
Abstract
Vascular endothelial growth factor (VEGF) is the principal regulator of tumor angiogenesis and is overexpressed in the majority of solid tumors. Therapeutic inhibition of VEGF and its main receptor (VEGFR2) has shown significant clinical efficacy in several human cancers. However, in unselected patient populations, often these agents have not offered sustainable clinical benefit. Some of the challenges with the clinical efficacy of anti-VEGF/VEGFR therapies may be explained by the heterogeneity of human tumor vessels and variation in their sensitivity to VEGF/VEGFR inhibition. However, the process of tumor angiogenesis is far more complex with frequent cross talk between VEGF/VEGFR and other signaling pathways. In addition to anti-angiogenic effects, anti-VEGF/VEGFR agents also cause “normalization” of tumor vessels and “pruning” of normal vessels. In order to achieve significant improvement in clinical efficacy of anti-VEGF/VEGFR therapies in the near future, it will be important to (1) better understand the complex biology of VEGF/VEGFR and non-VEGF/VEGFR signaling pathways in the context of pathologic (aberrant) angiogenesis in human cancer tissues, (2) translate such biologic concepts into a more comprehensive molecular profiling and pathologic disease state characterization, and (3) advance the much needed predictive biomarker science to drive rational patient-tailoring and combinatorial therapeutic strategies in next-generation clinical trials of anti-angiogenic therapies. It will also be critical to identify and address other clinical and scientific challenges, including various primary and acquired mechanisms of resistance to anti-angiogenic therapies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Ramucirumab
- Bevacizumab
- Aflibercept
- Tyrosine kinase inhibitor
- Vascular endothelial growth factor (VEGF)
- Lung cancer
- Breast cancer
- Colon cancer
- Gastric cancer
- Vascular endothelial growth factor receptor (VEGFR)
- Tumor angiogenesis
- Anti-angiogenic therapy
- Tumor endothelial cells
- Pericytes
- Vascular normalization
- Microvascular density
- Hypoxia
- Angiogenesis
- PlGF
- NRP1
- NRP2
- Biomarker
- Molecular profiling
- Drug resistance
Introduction
Several drugs targeting VEGF or its receptors (VEGFRs) have been approved for the treatment of various malignancies, and many more are in clinical trials [1]. Unfortunately, these agents, used as monotherapy or in combination with chemotherapy, have only provided modest survival benefits in some tumor types and have not been efficacious at all in others [1]. For example, bevacizumab (Avastin®, Roche/Genentech), a humanized antibody against VEGF-A, prolongs the life of patients with advanced colon cancer by 4–5 months when combined with triple chemotherapy (irinotecan, bolus fluorouracil, and leucovorin) [2]. In a recent meta-analysis of 9 randomized trials with 3710 mCRC patients, the OS/PFS benefit was observed only in the presence of irinotecan-based regimen (ILF or FOLFIRI) [3]. A recent editorial has questioned whether bevacizumab was “boon or bust” because of its limited effectiveness, serious (though uncommon) side effects, and high cost [4]. Other US Food and Drug Administration-approved drugs that bind VEGF-A such as aflibercept (Zaltrap), or that target VEGF receptors such as various tyrosine kinase inhibitors, have fared no better [5].
Tumors acquire blood supply via multiple mechanisms: angiogenesis (sprouting new vessels from existing vessels), cooption (tumor cells engulf host vessels in the normal surrounding tissue as the tumor invades), intussusception (new vessels are generated by the fission of existing vessels), vasculogenic mimicry (tumor cells directly form vascular channels that are perfused via connection to the host vasculature), and trans-differentiation of cancer cells into endothelial cells [6]. The original concept of anti-angiogenic therapy aimed to destroy (“starve”) tumor vessels was put forward by Judah Folkman [7]. It turned out that, in reality, anti-angiogenic drugs “normalize” tumor vasculature and as a result offer an improved delivery of chemotherapeutic agents to the tumor tissues [1, 8]. Furthermore, the initial idea that anti-angiogenic therapy would be resistance-free failed to materialize, and currently, we are faced with resistance to anti-angiogenic therapy as one of the major clinical challenges. Also, an increasing number of preclinical and clinical observations have shown that the process of angiogenesis is far from clearly understood. Apart from targeting the VEGF pathway, novel therapeutic strategies aim to influence other molecular factors that are involved in tumor angiogenesis.
Here I will review the clinically relevant aspects of biology of pathologic (aberrant) angiogenesis in human cancer, especially with reference to the principal angiogenic (VEGF-VEGFR) signaling pathway, cross talk between the main and alternative angiogenic pathways including an overview of the recent scientific and clinical advances in the field, and some of the challenges that we face in tailoring these agents to the right patients and also with reference to accurate prediction of response or resistance to these therapies, when administered in unselected cancer patient populations. In the end, I will summarize key patho-biologic and clinical learnings about tumor angiogenesis and anti-angiogenesis and outline some strategies to develop predictive biomarkers to improve clinical efficacy and the overall value of these drugs for cancer patients.
Angiogenic Signaling Pathways in Human Cancer
VEGF Signaling Pathway
Vascular endothelial growth factor (VEGF, VEGF-A) is the major player in the VEGF-driven angiogenic signaling pathway and principal regulator of physiological and pathological angiogenesis [9]. Discovery of VEGF (aka vascular permeability factor, VPF) as the primary tumor angiogenesis factor prompted the development of a number of drugs (e.g., bevacizumab, aflibercept, ramucirumab, and small-molecule tyrosine kinase inhibitors) that targeted this ligand or its receptors. These agents have often been successful in halting tumor angiogenesis and in regressing rapidly growing mouse tumors [10]. However, results in human cancer have been less impressive. Furthermore, while tumors induce their heterogeneous vasculature by secreting vascular endothelial growth factor (VEGF)-A, the underlying mechanisms how anti-VEGF/VEGF receptor (VEGFR) drugs treat cancer still remain unclear [11].
VEGF Ligands, Receptors, and Co-receptors
The complex process of angiogenesis is predominantly regulated by a single growth factor, VEGF (also known as VEGF-A), which is overexpressed in many human cancers. VEGF family consists of five members (VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PlGF (placental growth factor)), which transmit signals via three receptors (VEGFR-1, VEGFR-2, VEGFR-3) (Fig. 19.1).

Schematic representation of the mechanism of action of VEGF in regulating pathologic angiogenesis in human cancer. The VEGF signaling pathway with the three principal VEGF receptors (VEGFR1, VEGFR2, VEGFR3), co-receptors (neuropilin 1 (NRP1) and neuropilin 2 (NRP2)), and various ligands are illustrated. Redundancy of VEGF signaling in the form of cross talk among various receptors and ligands is obvious. In response to anti-VEGF therapies, hypoxia plays a major role in driving various proangiogenic factors to sustain proliferation and growth of tumor vessels and cells – forming the basis of acquired resistance to anti-VEGF therapy [14]. (Reprinted from Gacche and Assaraf [14]. With permission from Elsevier)
The most important factor is VEGF-A , which has been shown to stimulate endothelial cell mitogenesis and cell migration, leading to cancer progression and metastasis via binding to VEGFR-2 (also known as fetal liver kinase 1 (FLK1)). VEGF-B plays a role in the maintenance of newly formed blood vessels via VEGFR-1. VEGF-C and VEGF-D bind to VEGFR-3, predominately expressed in lymphatic vessels and play a role in lymphangiogenesis and metastatic spread to lymph nodes. PlGF is a multitasking cytokine that stimulates angiogenesis by direct or indirect mechanisms and also activates bone-marrow-derived endothelial progenitor and myeloid cells, as well as stromal cells, to create a nurturing “soil” for tumor cells, in addition to activating tumor cells [12]. By skewing the polarization of tumor-associated macrophages (TAMs), the loss of PlGF improves vessel perfusion and maturation and enhances responses to chemotherapy [13].
