
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease and its pathogenic mechanisms are poorly understood. The majority of PD cases are sporadic but a number of genes are associated with familial PD. Sporadic and familial PD have many molecular and cellular features in common, suggesting some shared pathogenic mechanisms. Induced pluripotent stem cells (iPSCs) have been derived from patients harboring a range of different mutations of PD-associated genes. PD patient-derived iPSCs have been differentiated into relevant cell types, in particular dopaminergic neurons and used as a model to study PD. In this review, we describe how iPSCs have been used to improve our understanding of the pathogenesis of PD. We describe what cellular and molecular phenotypes have been observed in neurons derived from iPSCs harboring known PD-associated mutations and what common pathways may be involved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although Parkinson’s disease (PD) was first described two centuries ago, our understanding of this disease is continually evolving. PD is an age-dependent complex neurodegenerative disorder. Clinical symptoms include classical motor abnormalities, such as resting tremor, bradykinesia and rigidity in addition to a wide range of non-motor symptoms (Trinh and Farrer 2013). PD is characterized by preferential loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the gold standard for diagnosis is the presence of Lewy body aggregates of α-synuclein (α-syn). At the cellular level, pathological mechanisms involve mitochondrial and lysosomal abnormalities, aberrant protein accumulation and dysfunctional clearance pathways, endoplasmic reticulum (ER) stress, increased oxidative stress, neuroinflammation and glial cell activation and possible involvement of apoptotic components (Levy et al. 2009). Most PD cases are sporadic, likely the result of a complex interplay between genetic susceptibility and environmental risk factors (Trinh and Farrer 2013). Approximately 10–20% of patients have a family history of PD (Schulte and Gasser 2011). Some PD-associated genes [e.g., α-synuclein (SNCA) and leucine-rich repeat kinase 2 (LRRK2)] cause autosomal dominant PD. Others [e.g., phosphatase and tensin homolog (PTEN)-induced novel kinase 1 (PINK1), parkin (PARK2) and parkinsonism-associated deglycase (DJ-1)] cause autosomal recessive PD. Mutations in the gene for β-glucocerebrosidase (GBA1) are an important risk factor for developing PD. Although the penetrance is relatively low, the high frequency of GBA1 mutations in the general population means that GBA1 mutations are responsible for more cases of PD than any other known genetic mutation. The prevalence of PD increases with age, with an estimated 1% of the population over the age of 65 years and 4–5% of the population over the age of 85 years affected (Trinh and Farrer 2013).
More accurate and predictive disease models are needed to understand the molecular pathways of PD and to develop effective treatment strategies. A decade ago, Yamanaka and colleagues made the groundbreaking discovery that adult somatic cells can be reprogrammed to generate induced pluripotent stem cells (iPSCs) by forced expression of several transcription factors, known as “Yamanaka factors” (Takahashi and Yamanaka 2006; Takahashi et al. 2007). Multiple strategies for reprogramming have been developed, each with advantages and disadvantages. Choosing one reprogramming method over another is usually dependent on multiple factors such as the nature of application (basic or clinical) and the efficiency and quality of reprogrammed cells (González et al. 2011; Malik and Rao 2013). Like embryonic stem cells, iPSCs have the capacity to be differentiated into almost any cell type. Fibroblasts and other somatic cells can be taken from a human patient, reprogrammed into iPSCs and differentiated into disease-relevant cell types, such as iPSC-derived neurons (ips-neurons), iPSC-derived DA (ips-DA) neurons, or iPSC-derived cortical neurons (ips-cortical). iPSCs offer the exciting potential to develop scalable, screenable models of disease in vitro using human disease-relevant cell types with patient-specific genetic backgrounds but without resorting to some of the non-physiological manipulations associated with the production of conventional transformed cell lines. This technology can be used for researching pathogenic disease mechanisms, high-throughput drug screening and therapeutics (Haston and Finkbeiner 2016).
In this review, we describe how iPSC-based models have been used to advance our understanding of the molecular and cellular pathogenesis of PD. We detail the results of studies using iPSC-based models derived from patients with several common forms of familial PD and explain some of the most commonly observed cellular abnormalities. We point out some recurring phenotypes that are shared among multiple forms of familial and sporadic PD ips-neurons, then discuss how they may relate to one another in overlapping pathways that converge, leading to neurodegeneration.
α-Synuclein
α-Syn, a member of the synuclein family, is a 14 kDa protein encoded by the SNCA gene. Although its function is not well understood, α-syn is associated with neuroprotective, synaptic and neuronal differentiation processes (Emamzadeh 2016). A pathological hallmark of PD is the presence of Lewy bodies, whose main component is α-syn (Spillantini et al. 1997). Mutations in SNCA, including rare missense mutations (e.g., A53T, A30P, E46K, H50Q, G51D, A53E), as well as multiplication mutations that result in increased dosage of wild-type (WT) α-syn, cause autosomal dominant PD (Xu and Pu 2016).
Synapse formation and function
α-Syn is enriched at presynaptic terminals and binds with lipids to modulate several synaptic functions, including promoting SNARE complex assembly and regulating neurotransmitter and synaptic vesicle release (Burré 2015). Downregulated mRNA expression in genes related to synapse formation and axon maintenance was observed in ips-DA neurons with A53T mutation. Impaired synapse formation in addition to α-syn and Tau-positive varicosities in axons preceding neurite degeneration were observed in A53T ips-DA neurons and reversed by small molecules specifically targeting α-syn (Kouroupi et al. 2017).
α-Syn aggregation mechanisms
Under normal physiological conditions, α-syn is flexible and adopts multiple conformations, partly influenced by cellular stress pathways (Lashuel et al. 2013). The prevailing view is that synuclein is an intrinsically disordered protein whose conformation is determined largely by the different protein and lipid partners with which synuclein can form a complex (Uversky et al. 2008; Tóth et al. 2014; Theillet et al. 2016). By contrast, Dettmer and colleagues developed a method to detect endogenous conformations of α-syn in intact brain tissue samples and reported observing α-syn tetramers (60 kDa) and monomers (14 kDa) in healthy samples. Ips-neurons with A53T SNCA had significantly lower ratios of α-syn60kDa:α-syn14kDa than isogenic controls, implicating a role of α-syn mutations in destabilizing the equilibrium between α-syn tetramers and monomers as a pathogenic mechanism in neurodegeneration (Dettmer et al. 2015).
Endosomal protein trafficking
WT and mutant forms of α-syn affect several components of the vesicular pathway, particularly the early secretory pathway involving ER-Golgi transport (Wang and Hay 2015). ER accumulation of lysosomal enzyme β-glucocerebrosidase (GCase) and glycoprotein nicastrin was observed in A53T SNCA ips-neurons. This phenotype was reversed by overexpressing Synoviolin (SYVN1), an E3 ubiqutin–protein ligase involved in ER-associated protein degradation, or by activating or overexpressing neuronal precursor cell-expressed developmentally down-regulated gene 4 (Nedd4), also an E3 ubiquitin ligase (Chung et al. 2013). Interestingly, overexpression of SYVN1 in rat primary neurons reduced α-syn-induced toxicity (Chung et al. 2013) and overexpression of Nedd4 has been shown to facilitate degradation of α-syn through the endosomal-lysosomal pathway in mammalian cell lines (Tofaris et al. 2011). A similar maturation defect in lysosomal GCase as a result of disrupted ER-Golgi trafficking was observed in ips-DA neurons with overexpressed WT α-syn as well as ips-DA neurons with SNCA triplication. This was associated with increased stabilization of α-syn oligomers and lysosomal impairment, resulting in further disruption of ER-Golgi GCase trafficking (Mazzulli et al. 2011, 2016a). Overexpression of Rab1a, a regulator of ER-Golgi traffic, reversed α-syn-induced lysosomal phenotypes and neuronal death in ips-DA neurons with SNCA triplication, confirming the pathogenic effects of α-syn on early steps in the secretory pathway (Mazzulli et al. 2016a).
Oxidative stress
Several pathways in PD, including DA metabolism, protein degradation pathways and neuroinflammation, are associated with increased oxidative stress as a potential mechanism for neurodegeneration (Blesa et al. 2015). Under pathological conditions, WT and mutant α-syn modulate oxidative stress through mitochondrial dysfunction, leading to cellular damage (Subramaniam and Chesselet 2013). iPSC-based models of both α-syn missense and multiplication mutations show an increased sensitivity to oxidative stressors. Ips-DA neurons harboring the A53T SNCA mutation have reduced relative mitochondrial respiration and increased sensitivity to apoptosis in the presence of mitochondrial toxins (Ryan et al. 2013). Ips-DA neurons from PD patients with SNCA triplication have increased expression of oxidative stress-related genes and are more vulnerable to neurodegeneration with exposure to oxidative stress (Byers et al. 2011). Higher basal reactive oxygen species (ROS) levels were observed in older ips-cortical neurons with SNCA triplication. The addition of α-syn oligomers increases ROS levels in ips-cortical neurons, with the levels being much higher with SNCA triplication (Deas et al. 2016). Associated with oxidative stress, nitrosative stress is also implicated in PD (Ischiropoulos and Beckman 2003). α-Syn is nitrated in patient brains with the A53T mutation (Good et al., 1998), as well as synucleinopathy brain lesions (Giasson et al. 2000) and induces neurodegeneration (Yu et al. 2010; Liu et al. 2011). A53T SNCA increases cytoplasmic nitrotyrosine staining and nitric oxide (NO) levels in ips-cortical neurons, resulting in ER stress (Chung et al. 2013). A similar increase in NO levels occurs in A53T SNCA ips-DA neurons, which are further increased by exposure to mitochondrial toxins. This increase in nitrosative stress results in higher levels of S-nitrosylated (SNO) forms of myocyte enhancer factor 2C (MEF2C), leading to decreased expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α), a regulator of mitochondrial bioenergetics, in A53T SNCA ips-DA neurons. Interestingly, mitochondrial toxins also increase SNO-MEF2C levels and decrease PGC1α expression in healthy ips-DA neurons, indicating that both A53T SNCA and pesticides cause similar nitrosative stress-induced phenotypes. Under basal conditions, apoptosis was unaltered in A53T SNCA ips-DA neurons; however, exposure to mitochondrial toxins caused a significant increase in neuron death in A53T SNCA ips-DA neurons. Nitrosative-stress-induced neuron death was rescued by overexpression of non-nitrosylatable MEF2C or a combination of NO synthesis inhibition with MEF2C overexpression, demonstrating that A53T SNCA provides a genetic susceptibility to nitrosative stress-induced neurodegeneration in the presence of environmental toxins (Ryan et al. 2013).
