
Abstract
The co-chaperone stress-inducible protein 1 (STI1) is released by astrocytes, and has important neurotrophic properties upon binding to prion protein (PrPC). However, STI1 lacks a signal peptide and pharmacological approaches pointed that it does not follow a classical secretion mechanism. Ultracentrifugation, size exclusion chromatography, electron microscopy, vesicle labeling, and particle tracking analysis were used to identify three major types of extracellular vesicles (EVs) released from astrocytes with sizes ranging from 20–50, 100–200, and 300–400 nm. These EVs carry STI1 and present many exosomal markers, even though only a subpopulation had the typical exosomal morphology. The only protein, from those evaluated here, present exclusively in vesicles that have exosomal morphology was PrPC. STI1 partially co-localized with Rab5 and Rab7 in endosomal compartments, and a dominant-negative for vacuolar protein sorting 4A (VPS4A), required for formation of multivesicular bodies (MVBs), impaired EV and STI1 release. Flow cytometry and PK digestion demonstrated that STI1 localized to the outer leaflet of EVs, and its association with EVs greatly increased STI1 activity upon PrPC-dependent neuronal signaling. These results indicate that astrocytes secrete a diverse population of EVs derived from MVBs that contain STI1 and suggest that the interaction between EVs and neuronal surface components enhances STI1–PrPC signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The co-chaperone stress-inducible protein 1 (STI1) was first described as a stress response protein, the expression of which increased after heat-shock, similar to classical heat shock proteins (Hsps) [1, 2]. STI1 binds to Hsp70 and Hsp90 in a complex responsible for the correct folding of client proteins [3, 4]. Elimination of STI1 does not affect growth in yeast, although STI1 mutants synergize with the effects of Hsp90 mutants to affect growth [5]. In Caenorhabditis elegans, lack of STI1 is not lethal, although it decreases life span and increases sensitivity to heat stress [6]. In mice, STI1 is expressed early in the embryo [7] indicating its key and non-redundant role in mammals.
In contrast to its classical intracellular localization, STI1 and its human homologue HOP (Hsp70/Hsp90 Organizing Protein) were found in the conditioned medium (CM) of several cell types [8–11]. Previous work from our group characterized the glycosylphosphatidylinositol (GPI)-anchored prion protein (PrPC) as a membrane receptor for STI1 [12–14]. Upon binding, STI1 triggers PrPC-dependent survival and differentiation in both neuronal cells [15–17] and astrocytes [18], and increases the proliferation of glioblastoma cells [10]. Secreted STI1 has also been described as a ligand for activin A receptor type II-like kinase 2 (ALK2), which induces SMAD-dependent proliferation of ovarian cancer cells [19].
Similar to STI1, Hsp70, Hsp60, and Hsp90 lack a consensus secretory signal peptide, but are also secreted [20, 21]. The secretion of Hsp70 and Hsp90 is reportedly mediated by exosomes [20, 22, 23], which are intraluminal vesicles formed in endocytic compartments known as multivesicular bodies (MVBs) [24–26]. However, the secretion of these proteins may also occur by a non-exosomal, lipid-raft-dependent mechanism [21].
Intercellular communication through small vesicles has recently been validated in several systems [27], but their functions are not fully understood. Extracellular vesicles (EVs) are found in physiologic conditions, and function both in signaling and in the transfer of membrane and/or cargo molecules. It is generally believed that EVs may include microparticles shed from the cell membrane, as well as exosomes [24–26]. Notwithstanding, there is little agreement regarding the characterization of these vesicles. The use of distinct protocols of isolation, together with the heterogeneity of these vesicles in size, and in phospholipid and protein composition [28–31] contribute to the lack of consensus in the area. In fact, such divergence is reflected in the multiple designations that appear in the literature: microparticles, microvesicles, exosomes, enlargeosomes, ectosomes, iccosomes, prostasomes, or prominosomes [32, 33]. Proteins such as chaperones, tetraspanins, adhesion molecules, Rabs, cytoskeletal components, metabolic enzymes, and PrPC are well-accepted markers for exosomes [25, 26, 34–36]; however, a consensus profile of markers for the different types of vesicles is still lacking.
The presence of Hsps in the extracellular space has been associated with neuroprotection, immunity, and cancer [37–40]. Thus, a deeper understanding, as well as the development of novel therapeutic approaches to various diseases, would greatly benefit from a thorough understanding of the mechanisms associated with the release of Hsps.
In this work, we examined the release of STI1 by astrocytes, and explored the properties of EVs secreted by this cell type. We found that astrocytes release STI1 among a heterogeneous population of multivesicular body-derived EVs, which contain classical exosome markers, and lead to the activation of PrPC-dependent neuronal signaling pathways.
Materials and methods
Reagents
All culture media components, Lipofectamine, and brefeldin A (BFA) were purchased from Invitrogen-Life Technologies (Carlsbad, CA, USA). His6–STI1, His6–PrPC, and GFP–STI1 vectors were constructed, and recombinant proteins were expressed and purified as previously described [12]. Vps4A–GFP and Vps4A E228Q–GFP constructs were a gift from Wes Sundquist (University of Utah School of Medicine). Polyclonal anti-STI1 antibodies raised in rabbit were produced by Bethyl Laboratories [12]. A polyclonal antibody against recombinant mouse PrPC was produced in PrPC-null (Prnp 0/0) mice [41]. A rabbit polyclonal Hsp70 antibody was purchased from Chemicon International (Temecula, CA, USA). A mouse monoclonal ApoE antibody, rat monoclonal LAMP1 antibody, mouse monoclonal Hsp90 antibody, and goat anti-GFP–HRP were obtained from Abcam (Cambridge, MA, USA). Mouse monoclonal anti-βII-microglobulin was from BD Biosciences. Mouse anti-human transferrin receptor (TfR) was purchased from Zymed Laboratories (Santiago, Chile). Mouse monoclonal anti-GAPDH was from AMBION/Life Technologies (Carlsbad, CA, USA). Mouse monoclonal anti-Vsp4/SKD1 was from Millipore (Billerica, MA, USA), and anti-rabbit Alexa 488 or anti-mouse Alexa 633 were purchased from Invitrogen (Carlsbad, CA, USA). Anti-phospho-ERK1/2 and anti-total ERK1/2 antibodies were from Cell Signaling (Danvers, MA, USA). Polyvinyl alcohol (PVA/MW 25,000, 88 mol% hydrolyzed was from Polysciences (Warrington, PA, USA). Poli-l-lysine, 5,5′-dithiobis (2-nitrobenzoic acid), acetylthiocholine, dibutyryl-cAMP, [9,10-3H] myristic acid, and monensin were purchased from Sigma-Aldrich (Saint Louis, MO, USA). The 96-well MICROLON 600 high binding plates were by Greiner Bio One (Frickenhausen, Germany). Superose 12 prep grade was from GE Healthcare (Amersham, UK) and the Amicon concentrator was from Millipore (Billerica, MA, USA). PVA was purchased from PolySciences (Warrington, PA, USA) and aldehyde-sulphate latex beads surfactant free, white, 4 μm were from Invitrogen (Carlsbad, CA, USA). [9,10-3H] myristic acid was purchased from PerkinElmer (Waltham, MA, USA).
Animals
All experiments followed the guidelines of the National Institutes of Health (The Principles of Laboratory Animal Care 8th edition, 2011 http://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=12910), and were approved by the local Animal Care and Use Committees at the A.C. Camargo Hospital and at the University of Western Ontario. ZrchI Prnp0/0 mice were provided by Dr. C. Weissmann (Scripps Florida, Jupiter, FL) [42], and the wild-type control mice (ZrchI Prnp+/+) were generated by crossing F1 descendants from 129/SV to C57BL/6J mating.
Plasmids
The pEGFP–C1 was purchased from Clontech (Mountain View, CA, USA), and the constructs GFP–STI1 [43], YFP–STI1 [43], mCherry–Rab7, mCherry–Rab5, GFP–VPS4, or GFP–VPS4 DN were previously described [44].
