2].
These data indicate that fibrinolytic factors or its receptors recruit MSC within the tumor microenvironment, which ultimately alters tumor growth.
Conclusions
The timely removal of blood clots and fibrin deposits is achieved by the fibrinolytic system, an enzymatic process that regulates the activation of plasminogen into plasmin. What has now come to light is that the fibrinolytic system, with plasmin as its main player, is not solely designed to eliminate fibrin. The fibrinolytic system can modify the cellular recruitment of inflammatory cells or MSC and alter the proteolytic activity in areas of wound healing and regenerative processes of mesodermal tissues such as bone, cartilage, and muscle, but also during cancer progression. MSC are in clinical use for the treatment of various diseases including ischemia, wound healing, and myocardial infarction, and have drawn attention because they can promote heart tissue recovery from massive myocardial infarction and improve symptoms in individuals with type 1 diabetes. Recent studies indicate that MSC are equipped with various proteases, including enzymes of the fibrinolytic system, e.g., PAI-1 and uPAR. Functionally, it seems that fibrinolytic factors ensure the ability of MSC to navigate through or adhere to the ECM of “regenerative” or ‘tumor” microenvironments. Fibrinolytic factors released from MSC set a proteolytic environment due to consecutive activation of other proteases like MMPs, which enable the release of local cytokines and chemokines. The receptor for uPA, uPAR, also represents an interesting cancer treatment target. MSC seem to be able to redirect the microenvironmental program and influence the outcome of the “tissue healing program”. Genetic lesions or certain disease conditions, like diabetes mellitus, that alter the function of MSC and the intrinsic fibrinolytic capacity of MSC may impinge on the ability of the body to mount a proper regenerative or immunological response.
These findings highlight the complexity of proteolytic interactions in the microenvironment, and show how microenvironmental cells like MSC impact tissue regeneration or cancer growth.
Abbreviations
- MSC:
-
Mesenchymal stem cells
- ECM:
-
Extracellular matrix
- BM:
-
Bone marrow
- IL:
-
Interleukin
- VEGF-A:
-
Vascular endothelial growth factor-A (VEGF-A)
- MMP:
-
Matrix metalloproteinase
- uPAR:
-
Urokinase-type plasminogen activator receptor
- PAI-1:
-
Plasminogen activator inhibitor-1
- uPA:
-
Urokinase-type plasminogen activator
- CD:
-
Cluster of differentiation
- FGF-2:
-
Fibroblast growth factor-2
- TGF-β:
-
Transforming growth factor-β
- tPA:
-
Tissue-type plasminogen activator
- HSC:
-
Hematopoietic stem cell
- MCP-1:
-
Monocyte chemoattractant protein-1
- suPAR:
-
Soluble uPAR
- PA:
-
Plasminogen activator
- HIF-1α:
-
Hypoxia inducible factor α
- HGF:
-
Hepatocyte growth factor
- CRC:
-
Colorectal cancer
- HER 2:
-
Human epidermal growth factor receptor 2
References
Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196:395–406
Gutova M, Najbauer J, Frank RT, Kendall SE, Gevorgyan A et al (2008) Urokinase plasminogen activator and urokinase plasminogen activator receptor mediate human stem cell tropism to malignant solid tumors. Stem Cells 26:1406–1413
Heissig B, Lund LR, Akiyama H, Ohki M, Morita Y et al (2007) The plasminogen fibrinolytic pathway is required for hematopoietic regeneration. Cell Stem Cell 1:658–670
Heissig B, Ohki M, Ishihara M, Tashiro Y, Nishida C et al (2009) Contribution of the fibrinolytic pathway to hematopoietic regeneration. J Cell Physiol 221:521–525
Tjwa M, Moura R, Moons L, Plaisance S, De Mol M et al (2009) Fibrinolysis-independent role of plasmin and its activators in the hematopoietic recovery after myeloablation. J Cell Mol Med 13:4587–4595
Gong Y, Fan Y, Hoover-Plow J (2011) Plasminogen regulates stromal cell-derived factor-1/CXCR4-mediated hematopoietic stem cell mobilization by activation of matrix metalloproteinase-9. Arterioscler Thromb Vasc Biol 31:2035–2043
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP (1968) Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 6:230–247