Several VEGF co-receptors have also been identified, including heparan sulfate proteoglycans, neuropilin 1 (NRP1), neuropilin 2, and CD146. Moreover, VEGF receptors can cross talk with additional cell surface molecules, including integrins and other growth factor receptors. Neuropilins such as NRP1 and NRP2 are VEGF co-receptors, which enhance the activity of VEGFR-2, but also signal independently [15].
Soluble VEGF isoforms promote vessel enlargement, whereas matrix-bound isoforms stimulate branching. Paracrine VEGF, released by tumor, myeloid, or other stromal cells, increases vessel branching and renders tumor vessels abnormal [16], whereas autocrine VEGF, released by endothelial cells, maintains vascular homeostasis [17]. Emerging evidence indicates that the biological effect of VEGFR-2 signaling depends on its subcellular localization – for example, for VEGF to induce arterial morphogenesis, VEGFR-2 must signal from intracellular compartments [18].
VEGF Receptors and Tumor Cells
Numerous studies have documented a role for VEGF signaling in tumor cells , but the data are conflicting [19]. Several studies have shown that cancer cell lines can express VEGFR1 or VEGFR2 and that signaling through these receptors in cancer cells can promote events associated with tumor progression, including cancer cell survival, proliferation, invasion, or metastasis [20,21,22,23]. The presence of functionally active VEGFR-2 has been shown on human ovarian cancer cells and suggests that the observed antitumor activity of VEGF-targeted therapies may be mediated by both anti-angiogenic and direct antitumor effects [23]. Based on these data, it has been proposed that inhibition of VEGF signaling in tumor cells may, at least in part, be mediated by direct activity against tumor cells [24]. In preclinical studies inhibition of VEGF signaling in CRC and glioblastoma cells made these cells more invasive [25, 26]. Further studies are required to determine the clinical significance of tumor cell expression of VEGF/VEGFR2.
Targeting VEGF Versus VEGFR2 May Have a Different Clinical Outcome
Because VEGFR2 is thought to be the main receptor conveying the proangiogenic signals downstream of VEGF, it is generally assumed that targeting VEGFR2 would have similar biological effects as targeting the ligand. However, this is not the case in some malignancies [1]. For example, although bevacizumab monotherapy has not improved overall survival in any phase III trial, the anti-VEGFR2 antibody ramucirumab led to an OS advantage of 1.4 months in advanced gastric or gastroesophageal junction (GEJ) adenocarcinomas. Interestingly, when added to paclitaxel, ramucirumab also increased OS by 2.3 months in patients with GEJ tumors. When combined with chemotherapy, both bevacizumab and ramucirumab failed to improve OS in metastatic breast cancer, but both improved survival in non-small cell lung cancer (NSCLC) (Table 19.1). It is tempting to assume that blood vessels of GEJ tumors are highly or even exclusively dependent on VEGFR2 signaling for their survival, and, hence, ramucirumab’s benefits result from starving these tumors, which is in support of the original anti-angiogenesis hypothesis [1]. However, the starvation hypothesis does not explain the failure of bevacizumab in the same tumor type [1].
VEGF-Independent Signaling Pathways
In addition to VEGF pathway, a series of VEGF-independent pathways like fibroblast growth factors 1 and 2 [27, 28], HGF/cMet pathway [29], angiopoietins [30], Delta-Notch signaling pathway [31], PDGF-C [32, 33], interleukins [34], ephrins [35], and epidermal growth factor [36] have been described as part of the anti-VEGF escape mechanisms. The abovementioned angiogenic factors and their redundant angiogenic signaling pathways are summarized in Fig. 19.2. These anti-VEGF/VEGFR escape mechanisms may contribute to acquired resistance to anti-angiogenic therapies and may contribute to subsequent recurrence and/or metastases.

VEGF-independent signaling pathways . In response to treatment of cancer patients with anti-angiogenic agents, cross talk and redundancy among various VEGF-independent compensatory proangiogenic signaling pathways can drive continued tumor angiogenesis and progression [14]. (Reprinted from Gacche and Assaraf [14]. With permission from Elsevier)
Cross Talk Between VEGFR2 and Other RTKs
VEGFR2 can also form direct complexes with other receptor tyrosine kinases. For example, stimulation of vascular smooth muscle cells with VEGF promotes the formation of a complex between VEGFR2 and the receptor tyrosine kinase PDGF-Rb [37]. This results in suppression of PDGF-Rb signaling and decreased pericyte coverage in tumors and may explain the observation that, in some experimental systems, inhibition of VEGF signaling leads to increased pericyte coverage of tumor vessels and increased maturation/normalization of the tumor vasculature [38]. In glioblastoma cells, VEGF stimulates the formation of a complex between VEGFR2 and the receptor tyrosine kinase, MET, which results in suppression of MET signaling and reduced tumor cell invasion [25]. Consequently, inhibition of VEGF has been shown to release MET from this inhibitory mechanism and allows for increased tumor invasion. Cross talk between VEGF and HER2 signaling pathways has been demonstrated in human breast cancer tissues in the form of higher VEGFR2 expression in HER2+ breast cancer [39]. Therefore, cross talk between VEGF/VEGF receptor signaling and other receptors may offer (1) plausible explanation for the diversity of clinical responses observed with VEGF-targeted therapies and (2) new opportunities to combine anti-angiogenic therapies with other targeted therapies to treat specific cancer types more effectively.
Pathologic Angiogenesis
Aberrant (pathologic) angiogenesis is one of the hallmarks of cancer. The imbalance of pro- and anti-angiogenic signaling within tumors creates an abnormal vascular network that is characterized by dilated, tortuous, and hyperpermeable vessels, which were elegantly described in Ad-VEGFA mouse model by Harold Dvorak as six different vessel types [40]. The physiological consequences of these vascular abnormalities include temporal and spatial heterogeneity in tumor blood flow and oxygenation and increased tumor interstitial fluid pressure. These abnormalities and the resultant microenvironment fuel tumor progression and also lead to a reduction in the efficacy of chemotherapy, radiotherapy, and immunotherapy. With the discovery of vascular endothelial growth factor (VEGF) as a major driver of tumor angiogenesis, efforts have focused on novel therapeutics aimed at inhibiting VEGF activity, with the goal of regressing tumors by starvation. Unfortunately, clinical trials of anti-VEGF monotherapy in patients with solid tumors have been largely negative or resulted in marginal clinical benefit. Intriguingly, the combination of anti-VEGF therapy with conventional chemotherapy has improved survival in cancer patients compared with chemotherapy alone. These seemingly paradoxical results could be explained by the concept of “normalization” of the tumor vasculature by anti-VEGF therapy. Preclinical studies have shown that anti-VEGF therapy changes tumor vasculature toward a more “mature” or “normal” phenotype. This “vascular normalization ” is characterized by attenuation of hyperpermeability, increased vascular pericyte coverage, and more normal vascular basement membrane, resulting in reduced tumor hypoxia and interstitial fluid pressure. These, in turn, can lead to an improvement in the metabolic profile of the tumor microenvironment, the delivery and efficacy of exogenously administered therapeutics, the efficacy of radiotherapy and of effector immune cells, and a reduction in number of metastatic cells shed by tumors into circulation in mice. These findings are consistent with data from clinical trials of anti-VEGF agents in patients with various solid tumors [41].
