
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, leading to a variety of motor and non-motor symptoms. Interestingly, non-motor symptoms often appear a decade or more before the first signs of motor symptoms. Some of these non-motor symptoms are remarkably similar to those observed in cases of impaired neurogenesis and several PD-related genes have been shown to play a role in embryonic or adult neurogenesis. Indeed, animal models deficient in Nurr1, Pitx3, SNCA and PINK1 display deregulated embryonic neurogenesis and LRRK2 and VPS35 have been implicated in neuronal development-related processes such as Wnt/β-catenin signaling and neurite outgrowth. Moreover, adult neurogenesis is affected in both PD patients and PD animal models and is regulated by dopamine and dopaminergic (DA) receptors, by chronic neuroinflammation, such as that observed in PD, and by differential expression of wild-type or mutant forms of PD-related genes. Indeed, an increasing number of in vivo studies demonstrate a role for SNCA and LRRK2 in adult neurogenesis and in the generation and maintenance of DA neurons. Finally, the roles of PD-related genes, SNCA, LRRK2, VPS35, Parkin, PINK1 and DJ-1 have been studied in NSCs, progenitor cells and induced pluripotent stem cells, demonstrating a role for some of these genes in stem/progenitor cell proliferation and maintenance. Together, these studies strongly suggest a link between deregulated neurogenesis and the onset and progression of PD and present strong evidence that, in addition to a neurodegenerative disorder, PD can also be regarded as a developmental disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD), first described in 1817 by James Parkinson [1], is the second most common neurodegenerative disease after Alzheimer’s disease and the first most common motor neurodegenerative disease, affecting approximately 2 % of the population aged 60 years and older [2, 3]. The disease is characterized by its locomotor phenotypes, which include resting tremors, difficulty initiating movement, postural instability, bradykinesia and rigidity as well as the formation of intracytoplasmic inclusions or protein aggregates called Lewy bodies, containing insoluble α-synuclein (SNCA) protein. Several non-motor symptoms of PD have also been described, including anhedonia [4–8], depression [9], anxiety [10], olfactory deficits [11–13], sleep disturbances [14, 15] and cognitive dysfunction [16–18]. These non-motor symptoms often precede the prototypical motor symptoms of PD by years or even decades [19, 20]. The PD-associated motor symptoms primarily result from the degeneration of DA neurons in the substantia nigra pars compacta (SNpc), although degeneration has also been observed in the striatum, hippocampus, and neocortex [21]. Interestingly, changes in DA innervation and DA concentration in the striatum can occur 20–30 years prior to the onset of any motor symptoms [22, 23].
Over 90 % of all PD cases appear to occur sporadically and most of them are idiopathic cases. Most plausibly, these cases arise from multiple factors acting simultaneously, including genetic susceptibility, environmental factors and, most importantly, age. Familial variants of PD result from genetic mutations leading to either autosomal dominant forms of PD, such as those caused by mutations in the SNCA, LRRK2, VPS35 or GBA gene, or autosomal recessive forms of PD, such as those caused by mutations in the PINK1, Parkin or DJ-1 gene. Several other genes have been described to confer an increased susceptibility to PD, including EIF4G1, ATP13A2, PLA2G6, FBXO7, DNAJC6, DNAJC13, SYNJ1, MAPT, Pitx3, Nurr1 and UCHL1. Although familial variants of PD account for up to only 10 % of all cases [24, 25], an induced pluripotent stem cells (iPSC)-based study has recently suggested that even in idiopathic cases of PD, the increased susceptibility of DA neurons is probably encoded in the genome [26]. Moreover, sporadic and genetic forms of PD have comparable motor phenotypes, implying that they share common neurodegenerative mechanisms [27].
No matter what the etiology, in PD, more than 50 % of SN neurons degenerate before the onset of motor symptoms and many of the non-motor symptoms of PD precede the occurrence of motor symptoms by years or even decades [19, 28–30]. Some of the prominent non-motor symptoms described during the premotor phase of PD such as hyposmia, depression and anxiety are also phenotypes observed in models of deregulated neurogenesis [31–34] and medications and treatments for depression and anxiety have been shown to increase adult neurogenesis (reviewed in [35]). Although a causative role for neurogenesis in depression and anxiety has yet to be demonstrated, it is likely that hyposmia is caused, at least in part, by deregulated neural stem cell (NSC) activity already occurring during development and/or in adult NSCs.
NSCs are self-renewing, multipotent progenitors that give rise to multiple types of neurons and glial cells during embryonic brain development as well as during adult neurogenesis (more detailed information in [36–51]). NSCs represent a potential endogenous source for neuronal replacement therapy in neurodegenerative disorders such as PD. Moreover, studies focusing on adult NSCs and neurogenesis in PD models offer the potential for earlier diagnosis, stratification and possibly therapeutic treatments for this debilitating disease.
Evidence for a developmental component of PD: deregulated embryonic neurogenesis
A number of PD-associated genes have been implicated in DA neuronal development during embryogenesis. Here, we discuss studies using mice deficient for Nurr1, Pitx3, and SNCA that have demonstrated deregulated embryonic neurogenesis of DA neurons in the midbrain as well as studies describing the role of PINK1, LRRK2 and VPS35 in neuronal development and the altered expression of the developmentally related brain-derived neurotrophic factor in PD.
The orphan nuclear receptor, Nurr1, is expressed principally in the limbic and ventral midbrain and plays an important role in the development and maintenance of DA neurons [52–55]. As previously mentioned, mutations in the Nurr1 transcription factor have been associated with a susceptibility to PD [56–60]. Moreover, Nurr1 gene and Nurr1 protein expression is reduced in DA neurons of the SN in PD patients [57, 61].
Given its role in the induction of differentiation of progenitor cells to tyrosine hydroxylase (TH)-positive DA neurons, it is likely that Nurr1 mutations contribute to PD pathology early in development, during the formation of the ventral midbrain DA neuron pool. Indeed, reduced Nurr1 expression in heterozygous mutant mice leads to decreased DA transporter expression [62], decreased TH-positive neurons in SNpc and less DA concentration in the dorsal striatum [63]. Moreover, these mice display morphological, biochemical and behavioral phenotypes similar to those observed in PD, reflecting the reduced number of DA neurons in the SNpc [62, 64, 65]. Nurr1 also inhibits expression of pro-inflammatory molecules involved in the neurotoxic response in microglia and astrocytes [66] and acts to transcriptionally downregulate SNCA [67]. Reduced expression of Nurr1 would also therefore lead to an increased inflammatory response, an unregulated increase in SNCA expression and possibly DA neuron cell death, further reducing the number of DA neurons in the developing brain, all factors that increase the risk to develop PD.
Another PD-susceptibility gene involved in neuronal development is the Pitx3 gene. The Pitx3 protein is a transcription factor necessary for terminal differentiation of TH-expressing neurons in the SN through the potentiation of Nurr1 transcription [68]. In mice lacking Pitx3, SN neuronal precursor numbers are dramatically decreased and these cells fail to produce TH, demonstrating that the Pitx3 protein is important for SNpc development and maintenance [69]. Moreover, overexpression of Pitx3 in ventral mesencephalon-derived NPCs improves motor function in a 6-OHDA PD model [70]. As previously mentioned, Pitx3 is necessary for Nurr1-mediated transcription, an important mechanism in DA neuron development and maintenance. As such, Pitx3 mutations may increase susceptibility to PD by a downstream effect on Nurr1 activity and the subsequent reduction in the numbers of DA neurons formed in the SN during brain development,
Endogenous α-synuclein has also been implicated in DA neuronal development [71]. In this study, Garcia-Reitboeck and colleagues describe a reduction in the number of DA neurons in the SN of mice with a spontaneous deletion of the SNCA gene, as well as in SNCA knockout (KO) mice, visible as early as E13.5. Their results demonstrate that α-synuclein is necessary for the embryonic development of at least a subpopulation of DA neurons and suggest that PD-related SNCA mutations may also lead to a reduction in the number of DA neurons in the developing brain, long before DA neuronal degeneration even starts. Interestingly, in this scenario, reduced levels of α-synuclein contribute to an increased PD risk, although more commonly, it is elevated α-synuclein levels that are associated to PD. This would suggest that only a balanced, optimal dosage of α-synuclein is beneficial for DA neuron formation and maintenance, while too high as well as too low levels are detrimental. Additionally, the function of α-synuclein as well as the dosage effects of this protein might be different during embryonic development or adulthood (see further discussion of SNCA below).
In the developing mouse brain, PINK1 protein expression is evident at E15 and increases significantly just before birth at E19 [72], coinciding with a period of increased neurogenesis in the brain [73]. PINK1 has recently been shown to interact with and phosphorylate the embryonic ectoderm development polycomb histone-methylation modulator, redistributing it to the mitochondria, resulting in a favorable change in transcription regulation for neuronal differentiation [74]. PINK1 downregulation in zebrafish results in delayed brain development, enlarged ventricles and a moderate decrease in TH-positive neurons in the diencephalon, likely due to an increase in apoptosis as evidenced by an increase in active Caspase 3 in these embryos [75].
LRRK2 expression in the developing mouse brain is particularly strong in the ventricular zone and SVZ of the telencephalon during the period of neurogenesis (E11.5 to E17.5) [76]. Furthermore, LRRK2 is expressed in NSCs isolated from both the DG and SVZ of E18.5 mice. The LRRK2 protein may interact with the regulator of neurite outgrowth during embryonic neurogenesis, CRMP2 [77] and numerous studies have demonstrated that mutant LRRK2 overexpression decreases neurite length/outgrowth, while LRRK2 deficits result in an increase of neurite length and arborization [78–94]. Moreover, LRRK2 has been shown to bind Wnt signaling components (the DVL proteins) and play a role in the canonical Wnt/β-catenin signaling pathway, an important pathway for neurogenesis and in particular the development of DA neurons [83, 95, 96].
Neuronal developmental defects are also visible in the absence of the PD-related gene, VPS35, discussed in more detail in the “Function of PD Genes in neural stem cells” section below. Briefly, VPS35 knockdown (KD) in mouse embryonic hippocampal neurons results in shortened apical dendrites, reduced dendritic spines and swollen commissural axons [97].
Lastly, brain-derived neurotrophic factor, known to regulate the differentiation and survival of midbrain DA neurons [98], displays reduced mRNA expression in the SNpc of PD models [99, 100].
Evidence for deregulated adult neural stem cell activity in PD
Neurogenesis in the PD brain
Adult neurogenesis has been described in the human brain in both the SVZ and the SGZ [46, 47, 101]. Unfortunately, few studies have been done to examine neurogenesis in the post-mortem PD brain and those that have been conducted present conflicting findings.
A first group of studies has demonstrated that post-mortem PD brains display deregulated adult neurogenesis. In these studies, cell proliferation appears to be decreased in the SGZ and SVZ of PD patients. The number of proliferating cell nuclear antigen (PCNA)-positive adult NSCs in the SVZ and Nestin-positive precursor cells in the OB are decreased in the PD-affected post-mortem brain [102] and a decrease in NSCs correlates with the progression of PD, while L-Dopa treatment appears to increase NSC numbers [103]. Moreover, EGF and EGFR levels are also decreased in the PD striatum and the prefrontal cortex of PD patients [104] and EGFR-positive neural stem/progenitor cell numbers are decreased in the SVZ of PD patients [105].
Other groups, however, have demonstrated no change in NSC proliferation in the PD-affected brain, such is the case in one study that examined NSC proliferation in the SVZ of PD patients and control brains [106]. NSCs have been identified in the SVZ of the aged human brain [107, 108] and in the SVZ of PD patients [106]. NPCs have been isolated from post-mortem human cortex and SN of PD patients [109, 110] and neurospheres have been obtained from the SVZ of a PD brain, indicating the presence of adult NSCs [106]. However, idiopathic human PD multipotent NPCs isolated from the SN appear to lack key factors required for neuronal differentiation as they must be co-cultured with embryonic stem cell-derived neural precursors to obtain neurons [110].
Neurogenesis defects in PD animal models
Mainly, two animal models for PD have been used to study neurogenesis: acute lesions formed by either 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration. Both experimental models replicate the DA neuron degeneration observed in PD and result in motor phenotypes. Although neurogenesis in both the SVZ and the SGZ has been shown to decline with age in rodents [111–113], whether this phenomenon is exacerbated, inversed or unchanged in PD animal models is still up for debate.
In one study, MPTP administration in the mouse brain decreased the number of proliferating cells in the SVZ and the corresponding DA neuron denervation decreased the number of PCNA-positive progenitor cells in the SGZ [102]. In later studies, MPTP administration was shown to induce apoptosis of migrating neuroblasts (NBs) from the SVZ [114, 115]. Conversely, MPTP treatment has also been shown to increase proliferation in the SVZ and the SGZ [116] and to increase neurogenesis in the SN and OB [116–119] of mice. Yet in another study, no change in proliferation or neurogenesis was observed. While MPTP treatment in mice induced DA denervation of the striatum, no change in proliferation of the SVZ progenitors was observed [106].
In macaque monkeys, MPTP treatment has been shown to deplete DA innervation in the SVZ and reduce the number of proliferating cells in this area [120]. In another study focusing on another brain region, MPTP treatment in the macaque revealed no increase in DA neurogenesis in the striatum [121].
In a number of studies, 6-OHDA treatment leads to decreased proliferation of neuronal precursors in the SVZ, visualized by a decrease in the number of PCNA-expressing cells in the SVZ of treated rats [102, 122] or a decrease in the number of BrdU-labeled cells in the SVZ of treated mice [123]. In the OB, 6-OHDA treatment results in decreased BrdU labeling in the granule cell layer (the site of integration for neural progenitors originating in the SVZ) but increased BrdU labeling in the glomerular cell layer with a concomitant increase in DA neurons in the OB, suggesting that DA differentiation is increased [122–124]. On the other hand, 6-OHDA treatments in rats have also increased proliferation in the SVZ [125–127]. It is noteworthy that, although proliferation appeared to be increased, these cells could only differentiate into astroglial cells and not neurons and thus neurogenesis would appear to be deregulated in these models as well [125]. Finally, examination of Dcx-positive NPCs in the SN of mice revealed no change after 6-OHDA injections [128].
Regulation of NSC activity by PD-associated neuroinflammation
Chronic neuroinflammation has been observed in both PD patients’ brains and in animal models of PD [129–132], but how this inflammation affects neurogenesis is only just being elucidated.
Inflammation-related proteins and molecules such as TNF-α, inflammatory cytokines and nitric oxide negatively regulate neurogenesis [133–136], while neurogenesis is increased in the hippocampus and SVZ by anti-inflammatory drug treatment in the focal traumatic brain injury and Japanese encephalitis models for acute neuroinflammation [137–139].