Leucine-rich repeat kinase 2
LRRK2 is a 285 kDa multidomain protein involved in a variety of cellular pathways. Dominant pathogenic mutations occur in several core domains of LRRK2, including G2019S and I2020T in the kinase domain, R1441G, R1441C, R1441H and N1437H in the GTPase domain and Y1699C in the carboxy-terminal of Roc (COR) domain that links the kinase and GTPase domains (Cookson 2010; Gómez-Suaga et al. 2014). Additionally, LRRK2 mutations are linked to sporadic PD (Schulte and Gasser 2011). The G2019S LRRK2 mutation is the most common known cause of familial PD, contributing to 2–7% of genetic cases and is also observed in 1% of sporadic PD cases (Schulte and Gasser 2011). The G2019S mutation results in an increase in kinase activity, and multiple other mutations reduce GTPase activity two molecular functions that may be related (Rudenko and Cookson 2014).
Survival
PD is characterized by the degeneration of DA neurons in the substantia nigra (Trinh and Farrer 2013). Longitudinal imaging of individual neurons shows that G2019S ips-DA neurons have reduced survival, a phenotype also observed in rat midbrain and cortical neurons overexpressing G2019S and Y1699C LRRK2 (Skibinski et al. 2014). An increase in basal levels of apoptosis in G2019S and I2020T ips-DA neurons has also been observed (Sánchez-Danés et al. 2012; Reinhardt et al. 2013; Ohta et al. 2015; Lin et al. 2016). This increase in apoptosis in G2019S ips-DA neurons is especially apparent in the presence of mitochondrial and proteasomal toxins, described in more detail below.
Neurites
Neurite defects due to the LRRK2 G2019S mutation or other pathogenic LRRK2 mutations occur in multiple models, including rodent neurons and neuroblastoma cell lines (MacLeod et al. 2006; Plowey et al. 2008; Ramonet et al. 2011; Winner et al. 2011) and dystrophic Lewy neurites and axonal degeneration are found in PD (Bellucci et al. 2016). As such, ips-DA neurons harboring the LRRK2 G2019S mutation have reduced neurite length and outgrowth (Sánchez-Danés et al. 2012; Reinhardt et al. 2013; Su and Qi 2013; Borgs et al. 2016; Lin et al. 2016). The G2019S mutation also leads to a reduction in neurite branching in mature ips-DA neurons (Sánchez-Danés et al. 2012), although neurite complexity is increased in early-stage ips-DA neuron cultures (Borgs et al. 2016), perhaps suggesting a developmental stage-dependent phenotype.
Cytoskeleton
An interaction between LRRK2 and cytoskeletal components may be important for maintaining normal neurite morphology (Parisiadou et al. 2009; Kawakami et al. 2012). Tau pathology is found in some LRRK2 PD patients (Martin et al. 2014). LRRK2 interacts with and phosphorylates the cytoskeletal components Tau and tubulin and pathogenic LRRK2 mutations can lead to hyperphosphorylation of these proteins (Gillardon 2009; Kawakami et al. 2012). LRRK2 alters the filopodia cytoskeletal structure and promotes phosphorylation of the cytoskeleton-associated Ezrin/Radixin/Moesin (ERM) proteins, a process disrupted by the G2019S mutation, possibly representing a mechanism by which G2019S LRRK2 reduces neurite outgrowth (Parisiadou et al. 2009). G2019S and I2020T ips-neurons display increased expression and phosphorylation levels of Tau and G2019S ips-DA neurons show misregulation of the microtubule-associated protein 7 (MAP7) (Reinhardt et al. 2013; Ohta et al. 2015). In PD patient iPSC-derived sensory neurons harboring the LRRK2 G2019S mutation, cytoskeletal aggregates composed of βIII tubulin, microtubule-associated protein 2 (MAP2), the intermediate filament protein peripherin and sometimes Tau occur in neurites, a phenotype that is partially rescued by LRRK2 kinase inhibition. However, neurite cytoskeletal aggregates were not observed in G2019S ips-DA neurons (Schwab and Ebert 2015). The importance of the interaction between LRRK2 and cytoskeletal components and the impact of this interaction on neuronal morphology in ips-DA neurons have yet to be fully understood.
α-Syn accumulation
An increase in α-syn levels is commonly seen in G2019S LRRK2 ips-DA neurons (Nguyen et al. 2011; Sánchez-Danés et al. 2012; Reinhardt et al. 2013; Skibinski et al. 2014; López de Maturana et al. 2016) but not in R1441G or I2020T ips-DA neurons (Ohta et al. 2015; López de Maturana et al. 2016), consistent with the observation that Lewy body pathology is usually present in G2019S but less frequently in R1441C, Y1699C, or I2020T patients (Martin et al. 2014). Interestingly, reducing α-syn levels rescues G2019S-mediated toxicity in ips-DA neurons and Y1699C- and G2019S-mediated toxicity in primary rat cortical neurons (Skibinski et al. 2014), suggesting that α-syn may be required for LRRK2-mediated toxicity.
Autophagy and endocytic sorting
Altered autophagy plays a role in the pathogenesis of PD and pathogenic LRRK2 contributes to the autophagy defect (Wang et al. 2016). At post-mortem, human brains from PD patients show an accumulation of autophagosomes and a reduction in lysosomes (Anglade et al. 1997; Chu et al. 2009; Dehay et al. 2010). An accumulation of autophagosomes is observed in LRRK2 I2020T ips-DA neurons (Ohta et al. 2015). In LRRK2 G2019S or sporadic ips-DA neurons, an accumulation of autophagosomes due to impaired autophagosome maturation and clearance occurs (Sánchez-Danés et al. 2012; Reinhardt et al. 2013) and autophagy genes are upregulated in early-stage G2019S ips-DA neurons (Borgs et al. 2016). Interestingly, studies in ips-DA neurons and differentiated neuroblastoma cells indicate that these defects in autophagy might contribute to the abnormal neurite morphology observed with the LRRK2 G2019S mutation (Plowey et al. 2008; Sánchez-Danés et al. 2012). Besides impacting macroautophagy, LRRK2 pathogenic mutations may also disrupt chaperone-mediated autophagy (CMA). One study suggested that LRRK2 itself may also be degraded by CMA, a process disrupted by pathogenic LRRK2 mutations. This LRRK2-induced disruption of CMA may in turn lead to the accumulation of CMA substrates, including α-syn, perhaps contributing to the pathogenicity in PD (Orenstein et al. 2013).
Rab GTPases, which play important roles in endocytic sorting, may be linked to PD pathogenesis. LRRK2 interacts with several Rab GTPases, which might help localize LRRK2 to various intracellular membranes and vesicular compartments (Madero-Pérez et al. 2017). LRRK2 directly phosphorylates several Rab GTPases and pathogenic LRRK2 mutations increase the phosphorylation of these substrates (Steger et al. 2016). Reducing levels of Rab7L1 causes lysosomal and Golgi apparatus abnormalities and DA neuron degeneration, similar to that observed with the LRRK2 G2019S mutation, while overexpression of Rab71L rescues these LRRK2 G2019S-induced defects (MacLeod et al. 2013), suggesting that pathogenic LRRK2 mediates endolysosomal abnormalities through a shared pathway with Rab71L. LRRK2 forms a complex with Rab7L1 in the trans-Golgi network (TGN), where it promotes Golgi-derived vesicle clearance through the autophagy-lysosome pathway (Beilina et al. 2014). LRRK2 regulates a number of other endolysosomal and vesicular pathways by modulating Rab GTPases, including lysosomal positioning, trafficking from early to late endosomes and autophagosome degradation (Esteves and Cardoso 2017; Shi et al. 2017). Further studies are needed to fully understand the mechanistic link between LRRK2, Rabs and endocytic sorting defects and whether these relationships are regulated similarly in PD ips-neurons.
Mitochondrial abnormalities
Mitochondrial dysfunction and oxidative stress likely contribute to the pathogenesis of PD (Zuo and Motherwell 2013). Oxidative damage is found post-mortem in PD brains (Dexter et al. 1994; Yoritaka et al. 1996; Alam et al. 1997). Substantia nigra neurons of patients with early-stage PD have increased numbers of mitochondrial DNA (mtDNA) mutations (Lin et al. 2012). Similarly, ips-neurons with G2019S or R1441C LRRK2 mutations have increased levels of mtDNA damage (Sanders et al. 2014). Morphological and functional mitochondrial abnormalities have been observed in LRRK2 G2019S or R1441C ips-neurons, including increased mitochondrial fragmentation, reduced basal oxygen consumption rate, reduced ATP levels, reduced mitochondrial membrane potential and increased mitochondrial reactive oxygen species (ROS) (Cooper et al. 2012; Su and Qi 2013; Hsieh et al. 2016). Oxidative damage occurs in I2020T LRRK2 ips-neurons (Ohta et al. 2015). Intriguingly, G2019S ips-DA neurons exhibit impaired mitophagy due to delayed removal of mitochondrial rho GTPase (Miro), an outer mitochondrial membrane protein that tethers mitochondria to microtubule motor proteins (Hsieh et al. 2016). WT but not G2019S LRRK2 interacts with Miro and is recruited to damaged mitochondria, suggesting a mechanism by which the G2019S mutation could impair mitophagy (Hsieh et al. 2016).