Evaluation of Pathologic Angiogenesis in Human Cancer
Although during last few decades there has been substantial research that contributed a great deal to our understanding of biology of tumor angiogenesis, including primary and acquired resistance mechanisms, due to several different factors, oncology biomarker research community has not been so successful in the development, optimization, and technical and clinical validation of biomarkers of pathologic angiogenesis in human cancer, especially in the context of histopathologic and molecular heterogeneity of cancers in various parts of the human body. This continues to be an area of unmet need, which if addressed appropriately by the biomarker and clinical trial teams, has the potential to further refine the current level of success of both the single agent and combinatorial anti-angiogenic therapies in clinical trials. Despite the urgent need for greater focus and investment in biomarker research and practice to support current and future trials of anti-angiogenic therapies, some of the ongoing efforts to investigate and advance tissue angiogenic biomarkers are outlined in the following sections.
Heterogeneity of Tumor Vessels in Experimental Models and Human Tumors
Therapies directed against VEGF-A and its receptors are effective in treating many mouse tumors but have been less so in treating human cancer patients. Such variation has been attributed to the nature of blood vessels that appear in human and mouse cancers and the tumor “surrogate” blood vessels that develop in immunodeficient mice in response to an adenovirus expressing the VEGF-A164 protein [40]. Both tumor and tumor surrogate blood vessels are heterogeneous and form by two distinct processes, angiogenesis and arterio-venogenesis [40].
The first new angiogenic blood vessels to form are mother vessels (MV); MV arise from preexisting venules and capillaries and evolve over time into glomeruloid microvascular proliferations (GMP) and subsequently into capillaries and vascular malformations (VM). Arterio-venogenesis results from the remodeling and enlargement of preexisting arteries and veins, leading to the formation of feeder arteries (FA) and draining veins (DV) that supply and drain angiogenic vessels. Among these, only MVs and GMPs were highly responsive to anti-VEGF therapy, whereas “late”-formed capillaries, VMs, FAs, and DVs, were relatively unresponsive. These findings were further supported by results of immunohistochemistry: early-forming MVs and GMPs, in which the lining endothelial cells expressed high levels of VEGFR-2, were highly susceptible to anti-VEGF blockade by VEGF-Trap (ziv-aflibercept, Zaltrap®, Sanofi-Aventis). In contrast, late-forming VMs, FAs, and DVs that expressed low levels of VEGFR-2 were largely resistant. Taken together, these findings may explain, at least in part, the relatively poor response of human cancers to anti-VEGF/VEGFR therapies, because human cancers, present for months or years prior to discovery, are expected to contain a large proportion of late-formed blood vessels [40]. Translating VEGFR2 IHC findings from Ad-VEGFA164 model to human cancer tissues will be an important consideration. As such high VEGFR2 expression levels in human tumor vessels are likely to correlate with tumor response to anti-VEGF/VEGFR therapies. Of course, in cancer patients the overall complexity of the angiogenic process and the redundancy of various signaling pathways can make such correlations less than straightforward and may need systematic evaluation of multiple biomarkers of pathologic angiogenesis.
VEGF/VEGF Receptor Expression in Human Tumor Tissues
In recent years progress has been made with regard to the evaluation of the clinical significance of VEGF/VEGF receptors in human cancer tissues by the development of robust methodologies with appropriate controls to accurately and reproducibly determine vascular and tumor cell expression levels of various angiogenic ligands and receptors. We and others have developed technically robust immunohistochemical assays [42, 43] to evaluate VEGFR2 and other VEGF receptors on archival tumor tissues. As part of the technical validation of the above assay, we carried out extensive optimization experiments and demonstrated comparable levels of VEGFR2 protein and in situ VEGFR2 RNA levels in serial sections of human (bladder) cancer and H441 (non-small cell lung cancer) xenograft tissues. In our solid tumor analyses, a frequent finding has been the heterogeneity of vascular and tumor cell expression of VEGFR2 in different areas of the same tumor (intra-tumor heterogeneity) and among different tumors (inter-tumor heterogeneity) (Fig. 19.3) and a degree of variation in subcellular localization of VEGFR2 in tumor cells [43, 45]. In our experience with different human cancer tissues, we came across many of the heterogeneous vessels described by Dvorak in Ad-VEGFA164 mouse model [44]. In survival analyses, tumor cell expression of VEGFR2 was found to be associated with adverse outcome in non-small cell lung cancer (NSCLC) [43] but favorable prognosis in bladder cancer [46]. In a more recent disease state characterization analysis, we have demonstrated significantly higher expression of the VEGFR2 protein in HER2+ breast cancer compared to other BRC subtypes [39]. Based on these findings, we hypothesized that compared to hormone receptor positive or triple negative subsets, HER2+ human breast cancers with high VEGFR2 expression might respond differently to anti-angiogenic therapies. Utilizing high-quality reagents, stringent method development strategies, well-optimized laboratory protocols, and “fit-for-purpose” yet sufficiently informative interpretation and scoring approaches, such methodologies are being evaluated in early- and late-phase clinical trials of anti-angiogenic agents.

Vascular endothelial cell and tumor cell expression of VEGFR2 on representative cases from a multi-tumor survey. Left panels H&E; right panels VEGFR2 IHC. (a) VEGFR2 IHC on renal cell carcinoma of the kidney showing endothelial cell immunoreactivity (X400). (b) VEGFR2 IHC on ADC of the colon showing endothelial cell immunoreactivity in the stromal blood vessels. Tumor cells are negative for VEGFR2 (X400). (c) VEGFR2 IHC on SCC of the lung showing endothelial cell and a range of tumor cell (nuclear cytoplasmic membranous) immunoreactivity (X200). (d) VEGFR2 IHC is showing vascular endothelial cell immunoreactivity and a range of tumor cell cytoplasmic and nuclear immunoreactivity on SCC of the cervix (X200). Immunoreactivity in endothelial cells lining vessels (white and black arrows). Slides were counterstained with hematoxylin. Scale bars: 50 μm. In renal and colonic cancer tissues, the VEGFR2 expression is restricted to tumor vessels. In squamous cell cancers, however, VEGFR2 expression was found both in tumor stromal vessels and tumor cells. Such variation in distribution and localization of VEGFR2 may result in part account for the differences in sensitivity of these cancer tissues to anti-VEGF/VEGFR2 therapies. (Reprinted from Holzer et al. [43]. With permission from Creative Commons License)
Microvascular Density (MVD)
In breast tumors, high baseline microvascular density (MVD) is considered a positive response to vascular normalization index induced by bevacizumab [47]. In the case of high MVD, the effect of anti-angiogenic drugs would be to remove some vessels and increase the functions of others, by inducing their normalization. In the case of low MVD, the anti-angiogenic therapy would reduce tumor microvasculature further and prevent their normalization. This can make the tumor insensitive to anti-angiogenic therapy. Therefore, baseline MVD can be a key factor in predicting the success of treatment with anti-angiogenic drugs [48].
Predictive Biomarkers
High variability in patient response to anti-angiogenic therapy across different indications exists, and this is coupled with the development of therapy resistance [49]. As with other targeted compounds, reliable biomarkers to identify patients with cancer who will benefit from anti-angiogenic therapy are still needed. One of the main challenges in identifying potential biomarkers for anti-angiogenic therapy is the complex nature of the angiogenic signaling process, which is characterized by multiple pathways that not only overlap but that continuously cross talk, making it difficult to eliminate an angiogenic stimulus [50]. Several types of biomarkers are being investigated across different indications: circulating biomarkers (e.g., concentrations of soluble angiogenic receptors/ligands), genetic biomarkers (e.g., single nucleotide polymorphisms), tissue biomarkers (e.g., immunohistochemical staining of angiogenic receptors), and physiologic biomarkers (e.g., hypertension) [49].