In the 6-OHDA PD model, treatment with anti-inflammatory drugs (minocycline or specific COX2 inhibitors) activates NSC proliferation in the SVZ and oligodendrogenesis and/or astrogliosis in the affected striatum and in the SN and even induces a functional regeneration as shown by rotational behavior experiments [128, 133].
Finally, the inflammatory response and microglia activation in a MPTP model of PD are partially mediated by the chemokine receptor CX3CR1 [140], a known regulator of adult neurogenesis [141].
Regulation of neurogenesis by dopamine
Dopamine appears to also play a role in the control of neurogenesis. Indeed, dopamine receptors have been observed on both neurospheres derived from rodent SVZ precursors [102, 142, 143] and in vivo on progenitor cells of the SVZ in mice and rats [102, 144, 145]. Moreover, DA fibers have been described in close proximity to EGFR-positive cells (progenitor cells) in the post-mortem human brain [102]. These DA fibers likely originate from SN cells, as has been described for non-human primates [120].
An experimentally reduced dopamine level in mice decreases precursor cell proliferation in both the subependymal zone and in the SGZ; this effect can be reversed by a selective D2-like receptor agonist (one of two subfamilies of DA receptors, including the D1, D2 and D3 DA receptors) [102]. Correspondingly, experimentally induced dopamine loss decreases SVZ proliferation and EGFR-positive adult NSCs via D2-like receptors [102, 105, 122, 123, 146]. It appears that dopamine can also exert its effects on neurogenesis, at least in part, via the D3 receptor. Activation of the D3 receptor in rats increases proliferation in the SVZ, RMS and possibly in the SN [147] and also stimulates OB neurogenesis [145]. In vitro studies have demonstrated that dopamine can also stimulate cell proliferation in neurospheres through the D3 receptor and may also stimulate neurogenesis [142].
Function of PD genes in neural stem cells
Studies of PD-related genes, in particular in NSCs, progenitor cells and iPSCs have suggested that these genes may be important in the regulation of stem/progenitor cell proliferation, maintenance and differentiation. Below, we outline the state of the art of this topic for the most common PD-related genes (reviewed in Table 1).
SNCA (PARK1/PARK4)
The SNCA gene (4q21), located at the PARK1/4 locus, encodes the protein α-synuclein. Several PD-related SNCA mutations have been identified. The A53T [148], A30P [149] and E46K [150] mutations as well as gene duplications [151] and triplications [152] are the SNCA genetic variations responsible for the onset of an autosomal dominant form of PD, while other PD-associated mutations such as the A18T, A29S, H50Q and G51D mutations may also contribute to PD progression [153–155].
α-Synuclein is a highly conserved protein that is abundantly expressed in the adult brain, but its precise function is still unknown [156, 157]. Certain studies have, however, described an involvement of α-synuclein in normal brain function [158], synaptic plasticity [159, 160] and the regulation of the presynaptic vesicular pool [161, 162], including DA release [163]. α-Synuclein has also been described as a chaperone and described to contribute to SNARE complex formation [164]. α-Synuclein is a natively unfolded monomer but it is able to switch from a helical to a β-sheet structure, aggregate and form fibrils [156, 157]. In PD brains, α-synuclein is the predominant protein found in Lewy Body inclusions [165, 166] and the misfolding and aggregation of this protein into neurotoxic species is considered central in PD pathogenesis [167].
An increasing number of studies reveal that α-synuclein plays an important role in neurogenesis. When the SNCA gene is differentially expressed or bears mutations or the α-synuclein protein forms aggregates, the neuronal stem cell pool is negatively regulated and the survival of newly generated neurons is decreased. The aforementioned suggests that there is a link between neurogenesis, SNCA and neurodegenerative diseases. If decoded, the details of this relationship could provide a valuable tool for designing potential therapies for patients with neurodegenerative diseases such as PD.
Overexpression of wild-type SNCA
Adult mice overexpressing human wild-type SNCA under the PDGF promoter were used to investigate adult NSC proliferation, migration and differentiation [168]. Although no difference in the number of PCNA-positive proliferating SVZ or DG cells could be observed between the transgenic and the control mice, a significant reduction of newly generated (BrdU positive) neurons was apparent in the OB and the DG. In particular, DA neurogenesis in the OB was severely impaired as was evident by the fact that the number of TH/BrdU double-positive cells was reduced more than half in the mice overexpressing SNCA. This effect observed in the OB was accompanied by increased cell death in the granule layer of the DG and in the OB [168].
In vitro, SNCA overexpression reduces the number of secondary neurospheres formed and affects NSC morphology and cell cycle progression, leading to their accelerated differentiation [169]. However, when the authors used the same virus to overexpress SNCA in vivo, no effect was seen on the proliferation of SVZ NSCs, an observation that agrees with previous study mentioned. Instead, they observed a delay in the migration of NSCs through the SVZ/RMS/OB system, evident by an increased number of NSCs that remained in the neurogenic niche [169].
A subsequent study using mice overexpressing human SNCA under the tet-off system investigated if impaired OB neurogenesis caused by SNCA overexpression could be rescued when the transgene is silenced [170]. As in the case of Winner et al., the authors demonstrated a decrease in NSC proliferation in the SVZ and in the generation of new neurons in the OB. When SNCA overexpression was ceased, the amount of new cells in the granule cell layer was increased but these cells did not manage to become neurons. On the other hand, glomerular layer neurogenesis was partially restored. It is worth mentioning that the amount of newly TH-positive cells was also reduced when the transgene was active and upon its silencing, the number of TH-positive cells was increased up to 89 % in the glomerular layer of the bulb [170].
A broader investigation concerning α-synuclein was performed in silico; mice overexpressing human wild-type SNCA were used to perform a transcriptome analysis of the striatal brain region [171]. The expression of many genes implicated in apoptosis, cell cycle progression, neurogenesis and synaptic function were impaired, demonstrating that indeed α-synuclein is involved in many different cell processes that, when deregulated due to differential SNCA expression, may influence neurogenesis. Indeed, rat hippocampal NPCs and mouse ESCs overexpressing SNCA display impaired neuronal differentiation and survival, an effect that is mediated by increased p53-mediated repression of Notch-1 signaling [172, 173].
Overexpression of mutated SNCA
OB neurogenesis during aging was investigated in mice that overexpress either human wild-type (WT) or A53T mutated SNCA [174]. Among these animals, only the aged mice bearing the SNCA mutation showed a significant decrease in proliferation of the SVZ NSCs. In addition, both transgenic mice revealed a reduction in the total number of newly generated neurons accompanied by an increase in apoptosis of neuronal progenitor cells in the OB. Newly generated TH-positive cells were decreased in both groups but the effect was more profound in the mice overexpressing the mutant form of SNCA [174]. This study demonstrates a correlation between aging and neurogenesis in the context of WT and mutant SNCA overexpression, showing that the A53T mutation produces more deleterious effects with respect to proliferation of NSCs and the survival of both stem cells and newly generated neurons.
Transgenic mice overexpressing the A30P mutant form of SNCA display no difference in proliferation in the SVZ, compared to control mice [175]. However, in the OB, the amount of newly generated DA neurons is decreased in the mutant mice. More recently, the same group investigated the effect of the mutation on hippocampus plasticity and found that the survival of new hippocampal neurons is dramatically decreased in these mice [176]. The cause of this decrease in newly formed neurons has not been investigated in this model. As such, it would be interesting to investigate if the decrease in neurons in this model is also due to decreased survival (increased apoptosis), as was the case for the A53T SNCA overexpression mouse model. In addition, the SNCA mutant mice present behavioral changes such as an increase in anxiety. Olfactory deficits and behavioral changes such as increased anxiety are also examples of non-motor symptoms of PD patients, suggesting a role for SNCA variations in non-motor deficits of PD.
Finally, mice expressing a truncated form of human SNCA (1–120 SNCA) under the control of the TH promoter in an endogenous null background developed inclusions in the SN, motor abnormalities, redistribution of SNARE proteins and impaired exocytosis [177, 178]. Recently, these mice were used to generate a new transgenic line by crossing them with WT mice. Compared to WT or to the 1–120 SNCA transgenic line, a significant increase in the number of SN DA neurons was observed [71]. The group, however, did not investigate the correlation between the increase of TH-positive neurons with increased neurogenesis nor did they investigate the proliferation, differentiation or survival potential of these cells, which would be an interesting point for future research. If these questions are answered, they might unveil an interesting mechanism or point to a certain direction concerning the specific function of α-synuclein in neurogenesis of the SN.
The sum of these studies demonstrates the deleterious effects of SNCA overexpression in the mouse brain. In vitro, WT SNCA overexpression leads to a decrease in proliferation. Overexpression of WT SNCA in vivo decreases the number of newly generated TH+ neurons via increased apoptosis of newly generated neurons and a decrease in cell migration through the RMS. Moreover, the decrease in the number of newly generated TH+ neurons is even more pronounced and is accompanied by a clear decrease in SVZ NSC proliferation when a mutated form of SNCA is expressed. Neurogenesis is a vital ongoing process in the adult brain and its disruption, via the deregulated expression or function (incurred by genetic mutations) of SNCA, could contribute to the non-motor deficits observed in patients with PD.
Interestingly, olfactory deficits have been described in mice overexpressing WT SNCA and olfaction impairment is an early non-motor symptom of PD. In one study, mice overexpressing SNCA under the Thy1 promoter displayed impairments in a number of olfactory functions [179]. In another study, mice overexpressing the human WT SNCA gene via the mouse SNCA promoter displayed olfactory deficits such as odor detection impairment, olfactory memory deficit and impaired odor discrimination [180, 181]. OB neurogenesis was also examined in the latter experiment but no difference in the number of newly generated neurons in the OB, based on BrdU labeling assays, was detected. Instead, it is likely that the olfaction deficits observed in SNCA transgenic mice are due, at least in part, to impaired growth and branching of dendrites and the subsequent failed stable integration into the OB of newly born DA neurons during adult neurogenesis, leading to a decrease in cell survival, as has been described for an A30P SNCA overexpressing mouse [182, 183].
Reduced expression of SNCA
Neurogenesis in the adult mammalian brain has also been investigated in the α/β–SNCA double knockout (DKO) mouse in the second neurogenic niche of the brain, the DG. These mice display increased neurogenesis in the DG but lack any apoptosis phenotype. When human SNCA is introduced (either as a transgene or a retrovirus), a substantial impairment in hippocampal neurogenesis and in dendrite development and morphology is observed [184]. In this study, the group investigated if there was an increase in the newly generated neurons by injecting the mice with BrdU. In addition, they used Caspase 3 staining to examine if the increase that they saw in the new neurons was balanced by increased apoptosis but they did not find any difference of cell death between the DKO mice and the wild type. These observations indicate that the results obtained indeed represent an increase in neurogenesis. It would be interesting to investigate the second adult neurogenic niche of this mouse to examine the effect of SNCA ablation on proliferation of the SVZ stem cells, their migration and the number of newly generated neurons in the OB.
Finally, in another study, surprisingly, the number of TH positive neurons is profoundly reduced in the SN of developing homozygous and heterozygous SNCA knockout embryos compared to WT embryos, suggesting that endogenous SNCA plays an important role in the embryonic development of DA neurons [71].
Altogether, the majority of the above-described experiments suggest that overexpression of SNCA (either WT or mutant forms) decreases the number of newly generated neurons in the DG, OB and SN of the adult brain, including DA neurons in the latter two brain regions. This decrease in DA neurogenesis has been attributed to a multitude of factors, depending on the study, including decreased NSC proliferation in the SVZ, decreased survival of newly generated neurons (increased apoptosis) in the DG and in the OB as well as decreased cell migration through the RMS. Moreover, the α/β–SNCA double KO mouse displays increased neurogenesis in the adult DG. By contrast, one study describes a decrease in the number of newly generated DA neurons in the embryonic DG in the SNCA KO mouse. These seemingly contradictory results hint at a differential effect of SNCA expression on neurogenesis, depending on the age of the animal (during embryonic development vs. in the adult neurogenic niches) or on the region of the brain being studied (SN vs. OB vs. DG). Moreover, it appears that SNCA expression levels need to be tightly regulated to avoid detrimental effects, as both increased and decreased expression of SNCA has been linked to an increase in PD risk.
It is obvious that there is a relation between cell proliferation, death and survival and that SNCA orchestrates, in an unknown way, cell fate decisions. In particular, it appears essential that SNCA expression and function be tightly regulated in the neuronal cells, as any changes in the expression levels (increased or decreased) or in the SNCA genetic sequence lead to defects in both embryonic and adult neurogenesis. In particular, the documented decrease in DA neurogenesis may suggest that individuals with SNCA mutations are born with less DA neurons and would therefore be more susceptible to PD. Still, PD symptoms would occur later in life due to the normal decline in neurogenesis with age. Mutant forms of SNCA would exasperate the neurogenesis decline; the accumulated decline in neurogenesis (from age and SNCA mutations) may bring it under a certain threshold, leading to the occurrence of non-motor symptoms.
LRRK2 (PARK8)
Mutations in the LRRK2 gene (12q12), located at the PARK8 locus, are the most common cause of autosomal dominant PD, accounting for 1–40 % of all the cases depending on the populations, and up to 5 % of apparently sporadic cases [185, 186]. PD-related LRRK2 gene mutations include R1441C, R1441G, Y1699C, G2019S and I2020T [187–191]. The G2019S and R1141G mutations, which increase kinase activity and decrease GTPase activity, respectively, are the two most common PD-associated mutations in LRRK2 [192–194].
The LRRK2 gene encodes for the leucine-rich repeat kinase (LRRK2) protein, a large and complex protein comprised of multiple domains, including a central catalytic tridomain with GTPase and kinase activities surrounded by several potential protein–protein interaction domains [187, 188]. Despite the knowledge regarding LRRK2 protein structure and its mRNA and protein expression, the precise physiological function of LRRK2 remains unclear.
LRRK2 shows widespread expression throughout the brain at both the mRNA and protein level [76]. In the mouse brain, LRRK2 mRNA is first detected at E16 and increases as development progresses through postnatal periods [195]. It is a ubiquitous protein expressed in various brain regions and various cell types, including neurons and glial cells. LRRK2 has been described to display robust expression in areas with “high proliferative and migratory activity as well as sites of differentiation and cell death” [76]. In particular, it is expressed in the main neurogenic niches of the mammalian brain: the ventricular zone, SVZ and hippocampus [195]. Moreover, LRRK2 co-localizes with the migrating NB marker PSA-NCAM [195]. Altogether, the expression pattern of LRRK2 suggests a role for this protein in CNS development and neurogenesis.