Primary hippocampal neuronal cultures
Primary hippocampal cultures were prepared as previously described. Briefly, the hippocampal structure was dissected in HBSS and treated with trypsin (0.06 %) in HBSS for 20 min at 37 °C. The protease was inactivated with 10 % fetal calf serum (FCS) in Neurobasal medium for 5 min. After three washes with HBSS, cells were mechanically dissociated in Neurobasal medium containing B-27 supplement, glutamine (2 mM), and penicillin/streptomycin (100 μg/ml). Cells (106) were plated onto 35-mm plates coated with 5 μg/ml poly-l-lysine.
Cultures of primary cortical astrocytes and SN56 cells
Astrocyte primary cultures were prepared as previously described [9, 16] from the cerebral hemispheres of embryonic day 17 (E17) wild-type and PrP C-null mice. Briefly, single cell suspensions were obtained by dissociating cerebral hemispheres in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glucose (33 mM), glutamine, penicillin/streptomycin, and sodium bicarbonate (3 mM). Cells were plated on pre-coated poly-l-lysine plates, and grown in DMEM enriched with 10 % FCS centrifuged for 16 h at 100,000 × g to remove endogenous vesicles. The medium was changed every 2 days. SN56 is a cell line derived from mouse septum neurons and was cultured as previously described [45, 46].
Transfection and imaging
For transfection experiments, confluent astrocytes were treated with 10 μl of Lipofectamine (1 mg/ml solution) and 5 μg DNA (pEGFP–C1, GFP–STI1, YFP–STI1, mCherry–Rab7, mCherry–Rab5, GFP–VPS4, or GFP–VPS4 DN) in Optimem. After 4 h, the medium was changed to DMEM enriched with 10 % FCS, and cells were further incubated for 48 h. Live cells were imaged on a Leica SP5 confocal microscope (for YFP–STI1 and GFP–VPS4 or GFP–VPS4 DN) or on a Leica TIRF microscope (for GFP–STI1, mCherry–Rab7, and mCherry–Rab5).
For ground-state depletion and individual molecule return (GSDIM), astrocyte primary cultures were plated on coverslips and at 70–80 % of confluence were fixed in a solution of 4 % paraformaldehyde/PBS for 20 min at room temperature. Cells were rinsed three times in PBS and unspecific binding sites were blocked for 1 h at room temperature with 2 % BSA and 2 % FBS in PBS 0.1 % Triton X-100. The cells were incubated overnight with anti-STI1 (1:200) [12] and anti-VPS4 (1:250) antibodies. Cells were washed five times with PBS and incubated for 1 h with anti-rabbit conjugated with Alexa 488 or anti-mouse conjugated with Alexa 633. Coverslips were coated with 50 μl of a solution of 1 % PVA pH 7.4 (polyvinyl alcohol/MW 25,000, 88 mol%), using a spincoater for 30 s at 3,000 rpm. The samples were mounted on depression slides, and GSDIM [16, 47] images were acquired using a Leica SR GSD system. Briefly, the images were acquired with a HCX PL APO 100x/1.47 Oil CORR objective. The fluorophores were pumped into a dark state using 75 % excitation laser power, and the molecules returning from the dark state were imaged using 30 % excitation laser and 10-ms exposures with an Andor iXon X3 897 back-illuminated EMCCD camera. The localization and construction of the final GSDIM super-resolution image was completed using a localization algorithm available within a GSD software module in the Leica Application Suite for Advanced Fluorescence (LAS AF).
Preparation and fractionation of astrocyte-conditioned media (CM)
Conditioned medium was prepared as previously described [9]. Briefly, confluent astrocytes grown in 100-mm culture dishes were washed three times with phosphate-buffered saline (PBS) and were covered with serum-free medium for 48 h. The CM was collected on ice and pre-cleared by centrifugation (300 × g for 10 min, 2,000 × g for 10 min, and 10,000 × g for 30 min) followed by filtering through gravity (0.22 μm pore size). Fractionation was done by sequential ultracentrifugation at 100,000 × g for 1 h, 2 h, and 16 h, in a SW40Ti rotor (Beckman-Coulter, Brea, CA, USA). The pellet from each ultracentrifugation was re-suspended in 100 μl of PBS. The final supernatant was concentrated 100-fold in an Amicon concentrator (Fig. 1a).

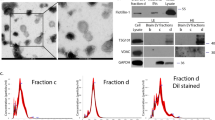
STI1 is found in both soluble and sedimented fractions after sequential ultracentrifugation of astrocyte CM, and is released by non-classical secretion. a Astrocyte CM was cleared of cell debris by centrifugation and concentrated 100×, or sequentially ultracentrifugated for 1, 2, and 16 h. After each round of centrifugation, the pellet and supernatant were separated, and the supernatant was taken to the next step of centrifugation. The final supernatant was concentrated 100×. b CM was sequentially ultracentrifugated for 1, 2, and 16 h, and the fractions were separated by SDS-PAGE followed by immunoblots for transferrin receptor (TfR), Hsp90, Hsp70, STI1, GAPDH, LAMP, flotillin, Tsg101, VPS36, and PrPc. For lysosomal-associated membrane protein 1 (LAMP) and GAPDH, additional cell extracts were used as a positive control. c Astrocyte medium conditioned for 12, 24, or 48 h was ultracentrifuged and the fractions (1, 2, or 16 h pellets and supernatants) were separated by SDS-PAGE followed by immunoblotting for STI1. d Astrocyte cultures were treated with brefeldin A (BFA, 1 μg/ml) and monensin (Mn, 5 μM). The CM was ultracentrifuged for 2 h, and the fractions (pellet and super) were separated by SDS-PAGE, followed by immunoblot for STI1 and β2-microglobulin
For isopycnic density centrifugation, pre-cleared CM was subjected to a 2-h centrifugation of 100,000 × g to deplete exosomes. To the remaining supernatant, 0.33 g/ml KBr was added followed by 48-h ultracentrifugation at 285,000 × g at 10 °C in a SW40Ti rotor. Twelve fractions were collected, and proteins from these fractions were TCA-precipitated and resolved via 10 % SDS-PAGE [9, 48]. For density determination, a standard curve was prepared using conductivity data and weight measures of six different concentrations of KBr in DMEM. Conductivity data from samples was then acquired continuously through the gradients and density values were obtained by reference to the standard curve [49].
For gel filtration chromatography, pre-cleared CM was concentrated tenfold in an Amicon concentrator. One milliliter of concentrated CM was fractionated by gel filtration using a 50-cm column packed with Superose 12 prep grade with 3,000-kDa exclusion (GE). Twenty-five fractions of 3.5 ml each were collected and analyzed.
For perturbations in intracellular protein trafficking, confluent astrocytes were pre-incubated with BFA (mg/ml) or monensin (5 μM) for 1 h, after which the medium was changed to serum-free medium containing the drugs. After 48 h, CM was subjected to fractionation.
Lipid analysis
For lipid disturbance, saponin (0.32 %), SDS (1 %), Triton X-100 (0.5 %) plus NP40 (0.5 %) or NaCl (800 mM) were added to the CM with gentle vortexing. The samples were then incubated on ice for 30 min before they were fractionated. For lipid formation, 60 % confluent astrocytes were labeled with 1 μCi/ml [9,10-3H] myristic acid. After 72 h, the cells were washed three times with PBS and covered with serum-free medium for 48 h. CM was fractionated by gel filtration chromatography. Lipids were extracted from the fractions by chloroform–methanol (1:1), dried, resuspended in 100 μl chloroform, and transferred to scintillation vials for measurement of [3H] incorporation.