Elevated levels of soluble VEGFR1 (sVEGFR1) prior to treatment were associated with a poor outcome from bevacizumab in rectal carcinoma, hepatocellular carcinoma (HCC), and metastatic colorectal carcinoma patients [51,52,53,54]. A retrospective analysis has shown that a genetic polymorphism in the VEGFR1 gene correlates with increased VEGFR1 expression and a poor outcome of bevacizumab treatment in metastatic renal cell carcinoma and pancreatic ductal adenocarcinoma patients [55]. Similarly, elevated levels of NRP1 were associated with a poor outcome in some trials [56]. It is possible that VEGFR1 and NRP1 function as endogenous VEGF-Traps. Therefore, adding an external anti-VEGF agent may not have significant biologic effects in patients with high sVEGFR1/NRP1 levels. Additionally, increased VEGFR1 levels may induce increased proangiogenic signaling by PlGF when VEGF is blocked [55].
In recent years, our laboratory developed technically robust immunohistochemical assays for localization of VEGFR2, VEGFR1, and VEGFR3 in archival human tissues. Large-scale biomarker prevalence and disease characterization analyses have shown significant variation in VEGF receptor profiles (VEGFR1, VEGFR2, VEGFR3) among NSCLC, BRC, and CRC tissues [43, 45, 57]. Since various anti-angiogenic therapies target one (VEGFR2 in case of ramucirumab) or multiple VEGF receptors (VEGFR1, VEGFR2, VEGFR3 in case of small-molecule tyrosine kinase inhibitors), the clinical significance of various VEGF receptor profiles may be determined by retrospective-prospective (VEGFR receptor profiling) analyses on tissue specimens from positive anti-angiogenic therapy trials. Just as assessments of circulating VEGF receptor levels are frequently performed in clinical trials of anti-angiogenic therapies (more for logistical reasons than true science, given the frequent uncertainty about the source of circulating receptors), it will also be prudent to evaluate all three VEGF receptors along with NRPs in pathologically well-characterized archival tumor tissues in order to determine their value in predicting response or resistance to anti-VEGF/VEGFR2/VEGFR1,2,3 (bevacizumab, ramucirumab, TKIs) agents.
Despite initial reports on predictive biomarkers, overall reproducibility of candidate biomarkers across indications is limited, and there is a paucity of studies comparing the same biomarkers for the same indication. The appropriate use of genomic and proteomic technologies will be key in improving our ability to match a target pathology with the efficacy of a specific anti-angiogenic therapy, although a lot of cross-platform validation work will be required to implement newly discovered candidate predictive biomarkers into clinical practice. Another area that needs urgent attention in clinical trials is an accurate diagnostic evaluation of clinical trial tissues, which can result in significant misclassification of clinical trial tissue specimens – with obvious negative impact on the quality of biomarker data from clinical trials. Central pathology review and even sub-specialty level histopathologic characterization of cancer tissue specimens from clinical trials and implementation of comprehensive angiogenic biomarker approaches in analyzing the clinical trial data sets need to be serious considerations in future trials of anti-angiogenic therapies.
Molecular Cancer Subtyping and Response to Anti-angiogenic Therapies
A recent analysis of patients with gastroesophageal carcinoma demonstrated that the ratio of progression-free survival (PFS) on the molecular profile (MP)-based treatment to PFS on treatment prior to molecular profiling exceeds 1.3, suggesting the potential value of MP in guiding selection of individualized therapy [58]. Biologic rationale for this clinical finding is evident by the presence of four molecular subtypes of human gastric cancer by TCGA [59]. The TCGA subgroup labeled “chromosomal instability (CIN)” is characterized by amplifications of several therapeutic targets including HER2, VEGF-A, MET, and others. In the current and future trials of anti-VEGF/VEGFR2 therapies, it will be interesting to see if this molecular subset responds better to such therapies compared to the other molecular subsets of GC. In a recent analysis of stromal gene signature in GC, we identified differentially expressed genes in various molecular subtypes of GC [60]. Such analyses can provide invaluable biologic insights and can help with the selection of promising predictive biomarker candidates for subsequent clinical validation in clinical trials of anti-angiogenic therapies. An important technical challenge in tailoring anti-angiogenic therapies to various molecular subgroups of human cancers will be that we still do not have robust, clinical grade tissue-based methodologies with clinically validated scoring cutoffs for pertinent solid tumors for many of the newer therapeutic targets, so that various biologically relevant combinatorial therapeutic approaches can be tested in appropriately selected cancer patient subsets.
Anti-angiogenic Therapies to Treat Human Cancer
Currently, there are four main approaches targeting angiogenesis in human cancer, which have been tested in clinical trials and approved for clinical practice: (1) neutralizing monoclonal antibody that binds circulating VEGF (bevacizumab, Avastin®, Roche/Genentech), (2) recombinant protein called decoy receptor or “VEGF-Trap” (Aflibercept, Zaltrap®, Sanofi Genzyme) that binds more than one proangiogenic growth factor, (3) small-molecule tyrosine kinase inhibitors (like sunitinib (SUTENT®, Pfizer), sorafenib (Nexavar®, Bayer)) that block tyrosine kinase activity of VEGFRs, and (4) therapeutic monoclonal antibodies targeting VEGF receptor 2 (ramucirumab, Cyramza®, Eli Lilly).
Anti-VEGF Therapy (Bevacizumab)
One of the first anti-angiogenic therapies was the monoclonal antibody neutralizing circulating VEGF. In 2004, the first phase III trial results showed that bevacizumab (Avastin®, Roche/Genentech), a humanized monoclonal antibody binding specifically to VEGF-A alone, when combined with chemotherapy in metastatic colorectal cancer improved progression-free survival (PFS) (10.6 vs. 6.2 months) and overall survival (OS) (23 vs. 15.3 months) compared to chemotherapy arm [2]. An improvement in PFS for the combination of bevacizumab plus chemotherapy was next shown in two phase III trials in non-squamous non-small cell lung cancer (NSCLC) [61,62,63], but only one study reported an improvement in OS [61]. Within the next few years, bevacizumab was approved as a monotherapy in second-line treatment of glioblastoma and in combination with interferon-α (INF- α) for renal cell carcinoma. There were some controversies in cases of using bevacizumab in the treatment of metastatic breast cancer. The ECOG-2100 trial showed that adding bevacizumab to paclitaxel improved PFS (11.8 vs. 5.9), as well as OS rates (36.9% vs. 21.2%) compared to paclitaxel alone. Based on those results, the US Food and Drug Administration (FDA) accelerated in 2008 approval of bevacizumab in combination with paclitaxel in metastatic breast cancer. Further trials, AVADO and RIBBON-1, confirmed the improvement of PFS by bevacizumab, but neither demonstrated any improvement of OS. In addition, bevacizumab-induced hypertension was reported as a clinically relevant adverse event in a phase III breast cancer trial (E5103) [64]. In 2011, FDA withdrew approval for bevacizumab in metastatic breast cancer. In 2014, bevacizumab was approved for the treatment of patients with platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan [65, 66] and for recurrent or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan [67, 68].
Aflibercept (Human Recombinant Fusion Protein)
Aflibercept (Zaltrap ®, Sanofi Genzyme) is a human recombinant fusion protein that acts as a decoy receptor of angiogenic factors. Unlike bevacizumab, it targets not only VEGF-A but also VEGF-B and placental growth factor (PlGF). This is a fusion protein of the second immunoglobulin domain of VEGFR1, third immunoglobulin domain of VEGFR2, and constant region Fc of human IgG1. In 2012, FDA approved aflibercept in the treatment of metastatic colorectal cancer (CRC) with infusional fluorouracil, leucovorin, and irinotecan, based on phase III trial results [69].