Reduced expression of LRRK2
Although LRRK2 KO mice do not appear to exhibit any alterations in neuronal function or survival [196], the different NSC gene expression profiles in the presence or absence of LRRK2 expression demonstrate that the absence of LRRK2 protein affects a number of different cellular processes, including genes implicated in cell cycle regulation, ribosome biosynthesis, proteasome function and mitochondrial oxidation or reduction processes [197].
LRRK2 loss of function does not affect NSC self-renewal in in vitro cultures, nor does it affect cell proliferation in the adult DG of LRRK2 KO mice [94, 197]. LRRK2 deficiency has, however, been linked to an increase in neuronal commitment in vitro and an increase in immature NBs due to delayed neuronal maturation in the adult DG [94, 197, 198]. In fact, neuronal differentiation appears to be closely linked to LRRK2 protein expression levels, as even a 50 % decrease of this protein’s expression levels in LRRK2 heterozygote ESCs appears to accelerate retinoic acid-induced neuronal differentiation [198]. This was visible through a differential gene expression analysis that revealed accelerated silencing of ESC pluripotency markers and downstream targets as well as increased expression of voltage-gated ion channels and neurotransmitter receptors or transporters in LRRK2 heterozygous cells 7 days after retinoic acid-induced neuronal differentiation. This effect may be mediated through the alleviation of let-7 miRNA (a key pro-differentiation miRNA, discussed further below) suppression via Lin28. Indeed, the reduction in Lin28 expression was also exaggerated in LRRK2 heterozygous cells compared to WT cells during differentiation.
The sum of these studies demonstrates that LRRK2 is implicated in early neuronal differentiation and appears to negatively regulate this process. Of note, another study utilizing siRNA-mediated KD of LRRK2 in NPCs did not observe a difference in the number of total neurons after neuronal differentiation but instead observed a difference in the number of TH+ neurons, suggesting a reduced capacity of these cells to differentiate into DA neurons [199]. Varying the expression levels of LRRK2 may, therefore, differentially affect the genesis of specific neuronal populations.
Overexpression of mutated LRRK2
Overexpression of PD-related mutant G2019S LRRK2 protein produces defects in adult neurogenesis, at the level of cell proliferation and generation of newborn neurons. Indeed, transgenic mice overexpressing the PD-associated human LRRK2 G2019S protein display a significant decrease in the number of proliferating (BrdU positive) cells in the adult DG, SVZ and RMS and decreased neurogenesis (BrdU and NeuN positive) and DA neurogenesis (BrdU, NeuN and TH positive) as well as decreased survival of newly generated neurons in the OB [86]. The LRRK2 G2019S mutation may contribute to the hippocampal and SVZ-related age-dependent non-motor symptoms via dysfunctions in the NSC pool and in their neural derivatives at early stages of maturation. Indeed, LRRK2 G2019S iPSC-derived NSCs in culture, especially at late passages, display increased susceptibility to proteasomal stress and a passage-dependent decrease in clonal expansion and neuronal differentiation capacities [200].
LRRK2 regulates microRNAs
microRNAs, also known as miRNAs, are short (20–25 nucleotides) endogenous RNAs that regulate gene expression at the mRNA level. Briefly, pre-miRNAs are recruited to the miRNA-induced silencing complex, containing at least the Dicer protein, the double-stranded RNA-binding domain protein transactivation-responsive RNA-binding protein and a member of the Argonaut family [201]. The pre-miRNA is then processed by Dicer [202] and the mature miRNA guides the miRNA-induced silencing complex to the target mRNA sequence, via complementary base pairing, to downregulate its expression through translational repression, mRNA degradation or mRNA cleavage [203]. Several miRNAs are specifically enriched in the brain and the necessity of miRNA regulation for normal brain development has been the subject of several recent reviews [204–206]. What is more, miRNAs are regulators of NSC proliferation and differentiation and several studies have linked deregulated miRNA expression or function in the brain or in NSCs with PD pathogenesis (reviewed in [207, 208]).
Although miRNAs were initially described to function upstream of protein expression by targeting the mRNA of disease-associated genes to direct their posttranscriptional repression, more recent studies have demonstrated that some disease-associated proteins are also involved in the regulation of miRNAs pathways. For example, in Drosophila, pathogenic LRRK2 antagonizes let-7 and miR-184*; transgenic flies carrying the equivalent of the human G2019S mutation displayed increased inhibition of let-7 and miR184* activity [209]. Furthermore, the effect of pathogenic LRRK2, at least on let-7 inhibition, was dependent on its kinase activity, evident by the fact that the introduction of kinase-inactivating mutations abolished the observed effect. let-7 and miR184* are implicated in the repression of E2F and DP, respectively. These transcription factors are involved in cell cycle control and survival. As such, their repression results in a reduction in locomotor activity and an enhanced neuronal degeneration phenotype, suggesting another mechanism of miRNA pathway regulation in LRRK2-associated PD.
Additionally, Bahnassawy and collaborators investigated miRNA regulation by the R1441G mutation, the second most common PD-associated LRRK2 mutation. They showed that expression of R1441G LRRK2 in murine NSCs leads to a downregulation of let-7a and miR-9, although LRRK2 deficiency did not affect microRNA expression levels [197]. Altogether, these studies point to an important role for the LRRK2 gene in the pathogenesis of PD, mediated through miRNA regulatory pathways and point to future possible targets in the search for new therapeutic strategies for PD.
LRRK2 is involved in Wnt signaling
In addition to its role in the regulation of NSC differentiation, LRRK2 has been suggested to play a role in neurogenesis. Correspondingly, Berwick and Harvey have proposed that LRRK2 functions as a scaffolding protein that plays a role in numerous signaling pathways, thus linking LRRK2 to neurogenesis and to the function of post-mitotic and mature neurons [210]. The canonical Wnt/β-catenin pathway is part of the signaling mechanisms involved in the regulation of embryonic and adult neurogenesis, and in particular in the development of DA neurons. It plays a role in the regulation of axonal guidance, dendritic morphogenesis and synapse formation. Wnt cascades have been implicated in PD and deregulated Wnt signaling represents a feasible initiation event in neurodegeneration [211].
LRRK2 has been associated to Wnt signaling via its interaction with key components of the Wnt signaling pathway, such as DVL, the β-catenin destruction complex and LRP6 [95, 96]. Moreover, it has been demonstrated that pathogenic mutations of LRRK2 modulate its interaction with different Wnt pathway molecules, including DVL and GSK3, a component of the β-catenin destruction complex [83, 95]. Importantly, KD of LRRK2 results in enhanced canonical Wnt signaling [96]. This data suggest that LRRK2 plays a central role in Wnt signaling, an essential pathway in NSC biology and neurogenesis.
Despite the growing body of knowledge concerning the LRRK2 gene and its PD-related mutations, the molecular and cellular implications of LRRK2 mutations during the onset and progression of PD still remain unknown. Given LRRK2’s implication in neurogenesis and the parallels between neurogenic niches and brain regions implicated in non-motor PD symptoms, it is likely that LRRK2 mutations are responsible for some of the non-motor symptoms related to PD.
VPS35 (PARK17)
The VPS35 gene (16q12) is located at the PARK17 locus and codes for the vacuolar protein sorting 35 (VPS35). Recently, the p.D620N mutation in the VPS35 gene was identified as a novel cause of autosomal dominant late-onset PD by two research groups [212, 213] and five subsequent studies have identified this mutation in PD patients [214–217]. Moreover, PD-associated defects in RAB7L1 or LRRK2 lead to a deficiency of the VPS35 component of the retromer complex [218].
VPS35, along with VPS26 and VPS29, makes up the cargo recognition and binding subcomplex of the retromer, an important complex for the trafficking and recycling of lipids and proteins [219–221]. The D620N PD-related mutation in VPS35 displays a dominant negative missorting phenotype of retrograde transport of proteins dependent on the WASH complex [218, 222]. WT VPS35 regulates mitochondrial-anchored protein ligase transport from the mitochondria to the peroxisomes [223], suggesting that VPS35 mutations may produce mitochondrial defects in patients. VPS35 is also responsible for divalent metal transporter 1 transport [224]. Mutated VPS35 may, therefore, lead to divalent metal transporter 1 missorting and iron accumulation, similar to the iron accumulation described in PD patients and in the 6-OHDA mouse model of PD [225]. Finally, VPS35 is also hypothesized to influence the Wnt signaling pathway via deficient sorting of the Wntless protein, which leads to Wntless degradation [226–228] and Wnt/β-catenin signaling is impaired in 6-OHDA-lesioned rats [229] and MPTP-induced mouse and monkey models [230].
As previously mentioned, Wnt signaling plays a role in embryonic and adult neurogenesis and has been linked to PD. As such, the PD-related VPS35 mutation (D620N) may also have an effect on neurogenesis in the PD brain. Little is known about the role of VPS35 in neurogenesis partly because homozygous KO mice die early during embryonic development, before E10 and before the onset of neurogenesis [231]. In one study focusing on Alzheimer’s disease by Wang et al. [97], VPS35 was shown to promote apical dendritic growth and maturation and axonal protein transport in developing mouse hippocampal neurons. The authors demonstrated that embryonic hippocampal CA1 neurons with reduced VPS35 expression (via electroporation with microRNAs) display shortened apical dendrites, less dendritic spines and swollen commissural axons. The authors suggest that these observations reflect a defective protein transport in developing mouse neurons via impaired retrograde trafficking of beta1-secretase and altered beta1-secretase distribution. Similarly, KD of VPS35 or overexpression of D620N VPS35 in rat primary cortical cultures leads to a reduction in neurite length [218]. It will be interesting in the future to see if this protein also plays a role in the development or maintenance of other neuronal types in the adult brain as well as during embryonic neurogenesis. One could hypothesize that the dominant negative VPS35 mutant may drastically decrease the survival of newly generated neurons by inhibiting proper retrograde transport of important trophic factors and by inhibiting normal neurite outgrowth.
Of note, in a non-neuronal system, the mouse intestinal epithelium, although depletion of VPS35 does reduce Wntless protein levels, it did not affect stem cell proliferation in intestinal epithelium-derived cell cultures but did slightly reduced the growth rate of in vitro organoids derived from these cultures [232]. Given its role in Wnt signaling, it is not surprising that the loss of normal VPS35 function results in a decreased proliferative phenotype. It will be interesting to see if this phenotype is reproduced or even exaggerated in other systems such as during development of the CNS, in which Wnt signaling is required for long-distance gradient formation to guide NB migration and to establish neuronal polarity. Recalling that Wnt signaling has been implicated in the development of midbrain DA neurons, we can then hypothesize that disruption of Wnt signaling via the D620N VPS35 mutation has the potential to decrease the number of DA neurons formed during embryonic development, resulting in fewer DA neurons at birth, thus increasing an individual’s susceptibility to PD. Moreover, defects in adult neurogenesis caused by a disruption in Wnt signaling are likely to contribute to the non-motor symptoms observed in PD.
Finally, the D620N VPS35 mutation has also been shown to impair the autophagic process via missorting of the autophagic protein, Atg9, thought to contribute to autophagosome formation [222]. Several studies using mice deficient for autophagy-related genes have outlined the importance of the autophagic machinery in embryonic development of the CNS [233–236]. In particular, autophagy is necessary for proper neuronal differentiation [237, 238]. It will be interesting in the future to evaluate if impaired autophagosome formation caused by the PD-related VPS35 mutation also leads to impaired neurogenesis via defects in neuronal differentiation.
PINK1 (PARK6)
More than 50 mutations in the PINK1 gene (1p36), located at the PARK6 locus, have been identified in PD-affected patients [239–243] and these mutations are believed to account for up to 1–8 % of sporadic cases with early onset [244].
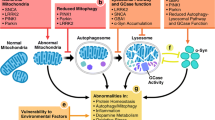
PTEN-induced putative kinase 1 (PINK1) is a serine/threonine kinase protein localized to the mitochondrial matrix and the intermembrane space [245, 246]. Under normal conditions, it is rapidly degraded but when the mitochondrial membrane is compromised, PINK1 is stabilized at the outer mitochondrial membrane where it recruits the E3-like ligase protein, Parkin, which in turn ubiquitinates other matrix surface proteins and targets the damaged mitochondria for autophagic degradation [247, 248].
PINK1 transcripts are ubiquitously expressed in the murine brain, including brain structures in which neurogenesis still occurs in the adult such as the hippocampus [249]. PINK1 expression has also been confirmed in human NSCs using RT-PCR. Interestingly, PINK1 expression increases 100-fold following differentiation to DA neurons [250]. In the latter study, the authors created a stable PINK1 KD human NSC line using RNAi methods to study this proteins’ function in NSC-derived mature DA neurons. Although PINK1 expression was reduced by >90 % in the selected clone, these NSCs displayed a similar DA neuron differentiation efficiency as the WT controls, suggesting that PINK1 is not necessary for DA neuronal differentiation.
In another study, the role of PINK1 was examined in human iPSCs derived from a patient with a PD-associated PINK1 mutation (Q456X) that results in a premature stop codon and results in a 80–90 % reduction in PINK1 mRNA levels compared to human iPSCs derived from a healthy family member [251, 252]. iPSCs derived from the fibroblasts of the PD patient were comparable to WT iPSCs; they displayed a high expression of the pluripotency markers OCT4a, Tra-1-60, NANOG and SSEA-4 at both protein and transcript levels; the iPSC-derived embryonic bodies were able to differentiate into the three embryonic germ layers; and the iPSCs displayed a normal karyotype. Once again, the decreased expression of PINK1 in the mutant iPSCs did not impede DA neuronal differentiation, as was demonstrated by a slightly higher number of TH-positive neurons in the PINK1 mutant-derived cells.
Similar observations were made in a third study done by Cooper and colleagues in 2012 in which human iPSCs were again derived from patients with the Q456X PINK1 mutation [253]. Supplementary results to validate the cellular model confirmed that the PINK1 mutant iPSCs are able to differentiate into all the neuronal lineages including DA neurons and display normal karyotyping. As only one PINK1 mutation has been studied with respect to neurogenesis defects, future studies will be needed to determine if other PINK1 mutations affect DA neurogenesis or DA neuronal differentiation.