ERK phosphorylation
Phosphorylation assays were done using the PhosphoPlus p44–42 MAPK (Thr202/Tyr204) antibody kit as previously described. Briefly, primary hippocampal cell cultures (106 cells) from either Prnp +/+ or Prnp 0/0 mice were plated on dishes pretreated with poly-l-lysine. The cells were stimulated with 100-fold concentrated CM, CM fractions from ultracentrifugation, or CM fractions from gel filtration. In some experiments, 50 μl of CM were pre-incubated with either anti-STI1 antibody (3 μg/ml) or with a peptide that mimics the binding site for STI1 (p10, 6 μg/ml—PrPC peptide amino acids 113-GAAAAGAVVGGLGGYMLGSA-128, NeoMPS France) for 30 min before stimulation. Thirty seconds to 10 min after stimulation, the cells were rinsed once with ice-cold PBS and lysed in Laemmli buffer. For ERK1/2 phosphorylation assays, cell extracts were subjected to SDS-PAGE followed by immunoblotting with anti-phospho-ERK1/2 and anti-total ERK1/2 antibodies. After washing, membranes were incubated with peroxidase conjugated anti-rabbit or anti-mouse IgGs. Reactions were developed using ECL (GE). The bands obtained after X-ray film exposure to the membranes were analyzed by densitometric scanning and quantified using the Scion (Frederick, MD, USA) Image software. Values represent the ratio between phospho-ERK (p42 plus p44) and total ERK (p42 plus p44) for each sample. Values obtained from untreated cultured neurons from Prnp +/+ or Prnp 0/0 mice were set at 1.0, and the other values were relative to it.
Immunoblot analyses
Proteins were subjected to SDS-PAGE and transferred to nitrocellulose membranes. Western blotting was conducted using rabbit anti-STI1 (1:5,000), mouse anti-PrPc (1:1,000), mouse anti-LAMP1 (1:1,000), mouse anti-GAPDH (1:1,000), mouse anti-Hsp90 (1:1,000), mouse anti-βII microglobulin (1:500), mouse anti-ApoE (1:1,000), mouse anti-TfR (1:500), rabbit anti-GFP, and rabbit anti-Hsp70 (1:200) antibodies. After washing, membranes were incubated with peroxidase conjugated anti-rabbit or anti-mouse IgGs. Reactions were developed using ECL (GE Life Sciences, Uppsala, Sweden), and membranes were exposed to X-ray film. The appropriate rabbit or mouse pre-immune sera were used as negative controls.
Release of GFP–STI1
A 100-mm confluent plate of astrocytes was transfected with 40 μl Lipofectamine and 20 μg DNA (GFP of GFP–STI1) in Optimem for 4 h. Cells were washed, incubated for 24 h, and serum starved for 48 h to produce CM. Cleared CM was fractionated by ultracentrifugation for 2 h. The pellets were resuspended in 50 μl PBS and the supernatant was concentrated. Pellets and supernatants were added to primary cultured neurons or SN-56 cells, and live cells were imaged after 30 min in a Leica SP5 confocal microscope. Alternatively, pellets and supernatants were immunoblotted for GFP or STI1.
ELISA
A 96-well plate was coated overnight at 4 °C with 50 μl of concentrated conditioned media or fraction. Afterwards, the wells were washed three times with wash buffer (0.3 % Triton X-100 in PBS). Each well was filled with 100 μl blocking buffer (5 % milk in PBS) and the plate was incubated for 2 h at 37 °C. After washing, 50 μl anti-STI1 [12] in PBS was added to the wells at a final dilution of 1:300. After incubation for 2 h at 37 °C, the solution was removed, the wells were washed, and 50 μl of anti-rabbit-HRP conjugated in PBS was added to the wells to a final dilution of 1:2,000. After incubation for 1 h at 37 °C, the wells were washed, and 50 μl of an orthophenylenediamine solution (0.33 mg/ml in 0.5 M citrate buffer, pH 5.2, and 0.4 % hydrogen peroxide) were added to each well. After 5 min at room temperature, protected from light, the enzymatic reaction was stopped by the addition of 50 μl of 4 M sulfuric acid. The absorbance (490 nm) was measured using a Bio-Rad microplate reader.
Silver staining
Fractionated CM was subjected to SDS-PAGE and the gel was silver stained. Briefly, gel was fixed in 50 % methanol and stained with 0.8 % AgNO3, 1.4 % NH4OH, and 0.0756 % NaOH. Color was developed with 0.005 % citric acid and 0.05 % formaldehyde, and the reaction was blocked by the addition of 45 % methanol and 10 % acetic acid.
Electron microscopy
Isolated extracellular vesicles
Fractions from ultracentrifugation or gel filtration chromatography were deposited onto Formvar-carbon-coated electron microscopy grids, fixed with a mixture of 2 % paraformaldehyde and 0.125 % of glutaraldehyde, and single or double immunogold-labeled with primary antibodies followed by addition of protein A-gold (PAG). Samples were contrasted end embedded in a mixture of methylcellulose and uranyl acetate, and observed under a Zeiss EM 900 transmission electron microscope.
Ultrathin sections
Astrocytes obtained from primary cultures were fixed in 2.5 % glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) for 2 h at room temperature, post-fixed in 1 % osmium tetroxide +0.8 % potassium ferricyanide +5 mM calcium chloride in 0.1 M cacodylate buffer (pH 7.2) on ice for 1 h, and blockstained (fixed) in 1 % uranyl acetate in maleate buffer at 4 °C for 1 h. After dehydration in a graded series in ethanol at 4 °C, samples were infiltrated with ethanol/epon mixtures and polymerized in pure epon at 60 °C for 48 h. Ultrathin sections were stained with uranyl acetate and lead citrate, and observed in a Zeiss EM 900 transmission electron microscope.
Flow cytometry
Conditioned medium from astrocytes was ultracentrifuged for 2 h, and the derived pellet was incubated with aldehyde-sulphate latex beads (surfactant free, white, 4 μm) overnight at 4 °C with gentle agitation in PBS. To block the remaining active sites, the beads were incubated for 30 min with 100 mM glycine. After two washes with 3 % BSA in PBS, vesicle-bead complexes were stained with antibodies against STI1, PrPC, Hsp90, and flotillin followed by anti-mouse or anti-rabbit IgG labeled with Alexa488. Specific staining was compared to isotype-matched antibodies by flow cytometry. Samples were analyzed on a FACSCalibur flow cytometer.
Microvesicle analysis and quantification
Conditioned medium was fractioned by sequential ultracentrifugation or gel filtration chromatography, and the number of particles and particle size was counted by a nanoparticle tracking analysis device (Nanosight LM20, coupled to a CCD camera and a laser emitting a 60-mW beam at 405-nm wavelength). The acquisitions were performed for 60 s using the following parameters: shutter of 604, gain of 100, and threshold of 10.
PK digestion
Extracellular vesicles isolated from CM after 16 h of centrifugation were resuspended in 100 μl of PBS or PBS plus 0.5 % Triton X-100 and incubated with 400 ng of PK for 10 min at 37 °C. Reaction was stopped with the addition of Laemmli buffer and samples were subjected to Western blot using antibodies against STI1.
Statistical analyses
Experiments were replicated at least three times. Statistical analyses were done by ANOVA followed by Tukey’s post hoc test.
Results
STI1 is found in both soluble and insoluble fractions from CM and its release is independent of the Golgi apparatus or secretory lysosomes
Stress-inducible protein 1 is known to be released by astrocytes [9], even though it does not have a signal peptide. We thus explored the possibility of an unconventional STI1 release through EVs. Given that the protocols to isolate EVs are quite variable in the literature, we chose to use distinct ultracentrifugation times to isolate CM fractions (Fig. 1a). Upon sequential ultracentrifugation of CM (100,000 × g), STI1 was found in the pellets obtained at 1, 2, and 16 h of centrifugation, and also in the final supernatant (Fig. 1b). STI1 was detected in the culture medium as early as 12 h after conditioning, and accumulated over time, suggesting that this co-chaperone is constitutively released from astrocytes in the conditions used here, and is stable in the CM (Fig. 1c). Quantification of STI1 release by ELISA indicates that after 48 h, STI1 concentration in the CM from 3 × 105 cells is 85.4 ± 13.9 pM.