Sensitivity to mitochondrial and proteasomal toxins
G2019S and I2020T LRRK2 ips-DA neurons show an increased susceptibility to death by oxidative stressors and mitochondrial toxins, such as H2O2, 6-hydroxydopamine (6-OHDA) and rotenone, as well as the proteasome inhibitor MG-132 (Nguyen et al. 2011; Reinhardt et al. 2013; Ohta et al. 2015) and upregulation of several oxidative response pathway genes (Nguyen et al. 2011). Interestingly, the toxin 6-OHDA reduces neurite length more in G2019S LRRK2 ips-DA neurons than in controls (Lin et al. 2016), perhaps suggesting a relationship between the sensitivity to oxidative stress and altered neurite morphology phenotypes observed in G2019S ips-DA neurons.
PINK1 and parkin
PINK1 is a mitochondrial kinase that plays a role in maintaining mitochondrial health. Parkin, an E3-ubiquitin protein ligase, promotes ubiquitination of a variety of substrates and targets them for degradation by the proteasome or lysosome (Dawson and Dawson 2010). Loss of mitochondrial membrane potential leads to the accumulation of PINK1 on the outer mitochondrial membrane. Active PINK1 phosphorylates both parkin and ubiquitin (Ub) conjugates, which activate parkin and promote its translocation to damaged mitochondria, where it ubiquitinates various mitochondrial substrates. This initiates a feed-forward loop of sequential rounds of parkin-mediated ubiquitination, PINK1 phosphorylation of parkin and Ub conjugates and further parkin recruitment to mitochondria. Thus, PINK1 and parkin function together to detect damaged mitochondria and target them for degradation (Durcan and Fon 2015). Mutations in PINK1 and parkin lead to an autosomal recessive early-onset form of PD (Kitada et al. 1998; Hatano et al. 2004; Valente et al. 2004). A large number of PD-associated mutations in PINK1 and parkin have been found, including missense, nonsense, insertions and deletions (indels) and single or multiple exon copy number variants. The majority of these mutations result in a loss of function of PINK1 and parkin (Nuytemans et al. 2010).
Survival and neurite degeneration
As in sporadic PD and other familial forms of PD, patients with mutations in PINK1 or parkin exhibit neuronal death in the substantia nigra (Farrer 2006; Samaranch et al. 2010; Steele et al. 2015; Takanashi et al. 2016). Attempts to model this phenotype in mice have largely been unsuccessful, as PINK1 knockout (KO) and parkin KO mice do not show death of DA neurons in the substantia nigra (Dawson et al. 2010). Similarly, ips-DA neurons from patients harboring loss-of-function mutations in PINK1 or parkin do not show increased cell death under normal culture conditions (Miller et al. 2013; Chang et al. 2016). Miller and colleagues suggested that a cell death phenotype could be age-dependent, as neuronal death and the onset of PD symptoms are not observed early in life. In support of this idea, they demonstrated that increased cell death in ips-DA neurons with PINK1 or parkin mutations was not present under normal culture conditions but become observable by overexpression of progerin, which is intended to accelerate cellular aging (Miller et al. 2013). Cell death in PINK1 or parkin mutant ips-DA neurons is increased in response to proteasome inhibition, mitochondrial toxins, or oxidative stressors (Cooper et al. 2012; Chang et al. 2016; Chung et al. 2016; Suzuki et al. 2017). A reduction of neurite length and complexity occurs in parkin ips-DA and tyrosine hydroxylase (TH)-negative ips-neurons under normal culture conditions, which is rescued by overexpression of WT parkin (Ren et al. 2015). However, others have only observed this phenotype in parkin or PINK1 ips-DA neurons in the presence of toxins (Miller et al. 2013; Lin et al. 2016). This suggests that PINK1 and parkin mutations confer a genetic susceptibility but additional genetic and/or environmental insults, such as proteasome inhibition, oxidative stress, or aging, may be required for a survival or neurite deficit in ips-DA neurons or in patients.
α-Syn accumulation
Most parkin PD patients do not display Lewy body pathology but some do (Pramstaller et al. 2005). One of three post-mortem studies performed on PD patients with PINK1 mutations revealed Lewy body pathology (Samaranch et al. 2010; Steele et al. 2015; Takanashi et al. 2016). Consistent with this, some studies did not observe altered levels of α-syn in ips-DA neurons from PD patients with PINK1 or parkin mutations (Imaizumi et al. 2012; Jiang et al. 2012) but others did see an increase in α-syn levels (Imaizumi et al. 2012; Chang et al. 2016; Chung et al. 2016). α-Syn accumulation is rescued by overexpressing WT PINK1 or parkin, showing that this phenotype is due to loss of function of these proteins (Chang et al. 2016; Chung et al. 2016). Imaizumi and colleagues found an accumulation of α-syn in ips-DA neurons from one patient with a deletion mutation in PARK2 but not in ips-DA neurons from a different patient with a different PARK2 deletion mutation. Interestingly, α-syn accumulation was also observed in the post-mortem brain of a patient whose ips-DA neurons showed α-syn accumulation, suggesting that ips-DA neuron phenotypes correlate with the patient phenotypes in this measure (Imaizumi et al. 2012). Parkin mutant ips-DA neurons show reduced proteasome activity, which is rescued by overexpression of WT parkin (Chang et al. 2016). Mutation of parkin also results in increased sensitivity to the proteasome inhibitor MG-132 in ips-DA neurons (Chang et al. 2016). Although the mechanism is unclear, both the ubiquitin-proteasome system and autophagy are implicated in the degradation of α-syn (Xilouri et al. 2013). Interestingly, parkin colocalizes with α-syn in Lewy bodies (Schlossmacher et al. 2002) and promotes its ubiquitination (Shimura et al. 2001). This functional relationship between the two proteins suggests the possibility that WT parkin promotes the formation of Lewy bodies, a process disturbed in parkin PD, explaining why Lewy body pathology is not present in most cases. Alternatively, loss of parkin function may lead to accumulation of non-ubiquitinated α-syn in parkin PD patients. The role of parkin localization to Lewy bodies and ubiquitination of α-syn in the pathology of PD is still not fully understood.
Mitochondria abnormalities
With the role of PINK1 and parkin in mitophagy, it is not surprising that abnormal mitochondrial morphology and function are observed in ips-DA neurons from PD patients with mutations in these genes. Morphologically abnormal enlarged mitochondria and increased mitochondrial superoxide levels are seen in ips-DA neurons from patients harboring mutations in either gene (Chung et al. 2016). Interestingly, in contrast to this study, reduced mitochondria volume in ips-DA neurons with PARK2 mutations has also been observed (Shaltouki et al. 2015). ips-DA neurons with parkin mutations display increased oxidative stress and ROS and impaired mitophagy (Imaizumi et al. 2012; Suzuki et al. 2017). In non-neuronal cells, PINK1 recruits parkin to damaged mitochondria and targets them for degradation by mitophagy and mutations of either protein disrupt this process (Narendra et al. 2008, 2010; Rakovic et al. 2010; Vives-Bauza et al. 2010). Consistent with this, PINK1 mutations in ips-DA neurons lead to impaired recruitment of parkin to damaged mitochondria, as well as a lack of reduction in mtDNA after mitochondria damage, suggesting impaired mitophagy. Both phenotypes are rescued by overexpressing WT PINK1 (Seibler et al. 2011). However, two studies did not report mitophagy even in control ips-neurons, perhaps due to variations in the experimental protocol (Jiang et al. 2012; Rakovic et al. 2013).
Dopamine regulation
Parkin also plays a role in DA transmission. Jiang and colleagues found that spontaneous DA release is increased in ips-DA neurons with mutant parkin. Furthermore, DA uptake decreased due to reduced dopamine transporter (DAT) binding sites (Jiang et al. 2012). It was demonstrated that parkin promotes ubiquitination and degradation of misfolded DAT (Jiang et al. 2004), so perhaps the reduced number of DAT binding sites in parkin ips-DA neurons is due to misfolding of DAT. In contrast, activity-dependent DA release is unchanged. Monoamine oxidases MAO-A and MAO-B, which catalyze the oxidative deamination of DA and increase ROS (Segura-Aguilar et al. 2014), have elevated expression levels and activity in parkin ips-DA neurons (Jiang et al. 2012), although a subsequent study observed no difference in expression of these transcripts (Imaizumi et al. 2012). This altered DA homoeostasis is also associated with increased DA-induced oxidative stress in parkin ips-neurons (Jiang et al. 2012). Interestingly, each of these phenotypes was rescued by overexpressing WT parkin but not mutant parkin, suggesting that loss of parkin function is responsible for these altered DA utilization phenotypes (Jiang et al. 2012). DA also regulates glutamatergic transmission differently in parkin ips-neurons than in controls. A recent study demonstrated that DA caused delayed enhanced spontaneous excitatory postsynaptic currents (sEPSC) amplitude and increased quantal content in ips-neurons from patients with parkin mutations but not in control ips-neurons. Miniature EPSCs were not affected. Additionally, the specific activation of D1-class dopamine receptor induced an oscillatory bursting of sEPSCs only in parkin ips-neurons, an effect that was reversed by the overexpression of WT parkin (Zhong et al. 2017). This demonstrates that mutations in parkin perturb dopaminergic transmission, as well as dopaminergic regulation of glutamatergic transmission, perhaps providing a mechanistic clue to the pathogenesis of PD.
GBA1
An unexpected link between Gaucher’s disease (GD), a rare inherited lysosomal storage disorder and PD was first discovered by clinicians who observed parkinsonism in GD patients (Neudorfer et al. 1996; Tayebi et al. 2001; Várkonyi et al. 2003). GD is caused by mutations in the β-glucocerebrosidase gene or GBA1, resulting in deficient production of lysosomal enzyme GCase and leading to the accumulation of GCase substrates in macrophages (also known as “Gaucher’s cells) and neurons and impairment in lysosomes and autophagosomes (Sun and Grabowski 2010; Sidransky and Lopez 2012). About 300 GBA1 mutations have been identified, several of which confer a high risk for PD (Migdalska-Richards and Schapira 2016), with N370S and L444P being the two most common GBA1 PD mutation sites (Schapira 2015). The extent to which mutations in GBA1 confer risk of PD through loss-of-function versus gain-of-function mechanisms is an area of active investigation (Futerman and Hardy 2016).