Anti-VEGFR2 Therapy (Ramucirumab)
Ramucirumab (Cyramza® Eli Lilly) is a human monoclonal antibody that inhibits angiogenesis by blocking binding of VEGF to the extracellular domain of VEGFR2. It is advantageous due to its receptor selectivity with minimal off-target activity. Preclinical studies showed that ramucirumab binds selectively to VEGFR2 with a greater efficacy than its natural ligand VEGF-A. It is approved for second-line treatment in gastric cancer, NSCLC, and colon cancer. Based on the RAISE study, ramucirumab was approved in combination with FOLFIRI (folinic acid, 5-fluorouracil, and irinotecan) in metastatic CRC patients, if disease progressed after therapy with bevacizumab, oxaliplatin, and fluoropyrimidine. In NSCLC, ramucirumab was approved in combination with docetaxel after platinum-based chemotherapy. In gastric cancer patients, FDA approved ramucirumab as a monotherapy in advanced or metastatic disease or in gastroesophageal junction carcinoma patients for whom first-line chemotherapy had failed [70, 71]. The FDA guidelines for the therapeutic applications of the approved anti-angiogenic drugs are summarized in Table 19.1 [11].
Small-Molecule Tyrosine Kinase Inhibitors
Tyrosine kinase inhibitors (TKIs) are small-molecular-weight drugs that inhibit the kinase activity of different receptors. The mechanism of action of TKIs relies on binding around the ATP-binding site of a given receptor, thus hindering phosphorylation of the tyrosine residue of that receptor and downstream signaling. There are several small-molecule kinase inhibitors, tyrosine kinase, serine/threonine kinase, or dual protein kinase inhibitors, approved by the FDA (sunitinib, sorafenib, axitinib, and pazopanib), some of which target VEGF receptors (VEGFRs) and are used to treat a number of different types of cancer (Table 19.1) [72, 73]. Compared to VEGF neutralizing antibodies, TKI does not interfere with the binding of VEGF to its receptors, and they usually target not only VEGFR but other kinases as well like PDGFR, FGFR, and c-KIT [41].
Bispecific Antibodies Targeting Both Tumor Cells and Angiogenesis
In recent years, monospecific antibodies targeting cell surface receptors have achieved remarkable success with cancer treatment. However, redundant signaling and cross talk between different pathways within tumor cells and between tumor cells and their microenvironment can limit the efficacy of receptor-targeted monospecific-based therapies [74]. During tumor progression, hypoxia and acidosis are known to induce angiogenesis within the tumor. Both tumor cells and tumor-associated endothelial cells express growth factors and their corresponding receptors, such as EGFR and VEGFR. In a mouse model of colon cancer [75], dual inhibition of EGFR and VEGFR by kinase inhibitors reduced tumor growth and metastasis, suggesting that the EGFR and VEGFR pathways have important roles in regulating tumor progression and neovascularization. More strikingly, an increase in EGFR expression and loss of ErbB3 expression has been identified in tumor vasculature and provides the rationale to target EGF-induced endothelial cell proliferation in tumor vasculature [76]. Co-inhibition of PDGFRβ and VEGFR has been shown to prevent new blood vessel growth better than VEGFR alone [77]. A number of tyrosine kinase inhibitors (axitinib, sorafenib, and sunitinib) targeting both VEGFR and PDGFR are effective in renal cell carcinoma and other types of human cancer [78]. Bispecific antibodies – such as single-chain variable fragments dual-targeting PDGFRβ and VEGF-A [79] and Ang-2-VEGF-A CrossMab, which targets angiopoietin 2 and VEGF-A simultaneously [80] – inhibit two distinct pathways targeting tumor angiogenesis. Bispecific antibodies against those targets that are shared by tumor cells and tumor-associated endothelial cells have the potential for an enhanced therapeutic efficacy.
Potential Mechanisms of Resistance to Anti-angiogenic Therapies
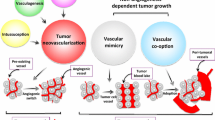
As illustrated by Vasudev and Reynolds [19] (Fig. 19.4), several different mechanisms are involved in lack of clinical response (resistance) to anti-angiogenic therapies. These include:
-
A.
Heterogeneity of tumor vessels in the form of therapy-sensitive and therapy-insensitive vessels. For example, some of the tumor vessels may be destroyed by the therapy, while others may survive.
-
B.
Alternative signaling pathways that can regulate the sensitivity of vessels to therapy.
-
C.
Stromal cells, such as immature myeloid cells or fibroblasts that can infiltrate the tumor and mediate resistance either by releasing pro-angiogenic growth factors or by physically incorporating into the tumor vessels.
-
D.
Tumor cells that can survive conditions of stress. For example, some tumor cells may have survived the loss of a vascular supply because they are adapted to survive conditions of hypoxia or nutrient shortage.
-
E.
Tumors may use alternative mechanisms of vascularization besides sprouting angiogenesis. In intussusceptive microvascular growth, new vessels are generated by the fission of existing vessels. Glomeruloid angiogenesis is characterized by tight nests of vessels that resemble the renal glomerulus. In vasculogenic mimicry, tumor cells directly form vascular channels (blue cells) that are perfused via connection to the host vasculature (red cells). In looping angiogenesis, contractile myofibroblasts (green) pull host vessels out of the normal surrounding tissue (pink region). In vessel co-option tumor cells engulf host vessels in the normal surrounding tissue (pink region) as the tumor invades.
-
F.
Increased tumor aggressiveness, i.e., therapy causes the tumor to become more invasive and/or accelerates the growth of metastases.

Potential mechanisms involved in resistance to VEGF-targeted therapy; see text. (Reprinted from Vasudev and Reynolds [19]. With permission from Springer Nature)
Emerging Trends in Anti-angiogenic Therapies
The anti-angiogenic (anti-VEGF/VEGFR) therapies, intended to block tumors’ blood supply, may cause hypoxia, which may fuel tumor progression and treatment resistance. Emerging clinical data suggest that patients whose tumor perfusion or oxygenation increases in response to these agents may actually survive longer. Hence, strategies aimed at alleviating tumor hypoxia while improving perfusion may enhance the outcome of radiotherapy, chemotherapy, and immunotherapy [1].
Vascular Normalization and Pruning After Anti-angiogenic Therapy
In normal tissue, the blood vessels have normal structure and function due to the balance of the signals downstream of the pro-angiogenic molecules (e.g., VEGF, Ang2) and anti-angiogenic molecules (e.g., sVEGFR1, thrombospondins, semaphorins). In contrast, tumor vessels are structurally and functionally abnormal due to an imbalance between pro- and anti-angiogenic signals. This creates an abnormal microenvironment in tumors – characterized by hypoxia, acidosis, and elevated fluid pressure – which fuels tumor progression and treatment resistance via multiple mechanisms [1]. Inhibiting pro-angiogenic signaling or enhancing anti-angiogenic signaling can prune some abnormal vessels and remodel the rest resulting in a “normalized vasculature .” Depending upon the extent of normalization versus pruning, tumor perfusion/oxygenation may increase, remain unchanged, or decrease. Some tumors might be intrinsically resistant to a given AA agent, and others may switch to non-sprouting mechanisms of vessel recruitment (e.g., vessel cooption) that are refractory to the given AA agent and continue to make abnormal vessels again.
Combining Anti-angiogenic Agents with Drugs That Target Oncogenic Pathways
Combining AA agents with agents targeting oncogenic pathways, similar to chemotherapeutic agents, has led to some unexpected results.