All the above studies focused on DA neuron differentiation in cells with reduced, but not absence of, PINK1 expression. By contrast, in drosophila neuroblasts (dNB) and mammalian NSCs, PINK1 deficiency is inversely related to dNB maintenance and cell proliferation [254]. The number of type II dNBs, which contain transit-amplifying intermediate progenitors and are similar to mammalian NSCs, was significantly reduced in PINK1-null Drosophila mutants. Moreover, RNAi KD of dPINK1 blocks activation of mTORC2 and formation of ectopic NBs. In human NSC cultures, shRNA KD of PINK1 significantly inhibited cell proliferation. This effect appeared to be regulated by a non-canonical Notch signaling involving PINK1, mTORC2 and phosphorylated AKT, in which Notch directly interacts with PINK1 at the mitochondria. Indeed, Notch signaling is implicated in neuronal precursor cell proliferation and loss of function mutations of Notch leads to early neuronal differentiation and a decrease in the number of neuronal precursor cells [255, 256].
As previously mentioned, the PINK1 protein is important for mitochondrial quality control via autophagic degradation of damaged mitochondria. Mitochondria may play a role in stem cell proliferation by reducing mitochondrial oxygen levels, a phenomenon that enhances both ESC and NSC proliferation and differentiation potential [257–259], and by reducing mitochondrial Ca2+ levels, that are increased, along with mitochondrial fusion, during neuronal differentiation [260]. Loss of normal mitochondrial function may also contribute to the observed decrease in proliferation in the PINK1 KD experiments.
Although little research has been done to date on the implication of the PINK1 protein in neurogenesis, the little data thus far would suggest that the PINK1 loss of function mutations results in a decrease of NSC proliferation. Moreover, conditional PINK1 KO mice display impaired olfaction [261], which is a non-motor symptom of PD and a phenotype that can be attributed to impaired neurogenesis.
Parkin (PARK2)
Parkin deletion mutations are a common cause of early-onset PD and over 100 mutations have been identified to date [262–266]. The Parkin gene, located at the PARK2 locus (6q25.2-q27), is mutated in up to 50 % of all familial PD cases and in 10–15 % of sporadic early-onset PD [267].
The Parkin gene, located at the PARK2 locus, codes for the protein Parkin, an E3 ubiquitin ligase enzyme that is involved in proteasome-mediated and autophagy-mediated degradation of several substrates, particularly mitochondria [268]. During membrane depolarization or in the presence of reactive oxygen species, Parkin is recruited to the mitochondrial membrane by PINK1 and ubiquitinates mitochondrial proteins, targeting them for autophagic degradation [269]. PD-related mutations in Parkin result in the loss of its ubiquitin ligase activity [268]. Parkin KO mice display decreased levels of proteins involved in mitochondrial function or oxidative stress [270] and Parkin KO Drosophila display a mitochondrial pathology [271]. Given its role in mitochondrial quality control, Parkin mutations may act similarly to PINK1 loss of function and affect neuronal differentiation, via abnormal mitochondrial activity. Moreover, the Parkin protein ubiquitinates the neuronal cell fate determinant TRIM32, which in turn regulates NSC differentiation [272–275]. However, the experimental evidence for a role for Parkin in neuronal differentiation, outlined below, is contradictory.
Parkin KO ESCs display the same efficiency of in vitro neuronal differentiation as the WT ESCs, in particular the expression of the DA neuronal marker, TH, was the same in both WT and KO ESC-derived neurons [276]. RT-PCR analysis of these neurons revealed similar amounts of all transcripts examined, including the transcription factors Nurr1 and Pitx3, the enzymatic proteins TH and AADC, and the DA receptor, D2R.
In other studies, iPSCs derived from Parkin mutation related PD patients with homozygous or heterozygous deletions for a variety of different exons were created and compared to control iPSCs. Again, the mutant iPSCs displayed the same human pluripotent stem cell markers as their WT counterparts and were capable of generating embryonic bodies as well as neurospheres and differentiating into DA neurons [277, 278].
On the other hand, the drosophila Parkin protein has been shown to interact with clueless [279], which regulates atypical protein kinase C (aPKC) activity, a protein that is involved in NB asymmetric division, giving rise to both NBs and newly generated neurons [280]. Interestingly, both clueless and park (drosophila Parkin gene) mutants display similar Parkinsonian phenotypes and mitochondrial defects [279] as well as a similar NB phenotype, namely mislocalized cell fate determinants [281]. Furthermore, aPKC has been previously shown to regulate the neuronal cell fate determinant TRIM32 [272]. Together, these results suggest that, in Drosophila, Parkin is involved in the same neurogenic pathway as clueless, although further studies will be necessary to confirm this hypothesis.
Finally, in vitro experiments have shown that, in SH-SY5Y cells, PINK1 phosphorylates Parkin and activates its E3 ubiquitin ligase activity, which in turn adds K63-linked polyubiquitination chains to nuclear factor-kappaB (NF-κB), activating NF-κB signaling [282], a known regulator of embryonic and adult neurogenesis and neuronal differentiation. PD-related mutations in PINK1 (G309D, L347P) disrupt PINK1 kinase function [283–285] and are unable to phosphorylate Parkin and therefore unable to activate NF-κB signaling, suggesting that neurogenesis may also be disrupted in these PD genetic models. These findings and the subsequent effect on neurogenesis will need to be confirmed in in vivo models of PINK1 and Parkin mutations.
Although very little evidence exists linking PD-related Parkin mutations to neurogenesis and non-motor symptoms of PD, clearly this protein is implicated in several neurogenesis-related pathways, including mitochondrial quality control in neuronal differentiation, TRIM32 ubiquitination in cell fate determination, regulation of aPKC in stem cell renewal regulation and NF-κB signaling in adult neurogenesis and neuronal differentiation.
DJ-1 (PARK7)
Over 10 different mutations in the DJ-1 gene (1p36.23), located at the PARK7 locus, are responsible for 1–2 % of early-onset cases of autosomal recessive PD [286–288].
Originally identified as an oncogene, DJ-1 is thought to act as a molecular chaperone protein [289–291], an oxidative sensor and antioxidant [292–294] and in the regulation of gene transcription [295]. DJ-1 mutations can reduce DJ-1 protein synthesis or increase DJ-1 protein degradation [296]. In the cell, DJ-1 is expressed in the nucleus and the cytosol [297, 298]. Nuclear DJ-1 forms a complex with RNA-binding proteins and DNA-binding proteins that regulate gene transcription [295], while cytosolic DJ-1 can be found in several pools, including in the intermembrane space and matrix of the mitochondria.
DJ-1 is expressed in the cytosol of Nestin-positive cortical NSCs cultured from E14 rats, as well as in vivo, in the same cortical region and in rat NSC-derived neurospheres during proliferation [298]. DJ-1 may protect NSCs during proliferation through its antioxidant activity. Indeed, DJ-1 plays a role in the protection against oxidative stress and oxygen–glucose deprivation in human NPCs in culture [299] and DJ-1 deficiency in murine ESCs causes increased sensitivity to oxidative stress (increased apoptotic cell death after H2O2 treatment) and to proteasomal inhibition (increased sensitivity to the proteasomal inhibitor lactacystin) [300]. These results underline the importance of this protein in the protection of these highly proliferative cells against accumulation of deleterious ROS or accumulating proteins.
DJ-1 protein expression has also been described in both midbrain- and hippocampal-derived NSC neurospheres, in cells within the neurospheres as well as in cells migrating from the spheres [301]. During differentiation of cultured NSCs, DJ-1 expression increases gradually although the percentage of neuron-specific enolase NSE/DJ-1 colocalisation in neurons was less than 5 %, while the percentage of glial fibrillary acidic protein/DJ-1 colocalisation in astrocytes increased gradually to 85 % at day 7 of differentiation [297]. This is in accordance with the fact that, in the brain, DJ-1 is expressed mainly in astrocytes [302]. By contrast, another study has demonstrated a high level of DJ-1 expression in ESCs and a downregulation of DJ-1 expression to null amounts in these cells as they differentiate to cardiomyocytes [303], suggesting that any role for this protein in differentiation is likely specific to neural lineages. DJ-1 may play a role in NSC differentiation by acting as a scavenger of ROS, since controlled generation of ROS can stimulate differentiation via regulation of redox-sensitive transcription factors important for neurogenesis such as NF-κB and AP-1 [304, 305].
Conclusions
The studies described in this review demonstrate a link between defects in NSC characteristics, neurogenesis and PD. It suggests that some of the non-motor symptoms observed early in PD are likely due to defects in embryonic neuronal development as well as defects in adult neurogenesis. Furthermore, they underscore that PD has a strong developmental component. A better understanding of the interplay between deficits in embryonic and adult neurogenesis and PD onset and progression will be key to the development of better treatments for this debilitating disease. In particular, the adult SVZ contains a source of endogenous NSCs that are capable of differentiating into DA and non-DA neurons. As such, treatments that can restore impaired neurogenesis in PD patients could provide a source of endogenous repair. It may be time to change the way we look at this neurodegenerative disease, to start considering the possibility that PD starts much earlier than DA neuron degeneration and to consider that developmental and adult neurogenesis are key players in onset and progression of this disease.
Abbreviations
- 6-OHDA:
-
6-Hydroxydopamine
- aPKC:
-
Atypical protein kinase C
- DA:
-
Dopaminergic
- DG:
-
Dentate gyrus
- DKO:
-
Double knockout
- dNB:
-
Drosophila neuroblasts
- ESC:
-
Embryonic stem cell
- iPSC:
-
Induced pluripotent stem cell
- KD:
-
Knockdown
- KO:
-
Knockout
- LRRK2:
-
Leucine-rich repeat kinase 2
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NB:
-
Neuroblast
- NF-κB:
-
Nuclear factor-kappaB
- NPC:
-
Neural progenitor cell
- NSC:
-
Neural stem cell
- OB:
-
Olfactory bulb
- PCNA:
-
Proliferating cell nuclear antigen
- PD:
-
Parkinson’s disease
- PINK1:
-
PTEN-induced putative kinase 1
- RMS:
-
Rostral migratory stream
- SN:
-
Substantia nigra
- SNpc:
-
Substantia nigra pars compacta
- SNCA:
-
α-Synuclein
- SGZ:
-
Subgranular zone
- SVZ:
-
Subventricular zone
- TH:
-
Tyrosine hydroxylase
- VPS35:
-
Vacuolar protein sorting 35
- WT:
-
Wild type
References
Parkinson J (2002) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 14(2):223–236 (discussion 222)
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39(6):889–909
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79(4):368–376. doi:10.1136/jnnp.2007.131045
Lemke MR, Brecht HM, Koester J, Kraus PH, Reichmann H (2005) Anhedonia, depression, and motor functioning in Parkinson’s disease during treatment with pramipexole. J Neuropsychiatry Clin Neurosci 17(2):214–220. doi:10.1176/appi.neuropsych.17.2.214
Pluck GC, Brown RG (2002) Apathy in Parkinson’s disease. J Neurol Neurosurg Psychiatry 73(6):636–642
Isella V, Iurlaro S, Piolti R, Ferrarese C, Frattola L, Appollonio I, Melzi P, Grimaldi M (2003) Physical anhedonia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 74(9):1308–1311
Fujiwara S, Kimura F, Hosokawa T, Ishida S, Sugino M, Hanafusa T (2011) Anhedonia in Japanese patients with Parkinson’s disease. Geriatr Gerontol Int 11(3):275–281. doi:10.1111/j.1447-0594.2010.00678.x
Miura S, Kida H, Nakajima J, Noda K, Nagasato K, Ayabe M, Aizawa H, Hauser M, Taniwaki T (2012) Anhedonia in Japanese patients with Parkinson’s disease: analysis using the Snaith–Hamilton Pleasure Scale. Clin Neurol Neurosurg 114(4):352–355. doi:10.1016/j.clineuro.2011.11.008
Slaughter JR, Slaughter KA, Nichols D, Holmes SE, Martens MP (2001) Prevalence, clinical manifestations, etiology, and treatment of depression in Parkinson’s disease. J Neuropsychiatry Clin Neurosci 13(2):187–196
Menza MA, Robertson-Hoffman DE, Bonapace AS (1993) Parkinson’s disease and anxiety: comorbidity with depression. Biol Psychiatry 34(7):465–470
Doty RL, Deems DA, Stellar S (1988) Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 38(8):1237–1244
Mesholam RI, Moberg PJ, Mahr RN, Doty RL (1998) Olfaction in neurodegenerative disease: a meta-analysis of olfactory functioning in Alzheimer’s and Parkinson’s diseases. Arch Neurol 55(1):84–90
Hawkes CH, Shephard BC, Daniel SE (1997) Olfactory dysfunction in Parkinson’s disease. J Neurol Neurosurg Psychiatry 62(5):436–446
Shulman LM, Taback RL, Bean J, Weiner WJ (2001) Comorbidity of the nonmotor symptoms of Parkinson’s disease. Mov Disord 16(3):507–510
Tandberg E, Larsen JP, Karlsen K (1999) Excessive daytime sleepiness and sleep benefit in Parkinson’s disease: a community-based study. Mov Disord 14(6):922–927
Buter TC, van den Hout A, Matthews FE, Larsen JP, Brayne C, Aarsland D (2008) Dementia and survival in Parkinson disease: a 12-year population study. Neurology 70(13):1017–1022. doi:10.1212/01.wnl.0000306632.43729.24
Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG (2008) The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord 23(6):837–844. doi:10.1002/mds.21956
Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P (2003) Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 60(3):387–392
Savica R, Rocca WA, Ahlskog JE (2010) When does Parkinson disease start? Arch Neurol 67(7):798–801. doi:10.1001/archneurol.2010.135