α-Syn accumulation
Patients with GBA1 mutations exhibit Lewy body pathology (Sidransky and Lopez 2012), indicating possible involvement of α-syn. Loss-of-function GBA1 mutations at N370S or L444P, resulting in reduced GCase levels and activity, increases levels of α-syn in ips-DA neurons from both GD and GBA1 PD patients (Mazzulli et al. 2011, 2016b; Schöndorf et al. 2014; Woodard et al. 2014; Aflaki et al. 2016). Additionally, a GBA1 N370S gain-of-function mutation resulting in misfolded GCase elevates extracellular α-syn levels in ips-DA neurons from GBA1 PD patients (Fernandes et al. 2016). In primary cortical neurons, GCase knockdown can enhance α-syn aggregation by augmenting polymerization-dependent mechanisms, leading to neurotoxicity. These α-syn aggregates, in turn, affect trafficking of GCase from ER-Golgi to lysosomes, which affects lysosomal function and leads to more α-syn aggregation, suggesting a positive feed-forward relationship between GCase and α-syn (Mazzulli et al. 2011).
DA metabolism
Some studies have reported alterations in DA reuptake in brains of GBA1 PD patients, with striking similarities to sporadic PD patient brains (Sidransky and Lopez 2012). It is not known how GBA1 mutations directly affect DA levels; however, DA dysfunction might be a downstream effect of α-syn accumulation. Loss-of-function GBA1 mutations decrease intracellular levels of DA and DA uptake in ips-DA neurons from patients with GD and GBA1 PD (Aflaki et al. 2016). Woodard and colleagues reported irregularity in DA metabolism in ips-DA neurons from monozygotic twins with heterozygous GBA1 N370S mutation discordant for PD. Interestingly, DA levels with upregulated expression of MAO-B were lower in the GBA1 PD than in a GBA1 non-PD twin. They restored normal DA metabolism in both twins by treating with an MAO-B inhibitor (rasagiline), while simultaneously overexpressing WT GBA1. Only combinatorial treatment restored normal DA metabolism in both twins, highlighting the complex interplay between multiple mechanisms, including genetic and non-genetic factors underlying the relationship between GBA1 mutations, α-syn, DA metabolism and onset of PD (Woodard et al. 2014).
Autophagy-lysosomal pathway (ALP)
GBA1 mutations have been associated with dysfunctional lysosomes and autophagy; it is not surprising that ips-DA neurons derived from GBA1 PD patients with mutations at N370S or L444P have several ALP defects, such as lysosomal enlargement and autophagosome accumulation (Mazzulli et al. 2011; Schöndorf et al. 2014; Fernandes et al. 2016), in addition to impaired lysosomal proteolysis of long-lived proteins such as α-syn (Mazzulli et al. 2011). A gain-of-function GBA1 N370S mutation also enhanced ER stress as a result of improper processing of GCase by the Golgi due to possible retention in the ER in ips-DA neurons from GBA1 PD patients (Fernandes et al. 2016).
Ips-neurons derived from iPSC patients with sporadic PD
The etiology of PD is multifactorial, involving a complex combination of genetic and environmental factors. Most PD cases are sporadic. Interestingly, many of the disease-associated phenotypes observed in ips-DA neurons from PD patients with familial mutations are also observed in ips-DA neurons from patients with sporadic PD, indicating that sporadic and familial PD patients may share common pathogenic mechanisms. For example, ips-neurons and ips-DA neurons from patients with sporadic PD show increased apoptosis, reduced neurites, impaired autophagy, impaired mitophagy and irregular DA metabolism (Sánchez-Danés et al. 2012; Woodard et al. 2014; Hsieh et al. 2016; Lin et al. 2016). However, accumulation of α-syn and mitochondrial abnormalities have not been observed (Sánchez-Danés et al. 2012; Woodard et al. 2014). Because the causes of sporadic PD are not fully understood, it is difficult to model.
Immune cells in PD
Neuroinflammation plays a role in multiple neurological diseases, including Parkinson’s disease (Tansey et al. 2007; Vivekanantham et al. 2015). PD patients show signs of increased immune dysregulation, such as activated microglia and increased expression of inflammatory signaling molecules (McGeer et al. 1988; Banati et al. 1998; Hunot et al. 1999; Gerhard et al. 2006; Duke et al. 2007; Tansey et al. 2007). Evidence suggests that α-syn promotes the activation of microglia, which in turn aid in clearing extracellular α-syn and that perhaps LRRK2 can regulate microglial function (Sanchez-Guajardo et al. 2015; Schapansky et al. 2015). Studies have suggested that LRRK2 modulates inflammatory signaling through the nuclear factor kappa-B (NF-κb) pathway in microglia, as well as ips-neurons (Kim et al. 2012; Russo et al. 2015; López de Maturana et al. 2016). While iPSC technology enables patient-derived cells to be differentiated into disease-relevant cells, such as neurons, the etiology of PD is complex, involving a variety of cell types, including immune cells. Protocols to differentiate iPSCs into immune cells, such as microglia, have been established and incorporation of these iPSC-derived microglia (ips-microglia) into co-cultures with neurons or brain organoids shows promise in the development of a culture system in which the interaction between these immune cells and neurons can be further studied (Muffat et al. 2016; Abud et al. 2017; Douvaras et al. 2017; Pandya et al. 2017).
Environmental factors in the pathogenesis of PD
Environmental factors are also important in PD pathology. Epidemiological data suggest that exposure to pesticides, such as rotenone, paraquat and maneb, correlates with an increased risk of developing PD. Treating animals with these compounds or with other neurotoxins, such as 6-OHDA, or the illicit drug contaminant 1-methyl-1,2,3,6-tetrahydropiridine (MPTP), induces PD behavioral symptoms, as well as degeneration of dopaminergic neurons (Moretto and Colosio 2013; Le et al. 2014). Many of these compounds are inhibitors of complex I of the electron transport chain in mitochondria and/or oxidative stressors. Post-mortem brains from PD patients show signs of oxidative stress and defects in complex I (Schapira et al. 1990; Hattori et al. 1991; Dexter et al. 1994; Yoritaka et al. 1996; Alam et al. 1997), possibly indicating that PD and the neurotoxin models of PD share common pathogenic mechanisms. Sensitivity to neurotoxins such as these has been observed in ips-DA neurons with a variety of different familial mutations (Byers et al. 2011; Nguyen et al. 2011; Cooper et al. 2012; Reinhardt et al. 2013; Ryan et al. 2013; Ohta et al. 2015; Chang et al. 2016; Chung et al. 2016; Suzuki et al. 2017), indicating that, while certain toxins may induce PD symptoms, genetic susceptibility increases this effect.
Common pathways
Several phenotypes recur in ips-neurons derived from PD patients with a variety of different familial mutations and patients with sporadic PD. Mitochondrial abnormalities, defects in mitophagy and autophagy, sensitivity to oxidative stressors and accumulation of α-syn are some of the most common phenotypes. Though the exact pathways differ, all pathogenic mutations likely converge at some point, ultimately culminating in neurodegeneration (Fig. 1).

Cellular pathways disrupted by familial PD mutations in ips-neurons. iPSC-based models from PD patients from multiple genetic backgrounds demonstrate several alterations, including a mitochondrial abnormalities, b reduced mitophagy, c ALP defects and reduced GCase function, d α-syn accumulation, and e vulnerability to environmental factors. Pathogenic mutations and pathways contributing to each cellular alteration are listed below as bullet points. Reduced ALP GCase function in (c) contributes to α-syn accumulation (d), which in turn may inhibit ALP and GCase function, suggesting a bidirectional relationship (f). Together, these and other cellular abnormalities (g) contribute to neurodegeneration
Abnormal mitochondrial morphology and function are observed in ips-DA neurons with pathogenic LRRK2, PINK1, parkin and α-syn mutations (see above). Adding to the insult, mitophagy is impaired in sporadic as well as LRRK2, PINK1 and parkin mutant ips-DA neurons (see above), leading to a buildup of damaged mitochondria. Studies in ips-DA neurons and other cell types suggest that, upon damage to mitochondria WT LRRK2, PINK1 and parkin interact with and promote the degradation of the outer mitochondrial membrane protein, Miro. This arrests the motility of damaged mitochondria, which occurs prior to their clearance by mitophagy. Pathogenic mutations in these proteins disrupt this process (Wang et al. 2011; Hsieh et al. 2016). Remarkably, Miro degradation, mitophagy and recruitment of LRRK2 and parkin to damaged mitochondria are disrupted in sporadic PD fibroblasts. This indicates a shared pathogenic convergence point at Miro among sporadic and multiple forms of familial PD. However, overexpression of parkin in LRRK2 mutant cells does not rescue the impaired mitochondria damage-induced degradation of Miro. A pathogenic LRRK2 mutation does not disrupt parkin recruitment to damaged mitochondria, nor do pathogenic PINK1 and parkin mutations prevent LRRK2 recruitment to damaged mitochondria nor its interaction with Miro in fibroblasts. These observations suggest that LRRK2 and PINK1/parkin act in separate pathways that converge at Miro (Hsieh et al. 2016).
LRRK2, PINK1, parkin, or α-syn mutations in ips-neurons cause a vulnerability to toxins. Many of these neurotoxins target mitochondria, increase ROS and inflammation and/or inhibit the proteasome (Bové and Perier 2012), suggesting that mitochondrial dysfunction, oxidative stress, inflammation and disrupted proteostasis might be common pathways by which these toxins induce neurodegeneration in multiple genetic backgrounds. In fact, PD-derived ips-neurons show defects in many of these pathways in the absence of toxin exposure. How certain pathogenic PD mutations confer vulnerability to toxins is unclear but perhaps the addition of toxins disrupts key cellular pathways past a certain tolerance, leading from neuron dysfunction to cell death.