Combining Anti-VEGF and Other Targeted Therapies
Despite promising preclinical results from combining VEGF- and EGFR-targeted agents in colorectal and NSCLC models, all phase III trials combining these targeted agents have failed [81]. Similarly, phase III trials combining VEGF- and HER2-targeted therapies in HER2+ breast cancer patients also failed [82]. A potential mechanism for these failures is that the dose of bevacizumab used may have decreased the size of pores in the tumor vessel walls and compromised the delivery of antibodies [83]. This hypothesis is consistent with elevated baseline plasma VEGF concentrations being associated with a greater bevacizumab benefit. It is also consistent with the recent randomized phase II trial showing the benefit of combining bevacizumab with a smaller drug, erlotinib, in EGFR-mutant NSCLC patients [84].
Combining Anti-VEGFR2 and Anti-HER2 Agents
Treatment of HER2+ breast tumors in the mouse brain with trastuzumab leads to increased VEGF production by host cells in the brain [85]. To this end, we combined HER2-targeted drugs (trastuzumab and lapatinib) with an anti-VEGFR2 antibody and demonstrated a significant improvement in survival of mice bearing HER2+ tumors in the brain [86]. Moreover, a phase II clinical trial with dual HER2 blockade and bevacizumab showed encouraging results in heavily pretreated HER2+ breast cancer patients with brain metastases [87]. Some of these clinical results are in line with our recent finding of higher VEGFR2 protein levels by IHC in HER2+ breast cancer [39], pointing toward potential clinical relevance of combining anti-VEGFR2 therapy (Ramucirumab: Cyramza®) with anti-HER2 therapies in HER2+ breast cancer.
Combining Anti-angiogenic and Immunotherapeutic Agents
Vascular Normalization Can Improve Benefit from Immunotherapy
The abnormal tumor vasculature can impede T effector cell infiltration into tumors and create a hypoxic and acidic tumor microenvironment that upregulates PD-L1 on myeloid-derived suppressor cells (MDSCs), dendritic cells, and cancer cells; increases the accumulation of regulatory T cells (Tregs); impairs T effector cells; and polarizes TAMs to the immune inhibitory M2-like phenotype to suppress T effector cell function.
Hypoxia can also upregulate multiple immune-suppressive growth factors and cytokines (e.g., VEGF and TGF-b). Vascular normalization with an appropriate dose and schedule of anti-angiogenic treatment can normalize the tumor vasculature and generate a more homogeneous distribution of perfused tumor vessels, facilitating the infiltration of T effector cells while reducing MDSC and regulatory T cell (Treg) accumulation. In addition, alleviation of hypoxia and acidity by improved vascular perfusion polarizes TAMs to an immunostimulatory M1-like phenotype [88].
Conclusions and Perspective
In recent years, significant advances in cancer treatment have been made with anti-angiogenic therapies, many of which have focused on inhibition of the vascular endothelial growth factor/VEGFR receptor (VEGF/VEGFR) pathway. VEGF/VEGFR targeting alone, however, has not been as efficacious as originally hoped. Based on recent advances in demystifying the complex biology of tumor angiogenesis, it has become clear that there are many redundant, compensatory signaling pathways that can overcome VEGF/VEGFR-targeted inhibition of tumor angiogenesis and may contribute to subsequent tumor progression. Therefore, refinement of the efficacy of various anti-angiogenic therapies will, at one end, require more focused approach rationalized by predictive biomarkers and, on the other, a rather broader therapeutic approach using biologically relevant combinatorial strategies or multitargeted anti-angiogenic agents, based on diverse molecular pathologic profiles of various human cancer types and subtypes.
Advanced histopathologic characterization and molecular classification of human cancer tissues from clinical trials will enable clinical trial teams to accurately interpret clinical efficacy data emerging from ongoing trials of anti-angiogenic therapies. As high-throughput technologies like NGS and more targeted sequencing approaches are becoming less and less cost-prohibitive, in addition to gold standard single marker methodologies like IHC and FISH, a great deal of progress can be made in terms of discovery and analytical and clinical validation of appropriate targeted panels of molecular biomarkers of response or resistance to AA-Rxs. Anatomic pathologists with sub-specialty expertise in molecular oncologic pathology will have a key role in designing and advancing predictive and prognostic biomarker science on well-characterized human cancer tissues. Acquisition of high-quality human tissue specimens and relevant clinicopathologic data will facilitate exploratory biomarker analyses at earlier stages of clinical development of AA-Rxs.
Although a great deal of scientific knowledge has accumulated about the highly complex biology of VEGF/VEGFR and non-VEGF/VEGFR signaling pathways with frequent cross talk and redundant mechanisms of primary and acquired resistance to anti-angiogenic therapies, there is an urgent need to incorporate and translate that scientific knowledge into patient-tailoring strategies in the current and future clinical trials of anti-angiogenic therapies, so that these agents can be offered to the right patients in order to maximize and sustain clinical benefit at the individual patient level. Because of its complexity, it will also be important to develop reliable methodologies to more fully characterize pathobiology of angiogenesis in the context of molecular pathology of various human cancer types. Such efforts will benefit from well-integrated, interdisciplinary clinical, translational, and basic research teamwork including the industry, academia, diagnostic, and biotechnology companies. Prioritization of well-established and innovative technologies to develop and standardize predictive biomarker assays, the definition of optimal scoring strategies/cutoffs, and accurate diagnostic classification of clinical trial cancer tissues will be important considerations for next-generation clinical trials of anti-angiogenic therapies.
Future Directions
To Improve Efficacy of Anti-angiogenic Therapies
While approved anti-angiogenic therapies have become a notable advance in targeted therapeutic options for patients with several different cancer types, in order to further improve clinical efficacy of these agents in the future, there is an urgent need to:
-
1.
Discover, validate, and qualify clinically relevant predictive biomarkers for various anti-angiogenic therapies.
-
2.
Develop and standardize technically robust tissue-based molecular methodologies that can quantify relevant molecular targets at protein, RNA, or DNA level, and provide an objective measure of the tumor biology to allow correlations with circulating angiogenic biomarker levels, which in isolation may not be uniformly representative of the biology of primary tumor or various metastases in an individual patient at a given time.
-
3.
Further develop and advance technically robust and cost-effective tissue-based methodologies for broader evaluation of the elegant concept of vascular heterogeneity put forward by Harold Dvorak from Harvard University (“early” and “late” tumor vessel phenotypes) and to systematically evaluate the clinical relevance of tumor vessel phenotypes as potential predictors of response or resistance to anti-VEGF/VEGFR therapies in various cancer indications.
-
4.
Carry out further clinical evaluation of tissue-based VEGF receptor profiling to determine its clinical significance and utility in the context of various anti-angiogenic therapies targeting one or more VEGF receptors.
-
5.
Consider implementing central sub-specialty level human tumor pathology reporting in clinical trials of anti-angiogenic therapies, so as to minimize (ideally eliminate) histologically misclassified tumor data submitted by global clinical trial sites and to improve reliability of tissue biomarker data analyses from clinical trials.
-
6.
Develop rational patient-tailoring hypotheses, based on patho-biologically relevant predictive biomarker/drug target profiles of tumor subsets, to be tested and refined in future clinical trials of anti-angiogenic therapies.
-
7.
Build effective collaborations among basic, translational, and clinical research teams in the industry led by experienced oncologists and pathologists, so that tailoring biomarker research can be implemented early along the drug development process.
-
8.
Generate biologically relevant retrospective biomarker data sets using optimal technologies to interrogate and advance promising single markers and carefully designed targeted marker panels representing broader biologic profile of a given human cancer type.