Transferrin receptor, Hsp90, Hsp70, flotillin, Tsg101, VPS36, and PrPC, which are found in exosomes released by several cell types [25], were present in all CM pellets (1, 2, and 16 h), but were absent in the supernatant (Fig. 1b). GAPDH was not detected in any fraction, indicating that STI1 release was not the result of membrane leakage caused by cell death. LAMP-1, a constituent of lysosomes that can be released through an unconventional pathway mediated by secretory lysosomes [50–52] was also absent from the CM fractions, indicating that in our experimental conditions, there was no contribution of this pathway to the final protein profile of the CM (Fig. 1b). To further test for the involvement of the classical secretory pathway in STI1 release, astrocytes were pre-treated with BFA or monensin (Mn), and CM was fractionated by ultracentrifugation. Treatment with either BFA or Mn had no effect on the release of STI1, whereas both drugs abolished the release of β-II microglobulin, a component of the MHC complex secreted by Golgi (Fig. 1d).
These results indicate that STI1 is released into compartments that can be differentially sedimented by ultracentrifugation (1, 2, and 16 h). The release of both non-sedimented and sedimented forms of STI1 is independent of both Golgi-mediated classical secretion and non-classical pathways mediated by secretory lysosomes.
Released STI1 is associated with lipids
The presence of astrocyte-derived STI1 in structures that can be sedimented by 100,000 × g centrifugation raises the possibility of its incorporation into either protein aggregates or membranous vesicles. To discriminate between these two possibilities, CM was treated with high salt or detergents and fractionated by sequential ultracentrifugation. Treatment with detergent (Triton X-100 plus NP40, SDS or saponin) shifted STI1 as well as PrPC from the pellet (100,000 × g for 1, 2, and 16 h) (Fig. 2a) to the supernatant (Fig. 2b). In contrast, high-salt treatments (NaCl 800 mM) did not alter the pattern of STI1 or PrPC association to the pelleted fraction (Fig. 2a, b). These results suggest that released, sedimentable STI1 might be associated with lipids. Recombinant His-tagged STI1 added either to the CM or to DMEM remained in the supernatant following ultracentrifugation, thus ruling out artifactual pelleting of released STI1 (Fig. 2c).

STI1 in ultracentrifugation pellets is associated with lipids. CM from astrocytes was treated with distinct detergents (triton 0.5 % + NP40 0.5 %; SDS 1 %; or saponin 0.32 %) and high salt (800 mM NaCl) for 30 min. After treatment, the CM was ultracentrifuged for 1, 2, and 16 h and the pellets (a) or supernatant (b) were separated by SDS-PAGE, followed by immunoblotting against STI1 and PrPC. c 200 ng of recombinant His6–STI1 was added to DMEM or to conditioned medium incubated for 30 min, followed by 2 h ultracentrifugation. SDS-PAGE and immunoblot for His6 was done for the pellets and supernatants
STI1 is released by astrocytes in three types of lipid vesicles
To further identify the vesicles or particles containing released STI1, a gel filtration chromatography assay was developed and used to separate the CM in 25 fractions according to molecular mass. These fractions were immunoblotted for STI1 and PrPC (Fig. 3a). STI1 was mainly found in fractions 1–2, 5–6, 11–12, 16–17, which may represent four types of vesicles of distinct sizes (Fig. 3a). In this chromatography, recombinant STI1 and other proteins of 60 kDa were found in fractions 16–17 (data not shown), indicating that endogenous STI1 in fractions 16–17 corresponded to the soluble STI1 that remained in the supernatant after ultracentrifugation (see Fig. 1). Serum withdrawal did not affect the amount or distribution of STI1 secretion (Supplementary Fig. 1). PrPC was found in fractions 5–6 (Fig. 3a), whereas recombinant PrPC segregated into fractions 21–22 (data not shown), suggesting that endogenous PrPC in the astrocyte-derived CM was associated with other proteins or vesicular structures. To confirm the presence of lipids associated with STI1 and PrPC, the CM was treated with SDS prior to gel filtration chromatography. In detergent-treated CM, STI1 shifted to fractions 16–20, and PrPC to fraction 21, suggesting that lipids are required for the differential distribution of these proteins in the chromatography (Fig. 3b). The relative levels of STI1 and PrPC in each fraction before and after detergent treatment show the shift of STI1 from fractions 1–2, 5–6 and 11–12 to fractions 16–22 and PrPC from fraction 5–6 to fractions 21–22 (Fig. 3c).

STI1 and PrPC are present in lipid compartments. a–c CM from astrocytes was untreated (a) or treated with SDS (b) followed by gel filtration chromatography (Superose 12 prep grade, GE Healthcare) and collection of 25 fractions. The proteins were precipitated with TCA and separated by SDS-PAGE, followed by immunoblotting with antibodies for STI1 and PrPC and c quantified by densitometry. d Semi-confluent astrocyte cultures were labeled with [9,10-3H] myristic acid. After 3 days, the cells were washed with PBS and the medium was conditioned for 48 h. The CM was run through gel filtration, lipids were extracted with methanol-chloroform, and the fractions were counted using liquid scintillography. The graph compares the contents of STI1 (absolute values) and lipids (cpm). e The number of vesicles in fractions 1–2, 5–6, and 11–12 were analyzed by nanoparticle tracking. f The number of vesicles in the pellets of 1, 2, or 16 h of centrifugation was analyzed by nanoparticle tracking. g Sizes of vesicles in fractions 1–2, 5–6, and 11–12 measured by particle tracking and compared with the sizes of vesicles in sedimentable fractions from 1, 2, and 16 h of centrifugation
We confirmed the presence of lipids in each fraction by labeling astrocytes with [3H]-myristic acid, a lipidic precursor. In agreement with the results in Fig. 3a, [3H] labeling in fractions 2, 5–6, and 11–12 closely corresponded with the fractions that contained sedimentable STI1 (Fig. 3d).
Chromatography fractions were also evaluated by nanoparticle tracking and two peaks containing 1.4 × 108 particles/ml were found in fractions 1–2 and 5–6. A third peak containing 0.6 × 108 particles/ml was found in fractions 11–12 (Fig. 3e). Pellets from ultracentrifugation were also evaluated by particle tracking in respect to number (Fig. 3f) and size distribution and compared with chromatography fractions (Fig. 3g). Particles from 1 h of centrifugation and chromatography fractions 1–2 had a similar average size of 300 nm. Particles from 2 h of centrifugation and chromatography fractions 5–6 had an average size of 137 and 195 nm, respectively. Particles from 16 h of centrifugation had an average size of 143 nm, while chromatography fractions 11–12 presented an average size of 64 nm. Thus, both techniques seem to segregate vesicles of three different sizes, although separation by chromatography showed a smaller standard deviation, consistent with better separation using this method. Additionally, our experiment demonstrates that after 1 h ultracentrifugation, a great portion of EVs remain on the supernatant, and thus studies that evaluate EVs using a centrifugation time of 1 h are indeed not working with the complete EVs population. These results clearly demonstrate that astrocytes secrete three major types of EVs, and that STI1 is carried in all three types, although an additional fraction of this protein is not associated with lipids.
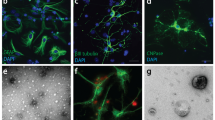
Electron microscopy also indicated that structures found in concentrated CM preparations had size variation (Fig. 4a). The micrographs contained vesicles with diameter ranging from 400 nm to smaller than 50 nm. Remarkably, electron micrographs of cultured astrocytes also showed MVBs containing vesicles of variable size (Fig. 4b). Pellets from ultracentrifugation and chromatography fractions from CM were also examined through electron microscopy. There was a predominance of vesicles sized in the range of 200–400 nm in fractions 1–2 and after 1 h of centrifugation (Fig. 4c, f, respectively). In chromatography fractions 5–6 and after 2 h of centrifugation (Fig. 4d, g, respectively), the majority of vesicles were sized in range of 100–200 nm and in fractions 11–12 and after 16 h of centrifugation (Fig. 4e, h, respectively) most of vesicles sized smaller than 100 nm. Immunolabeling showed STI1 in all three types of vesicles (Fig. 4i–k). Fractions 16–17 contained no structures distinguishable by electron microscopy (data not shown).