Accumulation of α-syn is common among familial PD ips-DA neurons. As defects in autophagy are also seen in LRRK2, GBA1 and sporadic PD ips-DA neurons (see above), it is interesting to speculate that autophagy defects might contribute to α-syn accumulation. Indeed, the ubiquitin–proteasome system (UPS) and ALP are both implicated in α-syn degradation (Xilouri et al. 2013). Interestingly, the interaction between α-syn and the ALP may be bidirectional, as modulating α-syn levels regulates lysosomal function (Mazzulli et al. 2016a).
Along with other familial mutations, GBA1 mutations lead to α-syn accumulation, possibly due to these lysosomal defects. Glucosylceramide, a GCase substrate that accumulates in the absence of GCase activity, promotes the pathogenic oligomerization α-syn (Mazzulli et al. 2011). Pharmacologically increasing GCase activity rescues pathological α-syn accumulation in ips-DA neurons from sporadic, SNCA, GBA1 and PARK9 PD patients (Aflaki et al. 2016; Mazzulli et al. 2016b), suggesting that α-syn accumulation may occur through disruption of GCase activity in PD patients with a range of genetic backgrounds. Evidence also suggests that the relationship between GCase and α-syn also works in the opposite direction. α-syn accumulation disrupts GCase maturation, reducing its activity in lysosomes, leading to lysosomal impairment in ips-DA neurons (Mazzulli et al. 2016a). Together, these studies indicate a bidirectional relationship in which loss of GCase activity leads to pathological α-syn accumulation that, in turn, disrupts GCase maturation and its activity in lysosomes and promotes further α-syn accumulation.
Defects in autophagy, abnormal mitochondria and reduced mitochondrial clearance, oxidative stress and altered proteostasis have emerged as common themes in the pathogenesis of PD. Diminished GCase activity and ALP function may promote the accumulation of α-syn. Further studies are needed to fully understand the mechanistic link between the multiple pathological abnormalities observed in PD ips-neurons and the pathways that lead to neurodegeneration.
Benefits and challenges of using iPSCs as a model for PD
As PD appears to result from a complex interplay of genetic and environmental risk factors that are not yet fully understood, modeling PD has been challenging. The major hallmarks of PD include the preferential degeneration of dopaminergic neurons in the substantia nigra and Lewy bodies composed of α-syn. An ideal model is expected to mimic these principal features of PD. Genetic animal models, in which PD-associated genes have been altered and neurotoxin-induced animal models have contributed widely to advances in our understanding of the molecular and cellular pathogenesis of PD. However, genetic animal models often do not display progressive loss of DA neurons or obvious α-syn inclusions and although many neurotoxin-induced animal models have successfully demonstrated DA neuron loss and in some cases α-syn inclusions, they generally represent an acute model rather than progressive degeneration (Bezard et al. 2013; Le et al. 2014). Importantly, clinical studies based on therapies tested in neurotoxin-based models have not yet yielded a successful treatment for PD, calling into question their predictive value (Dawson et al. 2010; Bezard et al. 2013). Perhaps this is due to the fact that neurotoxin-induced animal models act acutely rather than progressively, or because human neurons are uniquely sensitive or have different cellular mechanisms. The use of mouse models in preclinical testing has also not translated into successful clinical trials for a number of other neurodegenerative disorders, including Alzheimer’s disease, amyotrophic lateral sclerosis and Huntington’s disease (Mullane and Williams 2013; Perrin 2014; Haston and Finkbeiner 2016). Whether there are inherent mouse–human species-specific differences that are important and contribute to the failures to translate promising preclinical discoveries into clinical trial success is unknown. Nevertheless, the need to develop new models that can faithfully predict results in humans with neurodegenerative disorders, such as PD, seems clear.
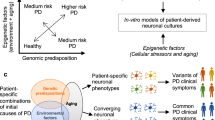
iPSCs offer several unique benefits as models for PD and other complex diseases. A major advantage is that they are human cells, avoiding the potential confounding factor of species–specific cellular mechanisms. Furthermore, as human cells derived from patients, they retain all of the genetic risk factors. However, to date, only a limited number of PD patient-derived iPSC lines are in use. To fully model the heterogeneity of PD, iPSC lines from a large number of PD patients harboring mutations in different PD-associated genes, as well as lines from a range of sporadic PD patients, are needed. Furthermore, with the exciting advancements in gene-editing techniques (Hendriks et al. 2016), iPSC lines can be made in which a pathogenic mutation has been corrected, thereby making control lines with a genetic composition that is otherwise similar (Soldner et al. 2011; Liu et al. 2012; Reinhardt et al. 2013). These gene-corrected controls minimize phenotypic variability due to differences in genetic background between patient and control cells. However, because gene-corrected controls cannot be made for iPSC lines from sporadic PD patients in which disease-associated variants have not been identified, the use of large numbers of both sporadic and healthy control iPSC lines will be needed to fully characterize PD-associated phenotypes from a wide diversity of patients with varying etiology. PD heritability estimates range widely, from approximately 4 to 27% (Do et al. 2011; Houlden and Singleton 2012) and so it may prove to be important to make human neurons in ways that retain more of the epigenetic signature, including the influences of aging, of the patient from whom the cells came (Mertens et al. 2015). It also remains to be seen whether there are common cellular phenotypes that are shared across all forms of sporadic and familial PD or whether the mechanisms of PD differ in important ways among subpopulations of PD patients. It will be important to carefully measure variation in phenotypic expression to determine whether it has a biological basis that is relevant to PD. If it does, it could be used for patient stratification and to guide the development of effective therapeutic strategies.
iPSCs offer another advantage in that they can be differentiated into disease-relevant cell types, such as DA neurons, cortical neurons, microglia, astrocytes, or enteric neurons. The transcriptomic profiles of purified ips-DA neurons are similar to human post-mortem DA neurons (Sandor et al. 2017), suggesting that ips-DA neurons are representative of mature human DA neurons in an intact brain. While some phenotypes are observed in multiple cell types, some phenotypes appear to be neuron-specific. For example, mtDNA damage, abnormal mitochondrial morphology and impaired mitophagy have been observed in patient ips-DA neurons but not in fibroblasts or iPSCs from the same patients (Imaizumi et al. 2012; Sanders et al. 2014). Compared to other neuronal cell types, DA neurons are particularly sensitive to stress (Subramaniam and Chesselet 2013; Zuo and Motherwell 2013). Evidence suggests that ips-DA neurons might be more sensitive to death induced by oxidative stress than peripheral sensory neurons derived from the same iPSCs or TH negative neurons in the same culture (Nguyen et al. 2011; Reinhardt et al. 2013). This observation suggests that DA neurons are particularly vulnerable to oxidative stress. Furthermore, familial PD ips-DA neurons show a greater sensitivity to oxidative stressors and MG-132 than controls but this is not seen in other neuron types from the same patients (Nguyen et al. 2011; Reinhardt et al. 2013), further reiterating the importance of using disease-relevant cell types to model PD.
While iPSCs offer the opportunity to use human, patient–specific and disease-relevant cell types to study the molecular and cellular pathogenesis of PD, the use of iPSCs for disease modeling is not without its challenges. Multiple different protocols are used to differentiate iPSCs into DA neurons (Perrier et al. 2004; Chambers et al. 2009; Zhang and Zhang 2010; Kriks et al. 2011; Studer 2012; Woodard et al. 2014), making direct comparisons between studies challenging. Importantly, one study found that several phenotypes were observed in PD patient-derived ips-DA neurons with one differentiation protocol but not another, highlighting the impact of differentiation technique (Chung et al. 2016). Inefficient DA neuron production is also common among many differentiation protocols, resulting in cultures of mixed neuronal subtypes. Enriching for the dopaminergic neuronal subtype resulted in more robust differences between PD and control ips-DA neuron phenotypes, underlining the importance of both the differentiation protocol and the yield of ips-DA neurons (Suzuki et al. 2017). Furthermore, although purified ips-DA neurons transcriptionally resemble mature human DA neurons (Sandor et al. 2017), ips-DA neurons do not display characteristic pace-making activity except in older cultures (Kriks et al. 2011; Studer 2012). Further optimization of differentiation protocols is needed to produce pure cultures of functionally mature ips-DA neurons without prolonged in vitro maturation.
PD is characterized by a loss of DA neurons in the substantia nigra and this principal phenotype has been recapitulated in a number of ips-neurons from PD patients (described above). However, not all patient-derived ips-DA neuron lines show a cell death phenotype under all conditions (Nguyen et al. 2011; Miller et al. 2013; Ohta et al. 2015; Chang et al. 2016). An increased susceptibility to cell death becomes apparent in some of these ips-DA neuron lines with the addition of various neurotoxins or by culturing for longer time periods (Nguyen et al. 2011; Ohta et al. 2015; Chang et al. 2016). Reprograming human somatic cells into iPSCs results in a cellular “rejuvenation”, in that age-related markers are lost (Cornacchia and Studer 2017). Compared to differentiated adult cells, iPSCs have DNA methylation patterns, nuclear morphology, telomere lengths, DNA damage and mitochondrial function and phenotype that resemble what is observed in young cells (Marion et al. 2009; Suhr et al. 2009, 2010; Prigione et al. 2010; Horvath 2013; Miller et al. 2013; Frobel et al. 2014). This points to a weakness in using iPSCs to model neurodegenerative disorders, in which symptoms often do not present until later in life. Overexpression of a progerin, an aberrantly shortened transcript of lamin A thought to accelerate aging in Hutchinson–Gilford progeria syndrome (HGPS) patients, was associated with the increased appearance of several PD-related phenotypes, including increased cell death, in patient-derived ips-DA neurons that were not observed under normal conditions (Miller et al. 2013). However, HGPS patients do not show premature neuronal aging and lamin A is not expressed in neurons, indicating that overexpression of progerin might not fully recapitulate normal aging in neurons (Jung et al. 2012; Nissan et al. 2012). One promising approach is transdifferentiation, or direct conversion from fibroblasts to other differentiated cell types, which can retain the age-related markers of the individual from whom they came (Vierbuchen et al. 2010; Yoo et al. 2011; Victor et al. 2014; Mertens et al. 2015).