Using high-quality tumor tissues as full sections or leveraging tissue microarray technology can help generate data-driven hypotheses that can provide clinical teams with the rationale to design monotherapy or combinatorial trials of anti-angiogenic therapies with other promising oncology drugs like anti-HER2 or immunotherapy agents.
Abbreviations
- BRC:
-
Breast carcinoma
- CIN:
-
Chromosomal instability
- CRC:
-
Colorectal carcinoma
- DV:
-
Draining vein
- ECOG:
-
Eastern Cooperative Oncology Group
- EGFR:
-
Epidermal growth factor receptor
- FA:
-
Feeder artery
- FDA:
-
Food and Drug Administration
- FGFR:
-
Fibroblast growth factor receptor
- FISH:
-
Fluorescent in situ hybridization
- FLK1:
-
Fetal liver kinase 1
- FOLFIRI:
-
Folinic acid, 5-fluorouracil, and irinotecan
- GEJ:
-
Gastroesophageal junction
- GIST:
-
Gastrointestinal stromal tumor
- GMP:
-
Glomeruloid microvascular proliferation
- HCC:
-
Hepatocellular carcinoma
- HER2:
-
Human epidermal growth factor receptor 2
- HGF:
-
Hepatocyte growth factor
- IgG:
-
Immunoglobulin G
- IHC:
-
Immunohistochemistry
- ILF:
-
Irinotecan, bolus fluorouracil, and leucovorin
- INF:
-
α Interferon-α
- MC:
-
Mast cell
- MDSCs:
-
Myeloid-derived suppressor cells
- MET:
-
Mesenchymal epithelial transition factor
- MTC:
-
Medullary thyroid carcinoma
- MVD:
-
Microvascular density
- NGS:
-
Next-generation sequencing
- NRP1:
-
Neuropilin 1
- NRP2:
-
Neuropilin 2
- NSCLC:
-
Non-small cell lung carcinoma
- OC:
-
Ovarian cancer
- OS:
-
Overall survival
- PACA:
-
Pancreatic cancer
- PDGFR:
-
Platelet-derived growth factor
- PDGF-Rb:
-
Platelet-derived growth factor receptor-beta
- PFS:
-
Progression-free survival
- PlGF:
-
Placental growth factor
- RCC:
-
Renal cell carcinoma
- RNA:
-
Ribonucleic acid
- TAM:
-
Tumor associated macrophage
- TAMs:
-
Tumor-associated macrophages
- TCGA:
-
The Cancer Genome Atlas
- TGF-b:
-
Transforming growth factor-beta
- TKIs:
-
Tyrosine kinase inhibitors
- Treg:
-
Regulatory T cells
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
- VPF:
-
Vascular permeability factor
References
Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–22.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42.
Jang HJ, Kim BJ, Kim JH, Kim HS. The addition of bevacizumab in the first-line treatment for metastatic colorectal cancer: an updated meta-analysis of randomized trials. Oncotarget. 2017;8(42):73009–16.
Hayes DF. Bevacizumab treatment for solid tumors: boon or bust? JAMA. 2011;305(5):506–8.
Jain RK. Lessons from multidisciplinary translational trials on anti-angiogenic therapy of cancer. Nat Rev Cancer. 2008;8(4):309–16.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. https://doi.org/10.1038/nature10144.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6.
Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31(17):2205–18.
Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol. 2009;29(6):789–91.
Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res. 2012;72(8):1909–14.
Sitohy B, Chang S, Sciuto TE, Masse E, Shen M, Kang PM, et al. Early actions of anti-vascular endothelial growth factor/vascular endothelial growth factor receptor drugs on angiogenic blood vessels. Am J Pathol. 2017;187(10):2337–47.
Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8(12):942–56.
Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44.
Gacche RN, Assaraf YG. Redundant angiogenic signaling and tumor drug resistance. Drug Resist Updat. 2018;36:47–76.
Neufeld G, Kessler O. The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat Rev Cancer. 2008;8(8):632–45.
Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456(7223):814–8.
Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703.
Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, et al. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18(5):713–24.
Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17(3):471–94.
Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, Bauer TW, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 2005;24(16):2647–53.
Dales JP, Garcia S, Bonnier P, Duffaud F, Carpentier S, Djemli A, et al. Prognostic significance of VEGF receptors, VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) in breast carcinoma. Ann Pathol. 2003;23(4):297–305.
Guo P, Fang Q, Tao HQ, Schafer CA, Fenton BM, Ding I, et al. Overexpression of vascular endothelial growth factor by MCF-7 breast cancer cells promotes estrogen-independent tumor growth in vivo. Cancer Res. 2003;63(15):4684–91.
Spannuth WA, Nick AM, Jennings NB, Armaiz-Pena GN, Mangala LS, Danes CG, et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int J Cancer. 2009;124(5):1045–53.
Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–91.
Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35.
Fan F, Samuel S, Gaur P, Lu J, Dallas NA, Xia L, et al. Chronic exposure of colorectal cancer cells to bevacizumab promotes compensatory pathways that mediate tumour cell migration. Br J Cancer. 2011;104(8):1270–7.
Ronca R, Benkheil M, Mitola S, Struyf S, Liekens S. Tumor angiogenesis revisited: regulators and clinical implications. Med Res Rev. 2017;37(6):1231–74.
Welti JC, Gourlaouen M, Powles T, Kudahetti SC, Wilson P, Berney DM, et al. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene. 2011;30(10):1183–93.
Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010;70(24):10090–100.
Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319(9):1271–80.
Li JL, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H, et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res. 2011;71(18):6073–83.
Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15(1):21–34.
di Tomaso E, London N, Fuja D, Logie J, Tyrrell JA, Kamoun W, et al. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS One. 2009;4(4):e5123.
Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70(3):1063–71.
Salvucci O, Tosato G. Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis. Adv Cancer Res. 2012;114:21–57.
Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest. 2011;121(4):1313–28.
Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456(7223):809–13.
Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–27.
Nasir A, Holzer TR, Chen M, Man MZ, Schade AE. Differential expression of VEGFR2 protein in HER2 positive primary human breast cancer: potential relevance to anti-angiogenic therapies. Cancer Cell Int. 2017;17:56.
Nagy JA, Dvorak HF. Heterogeneity of the tumor vasculature: the need for new tumor blood vessel type-specific targets. Clin Exp Metastasis. 2012;29(7):657–62.
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–121.
Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res. 2010;16(14):3548–61.
Holzer TR, Fulford AD, Nedderman DM, Umberger TS, Hozak RR, Joshi A, et al. Tumor cell expression of vascular endothelial growth factor receptor 2 is an adverse prognostic factor in patients with squamous cell carcinoma of the lung. PLoS One. 2013;8(11):e80292.
Sitohy B, Nagy JA, Jaminet SC, Dvorak HF. Tumor-surrogate blood vessel subtypes exhibit differential susceptibility to anti-VEGF therapy. Cancer Res. 2011;71(22):7021–8.
Nasir A, Reising LO, Nedderman DM, Fulford AD, Uhlik MT, Benjamin LE, et al. Heterogeneity of vascular endothelial growth factor receptors 1, 2, 3 in primary human colorectal carcinoma. Anticancer Res. 2016;36(6):2683–96.
Nasir A, Falcon B, Wang D, et al. Vascular and tumor cell expression of VEGFR2 and molecular subtyping: an innovative biomarker approach in bladder cancer. ASCO-GU Cancers Symposium. San Francisco; 2018.
Tolaney SM, Boucher Y, Duda DG, Martin JD, Seano G, Ancukiewicz M, et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc Natl Acad Sci U S A. 2015;112(46):14325–30.
Jeong HS, Jones D, Liao S, Wattson DA, Cui CH, Duda DG, et al. Investigation of the lack of angiogenesis in the formation of lymph node metastases. J Natl Cancer Inst. 2015;107(9):699.