Jellinger KA (2012) Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord 27(1):8–30. doi:10.1002/mds.23795
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8(12):1150–1157. doi:10.1016/s1474-4422(09)70238-8
Riederer P, Wuketich S (1976) Time course of nigrostriatal degeneration in Parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm 38(3–4):277–301
Scherman D, Desnos C, Darchen F, Pollak P, Javoy-Agid F, Agid Y (1989) Striatal dopamine deficiency in Parkinson’s disease: role of aging. Ann Neurol 26(4):551–557. doi:10.1002/ana.410260409
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31(7):763–780. doi:10.1002/humu.21277
Gasser T (2009) Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med 11:e22. doi:10.1017/s1462399409001148
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, Lopez-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A (2012) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4(5):380–395. doi:10.1002/emmm.201200215
Lesage S, Brice A (2012) Role of Mendelian genes in “sporadic” Parkinson’s disease. Parkinsonism Relat Disord 18(Suppl 1):S66–70. doi:10.1016/s1353-8020(11)70022-0
Tolosa E, Gaig C, Santamaria J, Compta Y (2009) Diagnosis and the premotor phase of Parkinson disease. Neurology 72(7 Suppl):S12–20. doi:10.1212/WNL.0b013e318198db11
Shiba M, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, Schaid DJ, Rocca WA (2000) Anxiety disorders and depressive disorders preceding Parkinson’s disease: a case–control study. Mov Disord 15(4):669–677
O’Sullivan SS, Williams DR, Gallagher DA, Massey LA, Silveira-Moriyama L, Lees AJ (2008) Nonmotor symptoms as presenting complaints in Parkinson’s disease: a clinicopathological study. Mov Disord 23(1):101–106. doi:10.1002/mds.21813
Revest JM, Dupret D, Koehl M, Funk-Reiter C, Grosjean N, Piazza PV, Abrous DN (2009) Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol Psychiatry 14(10):959–967. doi:10.1038/mp.2009.15
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20(24):9104–9110
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301(5634):805–809. doi:10.1126/science.1083328
Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C, Spitzer G, Santarelli L, Scharf B, Hen R, Rosoklija G, Sackeim HA, Dwork AJ (2007) Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci 27(18):4894–4901. doi:10.1523/jneurosci.0237-07.2007
Schoenfeld TJ, Cameron HA (2014) Adult Neurogenesis and mental illness. Neuropsychopharmacology. doi:10.1038/npp.2014.230
Morrison SJ (2001) Neuronal potential and lineage determination by neural stem cells. Curr Opin Cell Biol 13(6):666–672
Hartfuss E, Galli R, Heins N, Gotz M (2001) Characterization of CNS precursor subtypes and radial glia. Dev Biol 229(1):15–30. doi:10.1006/dbio.2000.9962
Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR (2002) Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci 22(8):3161–3173 (20026299)
Malatesta P, Hartfuss E, Gotz M (2000) Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development 127(24):5253–5263
Tarabykin V, Stoykova A, Usman N, Gruss P (2001) Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development 128(11):1983–1993
Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25(1):247–251. doi:10.1523/jneurosci.2899-04.2005
Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA (2004) Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol 479(2):168–180. doi:10.1002/cne.20322
Zimmer C, Tiveron MC, Bodmer R, Cremer H (2004) Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb Cortex 14(12):1408–1420. doi:10.1093/cercor/bhh102
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124(3):319–335
Curtis MA, Kam M, Nannmark U, Anderson MF, Axell MZ, Wikkelso C, Holtas S, van Roon-Mom WM, Bjork-Eriksson T, Nordborg C, Frisen J, Dragunow M, Faull RL, Eriksson PS (2007) Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science 315(5816):1243–1249. doi:10.1126/science.1136281
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4(11):1313–1317. doi:10.1038/3305
Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, Lawton MT, McDermott MW, Parsa AT, Manuel-Garcia Verdugo J, Berger MS, Alvarez-Buylla A (2004) Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427(6976):740–744. doi:10.1038/nature02301
Kam M, Curtis MA, McGlashan SR, Connor B, Nannmark U, Faull RL (2009) The cellular composition and morphological organization of the rostral migratory stream in the adult human brain. J Chem Neuroanat 37(3):196–205. doi:10.1016/j.jchemneu.2008.12.009
Winner B, Cooper-Kuhn CM, Aigner R, Winkler J, Kuhn HG (2002) Long-term survival and cell death of newly generated neurons in the adult rat olfactory bulb. Eur J Neurosci 16(9):1681–1689
Kempermann G, Wiskott L, Gage FH (2004) Functional significance of adult neurogenesis. Curr Opin Neurobiol 14(2):186–191. doi:10.1016/j.conb.2004.03.001
Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, Possnert G, Druid H, Frisen J (2014) Neurogenesis in the striatum of the adult human brain. Cell 156(5):1072–1083. doi:10.1016/j.cell.2014.01.044
Le W, Conneely OM, He Y, Jankovic J, Appel SH (1999) Reduced Nurr1 expression increases the vulnerability of mesencephalic dopamine neurons to MPTP-induced injury. J Neurochem 73(5):2218–2221
Backman C, Perlmann T, Wallen A, Hoffer BJ, Morales M (1999) A selective group of dopaminergic neurons express Nurr1 in the adult mouse brain. Brain Res 851(1–2):125–132
Zetterstrom RH, Williams R, Perlmann T, Olson L (1996) Cellular expression of the immediate early transcription factors Nurr1 and NGFI-B suggests a gene regulatory role in several brain regions including the nigrostriatal dopamine system. Brain Res Mol Brain Res 41(1–2):111–120
Sacchetti P, Mitchell TR, Granneman JG, Bannon MJ (2001) Nurr1 enhances transcription of the human dopamine transporter gene through a novel mechanism. J Neurochem 76(5):1565–1572
Liu H, Tao Q, Deng H, Ming M, Ding Y, Xu P, Chen S, Song Z, Le W (2013) Genetic analysis of NR4A2 gene in a large population of Han Chinese patients with Parkinson’s disease. Eur J Neurol 20(3):584–587. doi:10.1111/j.1468-1331.2012.03824.x
Chu Y, Le W, Kompoliti K, Jankovic J, Mufson EJ, Kordower JH (2006) Nurr1 in Parkinson’s disease and related disorders. J Comp Neurol 494(3):495–514. doi:10.1002/cne.20828
Grimes DA, Han F, Panisset M, Racacho L, Xiao F, Zou R, Westaff K, Bulman DE (2006) Translated mutation in the Nurr1 gene as a cause for Parkinson’s disease. Mov Disord 21(7):906–909. doi:10.1002/mds.20820
Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK (2003) Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet 33(1):85–89. doi:10.1038/ng1066
Xu PY, Liang R, Jankovic J, Hunter C, Zeng YX, Ashizawa T, Lai D, Le WD (2002) Association of homozygous 7048G7049 variant in the intron six of Nurr1 gene with Parkinson’s disease. Neurology 58(6):881–884
Moran LB, Croisier E, Duke DC, Kalaitzakis ME, Roncaroli F, Deprez M, Dexter DT, Pearce RK, Graeber MB (2007) Analysis of alpha-synuclein, dopamine and parkin pathways in neuropathologically confirmed parkinsonian nigra. Acta Neuropathol 113(3):253–263. doi:10.1007/s00401-006-0181-6
Jiang C, Wan X, He Y, Pan T, Jankovic J, Le W (2005) Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp Neurol 191(1):154–162. doi:10.1016/j.expneurol.2004.08.035
Zhang L, Le W, Xie W (1001) Dani JA (2012) Age-related changes in dopamine signaling in Nurr1 deficient mice as a model of Parkinson’s disease. Neurobiol Aging 33(5):e1007–1016. doi:10.1016/j.neurobiolaging.2011.03.022
Imam SZ, Jankovic J, Ali SF, Skinner JT, Xie W, Conneely OM, Le WD (2005) Nitric oxide mediates increased susceptibility to dopaminergic damage in Nurr1 heterozygous mice. FASEB J 19(11):1441–1450. doi:10.1096/fj.04-3362com
Le W, Conneely OM, Zou L, He Y, Saucedo-Cardenas O, Jankovic J, Mosier DR, Appel SH (1999) Selective agenesis of mesencephalic dopaminergic neurons in Nurr1-deficient mice. Exp Neurol 159(2):451–458. doi:10.1006/exnr.1999.7191
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137(1):47–59. doi:10.1016/j.cell.2009.01.038
Yang YX, Latchman DS (2008) Nurr1 transcriptionally regulates the expression of alpha-synuclein. NeuroReport 19(8):867–871. doi:10.1097/WNR.0b013e3282ffda48
Jacobs FM, van Erp S, van der Linden AJ, von Oerthel L, Burbach JP, Smidt MP (2009) Pitx3 potentiates Nurr1 in dopamine neuron terminal differentiation through release of SMRT-mediated repression. Development 136(4):531–540. doi:10.1242/dev.029769
Smidt MP, Smits SM, Bouwmeester H, Hamers FP, van der Linden AJ, Hellemons AJ, Graw J, Burbach JP (2004) Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development 131(5):1145–1155. doi:10.1242/dev.01022
O’Keeffe FE, Scott SA, Tyers P, O’Keeffe GW, Dalley JW, Zufferey R, Caldwell MA (2008) Induction of A9 dopaminergic neurons from neural stem cells improves motor function in an animal model of Parkinson’s disease. Brain 131(Pt 3):630–641. doi:10.1093/brain/awm340
Garcia-Reitboeck P, Anichtchik O, Dalley JW, Ninkina N, Tofaris GK, Buchman VL, Spillantini MG (2013) Endogenous alpha-synuclein influences the number of dopaminergic neurons in mouse substantia nigra. Exp Neurol 248:541–545. doi:10.1016/j.expneurol.2013.07.015
d’Amora M, Angelini C, Marcoli M, Cervetto C, Kitada T, Vallarino M (2011) Expression of PINK1 in the brain, eye and ear of mouse during embryonic development. J Chem Neuroanat 41(2):73–85. doi:10.1016/j.jchemneu.2010.11.004
Rice D, Barone S Jr (2000) Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 108(Suppl 3):511–533
Berthier A, Jimenez-Sainz J, Pulido R (2013) PINK1 regulates histone H3 trimethylation and gene expression by interaction with the polycomb protein EED/WAIT1. Proc Natl Acad Sci USA 110(36):14729–14734. doi:10.1073/pnas.1216844110
Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P, Rubinsztein DC (2008) Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci 28(33):8199–8207. doi:10.1523/jneurosci.0979-08.2008
Zechel S, Meinhardt A, Unsicker K, von Bohlen Und Halbach O (2010) Expression of leucine-rich-repeat-kinase 2 (LRRK2) during embryonic development. Int J Dev Neurosci 28(5):391–399. doi:10.1016/j.ijdevneu.2010.04.002
Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J 405(2):307–317. doi:10.1042/bj20070209
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52(4):587–593. doi:10.1016/j.neuron.2006.10.008
Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT (2008) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105(3):1048–1056. doi:10.1111/j.1471-4159.2008.05217.x
Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H (2009) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 29(44):13971–13980. doi:10.1523/jneurosci.3799-09.2009
Dachsel JC, Behrouz B, Yue M, Beevers JE, Melrose HL, Farrer MJ (2010) A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat Disord 16(10):650–655. doi:10.1016/j.parkreldis.2010.08.018
Heo HY, Kim KS, Seol W (2010) Coordinate regulation of neurite outgrowth by LRRK2 and its interactor, Rab5. Exp Neurobiol 19(2):97–105. doi:10.5607/en.2010.19.2.97
Lin CH, Tsai PI, Wu RM, Chien CT (2010) LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J Neurosci 30(39):13138–13149. doi:10.1523/jneurosci.1737-10.2010
Chan D, Citro A, Cordy JM, Shen GC, Wolozin B (2011) Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2). J Biol Chem 286(18):16140–16149. doi:10.1074/jbc.M111.234005
Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ (2011) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 6(4):e18568. doi:10.1371/journal.pone.0018568
Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH (2011) Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis 41(3):706–716. doi:10.1016/j.nbd.2010.12.008
Maekawa T, Mori S, Sasaki Y, Miyajima T, Azuma S, Ohta E, Obata F (2012) The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol Neurodegener 7:15. doi:10.1186/1750-1326-7-15
Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Solanoy HO, Drummond J, Zhang X, Ding X, Cai F, Song Q, Li X, Yue Z, van der Brug MP, Burdick DJ, Gunzner-Toste J, Chen H, Liu X, Estrada AA, Sweeney ZK, Scearce-Levie K, Moffat JG, Kirkpatrick DS, Zhu H (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 4(164):164ra161. doi:10.1126/scitranslmed.3004485
Stafa K, Trancikova A, Webber PJ, Glauser L, West AB, Moore DJ (2012) GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet 8(2):e1002526. doi:10.1371/journal.pgen.1002526
Biosa A, Trancikova A, Civiero L, Glauser L, Bubacco L, Greggio E, Moore DJ (2013) GTPase activity regulates kinase activity and cellular phenotypes of Parkinson’s disease-associated LRRK2. Hum Mol Genet 22(6):1140–1156. doi:10.1093/hmg/dds522
Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182(2):474–484. doi:10.1016/j.ajpath.2012.10.027
Cho HJ, Liu G, Jin SM, Parisiadou L, Xie C, Yu J, Sun L, Ma B, Ding J, Vancraenenbroeck R, Lobbestael E, Baekelandt V, Taymans JM, He P, Troncoso JC, Shen Y, Cai H (2013) MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet 22(3):608–620. doi:10.1093/hmg/dds470
Law BM, Spain VA, Leinster VH, Chia R, Beilina A, Cho HJ, Taymans JM, Urban MK, Sancho RM, Ramirez MB, Biskup S, Baekelandt V, Cai H, Cookson MR, Berwick DC, Harvey K (2014) A direct interaction between leucine-rich repeat kinase 2 and specific beta-tubulin isoforms regulates tubulin acetylation. J Biol Chem 289(2):895–908. doi:10.1074/jbc.M113.507913
Paus M, Kohl Z, Ben Abdallah NM, Galter D, Gillardon F, Winkler J (2013) Enhanced dendritogenesis and axogenesis in hippocampal neuroblasts of LRRK2 knockout mice. Brain Res 1497:85–100. doi:10.1016/j.brainres.2012.12.024
Sancho RM, Law BM, Harvey K (2009) Mutations in the LRRK2 Roc-COR tandem domain link Parkinson’s disease to Wnt signalling pathways. Hum Mol Genet 18(20):3955–3968. doi:10.1093/hmg/ddp337
Berwick DC, Harvey K (2012) LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane-localized LRP6. Hum Mol Genet 21(22):4966–4979. doi:10.1093/hmg/dds342
Wang CL, Tang FL, Peng Y, Shen CY, Mei L, Xiong WC (2012) VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol Open 1(12):1248–1257. doi:10.1242/bio.20122451
Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM (1994) Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol 342(3):321–334
Gash DM, Zhang Z, Ovadia A, Cass WA, Yi A, Simmerman L, Russell D, Martin D, Lapchak PA, Collins F, Hoffer BJ, Gerhardt GA (1996) Functional recovery in parkinsonian monkeys treated with GDNF. Nature 380(6571):252–255. doi:10.1038/380252a0
Skaper SD, Negro A, Facci L, Dal Toso R (1993) Brain-derived neurotrophic factor selectively rescues mesencephalic dopaminergic neurons from 2,4,5-trihydroxyphenylalanine-induced injury. J Neurosci Res 34(4):478–487. doi:10.1002/jnr.490340413
Curtis MA, Eriksson PS, Faull RL (2007) Progenitor cells and adult neurogenesis in neurodegenerative diseases and injuries of the basal ganglia. Clin Exp Pharmacol Physiol 34(5–6):528–532. doi:10.1111/j.1440-1681.2007.04609.x
Höglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, Hirsch EC (2004) Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat Neurosci 7(7):726–735. doi:10.1038/nn1265
O’Sullivan SS, Johnson M, Williams DR, Revesz T, Holton JL, Lees AJ, Perry EK (2011) The effect of drug treatment on neurogenesis in Parkinson’s disease. Mov Disord 26(1):45–50. doi:10.1002/mds.23340
Iwakura Y, Piao YS, Mizuno M, Takei N, Kakita A, Takahashi H, Nawa H (2005) Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: neurotrophic implication in nigrostriatal neurons. J Neurochem 93(4):974–983. doi:10.1111/j.1471-4159.2005.03073.x
O’Keeffe GC, Tyers P, Aarsland D, Dalley JW, Barker RA, Caldwell MA (2009) Dopamine-induced proliferation of adult neural precursor cells in the mammalian subventricular zone is mediated through EGF. Proc Natl Acad Sci USA 106(21):8754–8759. doi:10.1073/pnas.0803955106
van den Berge SA, van Strien ME, Korecka JA, Dijkstra AA, Sluijs JA, Kooijman L, Eggers R, De Filippis L, Vescovi AL, Verhaagen J, van de Berg WD, Hol EM (2011) The proliferative capacity of the subventricular zone is maintained in the parkinsonian brain. Brain 134(Pt 11):3249–3263. doi:10.1093/brain/awr256
van den Berge SA, Middeldorp J, Zhang CE, Curtis MA, Leonard BW, Mastroeni D, Voorn P, van de Berg WD, Huitinga I, Hol EM (2010) Longterm quiescent cells in the aged human subventricular neurogenic system specifically express GFAP-delta. Aging Cell 9(3):313–326. doi:10.1111/j.1474-9726.2010.00556.x
Leonard BW, Mastroeni D, Grover A, Liu Q, Yang K, Gao M, Wu J, Pootrakul D, van den Berge SA, Hol EM, Rogers J (2009) Subventricular zone neural progenitors from rapid brain autopsies of elderly subjects with and without neurodegenerative disease. J Comp Neurol 515(3):269–294. doi:10.1002/cne.22040
Walton NM, Sutter BM, Chen HX, Chang LJ, Roper SN, Scheffler B, Steindler DA (2006) Derivation and large-scale expansion of multipotent astroglial neural progenitors from adult human brain. Development 133(18):3671–3681. doi:10.1242/dev.02541
Wang S, Okun MS, Suslov O, Zheng T, McFarland NR, Vedam-Mai V, Foote KD, Roper SN, Yachnis AT, Siebzehnrubl FA, Steindler DA (2012) Neurogenic potential of progenitor cells isolated from postmortem human Parkinsonian brains. Brain Res 1464:61–72. doi:10.1016/j.brainres.2012.04.039
Kuzumaki N, Ikegami D, Imai S, Narita M, Tamura R, Yajima M, Suzuki A, Miyashita K, Niikura K, Takeshima H, Ando T, Ushijima T, Suzuki T (2010) Enhanced IL-1beta production in response to the activation of hippocampal glial cells impairs neurogenesis in aged mice. Synapse 64(9):721–728. doi:10.1002/syn.20800
Kuzumaki N, Ikegami D, Tamura R, Sasaki T, Niikura K, Narita M, Miyashita K, Imai S, Takeshima H, Ando T, Igarashi K, Kanno J, Ushijima T, Suzuki T (2010) Hippocampal epigenetic modification at the doublecortin gene is involved in the impairment of neurogenesis with aging. Synapse 64(8):611–616. doi:10.1002/syn.20768
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443(7110):448–452. doi:10.1038/nature05091
He XJ, Nakayama H, Dong M, Yamauchi H, Ueno M, Uetsuka K, Doi K (2006) Evidence of apoptosis in the subventricular zone and rostral migratory stream in the MPTP mouse model of Parkinson disease. J Neuropathol Exp Neurol 65(9):873–882. doi:10.1097/01.jnen.0000235115.29440.ce
He XJ, Yamauchi H, Uetsuka K, Nakayama H (2008) Neurotoxicity of MPTP to migrating neuroblasts: studies in acute and subacute mouse models of Parkinson’s disease. Neurotoxicology 29(3):413–420. doi:10.1016/j.neuro.2008.02.007
Peng J, Xie L, Jin K, Greenberg DA, Andersen JK (2008) Fibroblast growth factor 2 enhances striatal and nigral neurogenesis in the acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neuroscience 153(3):664–670. doi:10.1016/j.neuroscience.2008.02.063
Peng J, Andersen JK (2011) Mutant alpha-synuclein and aging reduce neurogenesis in the acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Aging Cell 10(2):255–262. doi:10.1111/j.1474-9726.2010.00656.x
Zhao M, Momma S, Delfani K, Carlen M, Cassidy RM, Johansson CB, Brismar H, Shupliakov O, Frisen J, Janson AM (2003) Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci USA 100(13):7925–7930. doi:10.1073/pnas.1131955100
Shan X, Chi L, Bishop M, Luo C, Lien L, Zhang Z, Liu R (2006) Enhanced de novo neurogenesis and dopaminergic neurogenesis in the substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease-like mice. Stem Cells 24(5):1280–1287. doi:10.1634/stemcells.2005-0487
Freundlieb N, Francois C, Tande D, Oertel WH, Hirsch EC, Hoglinger GU (2006) Dopaminergic substantia nigra neurons project topographically organized to the subventricular zone and stimulate precursor cell proliferation in aged primates. J Neurosci 26(8):2321–2325. doi:10.1523/jneurosci.4859-05.2006
Tande D, Hoglinger G, Debeir T, Freundlieb N, Hirsch EC, Francois C (2006) New striatal dopamine neurons in MPTP-treated macaques result from a phenotypic shift and not neurogenesis. Brain 129(Pt 5):1194–1200. doi:10.1093/brain/awl041
Winner B, Geyer M, Couillard-Despres S, Aigner R, Bogdahn U, Aigner L, Kuhn G, Winkler J (2006) Striatal deafferentation increases dopaminergic neurogenesis in the adult olfactory bulb. Exp Neurol 197(1):113–121. doi:10.1016/j.expneurol.2005.08.028
Baker SA, Baker KA, Hagg T (2004) Dopaminergic nigrostriatal projections regulate neural precursor proliferation in the adult mouse subventricular zone. Eur J Neurosci 20(2):575–579. doi:10.1111/j.1460-9568.2004.03486.x
Sui Y, Horne MK, Stanic D (2012) Reduced proliferation in the adult mouse subventricular zone increases survival of olfactory bulb interneurons. PLoS ONE 7(2):e31549. doi:10.1371/journal.pone.0031549
Aponso PM, Faull RL, Connor B (2008) Increased progenitor cell proliferation and astrogenesis in the partial progressive 6-hydroxydopamine model of Parkinson’s disease. Neuroscience 151(4):1142–1153. doi:10.1016/j.neuroscience.2007.11.036
Arias-Carrion O, Hernandez-Lopez S, Ibanez-Sandoval O, Bargas J, Hernandez-Cruz A, Drucker-Colin R (2006) Neuronal precursors within the adult rat subventricular zone differentiate into dopaminergic neurons after substantia nigra lesion and chromaffin cell transplant. J Neurosci Res 84(7):1425–1437. doi:10.1002/jnr.21068
Liu BF, Gao EJ, Zeng XZ, Ji M, Cai Q, Lu Q, Yang H, Xu QY (2006) Proliferation of neural precursors in the subventricular zone after chemical lesions of the nigrostriatal pathway in rat brain. Brain Res 1106(1):30–39. doi:10.1016/j.brainres.2006.05.111
Worlitzer MM, Viel T, Jacobs AH, Schwamborn JC (2013) The majority of newly generated cells in the adult mouse substantia nigra express low levels of Doublecortin, but their proliferation is unaffected by 6-OHDA-induced nigral lesion or Minocycline-mediated inhibition of neuroinflammation. Eur J Neurosci 38(5):2684–2692. doi:10.1111/ejn.12269
Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM (2008) Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci 28(30):7687–7698. doi:10.1523/jneurosci.0143-07.2008
Machado A, Herrera AJ, Venero JL, Santiago M, de Pablos RM, Villaran RF, Espinosa-Oliva AM, Arguelles S, Sarmiento M, Delgado-Cortes MJ, Maurino R, Cano J (2011) Inflammatory animal model for Parkinson’s disease: the intranigral injection of LPS induced the inflammatory process along with the selective degeneration of nigrostriatal dopaminergic neurons. ISRN Neurol 2011:476158. doi:10.5402/2011/476158
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38(8):1285–1291
Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O (2002) Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci 15(6):991–998
Worlitzer MM, Bunk EC, Hemmer K, Schwamborn JC (2012) Anti-inflammatory treatment induced regenerative oligodendrogenesis in parkinsonian mice. Stem Cell Res Ther 3(4):33. doi:10.1186/scrt124
Vallieres L, Campbell IL, Gage FH, Sawchenko PE (2002) Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci 22(2):486–492
Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302(5651):1760–1765. doi:10.1126/science.1088417
Zhang P, Liu Y, Li J, Kang Q, Tian Y, Chen X, Shi Q, Song T (2006) Cell proliferation in ependymal/subventricular zone and nNOS expression following focal cerebral ischemia in adult rats. Neurol Res 28(1):91–96. doi:10.1179/016164106x91942
Das S, Dutta K, Kumawat KL, Ghoshal A, Adhya D, Basu A (2011) Abrogated inflammatory response promotes neurogenesis in a murine model of Japanese encephalitis. PLoS ONE 6(3):e17225. doi:10.1371/journal.pone.0017225
Hoehn BD, Palmer TD, Steinberg GK (2005) Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke 36(12):2718–2724. doi:10.1161/01.STR.0000190020.30282.cc
Ng SY, Semple BD, Morganti-Kossmann MC, Bye N (2012) Attenuation of microglial activation with minocycline is not associated with changes in neurogenesis after focal traumatic brain injury in adult mice. J Neurotrauma 29(7):1410–1425. doi:10.1089/neu.2011.2188
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9(7):917–924. doi:10.1038/nn1715
Bachstetter AD, Morganti JM, Jernberg J, Schlunk A, Mitchell SH, Brewster KW, Hudson CE, Cole MJ, Harrison JK, Bickford PC, Gemma C (2011) Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging 32(11):2030–2044. doi:10.1016/j.neurobiolaging.2009.11.022
Coronas V, Bantubungi K, Fombonne J, Krantic S, Schiffmann SN, Roger M (2004) Dopamine D3 receptor stimulation promotes the proliferation of cells derived from the post-natal subventricular zone. J Neurochem 91(6):1292–1301. doi:10.1111/j.1471-4159.2004.02823.x
Kippin TE, Kapur S, van der Kooy D (2005) Dopamine specifically inhibits forebrain neural stem cell proliferation, suggesting a novel effect of antipsychotic drugs. J Neurosci 25(24):5815–5823. doi:10.1523/jneurosci.1120-05.2005
Diaz J, Ridray S, Mignon V, Griffon N, Schwartz JC, Sokoloff P (1997) Selective expression of dopamine D3 receptor mRNA in proliferative zones during embryonic development of the rat brain. J Neurosci 17(11):4282–4292
Kim Y, Wang WZ, Comte I, Pastrana E, Tran PB, Brown J, Miller RJ, Doetsch F, Molnar Z, Szele FG (2010) Dopamine stimulation of postnatal murine subventricular zone neurogenesis via the D3 receptor. J Neurochem 114(3):750–760. doi:10.1111/j.1471-4159.2010.06799.x
Winner B, Desplats P, Hagl C, Klucken J, Aigner R, Ploetz S, Laemke J, Karl A, Aigner L, Masliah E, Buerger E, Winkler J (2009) Dopamine receptor activation promotes adult neurogenesis in an acute Parkinson model. Exp Neurol 219(2):543–552. doi:10.1016/j.expneurol.2009.07.013
Van Kampen JM, Robertson HA (2005) A possible role for dopamine D3 receptor stimulation in the induction of neurogenesis in the adult rat substantia nigra. Neuroscience 136(2):381–386. doi:10.1016/j.neuroscience.2005.07.054
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18(2):106–108. doi:10.1038/ng0298-106
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55(2):164–173. doi:10.1002/ana.10795
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364(9440):1169–1171. doi:10.1016/s0140-6736(04)17104-3
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841. doi:10.1126/science.1090278
Hoffman-Zacharska D, Koziorowski D, Ross OA, Milewski M, Poznanski J, Jurek M, Wszolek ZK, Soto-Ortolaza A, Slawek J, Janik P, Jamrozik Z, Potulska-Chromik A, Jasinska-Myga B, Opala G, Krygowska-Wajs A, Czyzewski K, Dickson DW, Bal J, Friedman A (2013) Novel A18T and pA29S substitutions in alpha-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat Disord 19(11):1057–1060. doi:10.1016/j.parkreldis.2013.07.011
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ (2013) Alpha-synuclein p. H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 28(6):811–813. doi:10.1002/mds.25421
Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, Verny C, Brice A (2013) G51D alpha-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann Neurol 73(4):459–471. doi:10.1002/ana.23894
Vekrellis K, Rideout HJ, Stefanis L (2004) Neurobiology of alpha-synuclein. Mol Neurobiol 30(1):1–21. doi:10.1385/MN:30:1:001
Bendor JT, Logan TP, Edwards RH (2013) The function of alpha-synuclein. Neuron 79(6):1044–1066. doi:10.1016/j.neuron.2013.09.004
Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ (2002) A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci 22(8):3090–3099 (pii: 20026307 22/8/3090)
Clayton DF, George JM (1999) Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res 58(1):120–129. doi:10.1002/(SICI)1097-4547(19991001)58:1<120:AID-JNR12>3.0.CO;2-E
George JM, Jin H, Woods WS, Clayton DF (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15(2):361–372 (pii: 0896-6273(95)90040-3)
Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci 22(20):8797–8807 (pii: 22/20/8797)
Murphy DD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20(9):3214–3220
Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25(1):239–252
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123(3):383–396. doi:10.1016/j.cell.2005.09.028
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. doi:10.1038/42166
Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E (1998) Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol 152(2):367–372
Olanow CW, Brundin P (2013) Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord 28(1):31–40. doi:10.1002/mds.25373