Latsinik NV, Luria EA, Friedenstein AJ, Samoylina NL, Chertkov IL (1970) Colony-forming cells in organ cultures of embryonal liver. J Cell Physiol 75:163–165
Friedenstein AJ, Deriglasova UF, Kulagina NN, Panasuk AF, Rudakowa SF et al (1974) Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp Hematol 2:83–92
da Silva Meirelles L, Chagastelles PC, Nardi NB (2006) Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci 119:2204–2213
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R et al (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI et al (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279–4295
Bieback K, Kern S, Kluter H, Eichler H (2004) Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 22:625–634
Tsai MS, Lee JL, Chang YJ, Hwang SM (2004) Isolation of human multipotent mesenchymal stem cells from second-trimester amniotic fluid using a novel two-stage culture protocol. Hum Reprod 19:1450–1456
Kim JW, Kim SY, Park SY, Kim YM, Kim JM et al (2004) Mesenchymal progenitor cells in the human umbilical cord. Ann Hematol 83:733–738
Friedenstein AJ (1980) Stromal mechanisms of bone marrow: cloning in vitro and retransplantation in vivo. Haematol Blood Transfus 25:19–29
Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I et al (2005) Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy 7:393–395
Lv FJ, Tuan RS, Cheung KM, Leung VY (2014) Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells 32:1408–1419
Houlihan DD, Mabuchi Y, Morikawa S, Niibe K, Araki D et al (2012) Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-α. Nat Protocols 7:2103–2111
Park CW, Kim KS, Bae S, Son HK, Myung PK et al (2009) Cytokine secretion profiling of human mesenchymal stem cells by antibody array. Int J Stem Cells 2:59–68
Prockop DJ (1997) Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 276:71–74
Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK et al (2012) Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol 3:297
Fischer UM, Harting MT, Jimenez F, Monzon-Posadas WO, Xue H et al (2009) Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev 18:683–692
Aurich H, Sgodda M, Kaltwasser P, Vetter M, Weise A et al (2009) Hepatocyte differentiation of mesenchymal stem cells from human adipose tissue in vitro promotes hepatic integration in vivo. Gut 58:570–581
Boulland JL, Leung DS, Thuen M, Vik-Mo E, Joel M et al (2012) Evaluation of intracellular labeling with micron-sized particles of iron oxide (MPIOs) as a general tool for in vitro and in vivo tracking of human stem and progenitor cells. Cell Transplant 21:1743–1759
Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC et al (2007) Stem Cell Transplantation: the lung barrier. Transpl Proc 39:573–576
Shi XL, Gu JY, Han B, Xu HY, Fang L et al (2010) Magnetically labeled mesenchymal stem cells after autologous transplantation into acutely injured liver. World J Gastroenterol 16:3674–3679
Hu SL, Lu PG, Zhang LJ, Li F, Chen Z et al (2012) In vivo magnetic resonance imaging tracking of SPIO-labeled human umbilical cord mesenchymal stem cells. J Cell Biochem 113:1005–1012
Collen D, Lijnen HR (1991) Basic and clinical aspects of fibrinolysis and thrombolysis. Blood 78:3114–3124
Rijken DC, Lijnen HR (2009) New insights into the molecular mechanisms of the fibrinolytic system. J Thromb Haemost 7:4–13
Heissig B, Ohki-Koizumi M, Tashiro Y, Gritli I, Sato-Kusubata K et al (2012) New functions of the fibrinolytic system in bone marrow cell-derived angiogenesis. Int J Hematol 95:131–137
Fuchs H, Simon MM, Wallich R, Bechtel M, Kramer MD (1996) Borrelia burgdorferi induces secretion of pro-urokinase-type plasminogen activator by human monocytes. Infect Immun 64:4307–4312
Deryugina EI, Quigley JP (2012) Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol 2012:564259
Ploug M, Ronne E, Behrendt N, Jensen AL, Blasi F et al (1991) Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem 266:1926–1933
Santibanez JF (2013) Transforming growth factor-Beta and urokinase-type plasminogen activator: dangerous partners in tumorigenesis-implications in skin cancer. ISRN Dermatol 2013:597927