Wehland M, Bauer J, Magnusson NE, Infanger M, Grimm D. Biomarkers for anti-angiogenic therapy in cancer. Int J Mol Sci. 2013;14(5):9338–64.
Pilotto S, Bonomi M, Massari F, Milella M, Ciuffreda L, Brunelli M, et al. Anti-angiogenic drugs and biomarkers in non-small-cell lung cancer: a ‘hard days night’. Curr Pharm Des. 2014;20(24):3958–72.
Duda DG, Willett CG, Ancukiewicz M, di Tomaso E, Shah M, Czito BG, et al. Plasma soluble VEGFR-1 is a potential dual biomarker of response and toxicity for bevacizumab with chemoradiation in locally advanced rectal cancer. Oncologist. 2010;15(6):577–83.
Meyerhardt JA, Ancukiewicz M, Abrams TA, Schrag D, Enzinger PC, Chan JA, et al. Phase I study of cetuximab, irinotecan, and vandetanib (ZD6474) as therapy for patients with previously treated metastastic colorectal cancer. PLoS One. 2012;7(6):e38231.
Willett CG, Duda DG, di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J Clin Oncol. 2009;27(18):3020–6.
Zhu AX, Ancukiewicz M, Supko JG, Sahani DV, Blaszkowsky LS, Meyerhardt JA, et al. Efficacy, safety, pharmacokinetics, and biomarkers of cediranib monotherapy in advanced hepatocellular carcinoma: a phase II study. Clin Cancer Res. 2013;19(6):1557–66.
Lambrechts D, Claes B, Delmar P, Reumers J, Mazzone M, Yesilyurt BT, et al. VEGF pathway genetic variants as biomarkers of treatment outcome with bevacizumab: an analysis of data from the AViTA and AVOREN randomised trials. Lancet Oncol. 2012;13(7):724–33.
Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, Scherer SJ. Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol. 2013;31(9):1219–30.
Holzer TR, Fulford AD, Reising LO, Nedderman DM, Zhang X, Benjamin LE, et al. Profiling of vascular endothelial growth factor receptor heterogeneity identifies protein expression-defined subclasses of human non-small cell lung carcinoma. Anticancer Res. 2016;36(7):3277–88.
Kankeu Fonkoua L, Yee NS. Molecular characterization of gastric carcinoma: therapeutic implications for biomarkers and targets. Biomedicines. 2018;6(1).
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Uhlik MT, Liu J, Falcon BL, Iyer S, Stewart J, Celikkaya H, et al. Stromal-based signatures for the classification of gastric cancer. Cancer Res. 2016;76(9):2573–86.
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50.
Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAil. J Clin Oncol. 2009;27(8):1227–34.
Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL). Ann Oncol. 2010;21(9):1804–9.
Schneider BP, Li L, Shen F, Miller KD, Radovich M, O’Neill A, et al. Genetic variant predicts bevacizumab-induced hypertension in ECOG-5103 and ECOG-2100. Br J Cancer. 2014;111(6):1241–8.
Poveda AM, Selle F, Hilpert F, Reuss A, Savarese A, Vergote I, et al. Bevacizumab combined with weekly paclitaxel, pegylated liposomal doxorubicin, or topotecan in platinum-resistant recurrent ovarian cancer: analysis by chemotherapy cohort of the randomized phase III AURELIA trial. J Clin Oncol. 2015;33(32):3836–8.
Liu JF, Matulonis UA. Bevacizumab in newly diagnosed ovarian cancer. Lancet Oncol. 2015;16(8):876–8.
Krill LS, Tewari KS. Integration of bevacizumab with chemotherapy doublets for advanced cervical cancer. Expert Opin Pharmacother. 2015;16(5):675–83.
Crafton SM, Salani R. Beyond chemotherapy: an overview and review of targeted therapy in cervical cancer. Clin Ther. 2016;38(3):449–58.
Ciombor KK, Berlin J. Aflibercept – a decoy VEGF receptor. Curr Oncol Rep. 2014;16(2):368.
Aprile G, Rijavec E, Fontanella C, Rihawi K, Grossi F. Ramucirumab: preclinical research and clinical development. Oncol Targets Ther. 2014;7:1997–2006.
Ramucirumab TP. Boon or bane. J Egypt Natl Canc Inst. 2016;28(3):133–40.
Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36(7):422–39.
Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016;21(1):5–10.
Zhu Y, Choi SH, Shah K. Multifunctional receptor-targeting antibodies for cancer therapy. Lancet Oncol. 2015;16(15):e543–e54.
Yokoi K, Thaker PH, Yazici S, Rebhun RR, Nam DH, He J, et al. Dual inhibition of epidermal growth factor receptor and vascular endothelial growth factor receptor phosphorylation by AEE788 reduces growth and metastasis of human colon carcinoma in an orthotopic nude mouse model. Cancer Res. 2005;65(9):3716–25.
Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006;66(4):2173–80.
Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004;18(2):338–40.
Hojjat-Farsangi M. Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int J Mol Sci. 2014;15(8):13768–801.
Mabry R, Gilbertson DG, Frank A, Vu T, Ardourel D, Ostrander C, et al. A dual-targeting PDGFRbeta/VEGF-A molecule assembled from stable antibody fragments demonstrates anti-angiogenic activity in vitro and in vivo. MAbs. 2010;2(1):20–34.
Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D, et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res. 2013;19(24):6730–40.
Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360(6):563–72.
Gianni L, Romieu GH, Lichinitser M, Serrano SV, Mansutti M, Pivot X, et al. AVEREL: a randomized phase III trial evaluating bevacizumab in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive locally recurrent/metastatic breast cancer. J Clin Oncol. 2013;31(14):1719–25.
Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7(6):383–8.
Seto T, Kato T, Nishio M, Goto K, Atagi S, Hosomi Y, et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014;15(11):1236–44.
Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416(6878):279–80.
Kodack DP, Chung E, Yamashita H, Incio J, Duyverman AM, Song Y, et al. Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc Natl Acad Sci U S A. 2012;109(45):E3119–27.
Falchook GS, Moulder SL, Wheler JJ, Jiang Y, Bastida CC, Kurzrock R. Dual HER2 inhibition in combination with anti-VEGF treatment is active in heavily pretreated HER2-positive breast cancer. Ann Oncol. 2013;24(12):3004–11.
Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73(10):2943–8.
Acknowledgments
The author would like to acknowledge the dedication and hard work of Timothy Holzer, Angie Fulford, Beverly Falcon, Drew Nedderman, Leslie O’Neal Reising, James Alston, and Mia Chen in the lab in developing and optimizing technically robust immunohistochemical and bright-field in situ hybridization technologies for VEGFR2, VEGFR3, and VEGFR1 for archival human tissues. Thanks to Jeff Hanson for supporting pathologic angiogenesis and oncologic disease state characterization analyses. Thanks also to Andrew Schade, Kelly Credille, Aafia Chaudhry, Katherine Marie Bell-McGuinn, Mayukh Das, Richard Walgren, Laura Benjamin, Bronislaw Pytowsky, Mark Uhlik, and Jeremy Graff for great scientific and clinical collaboration on tumor angiogenesis and anti-angiogenesis projects over the years.
Disclaimer Expert scientific and clinical views expressed in this chapter are those of the author.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nasir, A. (2019). Angiogenic Signaling Pathways and Anti-angiogenic Therapies in Human Cancer. In: Badve, S., Kumar, G. (eds) Predictive Biomarkers in Oncology. Springer, Cham. https://doi.org/10.1007/978-3-319-95228-4_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-95228-4_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95227-7
Online ISBN: 978-3-319-95228-4
eBook Packages: MedicineMedicine (R0)