Winner B, Lie DC, Rockenstein E, Aigner R, Aigner L, Masliah E, Kuhn HG, Winkler J (2004) Human wild-type alpha-synuclein impairs neurogenesis. J Neuropathol Exp Neurol 63(11):1155–1166
Tani M, Hayakawa H, Yasuda T, Nihira T, Hattori N, Mizuno Y, Mochizuki H (2010) Ectopic expression of alpha-synuclein affects the migration of neural stem cells in mouse subventricular zone. J Neurochem 115(4):854–863. doi:10.1111/j.1471-4159.2010.06727.x
May VE, Nuber S, Marxreiter F, Riess O, Winner B, Winkler J (2012) Impaired olfactory bulb neurogenesis depends on the presence of human wild-type alpha-synuclein. Neuroscience 222:343–355. doi:10.1016/j.neuroscience.2012.07.001
Cabeza-Arvelaiz Y, Fleming SM, Richter F, Masliah E, Chesselet MF, Schiestl RH (2011) Analysis of striatal transcriptome in mice overexpressing human wild-type alpha-synuclein supports synaptic dysfunction and suggests mechanisms of neuroprotection for striatal neurons. Mol Neurodegener 6:83. doi:10.1186/1750-1326-6-83
Crews L, Mizuno H, Desplats P, Rockenstein E, Adame A, Patrick C, Winner B, Winkler J, Masliah E (2008) Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci 28(16):4250–4260. doi:10.1523/jneurosci.0066-08.2008
Desplats P, Spencer B, Crews L, Pathel P, Morvinski-Friedmann D, Kosberg K, Roberts S, Patrick C, Winner B, Winkler J, Masliah E (2012) Alpha-Synuclein induces alterations in adult neurogenesis in Parkinson disease models via p53-mediated repression of Notch1. J Biol Chem 287(38):31691–31702. doi:10.1074/jbc.M112.354522
Winner B, Rockenstein E, Lie DC, Aigner R, Mante M, Bogdahn U, Couillard-Despres S, Masliah E, Winkler J (2008) Mutant alpha-synuclein exacerbates age-related decrease of neurogenesis. Neurobiol Aging 29(6):913–925. doi:10.1016/j.neurobiolaging.2006.12.016
Marxreiter F, Nuber S, Kandasamy M, Klucken J, Aigner R, Burgmayer R, Couillard-Despres S, Riess O, Winkler J, Winner B (2009) Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur J Neurosci 29(5):879–890. doi:10.1111/j.1460-9568.2009.06641.x
Marxreiter F, Ettle B, May VE, Esmer H, Patrick C, Kragh CL, Klucken J, Winner B, Riess O, Winkler J, Masliah E, Nuber S (2013) Glial A30P alpha-synuclein pathology segregates neurogenesis from anxiety-related behavior in conditional transgenic mice. Neurobiol Dis 59:38–51. doi:10.1016/j.nbd.2013.07.004
Tofaris GK, Garcia Reitbock P, Humby T, Lambourne SL, O’Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG (2006) Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J Neurosci 26(15):3942–3950. doi:10.1523/JNEUROSCI.4965-05.2006
Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della Corte L, Spano P, Tofaris GK, Goedert M, Spillantini MG (2010) SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 133(Pt 7):2032–2044. doi:10.1093/brain/awq132
Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet MF (2008) Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur J Neurosci 28(2):247–256. doi:10.1111/j.1460-9568.2008.06346.x
Petit GH, Berkovich E, Hickery M, Kallunki P, Fog K, Fitzer-Attas C, Brundin P (2013) Rasagiline ameliorates olfactory deficits in an alpha-synuclein mouse model of Parkinson’s disease. PLoS ONE 8(4):e60691. doi:10.1371/journal.pone.0060691
Hansen C, Bjorklund T, Petit GH, Lundblad M, Murmu RP, Brundin P, Li JY (2013) A novel alpha-synuclein-GFP mouse model displays progressive motor impairment, olfactory dysfunction and accumulation of alpha-synuclein-GFP. Neurobiol Dis 56:145–155. doi:10.1016/j.nbd.2013.04.017
Neuner J, Filser S, Michalakis S, Biel M, Herms J (2014) A30P alpha-Synuclein interferes with the stable integration of adult-born neurons into the olfactory network. Sci Rep 4:3931. doi:10.1038/srep03931
Neuner J, Ovsepian SV, Dorostkar M, Filser S, Gupta A, Michalakis S, Biel M, Herms J (2014) Pathological alpha-synuclein impairs adult-born granule cell development and functional integration in the olfactory bulb. Nat Commun 5:3915. doi:10.1038/ncomms4915
Winner B, Regensburger M, Schreglmann S, Boyer L, Prots I, Rockenstein E, Mante M, Zhao C, Winkler J, Masliah E, Gage FH (2012) Role of alpha-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J Neurosci 32(47):16906–16916. doi:10.1523/JNEUROSCI.2723-12.2012
Kumari U, Tan EK (2009) LRRK2 in Parkinson’s disease: genetic and clinical studies from patients. FEBS J 276(22):6455–6463. doi:10.1111/j.1742-4658.2009.07344.x
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ, Wood NW (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case–control study. Lancet Neurol 7(7):583–590. doi:10.1016/s1474-4422(08)70117-0
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44(4):601–607. doi:10.1016/j.neuron.2004.11.005
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44(4):595–600. doi:10.1016/j.neuron.2004.10.023
Farrer M, Stone J, Mata IF, Lincoln S, Kachergus J, Hulihan M, Strain KJ, Maraganore DM (2005) LRRK2 mutations in Parkinson disease. Neurology 65(5):738–740. doi:10.1212/01.wnl.0000169023.51764.b0
Mata IF, Taylor JP, Kachergus J, Hulihan M, Huerta C, Lahoz C, Blazquez M, Guisasola LM, Salvador C, Ribacoba R, Martinez C, Farrer M, Alvarez V (2005) LRRK2 R1441G in Spanish patients with Parkinson’s disease. Neurosci Lett 382(3):309–311. doi:10.1016/j.neulet.2005.03.033
Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 76(4):672–680. doi:10.1086/429256
Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA (2006) LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci 29(5):286–293. doi:10.1016/j.tins.2006.03.006
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 102(46):16842–16847. doi:10.1073/pnas.0507360102
Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z (2007) Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem 103(1):238–247. doi:10.1111/j.1471-4159.2007.04743.x
Melrose HL, Kent CB, Taylor JP, Dachsel JC, Hinkle KM, Lincoln SJ, Mok SS, Culvenor JG, Masters CL, Tyndall GM, Bass DI, Ahmed Z, Andorfer CA, Ross OA, Wszolek ZK, Delldonne A, Dickson DW, Farrer MJ (2007) A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience 147(4):1047–1058. doi:10.1016/j.neuroscience.2007.05.027
Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM, Dawson VL (2009) Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J Neurosci 29(50):15846–15850. doi:10.1523/jneurosci.4357-09.2009
Bahnassawy L, Nicklas S, Palm T, Menzl I, Birzele F, Gillardon F, Schwamborn JC (2013) The Parkinson’s disease-associated LRRK2 mutation R1441G inhibits neuronal differentiation of neural stem cells. Stem Cells Dev. doi:10.1089/scd.2013.0163
Schulz C, Paus M, Frey K, Schmid R, Kohl Z, Mennerich D, Winkler J, Gillardon F (2011) Leucine-rich repeat kinase 2 modulates retinoic acid-induced neuronal differentiation of murine embryonic stem cells. PLoS ONE 6(6):e20820. doi:10.1371/journal.pone.0020820
Milosevic J, Schwarz SC, Ogunlade V, Meyer AK, Storch A, Schwarz J (2009) Emerging role of LRRK2 in human neural progenitor cell cycle progression, survival and differentiation. Mol Neurodegener 4:25. doi:10.1186/1750-1326-4-25
Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, Wagner U, Kim A, Ren B, Li Y, Goebl A, Kim J, Soligalla RD, Dubova I, Thompson J, Yates J 3rd, Esteban CR, Sancho-Martinez I, Izpisua Belmonte JC (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491(7425):603–607. doi:10.1038/nature11557
Meister G (2013) Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14(7):447–459. doi:10.1038/nrg3462
Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409(6818):363–366. doi:10.1038/35053110
Gu S, Kay MA (2010) How do miRNAs mediate translational repression? Silence 1(1):11. doi:10.1186/1758-907x-1-11
Follert P, Cremer H, Beclin C (2014) MicroRNAs in brain development and function: a matter of flexibility and stability. Front Mol Neurosci 7:5. doi:10.3389/fnmol.2014.00005
Nowak JS, Michlewski G (2013) miRNAs in development and pathogenesis of the nervous system. Biochem Soc Trans 41(4):815–820. doi:10.1042/bst20130044
Petri R, Malmevik J, Fasching L, Akerblom M, Jakobsson J (2014) miRNAs in brain development. Exp Cell Res 321(1):84–89. doi:10.1016/j.yexcr.2013.09.022
Palm T, Bahnassawy L, Schwamborn J (2012) MiRNAs and neural stem cells: a team to treat Parkinson’s disease? RNA Biol 9(6):720–730. doi:10.4161/rna.19984
Wang S, Xu J, Ye R, Wang J, Chen Z, Huang R, Peng Q, Xu Y, Cai X (2014) Emerging roles of microRNAs in neural stem cells. Curr Stem Cell Res Ther 9(3):234–243
Gehrke S, Imai Y, Sokol N, Lu B (2010) Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 466(7306):637–641. doi:10.1038/nature09191
Berwick DC, Harvey K (2013) LRRK2: an eminence grise of Wnt-mediated neurogenesis? Front Cell Neurosci 7:82. doi:10.3389/fncel.2013.00082
Rawal N, Corti O, Sacchetti P, Ardilla-Osorio H, Sehat B, Brice A, Arenas E (2009) Parkin protects dopaminergic neurons from excessive Wnt/beta-catenin signaling. Biochem Biophys Res Commun 388(3):473–478. doi:10.1016/j.bbrc.2009.07.014
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ, Wszolek ZK, Vingerhoets F, Farrer MJ (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89(1):162–167. doi:10.1016/j.ajhg.2011.06.001
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brucke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89(1):168–175. doi:10.1016/j.ajhg.2011.06.008
Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Bertram L, Bozi M, Barcikowska M, Crosiers D, Clarke CE, Facheris MF, Farrer M, Garraux G, Gispert S, Auburger G, Vilarino-Guell C, Hadjigeorgiou GM, Hicks AA, Hattori N, Jeon BS, Jamrozik Z, Krygowska-Wajs A, Lesage S, Lill CM, Lin JJ, Lynch T, Lichtner P, Lang AE, Libioulle C, Murata M, Mok V, Jasinska-Myga B, Mellick GD, Morrison KE, Meitnger T, Zimprich A, Opala G, Pramstaller PP, Pichler I, Park SS, Quattrone A, Rogaeva E, Ross OA, Stefanis L, Stockton JD, Satake W, Silburn PA, Strom TM, Theuns J, Tan EK, Toda T, Tomiyama H, Uitti RJ, Van Broeckhoven C, Wirdefeldt K, Wszolek Z, Xiromerisiou G, Yomono HS, Yueh KC, Zhao Y, Gasser T, Maraganore D, Kruger R (2012) A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet 49(11):721–726. doi:10.1136/jmedgenet-2012-101155
Ando M, Funayama M, Li Y, Kashihara K, Murakami Y, Ishizu N, Toyoda C, Noguchi K, Hashimoto T, Nakano N, Sasaki R, Kokubo Y, Kuzuhara S, Ogaki K, Yamashita C, Yoshino H, Hatano T, Tomiyama H, Hattori N (2012) VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov Disord 27(11):1413–1417. doi:10.1002/mds.25145
Kumar KR, Weissbach A, Heldmann M, Kasten M, Tunc S, Sue CM, Svetel M, Kostic VS, Segura-Aguilar J, Ramirez A, Simon DK, Vieregge P, Munte TF, Hagenah J, Klein C, Lohmann K (2012) Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch Neurol 69(10):1360–1364. doi:10.1001/archneurol.2011.3367
Lesage S, Condroyer C, Klebe S, Honore A, Tison F, Brefel-Courbon C, Durr A, Brice A (2012) Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology 78(18):1449–1450. doi:10.1212/WNL.0b013e318253d5f2
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77(3):425–439. doi:10.1016/j.neuron.2012.11.033
McGough IJ, Cullen PJ (2011) Recent advances in retromer biology. Traffic 12(8):963–971. doi:10.1111/j.1600-0854.2011.01201.x
Seaman MN (2005) Recycle your receptors with retromer. Trends Cell Biol 15(2):68–75. doi:10.1016/j.tcb.2004.12.004
Bonifacino JS, Hurley JH (2008) Retromer. Curr Opin Cell Biol 20(4):427–436. doi:10.1016/j.ceb.2008.03.009
Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, Rubinsztein DC (2014) Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun 5:3828. doi:10.1038/ncomms4828
Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM (2010) Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol 20(14):1310–1315. doi:10.1016/j.cub.2010.05.066
Tabuchi M, Yanatori I, Kawai Y, Kishi F (2010) Retromer-mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. J Cell Sci 123(Pt 5):756–766. doi:10.1242/jcs.060574
Jiang H, Song N, Xu H, Zhang S, Wang J, Xie J (2010) Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res 20(3):345–356. doi:10.1038/cr.2010.20
Belenkaya TY, Wu Y, Tang X, Zhou B, Cheng L, Sharma YV, Yan D, Selva EM, Lin X (2008) The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev Cell 14(1):120–131. doi:10.1016/j.devcel.2007.12.003
Coudreuse DY, Roel G, Betist MC, Destree O, Korswagen HC (2006) Wnt gradient formation requires retromer function in Wnt-producing cells. Science 312(5775):921–924. doi:10.1126/science.1124856
George A, Leahy H, Zhou J, Morin PJ (2007) The vacuolar-ATPase inhibitor bafilomycin and mutant VPS35 inhibit canonical Wnt signaling. Neurobiol Dis 26(1):125–133. doi:10.1016/j.nbd.2006.12.004
Dun Y, Li G, Yang Y, Xiong Z, Feng M, Wang M, Zhang Y, Xiang J, Ma R (2012) Inhibition of the canonical Wnt pathway by Dickkopf-1 contributes to the neurodegeneration in 6-OHDA-lesioned rats. Neurosci Lett 525(2):83–88. doi:10.1016/j.neulet.2012.07.030
Ohnuki T, Nakamura A, Okuyama S, Nakamura S (2010) Gene expression profiling in progressively MPTP-lesioned macaques reveals molecular pathways associated with sporadic Parkinson’s disease. Brain Res 1346:26–42. doi:10.1016/j.brainres.2010.05.066
Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, Zhang QG, Brann D, Kim TW, Yan R, Mei L, Xiong WC (2011) VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J Cell Biol 195(5):765–779. doi:10.1083/jcb.201105109
de Groot RE, Farin HF, Macurkova M, van Es JH, Clevers HC, Korswagen HC (2013) Retromer dependent recycling of the Wnt secretion factor Wls is dispensable for stem cell maintenance in the mammalian intestinal epithelium. PLoS ONE 8(10):e76971. doi:10.1371/journal.pone.0076971
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095):880–884. doi:10.1038/nature04723
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889. doi:10.1038/nature04724
Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447(7148):1121–1125. doi:10.1038/nature05925
Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME (1999) A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron 24(4):833–846
Vazquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F (2012) Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 8(2):187–199. doi:10.4161/auto.8.2.18535
Zhao Y, Huang Q, Yang J, Lou M, Wang A, Dong J, Qin Z, Zhang T (2010) Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res 1313:250–258. doi:10.1016/j.brainres.2009.12.004
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304(5674):1158–1160. doi:10.1126/science.1096284
Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N (2004) Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol 56(3):424–427. doi:10.1002/ana.20251
Ibanez P, Lesage S, Lohmann E, Thobois S, De Michele G, Borg M, Agid Y, Durr A, Brice A (2006) Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 129(Pt 3):686–694. doi:10.1093/brain/awl005
Kumazawa R, Tomiyama H, Li Y, Imamichi Y, Funayama M, Yoshino H, Yokochi F, Fukusako T, Takehisa Y, Kashihara K, Kondo T, Elibol B, Bostantjopoulou S, Toda T, Takahashi H, Yoshii F, Mizuno Y, Hattori N (2008) Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch Neurol 65(6):802–808. doi:10.1001/archneur.65.6.802
Li Y, Tomiyama H, Sato K, Hatano Y, Yoshino H, Atsumi M, Kitaguchi M, Sasaki S, Kawaguchi S, Miyajima H, Toda T, Mizuno Y, Hattori N (2005) Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 64(11):1955–1957. doi:10.1212/01.wnl.0000164009.36740.4e
Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, Tavella A, Marconi R, Nicholl DJ, Chien HF, Fincati E, Abbruzzese G, Marini P, De Gaetano A, Horstink MW, Maat-Kievit JA, Sampaio C, Antonini A, Stocchi F, Montagna P, Toni V, Guidi M, Dalla Libera A, Tinazzi M, De Pandis F, Fabbrini G, Goldwurm S, de Klein A, Barbosa E, Lopiano L, Martignoni E, Lamberti P, Vanacore N, Meco G, Oostra BA (2005) Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65(1):87–95. doi:10.1212/01.wnl.0000167546.39375.82
Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W (2012) Pink1 kinase and its membrane potential (Deltapsi)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem 287(27):22969–22987. doi:10.1074/jbc.M112.365700
Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S (2008) The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA 105(33):12022–12027. doi:10.1073/pnas.0802814105
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189(2):211–221. doi:10.1083/jcb.200910140
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8(1):e1000298. doi:10.1371/journal.pbio.1000298
Taymans JM, Van den Haute C, Baekelandt V (2006) Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem 98(3):951–961. doi:10.1111/j.1471-4159.2006.03919.x
Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AY, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E, Downward J, Mansfield L, Jat P, Taylor J, Heales S, Duchen MR, Latchman D, Tabrizi SJ, Wood NW (2008) PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3(6):e2455. doi:10.1371/journal.pone.0002455
Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31(16):5970–5976. doi:10.1523/jneurosci.4441-10.2011
Grunewald A, Breedveld GJ, Lohmann-Hedrich K, Rohe CF, Konig IR, Hagenah J, Vanacore N, Meco G, Antonini A, Goldwurm S, Lesage S, Durr A, Binkofski F, Siebner H, Munchau A, Brice A, Oostra BA, Klein C, Bonifati V (2007) Biological effects of the PINK1 c.1366C>T mutation: implications in Parkinson disease pathogenesis. Neurogenetics 8(2):103–109. doi:10.1007/s10048-006-0072-y
Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, Romero N, Jiang H, Uitti RJ, Huang Z, Opala G, Scarffe LA, Dawson VL, Klein C, Feng J, Ross OA, Trojanowski JQ, Lee VM, Marder K, Surmeier DJ, Wszolek ZK, Przedborski S, Krainc D, Dawson TM, Isacson O (2012) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med 4(141):141ra190. doi:10.1126/scitranslmed.3003985
Lee KS, Wu Z, Song Y, Mitra SS, Feroze AH, Cheshier SH, Lu B (2013) Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev 27(24):2642–2647. doi:10.1101/gad.225169.113
Bolos V, Grego-Bessa J, de la Pompa JL (2007) Notch signaling in development and cancer. Endocr Rev 28(3):339–363. doi:10.1210/er.2006-0046
Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442(7104):823–826. doi:10.1038/nature04940
Santilli G, Lamorte G, Carlessi L, Ferrari D, Rota Nodari L, Binda E, Delia D, Vescovi AL, De Filippis L (2010) Mild hypoxia enhances proliferation and multipotency of human neural stem cells. PLoS ONE 5(1):e8575. doi:10.1371/journal.pone.0008575
Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, Gil J, Beach D (2007) A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal 9(3):293–299. doi:10.1089/ars.2007.9.ft-14
Rodrigues CA, Diogo MM, da Silva CL, Cabral JM (2010) Hypoxia enhances proliferation of mouse embryonic stem cell-derived neural stem cells. Biotechnol Bioeng 106(2):260–270. doi:10.1002/bit.22648
Voccoli V, Colombaioni L (2009) Mitochondrial remodeling in differentiating neuroblasts. Brain Res 1252:15–29. doi:10.1016/j.brainres.2008.11.026
Glasl L, Kloos K, Giesert F, Roethig A, Di Benedetto B, Kuhn R, Zhang J, Hafen U, Zerle J, Hofmann A, de Angelis MH, Winklhofer KF, Holter SM, Vogt Weisenhorn DM, Wurst W (2012) Pink1-deficiency in mice impairs gait, olfaction and serotonergic innervation of the olfactory bulb. Exp Neurol 235(1):214–227. doi:10.1016/j.expneurol.2012.01.002
Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, Woodward H, Castellan CC, Scherer M, Vieregge P, Breakefield XO, Kramer PL, Ozelius LJ (2000) Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol 48(1):65–71
Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A (2000) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 342(21):1560–1567. doi:10.1056/nejm200005253422103
Maruyama M, Ikeuchi T, Saito M, Ishikawa A, Yuasa T, Tanaka H, Hayashi S, Wakabayashi K, Takahashi H, Tsuji S (2000) Novel mutations, pseudo-dominant inheritance, and possible familial affects in patients with autosomal recessive juvenile parkinsonism. Ann Neurol 48(2):245–250
Kobayashi T, Wang M, Hattori N, Matsumine H, Kondo T, Mizuno Y (2000) Exonic deletion mutations of the Parkin gene among sporadic patients with Parkinson’s disease. Parkinsonism Relat Disord 6(3):129–131
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608. doi:10.1038/33416
Djarmati A, Hedrich K, Svetel M, Schafer N, Juric V, Vukosavic S, Hering R, Riess O, Romac S, Klein C, Kostic V (2004) Detection of Parkin (PARK2) and DJ1 (PARK7) mutations in early-onset Parkinson disease: Parkin mutation frequency depends on ethnic origin of patients. Hum Mutat 23(5):525. doi:10.1002/humu.9240
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25(3):302–305. doi:10.1038/77060
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803. doi:10.1083/jcb.200809125
Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279(18):18614–18622. doi:10.1074/jbc.M401135200
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100(7):4078–4083. doi:10.1073/pnas.0737556100
Hillje AL, Worlitzer MM, Palm T, Schwamborn JC (2011) Neural stem cells maintain their stemness through protein kinase C zeta-mediated inhibition of TRIM32. Stem Cells 29(9):1437–1447. doi:10.1002/stem.687
Hillje AL, Pavlou MA, Beckmann E, Worlitzer MM, Bahnassawy L, Lewejohann L, Palm T, Schwamborn JC (2013) TRIM32-dependent transcription in adult neural progenitor cells regulates neuronal differentiation. Cell Death Dis 4:e976. doi:10.1038/cddis.2013.487
Schwamborn JC, Berezikov E, Knoblich JA (2009) The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136(5):913–925. doi:10.1016/j.cell.2008.12.024
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496(7445):372–376. doi:10.1038/nature12043
Hwang M, Lee JM, Kim Y, Geum D (2014) Functional role of Parkin against oxidative stress in neural cells. Endocrinol Metab (Seoul) 29(1):62–69. doi:10.3803/EnM.2014.29.1.62
Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S, Takanashi M, Funayama M, Hirayama A, Soga T, Hishiki T, Suematsu M, Yagi T, Ito D, Kosakai A, Hayashi K, Shouji M, Nakanishi A, Suzuki N, Mizuno Y, Mizushima N, Amagai M, Uchiyama Y, Mochizuki H, Hattori N, Okano H (2012) Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 5:35. doi:10.1186/1756-6606-5-35
Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, Azabdaftari G, Nakaso K, Yan Z, Feng J (2012) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun 3:668. doi:10.1038/ncomms1669
Cox RT, Spradling AC (2009) Clueless, a conserved Drosophila gene required for mitochondrial subcellular localization, interacts genetically with parkin. Dis Model Mech 2(9–10):490–499. doi:10.1242/dmm.002378
Chia W, Somers WG, Wang H (2008) Drosophila neuroblast asymmetric divisions: cell cycle regulators, asymmetric protein localization, and tumorigenesis. J Cell Biol 180(2):267–272. doi:10.1083/jcb.200708159
Goh LH, Zhou X, Lee MC, Lin S, Wang H, Luo Y, Yang X (2013) Clueless regulates aPKC activity and promotes self-renewal cell fate in Drosophila lgl mutant larval brains. Dev Biol 381(2):353–364. doi:10.1016/j.ydbio.2013.06.031
Sha D, Chin LS, Li L (2010) Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet 19(2):352–363. doi:10.1093/hmg/ddp501
Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR (2005) Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA 102(16):5703–5708. doi:10.1073/pnas.0500617102
Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14(22):3477–3492. doi:10.1093/hmg/ddi377
Sim CH, Lio DS, Mok SS, Masters CL, Hill AF, Culvenor JG, Cheng HC (2006) C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum Mol Genet 15(21):3251–3262. doi:10.1093/hmg/ddl398
Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, Brice A, van Duijn CM, Oostra B, Meco G, Heutink P (2003) DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci 24(3):159–160. doi:10.1007/s10072-003-0108-0
van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P (2001) Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet 69(3):629–634. doi:10.1086/322996
Healy DG, Abou-Sleiman PM, Valente EM, Gilks WP, Bhatia K, Quinn N, Lees AJ, Wood NW (2004) DJ-1 mutations in Parkinson’s disease. J Neurol Neurosurg Psychiatry 75(1):144–145
Lee SJ, Kim SJ, Kim IK, Ko J, Jeong CS, Kim GH, Park C, Kang SO, Suh PG, Lee HS, Cha SS (2003) Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J Biol Chem 278(45):44552–44559. doi:10.1074/jbc.M304517200
Tao X, Tong L (2003) Crystal structure of human DJ-1, a protein associated with early onset Parkinson’s disease. J Biol Chem 278(33):31372–31379. doi:10.1074/jbc.M304221200
Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A (2004) DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol 2(11):e362. doi:10.1371/journal.pbio.0020362
Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, Li L (2006) Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem 281(16):10816–10824. doi:10.1074/jbc.M509079200
Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H (2003) Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem Biophys Res Commun 312(4):1342–1348
Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H (2004) DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep 5(2):213–218. doi:10.1038/sj.embor.7400074
Takahashi K, Taira T, Niki T, Seino C, Iguchi-Ariga SM, Ariga H (2001) DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx alpha to the receptor. J Biol Chem 276(40):37556–37563. doi:10.1074/jbc.M101730200
Kahle PJ, Waak J, Gasser T (2009) DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med 47(10):1354–1361. doi:10.1016/j.freeradbiomed.2009.08.003
Li S, Sun Y, Zhao X, Pu XP (2012) Expression of the Parkinson’s disease protein DJ-1 during the differentiation of neural stem cells. Brain Res 1468:84–93. doi:10.1016/j.brainres.2012.05.022
Yan H, Pu XP (2010) Expression of the Parkinson’s disease-related protein DJ-1 during neural stem cell proliferation. Biol Pharm Bull 33(1):18–21
Kaneko Y, Shojo H, Burns J, Staples M, Tajiri N, Borlongan CV (2014) DJ-1 ameliorates ischemic cell death in vitro possibly via mitochondrial pathway. Neurobiol Dis 62:56–61. doi:10.1016/j.nbd.2013.09.007
Martinat C, Shendelman S, Jonason A, Leete T, Beal MF, Yang L, Floss T, Abeliovich A (2004) Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES- derived cell model of primary Parkinsonism. PLoS Biol 2(11):e327. doi:10.1371/journal.pbio.0020327
Ding Y-X, Wei L-C, Liu Y-H, Duan L, Jiao X-Y, Xia Y, Chen L-W (2012) Midbrain neural stem cells show unique cell survival, neuronal commitment and neurotrophic properties with therapeutic potential for Parkinson’s disease. J Alzheimers Dis Parkinsonism
Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, Pittman AM, Lashley T, Canet-Aviles R, Miller DW, McLendon C, Strand C, Leonard AJ, Abou-Sleiman PM, Healy DG, Ariga H, Wood NW, de Silva R, Revesz T, Hardy JA, Lees AJ (2004) The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 127(Pt 2):420–430. doi:10.1093/brain/awh054
Osman AM, van Dartel DA, Zwart E, Blokland M, Pennings JL, Piersma AH (2010) Proteome profiling of mouse embryonic stem cells to define markers for cell differentiation and embryotoxicity. Reprod Toxicol 30(2):322–332. doi:10.1016/j.reprotox.2010.05.084
Suzukawa K, Miura K, Mitsushita J, Resau J, Hirose K, Crystal R, Kamata T (2000) Nerve growth factor-induced neuronal differentiation requires generation of Rac1-regulated reactive oxygen species. J Biol Chem 275(18):13175–13178
Piao YJ, Seo YH, Hong F, Kim JH, Kim YJ, Kang MH, Kim BS, Jo SA, Jo I, Jue DM, Kang I, Ha J, Kim SS (2005) Nox 2 stimulates muscle differentiation via NF-kappaB/iNOS pathway. Free Radic Biol Med 38(8):989–1001. doi:10.1016/j.freeradbiomed.2004.11.011
Acknowledgments
This work was supported by the Fonds National de la Recherche Luxembourg (FNR). Jaclyn Nicole Le Grand and Laura Gonzalez-Cano are supported by fellowships from the FNR (AFR, Aides à la Formation-Recherche).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Le Grand, J.N., Gonzalez-Cano, L., Pavlou, M.A. et al. Neural stem cells in Parkinson’s disease: a role for neurogenesis defects in onset and progression. Cell. Mol. Life Sci. 72, 773–797 (2015). https://doi.org/10.1007/s00018-014-1774-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1774-1