Astrocytes secrete three distinct populations of vesicles that contain STI1. a Electron micrographs of a cultured astrocyte. Inset higher magnification of a MVB. b Electron micrographs of CM. c–e CM was fractionated through gel filtration chromatography and fractions 1–2 (c), 5–6 (d), and 11–12 (e) were examined by electron microscopy. CM was also fractioned by 1-h (f), 2-h (g), or 16-h (h) ultracentrifugation and pellets were examined by electron microscopy. i–k CM was immunolabeled with rabbit anti-STI1 followed by protein A coupled to gold particles observed by transmission electron microscopy
The data thus indicate that astrocytes release three types of EVs, of which only the medium-sized subpopulation may correspond to the classically defined exosomes, as observed by the characteristic size and cup-shaped form in electron microscopy. All three types of EVs carry STI1.
Evidence that EVs containing STI1 originate from multivesicular bodies
To identify the nature of astrocyte-secreted EVs containing STI1, it was tested whether STI1 was present in lipoprotein particles using an isopycnic density centrifugation [48]. Under these conditions, lipoprotein particles move to the top low-density fraction, whereas other particles remain in higher-density fractions. Apolipoprotein E, mainly produced and secreted by astrocytes as part of lipoprotein particles, was found entirely in the low-density fraction [53, 54], while STI1 was found in high-density fractions (Fig. 5a). These results indicate that sedimentable STI1 secreted by astrocytes is not associated with lipoproteins.

EVs from astrocytes are originated from MVBs. a Twelve fractions of isopycnic density centrifugation of CM were collected, separated by SDS-PAGE, and immunoblotted for STI1 (upper panel) and ApoE (lower panel). Densitometry of the bands is shown on the graph along with a density plot of the entire gradient. b After gel filtration chromatography of fractionated CM fractions 1–2, 5–6, 11–12, and 16–17 were immunoblotted for transferrin receptor (TfR), HSP90, HSP70, STI1, flotillin, Tgs101, Vps36, PrPC, and caveolin. c Astrocytes were co-transfected with STI1-GFP and mCherry Rab5 or Rab7 and examined by TIRF microscopy. Scale bar 20 μm. d Astrocytes were co-transfected with STI1–CFP and wild-type VPS4A-EGFP (VPS4–WT–EGFP) or its dominant-negative mutant E228Q (VPS4–Dn–eGFP), and examined by confocal microscopy. e Total number of vesicles in CM was counted by nanoparticle tracking and f levels of STI1 released were measured by ELISA in CM from astrocytes transfected with wild-type pEGFP–VPS4A (VPS4 WT) or its dominant-negative form pEGFP–VPS4A-E228Q (VPS4DN). Data represent mean ± SD of three independent experiments in triplicate. * p < 0.05. g Astrocyte cultures immunostained for VPS4A (red) and STI1 (green) were analyzed by ground-state depletion and individual molecule return (GSDIM) microscopy. Scale bars are 7.5 μm for wide-field and 2.5 μm for GSD super-resolution images
We then examined whether astrocyte EVs isolated using gel chromatography fractions carry markers of endocytic pathways or MVBs (Fig. 5b). Immunoblotting assays showed that fractions 1–2 contained TfR, HSP90, STI1, and the lipid-raft marker, flotillin. These fractions also contained VPS36 and low levels of Tsg101, two components of the ESCRT-I complex that are required for MVB formation and sorting of endosomal cargo proteins [55]. Fractions 5–6 contained all the markers mentioned above and also PrPC. In cells infected with murine leukemia virus, EVs secretion is increased and PrPC can be secreted in association with virus-like particles (VLP), which can co-purify with EVs [56]. Since our primary cultures are free of viral particles, we believe this is not the case in our system. It is worth mentioning that PrPC is detected only in these fractions and may represent the best maker for the classically defined exosome. Fractions 11–12 contained STI1, HSP70, and HSP90, the TfR, flotillin, and Tsg101 but had lower levels of VPS36 and caveolin. Fractions 16–17 contained only STI1, HSP70, and HSP90, and as noted above (Fig. 3), likely correspond to non-vesicular fractions of these released proteins (Fig. 5b).
To further test whether STI1 is carried along the endocytic pathway that contributes to MVBs formation, astrocytes were co-transfected with eGFP–STI1 and Rab5 or Rab7 fused to mCherry (Rab5–mCherry and Rab7–mCherry) as markers for early and late endosomes, respectively. Using TIRF microscopy, we observed several STI1-containing endosomal vesicles (denoted by the co-localization with the Rab proteins) that move along the cell, suggesting that STI1 can traffic along the endosomal system (Fig. 5c and Supplementary movies 1, 2). Astrocytes were also co-transfected with CFP–STI1 and the protein VPS4A–GFP, an AAA-ATPase essential for MVBs biogenesis [57] or with its dominant-negative mutant E228Q deficient for ATP hydrolysis [58]. Confocal microscopy showed that in cells transfected with VPS4A, STI1 displayed a punctate pattern with partial co-localization with VPS4A. In cells transfected with the dominant-negative form of VPS4, STI1 was recruited to enlarged endosomes (Fig. 5d). Remarkably, the expression of this mutant VPS4A reduced the release of EVs in CM by 85 % (Fig. 5e), while STI1 release decreased by 68 % (Fig. 5f). Transfection efficiency in these cultures was around 60 % (data not shown), suggesting that more efficient inhibition of EVs and STI1 could be achieved if all cells expressed the dominant negative VPS4A.
Using GSDIM microscopy (Fig. 5g), we confirmed the punctate pattern of endogenous STI1 in untransfected cells, which is normally masked by high levels of cytosolic protein. Interestingly, at super-resolution, most STI1 puncta were in very close proximity to VPS4A, suggesting that a fraction of this protein is present in VPS4A-containing organelles (Fig. 5g).
STI is found at the surface of EVs
Flow cytometry assays were employed to assess the localization of STI1 in EVs. Isolated EVs from CM centrifuged for 2 h at 100,000 × g were coupled to aldehyde-sulphate latex beads [59]. The expression of surface molecules was analyzed by flow-cytometry of the EV-bead complexes (Fig. 6a). Positive staining for STI1, as well as for PrPC and flotillin (surface markers), indicated that at least part of the STI1 was present at the outer leaflet of EVs. However, Hsp90 was not detected at the surface of the vesicles. To confirm the presence of STI1 at the surface of EVs, intact EVs exposed to PK digestion (Fig. 6b) show a 50 % reduction of STI1 expression when compared to non-digested samples. As a control, when EVs were lysed by detergent addition, no STI1 was recovered (Fig. 6b).

STI1 was detected at the surface of EVs. a Isolated EVs were immobilized onto aldehyde-sulphate latex beads, incubated with anti-STI1, anti-flotillin, anti-PrPC, or anti-HSP90 antibodies followed by anti-mouse or anti-rabbit IgG-Alexa488, and examined by flow cytometry. Filled histograms show EVs-bead complexes stained with isotype-matched antibody; unfilled histogram shows relevant antibody staining. b Isolated EVs were incubated with protease K (PK) or protease K plus Triton X-100 for 10 min. Samples were subjected to Western blot with anti-STI and quantification was performed by densitometry of the bands (n = 2)
STI1-containing EVs derived from astrocytes bind to neurons
To address whether astrocyte-derived EVs containing STI1 may be taken up and utilized by neurons, astrocytes were transfected with either GFP–STI1 or GPF (control) expression vectors, and CM was fractionated by sequential ultracentrifugation. GFP–STI1 was found in both the pellet and supernatant of the CM, whereas GFP alone was not detected in CM (Fig. 7a). EVs obtained from CM ultracentrifuged for 2 h were added to primary neurons. EVs containing GFP–STI1 formed puncta on neurons, whereas the supernatant did not (Fig. 7b). As expected, neither the pellet nor the supernatant of the CM from GFP-transfected astrocytes formed puncta (Fig. 7b). Similar results were obtained with the neuronal cell line SN-56 (Supplementary Fig. 2). These data demonstrate that EVs containing STI1 interact with the neuronal cell surface.