Smith HW, Marshall CJ (2010) Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol 11:23–36
Cesarman-Maus G, Hajjar KA (2005) Molecular mechanisms of fibrinolysis. Br J Haematol 129:307–321
Nielsen LS, Hansen JG, Skriver L, Wilson EL, Kaltoft K et al (1982) Purification of zymogen to plasminogen activator from human glioblastoma cells by affinity chromatography with monoclonal antibody. Biochemistry 21:6410–6415
Ellis V, Behrendt N, Dano K (1991) Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem 266:12752–12758
Solberg H, Ploug M, Hoyer-Hansen G, Nielsen BS, Lund LR (2001) The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J Histochem Cytochem 49:237–246
Loskutoff DJ, van Mourik JA, Erickson LA, Lawrence D (1983) Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc Natl Acad Sci USA 80:2956–2960
Kruithof EK, Baker MS, Bunn CL (1995) Biological and clinical aspects of plasminogen activator inhibitor type 2. Blood 86:4007–4024
Heeb MJ, Espana F, Geiger M, Collen D, Stump DC et al (1987) Immunological identity of heparin-dependent plasma and urinary protein C inhibitor and plasminogen activator inhibitor-3. J Biol Chem 262:15813–15816
Schleef RR, Loskutoff DJ (1988) Fibrinolytic system of vascular endothelial cells. Role of plasminogen activator inhibitors. Haemostasis 18:328–341
Zorio E, Gilabert-Estelles J, Espana F, Ramon LA, Cosin R et al (2008) Fibrinolysis: the key to new pathogenetic mechanisms. Curr Med Chem 15:923–929
Tashiro Y, Nishida C, Sato-Kusubata K, Ohki-Koizumi M, Ishihara M et al (2012) Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood 119:6382–6393
Loskutoff DJ, Samad F (1998) The adipocyte and hemostatic balance in obesity: studies of PAI-1. Arterioscler Thromb Vasc Biol 18:1–6
Werb Z (1997) ECM and cell surface proteolysis: regulating cellular ecology. Cell 91:439–442
Carmeliet P, Moons L, Lijnen R, Baes M, Lemaitre V et al (1997) Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet 17:439–444
Lijnen HR, Silence J, Lemmens G, Frederix L, Collen D (1998) Regulation of gelatinase activity in mice with targeted inactivation of components of the plasminogen/plasmin system. Thromb Haemost 79:1171–1176
Lijnen HR, Silence J, Van Hoef B, Collen D (1998) Stromelysin-1 (MMP-3)-independent gelatinase expression and activation in mice. Blood 91:2045–2053
Lijnen HR, Van Hoef B, Collen D (2001) Inactivation of the serpin alpha(2)-antiplasmin by stromelysin-1. Biochim Biophys Acta 1547:206–213
Watt FM, Huck WTS (2013) Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol 14:467–473
Ehninger A, Trumpp A (2011) The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med 208:421–428
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD et al (2010) Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466:829–834
Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ et al (2006) Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124:407–421
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS (2008) Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452:442–447
Gale RP (1985) Antineoplastic chemotherapy myelosuppression: mechanisms and new approaches. Exp Hematol 13(Suppl 16):3–7
Heissig B, Rafii S, Akiyama H, Ohki Y, Sato Y et al (2005) Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J Exp Med 202:739–750
Ibrahim AA, Yahata T, Onizuka M, Dan T, Strihou De, Van Ypersele De Strihou C et al (2014) Inhibition of plasminogen activator inhibitor type-1 activity enhances rapid and sustainable hematopoietic regeneration. Stem Cells 32:946–958
Kjoller L, Kanse SM, Kirkegaard T, Rodenburg KW, Ronne E et al (1997) Plasminogen activator inhibitor-1 represses integrin- and vitronectin-mediated cell migration independently of its function as an inhibitor of plasminogen activation. Exp Cell Res 232:420–429
Waltz DA, Natkin LR, Fujita RM, Wei Y, Chapman HA (1997) Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J Clin Invest 100:58–67
Czekay R-P, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet JM et al (2011) PAI-1: an Integrator of Cell Signaling and Migration. Int J Cell Biol 2011:9
Stefansson S, Lawrence DA (1996) The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature 383:441–443