Future directions
As the molecular pathogenesis of PD is not yet fully understood, developing sensitive methods to correlate disease-related phenotypes to causes of neurodegeneration is becoming increasingly important. For example, while α-syn inclusions are a hallmark pathogenic feature in PD, the role of these aggregates in pathogenicity is unclear (Ross and Poirier 2005; Cookson 2009). We developed an automated longitudinal imaging platform called robotic microscopy that provides increased sensitivity and resolution at the single-cell level to monitor the health of individual neurons over time. By implementing survival models and powerful statistical methods, we can quantify the prognostic value of biological factors for degenerative phenotypes, such as neuron death and neurite degeneration and thereby distinguish pathogenic changes from those that are incidental or are coping mechanisms (Arrasate and Finkbeiner 2005; Finkbeiner et al. 2015). Using this approach, we addressed a long-standing controversy about the role of inclusions in Huntington’s disease by showing that cells that form inclusions survive longer, indicating that they mitigate toxicity (Arrasate et al. 2004). Similarly, we discovered that intracellular levels of mutant LRRK2 and not its kinase activity or inclusion bodies, predicted neurodegeneration, which depended on α-syn (Skibinski et al. 2014) and identified a role for nuclear factor erythroid 2-related factor (Nrf2), a transcription factor regulating stress-response pathways, in mitigating α-syn-mediated neurotoxicity by modulating proteostasis (Skibinski et al. 2016). By using robust methods, such as robotic microscopy, we can accurately detect cellular phenotypes and understand the sequence of events leading up to neurodegeneration in PD.
PD patient-derived ips-neurons can be used in validating genetic (with knockdown or overexpression approaches) or small molecules that modify disease-relevant cellular phenotypes. Targets identified through high-throughput screens can be validated with molecular and mechanistic studies in ips-neurons. For example, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-edited ips-neurons have been used to validate an SNCA enhancer variant associated with PD (Soldner et al. 2016). Similarly, iPSC-based models themselves can be used as platforms for high-throughput screening of PD modifiers or therapeutics (Haggarty and Perlis 2014). High-throughput screening of compounds in human ips-neurons led to the identification of Isoxasole, specifically targeting the MEF2C-PCG1α pathway, which contributed to neuronal damage (Ryan et al. 2013). Traditionally, murine models and human cancer cell lines, such as the neuroblastoma cell line SH-SY5Y, have been frequently used in preclinical studies. However, species-specific differences in murine models and the lack of patient-specific genetic background and differentiation potential, limit these models (Schüle et al. 2009; Ko and Gelb 2014). Although iPSCs have limitations of their own, as described above, the ability of iPSCs to provide PD patient-specific, disease-relevant cell types has the potential to more accurately model human PD. iPSC-derived models can be used to bridge the gap between preclinical studies and clinical trials, resulting in increased translation of drug candidates and higher rates of clinical success.
Capturing the pathogenic diversity observed in patients is another significant challenge, as PD is a multifaceted disease with a high degree of heterogeneity, where patients can be classified into multiple subgroups based on their clinical features (Vidailhet 2003; Lewis et al. 2005). To what extent these diverse clinical presentations reflect underlying differences in molecular pathogenesis is unknown and so the response of different PD patients to a certain therapy is difficult to predict. Because there are a wide variety of genetic and environmental factors that influence the pathogenesis of PD, it is possible that different subpopulations of patients will respond differently to certain therapies. iPSC-based models derived from a range of PD patients have the potential to determine if a specific therapy might work in subpopulations of patients but not in all patients. This could potentially change the way that clinical trials are designed and improve their chances of success by allowing them to account for the genetic heterogeneity among PD patients and predict in what genetic background a particular therapy will be successful. In addition to improving the success of clinical trials by providing more accurate pre-clinical models of PD, transplantation of iPSC-derived neurons is also being considered as a potential therapy for PD (Lindvall 2016).
While improvements still need to be made in iPSC-based disease modeling, this technology offers an unprecedented ability to mimic disease in vitro with patient-specific disease-relevant cell types. Human iPSC technology has the potential to deepen our understanding of the pathogenesis of disease, provide a more predictive platform for pre-clinical studies and improve the success of clinical trials.
References
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, Caraway CA, Fote GM, Madany AM, Agrawal A, Kayed R, Gylys KH, Cahalan MD, Cummings BJ, Antel JP, Mortazavi A, Carson MJ, Poon WW, Blurton-Jones M (2017) iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94:278–293
Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, Schoenen FJ, Westbroek W, Zheng W, Sullivan P, Fujiwara H, Sidhu R, Khaliq M, Lopez GJ, Goldstein DS, Ory DS, Marugan J, Sidransky E (2016) A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with gaucher disease and parkinsonism. J Neurosci 36:7441–7452
Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B (1997) A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J Neurochem 69:1326–1329
Anglade P, Vyas S, Herrero MT, Michel PP, Marquez J, Ruberg M, Hirsch EC, Agid, Y. (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12:25–31
Arrasate M, Finkbeiner S (2005) Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A 102:3840–3845
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Banati RB, Daniel SE, Blunt SB (1998) Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord 13:221–227
Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, Ding J, Nalls MA, Olszewski M, Hauser DN, Kumaran R, Lozano AM, Baekelandt V, Greene LE, Taymans JM, Greggio E, Cookson MR (2014) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A 111:2626–2631
Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, Pizzi M, Missale C, Spano P (2016) Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol 42:77–94
Bezard E, Yue Z, Kirik D, Spillantini MG (2013) Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Mov Disord 28:61–70
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson’s disease. Front Neuroanat 9:91
Borgs L, Peyre E, Alix P, Hanon K, Grobarczyk B, Godin JD, Purnelle A, Krusy N, Maquet P, Lefebvre P, Seutin V, Malgrange B, Nguyen L (2016) Dopaminergic neurons differentiating from LRRK2 G2019S induced pluripotent stem cells show early neuritic branching defects. Sci Rep 6:33377
Bové J, Perier C (2012) Neurotoxin-based models of Parkinson’s disease. Neuroscience 211:51–76
Burré J (2015) The synaptic function of α-synuclein. J Parkinsons Dis 5:699–713
Byers B, Cord B, Nguyen HN, Schüle B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, Palmer TD (2011) SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 6:e26159
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280
Chang KH, Lee-Chen GJ, Wu YR, Chen YJ, Lin JL, Li M, Chen IC, Lo YS, Wu HC, Chen CM (2016) Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson’s disease. Park Relat Disord 24:81–88
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–398
Chung CY, Khurana V, Auluck PK, Tardiff, Daniel F, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schüle B, Lippard SJ, Tsai L, Krainc D, Buchwald SL, Jaenisch R, Lindquist S (2013) Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342:983–987
Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, Puspita L, Valiulahi P, Sulzer D, Milner TA, Taldone T, Krainc D, Studer L, Shim J (2016) Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-synuclein accumulation. Stem Cell Reports 7:664–677
Cookson MR (2009) α-Synuclein and neuronal cell death. Mol Neurodegener 4:9
Cookson MR (2010) The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci 11:791–797
Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, Romero N, Jiang H, Uitti RJ, Huang Z, Opala G, Scarffe LA, Dawson VL, Klein C, Feng J, Ross OA, Trojanowski JQ, Lee VM, Marder K, Surmeier DJ, Wszolek ZK, Przedborski S, Krainc D, Dawson TM, Isacson O. (2012) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med 4:141ra90
Cornacchia D, Studer L (2017) Back and forth in time: directing age in iPSC-derived lineages. Brain Res 1656:14–26
Dawson TM, Dawson VL (2010) The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord 25:S32–S39
Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson’s disease. Neuron 66:646–661
Deas E, Cremades N, Angelova PR, Ludtmann MHR, Yao Z, Chen S, Horrocks MH, Banushi B, Little D, Devine MJ, Gissen P, Klenerman D, Dobson CM, Wood NW, Gandhi S, Abramov AY (2016) Alpha-Synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid Redox Signal 24:376–391
Dehay B, Bove J, Rodríguez-muela N, Perier C, Recasens A, Boya P, Vila M (2010) Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 30:12535–12544
Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D (2015) Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 6:7314
Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD (1994) Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord 9:92–97
Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, Wojcicki A, Eriksson N (2011) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 7:e1002141
Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, Terrenoire C, Zhang B, Gandy S, Schadt E, Freytes DO, Noggle S, Fossati V (2017) Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep 8:1516–1524
Duke DC, Moran LB, Pearce RKB, Graeber MB (2007) The medial and lateral substantia nigra in Parkinson’s disease: mRNA profiles associated with higher brain tissue vulnerability. Neurogenetics 8:83–94
Durcan TM, Fon EA (2015) The three “P”s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev 29:989–999
Emamzadeh FN (2016) Alpha-synuclein structure, functions, and interactions. J Res Med Sci 21:29
Esteves AR, Cardoso SM (2017) LRRK2 at the crossroad between autophagy and microtubule trafficking: insights into Parkinson’s disease. Neuroscience. https://doi.org/10.1177/1073858415616558
Farrer MJ (2006) Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 7:306–318
Fernandes HJR, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, Bogetofte H, Lang C, Ryan BJ, Sardi SP, Badger J, Vowles J, Evetts S, Tofaris GK, Vekrellis K, Talbot K, Hu MT, James W, Cowley SA, Wade-Martins R (2016) ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep 6:342–356