EV-associated GFP–STI1 binds to neurons. Astrocyte cultures were transfected with either GFP or GFP–STI1 and the conditioned medium was ultracentrifuged for 2 h. a Pellets and supernatants were separated by SDS-PAGE followed by immunoblot for GFP or STI1. b Pellets and supernatant from ultracentrifugation of CM from astrocytes transfected with GFP or GFP–STI1 were added to primary hippocampal neurons and cells were live imaged. Scale bar 10 μm
STI1 present in EVs induces ERK1/2 activation
Our previous studies demonstrated that recombinant STI1 induces PrPC-dependent ERK1/2 activation, which promotes neuronal differentiation [16]. Fractions obtained by ultracentrifugation were tested for their ability to induce ERK1/2 activation in neurons. All the vesicular fractions were able to stimulate ERK1/2 in wild-type neurons whereas the supernatant was not. However, when PrPC knockout neurons were treated with the EV fractions, no ERK1/2 activation was observed, demonstrating the importance of PrPC on the neuronal surface (Fig. 8a).

Astrocyte CM and EVs containing STI1 activate PrPC-dependent ERK 1/2 signaling in neurons. a CM was fractioned through ultracentrifugation and 1-, 2-, or 16-h pellets or supernatant (s) were used to treat primary cultured wild-type (Prnp +/+) or PrPC knockout neurons (Prnp 0/0) for 30 s. b CM was fractionated through gel filtration chromatography and fractions 1–2, 5–6, 11–12, and 16–17 were used to treat neurons for 30 s. c CM was fractionated through gel filtration chromatography and fractions 1–2 and 16–17 were used to treat neurons for 30 s. Alternatively, fraction 1–2 was pre-treated with anti-STI1 antibodies (α-STI1) or with the peptide \({\text{PrP}}_{{ 1 0 6 { - 126}}}^{\text{C}} \; ( {\text{P10)}}\), before addition to the cells. Cells were also treated with three times more material from fractions 16–17 (3 × 16–17). After treatment, neurons were lysed and their extracts were immunoblotted for phospho-ERK1/2 and ERK1/2. Phospho-ERK/total ERK ratios were calculated from densitometric measurements. The control was set as one and values represent relative levels. *p < 0.05, One-way ANOVA followed by Tukey’s post hoc test
Fractions obtained by size exclusion chromatography were also tested for their ability to induce ERK1/2 activation in neurons. Fractions 1–2, 5–6, and 11–12 containing 1.5 nM of STI1 induced activation of ERK1/2 (Fig. 8a, b). This effect was inhibited when EVs derived from fractions 1–2 were pre-incubated either with a specific antibody against STI1 (α-STI1) or with a PrPC peptide (p10), that mimics the STI1 binding site (PrPC aas 113–128), and blocks STI1 interaction with PrPC [12] (Fig. 8b).
In contrast to the effect of all three EV fractions, ERK1/2 was not activated by fractions 16–17 containing 1.5 nM of STI1 (Fig. 8a, b). However, activation of ERK1/2 was observed when neurons were treated with concentrated fractions 16–17 containing 4.5 nM of STI1 (Fig. 8b). Together, these results suggest that the insertion of STI1 in astrocyte-derived EVs potentiates its activity upon PrPC-dependent ERK1/2 signaling in neurons.
Discussion
Stress-inducible protein 1 is released by astrocytes and binds with high affinity to the PrPC in neurons [9, 12, 43]. This interaction leads to signaling by calcium influx through alfa-7 nicotinic acetylcholine receptors, activation of PKA, ERK1/2 [16, 17], and PI3K [15], thus modulating neuronal survival and differentiation in vitro and memory formation [60].
Our current results show that the release of STI1 from astrocytes occurs through a mechanism independent of classical secretion via the Golgi network. Most of the released STI1 was associated with three types of EVs of distinct sizes, one of which had the typical morphology of classical exosomes. Nonetheless, proteins previously identified as exosomal markers were detected in all three EV types, which appeared to be derived from MVBs. The released vesicles carried at least part of the STI1 in their outer leaflet, bound to neuronal surfaces, and activated PrPC-dependent specific signals with a higher activity than soluble STI1.
Hsps have major chaperone roles in proteostasis [61], and despite their intracellular abundance, can also be released to the extracellular space and play a role in cell-to-cell communication [38] among immune, neuronal, cardiac, and cancer cells [37, 52, 62–65]. Hsps were reportedly transferred from glial cells to neuronal axons [66] by unconventional secretion [67]. Similarly to most Hsps, the co-chaperone STI1 lacks consensus signal peptides for ER-Golgi classical secretion, consistent with our findings that neither BFA nor Mn blocked the release of STI1. This corroborates a previous report that identified STI1 in a proteomics screening for non-conventional protein secretion [68].
Unconventional secretion has been attributed to at least two distinct types of EVs: budding vesicles shed from plasma membrane, and exosomes derived from exocytosis of MVBs [33, 69]. A large variety of protocols have been used to isolate EVs, and their nomenclature is confusing. For example, exosomal preparations have been described using ultracentrifugation at 100,000 × g for periods that vary from 1 to 16 h [20, 70–74]. Here, we found that pelleted material from various ultracentrifugation times contained exosomal makers, such as Hsp70, Hsp90 [27, 75], as well as STI1. Nonetheless, both nanoparticle tracking and electron microscopy were consistent with a heterogeneous set of vesicles.
To gain insight into the composition of the various EVs, we developed a protocol using size exclusion chromatography that allowed the isolation of four different fractions containing STI1, three of which were further characterized as EVs by their size, lipid composition, and morphology by electron microscopy. The three distinct fractions contained, respectively, vesicles between 200 and 400 nm, intermediate-sized vesicles that fit the classical description of exosomes, or small vesicles between 20 and 50 nm. All three fractions contained exosomal markers [27, 75], except PrPC, which was only detected in fractions containing typical exosomes as previously described [35]. These data show that astrocytes release a heterogeneous population of EVs including exosomes. Examination by TIRF and confocal microscopy showed that STI1 partially co-localized with Rab7, Rab5, and VPS4A, which are respectively associated with MVB formation and genesis of exosomes [27, 75]. These experiments were mostly done with overexpressed proteins, because a large amount of endogenous STI1 is present in the cytoplasm and making it difficult to localize endogenous protein with MVB markers. Nonetheless, by using super-resolution microscopy, we were able to observe endogenous STI1 present in puncta in close proximity to VPS4A.
The expression of a dominant-negative isoform of VPS4A inhibited both the release of STI1 and overall EV secretion, further suggesting an endocytic and MVB origin for the STI1-containing vesicles. Thus, our data is consistent with the hypothesis that a variety of EVs from 20 to 400 nm, which contain STI1, are derived from endocytic compartments, in particular from MVBs, and the classical exosome comprises one type of such vesicles. The origin of the soluble fraction of STI1 remains undetermined. It is possible that intravesicular STI1 is released in a soluble form upon EV rupture, similar to a mechanism proposed for HSP70 and HSP90 [76].
In addition to the expected intra-vesicular content, STI1 was detected at the surface of EVs. The inability to remove STI1 from vesicles using high salt concentrations, and the lack of binding of recombinant STI1 to EVs, agrees with previous findings of STI1 in the cell membrane membranes [77] and its co-immunoprecipitation with PrPC [12, 16]. This suggests a specific mechanism of STI1 transport to the EV surface. Interestingly, chaperones such as Hsp72 and Hsp60, have been reported at the cell membrane or exosome surface, in particular those from tumor cells [22, 64, 76]. Hsp70 can also be inserted in the lipid bilayer [78–81] and balance membrane fluidity upon heat-shock [82]. Further work is necessary to ascertain the mechanisms involved in the exposure of STI1 at the surface of astrocyte-derived EVs.
Notably, our data demonstrated that STI1 association with vesicles enhanced its effects upon PrPC-dependent activation of ERK1/2 in neurons. Indeed, to activate ERK1/2, EV-associated STI1 was effective at concentrations three times lower than those required for STI1 in the soluble fraction (Fig. 8). Remarkably, the concentration of recombinant STI1 necessary to activate ERK1/2 is 100 times higher than that present in EVs [16, 43]. It is possible that the binding affinity of PrPC to EV-associated STI1 is increased either by conformational changes of the lipid-associated co-chaperone, or by fusion between the EV and the target cell membranes, which may change local lipid content around the STI1-PrPC complex. In addition, PrPC is believed to scaffold multiprotein signaling complexes at the cell surface [83], thus its binding to EV-associated STI1 may bring together additional interacting partners from both EVs and neuronal membrane, and potentiate cell signaling through allosteric interactions [84]. Regardless of the mechanism, the enhanced effect of EV-associated STI1 upon cell signaling is consistent with analogous findings for the macrophage-activating activity of extracellular HSP70 [82].