Okumura Y, Kamikubo Y, Curriden SA, Wang J, Kiwada T et al (2002) Kinetic analysis of the interaction between vitronectin and the urokinase receptor. J Biol Chem 277:9395–9404
Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ (2003) Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol 160:781–791
Estreicher A, Muhlhauser J, Carpentier JL, Orci L, Vassalli JD (1990) The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J Cell Biol 111:783–792
Pluskota E, Soloviev DA, Plow EF (2003) Convergence of the adhesive and fibrinolytic systems: recognition of urokinase by integrin alpha Mbeta 2 as well as by the urokinase receptor regulates cell adhesion and migration. Blood 101:1582–1590
Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF (1998) Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 91:2005–2009
Gong Y, Hart E, Shchurin A, Hoover-Plow J (2008) Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest 118:3012–3024
Hoyer-Hansen G, Ronne E, Solberg H, Behrendt N, Ploug M et al (1992) Urokinase plasminogen activator cleaves its cell surface receptor releasing the ligand-binding domain. J Biol Chem 267:18224–18229
Andolfo A, English WR, Resnati M, Murphy G, Blasi F et al (2002) Metalloproteases cleave the urokinase-type plasminogen activator receptor in the D1-D2 linker region and expose epitopes not present in the intact soluble receptor. Thromb Haemost 88:298–306
Resnati M, Pallavicini I, Wang JM, Oppenheim J, Serhan CN et al (2002) The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc Natl Acad Sci USA 99:1359–1364
de Paulis A, Montuori N, Prevete N, Fiorentino I, Rossi FW et al (2004) Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J Immunol 173:5739–5748
Tjwa M, Sidenius N, Moura R, Jansen S, Theunissen K et al (2009) Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. J Clin Invest 119:1008–1018
Selleri C, Montuori N, Ricci P, Visconte V, Carriero MV et al (2005) Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood 105:2198–2205
Fietz T, Hattori K, Thiel E, Heissig B (2006) Increased soluble urokinase plasminogen activator receptor (suPAR) serum levels after granulocyte colony-stimulating factor treatment do not predict successful progenitor cell mobilization in vivo. Blood 107:3408–3409
Lazarus HM, Haynesworth SE, Gerson SL, Caplan AI (1997) Human bone marrow-derived mesenchymal (stromal) progenitor cells (MPCs) cannot be recovered from peripheral blood progenitor cell collections. J Hematother 6:447–455
Hoogduijn MJ, Verstegen MM, Engela AU, Korevaar SS, Roemeling-van Rhijn M et al (2014) No evidence for circulating mesenchymal stem cells in patients with organ injury. Stem Cells Dev 23:2328–2335
Vallabhaneni KC, Tkachuk S, Kiyan Y, Shushakova N, Haller H et al (2011) Urokinase receptor mediates mobilization, migration, and differentiation of mesenchymal stem cells. Cardiovasc Res 90:113–121
Syrovets T, Lunov O, Simmet T (2012) Plasmin as a proinflammatory cell activator. J Leukoc Biol 92:509–519
Gaestel M, Kotlyarov A, Kracht M (2009) Targeting innate immunity protein kinase signalling in inflammation. Nat Rev Drug Discov 8:480–499
Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L (2001) Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell 12:863–879
Nykjaer A, Conese M, Christensen EI, Olson D, Cremona O et al (1997) Recycling of the urokinase receptor upon internalization of the uPA:serpin complexes. EMBO J 16:2610–2620
Sturge J, Wienke D, East L, Jones GE, Isacke CM (2003) GPI-anchored uPAR requires Endo180 for rapid directional sensing during chemotaxis. J Cell Biol 162:789–794
Brizzi MF, Tarone G, Defilippi P (2012) Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr Opin Cell Biol 24:645–651
Hynes RO (2009) The Extracellular Matrix: not Just Pretty Fibrils. Science 326:1216–1219
Munakata S, Tashiro Y, Nishida C, Sato A, Komiyama H et al (2015) Inhibition of plasmin protects against colitis in mice by suppressing matrix metalloproteinase 9-mediated cytokine release from myeloid cells. Gastroenterology 148(565–578):e564
Rifkin DB, Mazzieri R, Munger JS, Noguera I, Sung J (1999) Proteolytic control of growth factor availability. APMIS 107:80–85