Finkbeiner S, Frumkin M, Kassner PD (2015) Cell-based screening: extracting meaning from complex data. Neuron 86:160–174
Frobel J, Hemeda H, Lenz M, Abagnale G, Joussen S, Denecke B, Šarić T, Zenke M, Wagner W (2014) Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep 3:414–422
Futerman AH, Hardy J (2016) Perspective: finding common ground. Nature 537:S160–S161
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412
Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VMY (2000) Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290:985–989
Gillardon F (2009) Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability - a point of convergence in parkinsonian neurodegeneration? J Neurochem 110:1514–1522
Gómez-Suaga P, Fdez E, Fernández B, Martínez-Salvador M, Blanca Ramírez M, Madero-Pérez J, Rivero-Ríos P, Fuentes JM, Hilfiker S (2014) Novel insights into the neurobiology underlying LRRK2-linked Parkinson’s disease. Neuropharmacology 85:45–56
González F, Stéphanie B, Izpisúa Belmonte JC (2011) Methods for making induced pluripotent stem cells: reprogramming à la carte. Nat Rev Genet 12:231–242
Good PF, Hsu A, Werner P, Perl DP, Olanow W (1998) Protein nitration in Parkinson’s disease. J Neuropathol Exp Neurol 57:338–342
Haggarty SJ, Perlis RH (2014) Translation: screening for novel therapeutics with disease-relevant cell types derived from human stem cell models. Biol Psychiatry 75:952–960
Haston KM, Finkbeiner S (2016) Clinical trials in a dish: the potential of pluripotent stem cells to develop therapies for neurodegenerative diseases. Annu Rev Pharmacol Toxicol 56:489–510
Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N (2004) Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol 56:424–427
Hattori N, Tanaka M, Ozawa T, Mizuno Y (1991) Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann Neurol 30:563–571
Hendriks WT, Warren CR, Cowan CA (2016) Genome editing in human pluripotent stem cells: approaches, pitfalls, and solutions. Cell Stem Cell 18:53–65
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14:R115
Houlden H, Singleton AB (2012) The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol 124:325–338
Hsieh CH, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St. Lawrence E, Schüle B, Krainc D, Palmer TD, Wang X (2016) Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19:709–724
Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC (1999) Fcepsilon RII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci 19:3440–3447
Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S, Takanashi M, Funayama M, Hirayama A, Soga T, Hishiki T, Suematsu M, Yagi T, Ito D, Kosakai A, Hayashi K, Shouji M, Nakanishi A, Suzuki N, Mizuno Y, Mizushima N, Amagai M, Uchiyama Y, Mochizuki H, Hattori N, Okano H (2012) Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 5:35
Ischiropoulos H, Beckman JS (2003) Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest 111:163–169
Jiang H, Jiang Q, Feng J (2004) Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J Biol Chem 279:54380–54386
Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, Azabdaftari G, Nakaso K, Yan Z, Feng J (2012) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun 3:668
Jung H-J, Coffinier C, Choe Y, Beigneux AP, Davies BSJ, Yang SH, Barnes RH, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG (2012) Regulation of prelamin a but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A 109:E423–E431
Kawakami F, Yabata T, Ohta E, Maekawa T, Shimada N, Maruyama H, Ichikawa T, Obata F (2012) LRRK2 phosphorylates tubulin-associated tau but not the free molecule : LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS ONE 7:e30834
Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe E (2012) Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS ONE 7:e34693
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Ko HC, Gelb BD (2014) Concise review: drug discovery in the age of the induced pluripotent stem cell. Stem Cells Transl Med 3:500–509
Kouroupi G, Taoufik E, Vlachos IS, Tsioras K, Antoniou N, Papastefanaki F, Chroni-Tzartou D, Wrasidlo W, Bohl D, Stellas D, Politis PK, Vekrellis K, Papadimitriou D, Stefanis L, Bregestovski P, Hatzigeorgiou AG, Masliah E, Matsas R (2017) Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc Natl Acad Sci U S A 114:E3679–E3688
Kriks S, Shim J-W, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480:547–551
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci:38–48
Le W, Sayana P, Jankovic J (2014) Animal models of Parkinson’s disease: a gateway to therapeutics? Neurotherapeutics 11:92–110
Levy OA, Malagelada C, Greene LA (2009) Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis 14:478–500
Lewis SJG, Foltynie T, Blackwell AD, Robbins TW, Owen AM, Barker RA (2005) Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry 76:343–348
Lin MT, Cantuti-castelvetri I, Zheng K, Jackson KE, Tan YB, Arzberger T, Lees AJ, Betensky RA, Beal MF, Simon DK (2012) Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol 71:850–854
Lin L, Gö J, Cukuroglu E, Dranias MR, Vandongen AMJ, Stanton LW (2016) Molecular features underlying neurodegeneration identified through in vitro modeling of genetically diverse Parkinson’s disease patients. Cell Rep 15:2411–2426
Lindvall O (2016) Clinical translation of stem cell transplantation in Parkinson’s disease. J Intern Med 279:30–40
Liu Y, Qiang M, Wei Y, He R (2011) A novel molecular mechanism for nitrated α-synuclein-induced cell death. J Mol Cell Biol 3:239–249
Liu G-H, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, Wagner U, Kim A, Ren B, Li Y, Goebl A, Kim J, Soligalla RD, Dubova I, Thompson J, Yates Iii J, Rodriguez Esteban C, Sancho-Martinez I, Izpisua Belmonte JC (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491:603–607
López de Maturana R, Lang V, Zubiarrain A, Sousa A, Vázquez N, Gorostidi A, Águila J, López de Munain A, Rodríguez M, Sánchez-Pernaute R (2016) Mutations in LRRK2 impair NF-κB pathway in iPSC-derived neurons. J Neuroinflammation 13:295
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A (2006) The familial parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52:587–593
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, MacCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77:425–439
Madero-Pérez J, Fdez E, Fernández B, Lara Ordóñez AJ, Blanca Ramírez M, Romo Lozano M, Rivero-Ríos P, Hilfiker S (2017) Cellular effects mediated by pathogenic LRRK2: homing in on Rab-mediated processes. Biochem Soc Trans 45:147–154
Malik N, Rao MS (2013) A review of the methods for human iPSC derivation. Methods Mol Biol 997:23–33
Marion RM, Strati K, Li H, Tejera A, Schoeftner S, Ortega S, Serrano M, Blasco MA (2009) Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4:141–154
Martin I, Kim JW, Dawson VL, Dawson TM (2014) LRRK2 pathobiology in Parkinson’s disease. J Neurochem 131:554–565
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016a) α-Synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 113:1931–1936
Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, Patnaik S, Sidransky E, Marugan JJ, Sue CM, Krainc D (2016b) Activation of β-Glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J Neurosci 36:7693–7706
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1285
Mertens J, Paquola ACM, Ku M, Hatch E, Böhnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, Gonçalves JT, Toda T, Kim Y, Winkler J, Yao J, Hetzer MW, Gage FH (2015) Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17:705–718
Migdalska-Richards A, Schapira AHV (2016) The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem 139:77–90
Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, Taldone T, Fusaki N, Tomishima MJ, Krainc D, Milner TA, Rossi DJ, Studer L (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13:691–705
Moretto A, Colosio C (2013) The role of pesticide exposure in the genesis of Parkinson’s disease: epidemiological studies and experimental data. Toxicology 307:24–34
Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai L, Aubourg P, Ransohoff RM, Jaenisch R (2016) Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 22:1358–1367
Mullane K, Williams M (2013) Alzheimer’s therapeutics: continued clinical failures question the validity of the amyloid hypothesis - but what lies beyond? Biochem Pharmacol 85:289–305
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298
Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A (1996) Occurrence of Parkinson’s syndrome in type I Gaucher disease. Q J Med 89:691–694
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schüle B, Dolmetsch RE, Langston W, Palmer TD, Pera RR (2011) LRRK2 mutant iPSC-derived da neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8:267–280
Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M (2012) Unique preservation of neural cells in Hutchinson-Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep 2:1–9
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780
Ohta E, Nihira T, Uchino A, Imaizumi Y, Okada Y, Akamatsu W, Takahashi K, Hayakawa H, Nagai M, Ohyama M, Ryo M, Ogino M, Murayama S, Takashima A, Nishiyama K, Mizuno Y, Mochizuki H, Obata F, Okano H (2015) I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased tau phosphorylation through the AKT/GSK-3 β signaling pathway. Hum Mol Genet 24:4879–4900
Orenstein SJ, Kuo S-H, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16:394–406
Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, Kim G, Brown MA, Elkahloun AG, Maric D, Sweeney CL, Gossa S, Malech HL, McGavern DB, Park K (2017) Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci 20:753–759
Parisiadou L, Xie C, Cho HJ, Lin X, Gu X, Long C, Lobbestael E, Baekelandt V, Taymans J, Sun L, Cai H (2009) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 29:13971–13980
Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, Studer L (2004) Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A 101:12543–12548
Perrin S (2014) Preclinical research: make mouse studies work. Nature 507:423–425
Plowey ED, Cherra SJ, Liu YJ, Chu CT (2008) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105:1048–1056
Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Adel S, Gonzales-McNeal M, Hilker R, Kramer PL, Klein C (2005) Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 58:411–422
Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J (2010) The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28:721–733
Rakovic A, Grünewald A, Seibler P, Ramirez A, Kock N, Orolicki S, Lohmann K, Klein C (2010) Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum Mol Genet 19:3124–3137
Rakovic A, Shurkewitsch K, Seibler P, Grünewald A, Zanon A, Hagenah J, Krainc D, Klein C (2013) Phosphatase and Tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy. J Biol Chem 288:2223–2237
Ramonet D, Daher JPL, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ (2011) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 6:e18568
Reinhardt P, Schmid B, Burbulla LF, Schöndorf DC, Wagner L, Glatza M, Höing S, Hargus G, Heck SA, Dhingra A, Wu G, Müller S, Brockmann K, Kluba T, Maisel M, Krüger R, Berg D, Tsytsyura Y, Thiel CS, Psathaki OE, Klingauf J, Kuhlmann T, Klewin M, Müller H, Gasser T, Schöler HR, Sterneckert J (2013) Genetic correction of a lrrk2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12:354–367
Ren Y, Jiang H, Hu Z, Fan K, Wang J, Janoschka S, Wang X, Ge S, Feng J (2015) Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cell Rev Rep 33:68–78
Ross CA, Poirier MA (2005) Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol 6:891–898
Rudenko IN, Cookson MR (2014) Heterogeneity of leucine-rich repeat kinase 2 mutations: genetics, mechanisms and therapeutic implications. Neurotherapeutics 11:738–750
Russo I, Berti G, Plotegher N, Bernardo G, Filograna R, Bubacco L, Greggio E (2015) Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J Neuroinflammation 12:230
Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, Lee B, Lopez K, Nutter A, Shan B, Molokanova E, Zhang Y, Han X, Nakamura T, Masliah E, Yates JR, Nakanishi N, Andreyev AY, Okamoto SI, Jaenisch R, Ambasudhan R, Lipton SA (2013) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155:1351–1364
Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, Pastor P (2010) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133:1128–1142
Sánchez-Danés A, Richaud-Patin Y, Carballo-Carbajal I, Jiménez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, López-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A (2012) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4:380–395
Sanchez-Guajardo V, Tentillier N, Romero-Ramos M (2015) The relation between α-synuclein and microglia in Parkinson’s disease: recent developments. Neuroscience 302:47–58
Sanders LH, Laganière J, Cooper O, Mak K, Vu BJ, Huang YA, Paschon DE, Vangipuram M, Sundararajan R, Urnov FD, Langston JW, Gregory PD, Zhang HS, Greenamyre JT, Isacson O, Schüle B (2014) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis 62:381–386
Sandor C, Robertson P, Lang C, Heger A, Booth H, Vowles J, Witty L, Bowden R, Hu M, Cowley SA, Wade-Martins R, Webber C (2017) Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum Mol Genet 26:552–566
Schapansky J, Nardozzi JD, Lavoie MJ (2015) The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience 302:74–88
Schapira AHV (2015) Glucocerebrosidase and Parkinson disease: recent advances. Mol Cell Neurosci 66:37–42
Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–827
Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS (2002) Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol 160:1655–1667
Schöndorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, Sardi SP, Valsecchi M, Hoffmann S, Schwarz LK, Hedrich U, Berg D, Shihabuddin LS, Hu J, Pruszak J, Gygi SP, Sonnino S, Gasser T, Deleidi M (2014) iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5:4028
Schüle B, Pera RAR, Langston JW (2009) Can cellular models revolutionize drug discovery in Parkinson’s disease? Biochim Biophys Acta 1792:1043–1051
Schulte C, Gasser T (2011) Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl Clin Genet 4:67–80
Schwab AJ, Ebert AD (2015) Neurite aggregation and calcium dysfunction in iPSC-derived sensory neurons with Parkinson’s disease-related LRRK2 G2019S mutation. Stem Cell Rep 5:1039–1052
Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA (2014) Protective and toxic roles of dopamine in Parkinson’s disease. J Neurochem 129:898–915
Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D (2011) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31:5970–5976
Shaltouki A, Sivapatham R, Pei Y, Gerencser AA, Momčilović O, Rao MS, Zeng X (2015) Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Rep 4:847–859
Shi M, Shi C, Xu Y (2017) Rab GTPases: the key players in the molecular pathway of Parkinson’s disease. Front Cell Neurosci 11:81
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of α-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293:263–269
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11:986–998
Skibinski G, Hwang V, Ando DM, Daub A, Lee AK, Ravisankar A, Modan S, Finucan MM, Shaby BA, Finkbeiner S (2016) Nrf2 mitigates LRRK2- and α-synuclein–induced neurodegeneration by modulating proteostasis. Proc Natl Acad Sci U S A 114:1165–1170
Skibinski G, Nakamura K, Cookson MR, Finkbeiner S (2014) Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci 34:418–433
Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, Zhang L, Guschin D, Fong LK, Vu BJ, Meng X, Urnov FD, Rebar EJ, Gregory PD, Zhang HS, Jaenisch R (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 146:318–331
Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533:95–99
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) α-synuclein in Lewy bodies. Nature 388:839–840
Steele JC, Guella I, Szu-Tu C, Lin MK, Thompson C, Evans DM, Sherman HE, Vilariño-Güell C, Gwinn K, Morris H, Dickson DW, Farrer MJ (2015) Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol 77:458–468
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M (2016) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. elife 5:e12813
Studer L (2012) Derivation of dopaminergic neurons from pluripotent stem cells In: Progress in Brain Research, vol 200. Elsevier, Amsterdam, pp 243–263
Su YC, Qi X (2013) Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet 22:4545–4561
Subramaniam SR, Chesselet M-F (2013) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 106–107:17–32
Suhr ST, Chang EA, Rodriguez RM, Wang K, Ross PJ, Beyhan Z, Murthy S, Cibelli JB (2009) Telomere dynamics in human cells reprogrammed to pluripotency. PLoS ONE 4:e8124
Suhr ST, Chang EA, Tjong J, Alcasid N, Perkins GA, Goissis MD, Ellisman MH, Perez GI, Cibell JB (2010) Mitochondrial rejuvenation after induced pluripotency. PLoS ONE 5:e14095
Sun Y, Grabowski GA (2010) Impaired autophagosomes and lysosomes in neuronopathic Gaucher disease. Autophagy 6:648–649
Suzuki S, Akamatsu W, Kisa F, Sone T, Ishikawa KI, Kuzumaki N, Katayama H, Miyawaki A, Hattori N, Okano H (2017) Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem Biophys Res Commun 483:88–93
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Takanashi M, Li Y, Hattori N (2016) Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology 86:2212–2213
Tansey MG, McCoy MK, Frank-Cannon TC (2007) Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208:1–25
Tayebi N, Callahan M, Madike V, Stubblefield BK, Orvisky E, Krasnewich D, Fillano JJ, Sidransky E (2001) Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab 73:313–321
Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P (2016) Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530:45–50
Tofaris GK, Tae H, Hourez R, Jung J, Pyo K, Goldberg AL (2011) Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal – lysosomal pathway. Proc Natl Acad Sci U S A 108:17004–17009
Tóth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, Tambe MA, Rochet JC, Galvagnion C, Skibinski G, Finkbeiner S, Bova M, Regnstrom K, Chiou SS, Johnston J, Callaway K, Anderson JP, Jobling MF, Buell AK, Yednock TA, Knowles TPJ, Vendruscolo M, Christodoulou J, Dobson CM, Schenk D, McConlogue L (2014) Targeting the intrinsically disordered structural ensemble of α-Synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS ONE 9:e87133
Trinh J, Farrer M (2013) Advances in the genetics of Parkinson disease. Nat Rev Neurol 9:445–454
Uversky VN, Oldfield CJ, Dunker AK (2008) Intrinsically disordered proteins in human diseases: introducing the D 2 concept. Annu Rev Biophys 37:215–246
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Turco DD, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Várkonyi J, Rosenbaum H, Baumann N, MacKenzie JJ, Simon Z, Aharon-Peretz J, Walker JM, Tayebi N, Sidransky E (2003) Gaucher disease associated with parkinsonism: four further case reports. Am J Med Genet A 116A:348–351
Victor MB, Richner M, Hermanstyne TO, Ransdell JL, Sobieski C, Deng PY, Klyachko VA, Nerbonne JM, Yoo AS (2014) Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 84:311–323
Vidailhet M (2003) Heterogeneity of Parkinson’s disease. Bull Acad Natl Med 187:256–259
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463:1035–1042
Vivekanantham S, Shah S, Dewji R, Dewji A, Khatri C, Ologunde R (2015) Neuroinflammation in Parkinson’s disease: role in neurodegeneration and tissue repair. Int J Neurosci 125:717–725
Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RLA, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107:378–383
Wang T, Hay JC (2015) Alpha-synuclein toxicity in the early secretory pathway: how it drives neurodegeneration in Parkinsons disease. Front Neurosci 9:433
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, Lavoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906
Wang B, Abraham N, Gao G, Yang Q (2016) Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl Neurodegener 5:19
Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH (2011) Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis 41:706–716
Woodard CM, Campos BA, Kuo S-H, Nirenberg MJ, Nestor MW, Zimmer M, Mosharov EV, Sulzer D, Zhou H, Paull D, Clark L, Schadt EE, Sardi SP, Rubin L, Eggan K, Brock M, Lipnick S, Rao M. (2014) iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease.Cell Rep 9:1173–1182
Xilouri M, Brekk OR, Stefanis L (2013) Alpha-synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol 47:537–551
Xu L, Pu J (2016) Alpha-Synuclein in Parkinson’s disease: From pathogenetic dysfunction to potential clinical application. Parkinsons Dis 1720621
Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR (2011) MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476:228–232
Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y (1996) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A 93:2696–2701
Yu Z, Xu X, Xiang Z, Zhou J, Zhang Z, Hu C, He C (2010) Nitrated α-Synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS ONE 5:e9956
Zhang XQ, Zhang SC (2010) Differentiation of neural precursors and dopaminergic neurons from human embryonic stem cells. Methods Mol Biol 584:355–366
Zhong P, Hu Z, Jiang H, Yan Z, Feng J (2017) Dopamine induces oscillatory activities in human midbrain neurons with parkin mutations. Cell Rep 19:1033–1044
Zuo L, Motherwell MS (2013) The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 532:18–23
Author information
Authors and Affiliations
Corresponding author
Additional information
This review is supported by the Michael J Fox Foundation, Gladstone Institutes, NIH U54 HG008105, R01 NS039074 and R01 NS083390
Rights and permissions
About this article

Cite this article
Cobb, M.M., Ravisankar, A., Skibinski, G. et al. iPS cells in the study of PD molecular pathogenesis. Cell Tissue Res 373, 61–77 (2018). https://doi.org/10.1007/s00441-017-2749-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2749-y