Fredriksson L, Li H, Fieber C, Li X, Eriksson U (2004) Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J 23:3793–3802
Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE et al (2008) Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR + CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112:3455–3464
Syrovets T, Simmet T (2004) Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci 61:873–885
Sato A, Nishida C, Sato-Kusubata K, Ishihara M, Tashiro Y et al (2015) Inhibition of plasmin attenuates murine acute graft-versus-host disease mortality by suppressing the matrix metalloproteinase-9-dependent inflammatory cytokine storm and effector cell trafficking. Leukemia 29:145–156
Yao Y, Tsirka SE (2010) The C terminus of mouse monocyte chemoattractant protein 1 (MCP1) mediates MCP1 dimerization while blocking its chemotactic potency. J Biol Chem 285:31509–31516
Ishihara M, Nishida C, Tashiro Y, Gritli I, Rosenkvist J et al (2012) Plasmin inhibitor reduces T-cell lymphoid tumor growth by suppressing matrix metalloproteinase-9-dependent CD11b(+)/F4/80(+) myeloid cell recruitment. Leukemia 26:332–339
Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM et al (2012) Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J Clin Investig 122:974–986
Song J, Wu C, Zhang X, Sorokin LM (2013) In vivo processing of CXCL5 (LIX) by matrix metalloproteinase (MMP)-2 and MMP-9 promotes early neutrophil recruitment in IL-1beta-induced peritonitis. J Immunol 190:401–410
Okaji Y, Tashiro Y, Gritli I, Nishida C, Sato A et al (2012) Plasminogen deficiency attenuates postnatal erythropoiesis in male C57BL/6 mice through decreased activity of the LH-testosterone axis. Exp Hematol 40:143–154
Lund LR, Green KA, Stoop AA, Ploug M, Almholt K et al (2006) Plasminogen activation independent of uPA and tPA maintains wound healing in gene-deficient mice. EMBO J 25:2686–2697
Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ et al (1996) Plasminogen and wound healing. Nat Med 2:725
Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y et al (2004) Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 53:336–346
Tamura Y, Kawao N, Okada K, Yano M, Okumoto K et al (2013) Plasminogen activator inhibitor-1 is involved in streptozotocin-induced bone loss in female mice. Diabetes 62:3170–3179
Fadini GP, Albiero M, Vigili de Kreutzenberg S, Boscaro E, Cappellari R et al (2013) Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care 36:943–949
Schaffer M, Witte M, Becker HD (2002) Models to study ischemia in chronic wounds. Int J Low Extrem Wounds 1:104–111
Clark RA, Lanigan JM, DellaPelle P, Manseau E, Dvorak HF et al (1982) Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound reepithelialization. J Invest Dermatol 79:264–269
Bensaid W, Triffitt JT, Blanchat C, Oudina K, Sedel L et al (2003) A biodegradable fibrin scaffold for mesenchymal stem cell transplantation. Biomaterials 24:2497–2502
Basiouny HS, Salama NM, Maadawi ZM, Farag EA (2013) Effect of bone marrow derived mesenchymal stem cells on healing of induced full-thickness skin wounds in albino rat. Int J Stem Cells 6:12–25
Chiellini C, Cochet O, Negroni L, Samson M, Poggi M et al (2008) Characterization of human mesenchymal stem cell secretome at early steps of adipocyte and osteoblast differentiation. BMC Mol Biol 9:26
Copland IB, Lord-Dufour S, Cuerquis J, Coutu DL, Annabi B et al (2009) Improved autograft survival of mesenchymal stromal cells by plasminogen activator inhibitor 1 inhibition. Stem Cells 27:467–477
Tsai MS, Hwang SM, Chen KD, Lee YS, Hsu LW et al (2007) Functional network analysis of the transcriptomes of mesenchymal stem cells derived from amniotic fluid, amniotic membrane, cord blood, and bone marrow. Stem Cells 25:2511–2523
Lin MT, Kuo IH, Chang CC, Chu CY, Chen HY et al (2008) Involvement of hypoxia-inducing factor-1alpha-dependent plasminogen activator inhibitor-1 up-regulation in Cyr61/CCN1-induced gastric cancer cell invasion. J Biol Chem 283:15807–15815
Tamama K, Kawasaki H, Kerpedjieva SS, Guan J, Ganju RK et al (2011) Differential roles of hypoxia inducible factor subunits in multipotential stromal cells under hypoxic condition. J Cell Biochem 112:804–817
Neuss S, Becher E, Woltje M, Tietze L, Jahnen-Dechent W (2004) Functional expression of HGF and HGF receptor/c-met in adult human mesenchymal stem cells suggests a role in cell mobilization, tissue repair, and wound healing. Stem Cells 22:405–414
Muller-Ehmsen J, Krausgrill B, Burst V, Schenk K, Neisen UC et al (2006) Effective engraftment but poor mid-term persistence of mononuclear and mesenchymal bone marrow cells in acute and chronic rat myocardial infarction. J Mol Cell Cardiol 41:876–884
Lane SW, Williams DA, Watt FM (2014) Modulating the stem cell niche for tissue regeneration. Nat Biotech 32:795–803
Mooney DJ, Vandenburgh H (2008) Cell delivery mechanisms for tissue repair. Cell Stem Cell 2:205–213
Daley GQ, Scadden DT (2008) Prospects for stem cell-based therapy. Cell 132:544–548
Herren T, Swaisgood C, Plow EF (2003) Regulation of plasminogen receptors. Front Biosci 1:d1–d8
Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M et al (2012) Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood 119:5879–5887
Neuss S, Schneider RK, Tietze L, Knuchel R, Jahnen-Dechent W (2010) Secretion of fibrinolytic enzymes facilitates human mesenchymal stem cell invasion into fibrin clots. Cells Tissues Organs 191:36–46
Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ (1996) Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor-mediated cell adhesion and release? J Cell Biol 134:1563–1571
Bajou K, Masson V, Gerard RD, Schmitt PM, Albert V et al (2001) The plasminogen activator inhibitor PAI-1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin. Implications for antiangiogenic strategies. J Cell Biol 152:777–784
Myohanen H, Vaheri A (2004) Regulation and interactions in the activation of cell-associated plasminogen. Cell Mol Life Sci 61:2840–2858
Menon LG, Picinich S, Koneru R, Gao H, Lin SY et al (2007) Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells 25:520–528
Dvorak HF (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315:1650–1659
Hogan NM, Joyce MR, Murphy JM, Barry FP, O’Brien T et al (2013) Impact of Mesenchymal Stem Cell secreted PAI-1 on colon cancer cell migration and proliferation. Biochem Biophys Res Commun 435:574–579
Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Kodama M et al (2010) Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer 127:2323–2333
Pulukuri SMK, Gorantla B, Dasari VR, Gondi CS, Rao JS (2010) Epigenetic Upregulation of Urokinase Plasminogen Activator Promotes the Tropism of Mesenchymal Stem Cells for Tumor Cells. Mol Cancer Res 8:1074–1083
De Boeck A, Pauwels P, Hensen K, Rummens JL, Westbroek W et al (2013) Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut 62:550–560
Chandran VI, Eppenberger-Castori S, Venkatesh T, Vine KL, Ranson M (2015) HER2 and uPAR cooperativity contribute to metastatic phenotype of HER2-positive breast cancer. Oncoscience 2:207–224
Sier CF, Verspaget HW, Griffioen G, Verheijen JH, Quax PH et al (1991) Imbalance of plasminogen activators and their inhibitors in human colorectal neoplasia. Implications of urokinase in colorectal carcinogenesis. Gastroenterology 101:1522–1528
Acknowledgments
We thank Stephanie C Napier for proofreading of the manuscript. This work was supported in part by grants from the Japan Society for the Promotion of Science and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (K.H.) Grant-in-Aid for Scientific Research on Priority Areas from the MEXT (K.H.), Mitsubishi Pharma Research Foundation (K.H), Naito Grant (B.H.), Grant-in-Aid for Scientific Research on Innovative Areas from the MEXT (B.H.), Program for Improvement of the Research Environment for Young Researchers funded by the Special Coordination Funds for Promoting Science and Technology of the MEXT, Japan (B.H.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Heissig, B., Dhahri, D., Eiamboonsert, S. et al. Role of mesenchymal stem cell-derived fibrinolytic factor in tissue regeneration and cancer progression. Cell. Mol. Life Sci. 72, 4759–4770 (2015). https://doi.org/10.1007/s00018-015-2035-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2035-7