In conclusion, the current data on the release of STI1 from astrocytes unraveled a heterogeneous population of MVB-derived EVs that may have been ignored in previous studies for the lack of proper isolation methods. The various EV types may, nonetheless, have distinct properties, which deserve further examination. Finally, due to the roles of STI1 in neuroprotection and tumor cell proliferation, the control of the release of EV-associated STI1 may be particularly relevant in the context of acute brain injury, neurodegenerative diseases and cancer.
References
Blatch GL, Lassle M, Zetter BR, Kundra V (1997) Isolation of a mouse cDNA encoding mSTI1, a stress-inducible protein containing the TPR motif. Gene 194:277–282 (pii: S0378-1119(97)00206-0)
Lassle M, Blatch GL, Kundra V, Takatori T, Zetter BR (1997) Stress-inducible, murine protein mSTI1. Characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J Biol Chem 272:1876–1884
Lee CT, Graf C, Mayer FJ, Richter SM, Mayer MP (2012) Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J 31:1518–1528. doi:10.1038/emboj.2012.37
Schmid AB, Lagleder S, Grawert MA, Rohl A, Hagn F, Wandinger SK, Cox MB, Demmer O, Richter K, Groll M, Kessler H, Buchner J (2012) The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J 31:1506–1517. doi:10.1038/emboj.2011.472
Chang HC, Nathan DF, Lindquist S (1997) In vivo analysis of the Hsp90 cochaperone Sti1 (p60). Mol Cell Biol 17:318–325
Song HO, Lee W, An K, Lee HS, Cho JH, Park ZY, Ahnn J (2009) C. elegans STI-1, the homolog of Sti1/Hop, is involved in aging and stress response. J Mol Biol 390:604–617. doi:10.1016/j.jmb.2009.05.035
Hajj GN, Santos TG, Cook ZS, Martins VR (2009) Developmental expression of prion protein and its ligands stress-inducible protein 1 and vitronectin. J Comp Neurol 517:371–384. doi:10.1002/cne.22157
Eustace BK, Jay DG (2004) Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 3:1098–1100 (pii: 1088)
Lima FR, Arantes CP, Muras AG, Nomizo R, Brentani RR, Martins VR (2007) Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J Neurochem 103:2164–2176. doi:10.1111/j.1471-4159.2007.04904.x
Erlich RB, Kahn SA, Lima FR, Muras AG, Martins RA, Linden R, Chiarini LB, Martins VR, Moura NV (2007) STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 55:1690–1698
Wang TH, Chao A, Tsai CL, Chang CL, Chen SH, Lee YS, Chen JK, Lin YJ, Chang PY, Wang CJ, Chao AS, Chang SD, Chang TC, Lai CH, Wang HS (2010) Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol Cell Proteomics 9:1873–1884. doi:10.1074/mcp.M110.000802
Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de OE, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 21:3307–3316. doi:10.1093/emboj/cdf325
Martins VR, Graner E, Garcia-Abreu J, de Souza SJ, Mercadante AF, Veiga SS, Zanata SM, Neto VM, Brentani RR (1997) Complementary hydropathy identifies a cellular prion protein receptor. Nat Med 3:1376–1382
Hajj GN, Santos TG, Landenberger MC, Lopes MH (2012) Transmissible spongiform encephalopathies. In: Quevedo A (ed) Brain damage—bridging between basic research and clinics. InTechOpen, Rijeka, pp 197–220
Roffe M, Beraldo FH, Bester R, Nunziante M, Bach C, Mancini G, Gilch S, Vorberg I, Castilho BA, Martins VR, Hajj GN (2010) Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc Natl Acad Sci USA 107:13147–13152. doi:10.1073/pnas.1000784107
Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR (2005) Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci 25:11330–11339. doi:10.1523/JNEUROSCI.2313-05.2005
Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA, Martins VR (2010) Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem 285:36542–36550. doi:10.1074/jbc.M110.157263
Arantes C, Nomizo R, Lopes MH, Hajj GN, Lima FR, Martins VR (2009) Prion protein and its ligand stress-inducible protein 1 regulate astrocyte development. Glia 57(13):1439–1449
Tsai CL, Tsai CN, Lin CY, Chen HW, Lee YS, Chao A, Wang TH, Wang HS, Lai CH (2012) Secreted stress-induced phosphoprotein 1 activates the ALK2-SMAD signaling pathways and promotes cell proliferation of ovarian cancer cells. Cell Rep 2:283–293. doi:10.1016/j.celrep.2012.07.002
Lancaster GI, Febbraio MA (2005) Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 280:23349–23355. doi:10.1074/jbc.M502017200
Evdokimovskaya Y, Skarga Y, Vrublevskaya V, Morenkov O (2010) Secretion of the heat shock proteins HSP70 and HSC70 by baby hamster kidney (BHK-21) cells. Cell Biol Int 34:985–990. doi:10.1042/CBI20100147
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De TA, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rebe C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120:457–471. doi:10.1172/JCI40483
McCready J, Sims JD, Chan D, Jay DG (2010) Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10:294. doi:10.1186/1471-2407-10-294
Thery C, Zitvogel L, Amigorena S (2002) Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:569–579. doi:10.1038/nri855
Fevrier B, Raposo G (2004) Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 16:415–421. doi:10.1016/j.ceb.2004.06.003
Simons M, Raposo G (2009) Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 21:575–581. doi:10.1016/j.ceb.2009.03.007
Thery C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9:581–593. doi:10.1038/nri2567
Piccin A, Murphy WG, Smith OP (2007) Circulating microparticles: pathophysiology and clinical implications. Blood Rev 21:157–171. doi:10.1016/j.blre.2006.09.001
Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C (2005) Exosomal-like vesicles are present in human blood plasma. Int Immunol 17:879–887. doi:10.1093/intimm/dxh267
Toth B, Lok CA, Boing A, Diamant M, van der Post JA, Friese K, Nieuwland R (2007) Microparticles and exosomes: impact on normal and complicated pregnancy. Am J Reprod Immunol 58:389–402. doi:10.1111/j.1600-0897.2007.00532.x
Keller S, Sanderson MP, Stoeck A, Altevogt P (2006) Exosomes: from biogenesis and secretion to biological function. Immunol Lett 107:102–108. doi:10.1016/j.imlet.2006.09.005
Cocucci E, Racchetti G, Rupnik M, Meldolesi J (2008) The regulated exocytosis of enlargeosomes is mediated by a SNARE machinery that includes VAMP4. J Cell Sci 121:2983–2991. doi:10.1242/jcs.032029
Cocucci E, Racchetti G, Meldolesi J (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51. doi:10.1016/j.tcb.2008.11.003
Vella LJ, Sharples RA, Lawson VA, Masters CL, Cappai R, Hill AF (2007) Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J Pathol 211:582–590. doi:10.1002/path.2145
Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci USA 101:9683–9688. doi:10.1073/pnas.0308413101
Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, Leblanc P (2008) Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 100:603–615. doi:10.1042/BC20080025
Tytell M (2005) Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int J Hyperth 21:445–455. doi:10.1080/02656730500041921
Calderwood SK, Mambula SS, Gray PJ Jr, Theriault JR (2007) Extracellular heat shock proteins in cell signaling. FEBS Lett 581:3689–3694. doi:10.1016/j.febslet.2007.04.044
Sherman M, Multhoff G (2007) Heat shock proteins in cancer. Ann NY Acad Sci 1113:192–201. doi:10.1196/annals.1391.030
Joly AL, Wettstein G, Mignot G, Ghiringhelli F, Garrido C (2010) Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J Innate Immun 2:238–247. doi:10.1159/000296508
Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R (2002) Cellular prion protein transduces neuroprotective signals. EMBO J 21:3317–3326
Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356:577–582
Caetano FA, Lopes MH, Hajj GN, Machado CF, Pinto AC, Magalhaes AC, Vieira MP, Americo TA, Massensini AR, Priola SA, Vorberg I, Gomez MV, Linden R, Prado VF, Martins VR, Prado MA (2008) Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J Neurosci 28:6691–6702. doi:10.1523/JNEUROSCI.1701-08.2008
Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65 (pii: S0092-8674(01)00506-2)
Lee HJ, Hammond DN, Large TH, Wainer BH (1990) Immortalized young adult neurons from the septal region: generation and characterization. Brain Res Dev Brain Res 52:219–228
Lee KS, Magalhaes AC, Zanata SM, Brentani RR, Martins VR, Prado MA (2001) Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J Neurochem 79:79–87
Sydor AM, Su AL, Wang FS, Xu A, Jay DG (1996) Talin and vinculin play distinct roles in filopodial motility in the neuronal growth cone. J Cell Biol 134:1197–1207
Panakova D, Sprong H, Marois E, Thiele C, Eaton S (2005) Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 435:58–65. doi:10.1038/nature03504
Fernandez-Higuero JA, Salvador AM, Arrondo JL, Milicua JC (2011) Low-density lipoprotein density determination by electric conductivity. Anal Biochem 417:283–285. doi:10.1016/j.ab.2011.06.004
Liu D, Bryceson YT, Meckel T, Vasiliver-Shamis G, Dustin ML, Long EO (2009) Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity 31:99–109. doi:10.1016/j.immuni.2009.05.009
Safaei R, Larson BJ, Cheng TC, Gibson MA, Otani S, Naerdemann W, Howell SB (2005) Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol Cancer Ther 4:1595–1604. doi:10.1158/1535-7163.MCT-05-0102
Mambula SS, Stevenson MA, Ogawa K, Calderwood SK (2007) Mechanisms for Hsp70 secretion: crossing membranes without a leader. Methods 43:168–175. doi:10.1016/j.ymeth.2007.06.009
Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, Getz GS, Reardon CA, Lukens J, Shah JA, LaDu MJ (1999) Unique lipoproteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem 274:30001–30007
Lu H, Daugherty A (2009) Atherosclerosis: cell biology and lipoproteins. Curr Opin Lipidol 20:528–529. doi:10.1097/MOL.0b013e328332c3bc;00041433-200912000-00014
Henne WM, Buchkovich NJ, Emr SD (2011) The ESCRT pathway. Dev Cell 21:77–91. doi:10.1016/j.devcel.2011.05.015
Leblanc P, Alais S, Porto-Carreiro I, Lehmann S, Grassi J, Raposo G, Darlix JL (2006) Retrovirus infection strongly enhances scrapie infectivity release in cell culture. EMBO J 25:2674–2685. doi:10.1038/sj.emboj.7601162
Babst M, Davies BA, Katzmann DJ (2011) Regulation of Vps4 during MVB sorting and cytokinesis. Traffic 12:1298–1305. doi:10.1111/j.1600-0854.2011.01230.x
Bishop N, Woodman P (2000) ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol Biol Cell 11:227–239
Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, Le Pecq JB (2002) Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods 270:211–226 (pii: S0022175902003307)
Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MM, Linden R (2010) Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol 12:63–86 (pii: v12/63)
Preissler S, Deuerling E (2012) Ribosome-associated chaperones as key players in proteostasis. Trends Biochem Sci 37:274–283. doi:10.1016/j.tibs.2012.03.002
Henderson B (2010) Integrating the cell stress response: a new view of molecular chaperones as immunological and physiological homeostatic regulators. Cell Biochem Funct 28:1–14. doi:10.1002/cbf.1609
Luo X, Zuo X, Zhang B, Song L, Wei X, Zhou Y, Xiao X (2008) Release of heat shock protein 70 and the effects of extracellular heat shock protein 70 on the production of IL-10 in fibroblast-like synoviocytes. Cell Stress Chaperones 13:365–373. doi:10.1007/s12192-008-0036-2
Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina BP, Meldolesi J (2009) The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem 110:284–294. doi:10.1111/j.1471-4159.2009.06130.x
Woodley DT, Fan J, Cheng CF, Li Y, Chen M, Bu G, Li W (2009) Participation of the lipoprotein receptor LRP1 in hypoxia-HSP90alpha autocrine signaling to promote keratinocyte migration. J Cell Sci 122:1495–1498. doi:10.1242/jcs.047894
Tytell M, Greenberg SG, Lasek RJ (1986) Heat shock-like protein is transferred from glia to axon. Brain Res 363:161–164 (pii: 0006-8993(86)90671-2)
Hightower LE, Guidon PT Jr (1989) Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J Cell Physiol 138:257–266. doi:10.1002/jcp.1041380206
Keller M, Ruegg A, Werner S, Beer HD (2008) Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818–831. doi:10.1016/j.cell.2007.12.040
Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, Raposo G, Amigorena S (1999) Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol 147:599–610
Savina A, Vidal M, Colombo MI (2002) The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci 115:2505–2515
Keller S, Rupp C, Stoeck A, Runz S, Fogel M, Lugert S, Hager HD, Abdel-Bakky MS, Gutwein P, Altevogt P (2007) CD24 is a marker of exosomes secreted into urine and amniotic fluid. Kidney Int 72:1095–1102. doi:10.1038/sj.ki.5002486
Gauvreau ME, Cote MH, Bourgeois-Daigneault MC, Rivard LD, Xiu F, Brunet A, Shaw A, Steimle V, Thibodeau J (2009) Sorting of MHC class II molecules into exosomes through a ubiquitin-independent pathway. Traffic 10:1518–1527. doi:10.1111/j.1600-0854.2009.00948.x
Savina A, Furlan M, Vidal M, Colombo MI (2003) Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem 278:20083–20090. doi:10.1074/jbc.M301642200;M301642200
Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. (Chapter 3, Unit 3.22). doi: 10.1002/0471143030.cb0322s30
Stoorvogel W, Kleijmeer MJ, Geuze HJ, Raposo G (2002) The biogenesis and functions of exosomes. Traffic 3:321–330
De MA (2011) Extracellular heat shock proteins, cellular export vesicles, and the stress observation system: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones 16:235–249. doi:10.1007/s12192-010-0236-4
Faca VM, Ventura AP, Fitzgibbon MP, Pereira-Faca SR, Pitteri SJ, Green AE, Ireton RC, Zhang Q, Wang H, O’Briant KC, Drescher CW, Schummer M, McIntosh MW, Knudsen BS, Hanash SM (2008) Proteomic analysis of ovarian cancer cells reveals dynamic processes of protein secretion and shedding of extra-cellular domains. PLoS ONE 3:e2425. doi:10.1371/journal.pone.0002425
Arispe N, Doh M, Simakova O, Kurganov B, De MA (2004) Hsc70 and Hsp70 interact with phosphatidylserine on the surface of PC12 cells resulting in a decrease of viability. FASEB J 18:1636–1645. doi:10.1096/fj.04-2088com
Horvath I, Multhoff G, Sonnleitner A, Vigh L (2008) Membrane-associated stress proteins: more than simply chaperones. Biochim Biophys Acta 1778:1653–1664. doi:10.1016/j.bbamem.2008.02.012
Gehrmann M, Liebisch G, Schmitz G, Anderson R, Steinem C, De MA, Pockley G, Multhoff G (2008) Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 3:e1925. doi:10.1371/journal.pone.0001925
Sugawara S, Kawano T, Omoto T, Hosono M, Tatsuta T, Nitta K (2009) Binding of Silurus asotus lectin to Gb3 on Raji cells causes disappearance of membrane-bound form of HSP70. Biochim Biophys Acta 1790:101–109. doi:10.1016/j.bbagen.2008.10.005
Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, Multhoff G, Arispe N, De MA (2008) Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol 180:4299–4307 (pii: 180/6/4299)
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiology of the prion protein. Physiol Rev 88:673–728
Linden R, Cordeiro Y, Lima LM (2012) Allosteric function and dysfunction of the prion protein. Cell Mol Life Sci 69:1105–1124. doi:10.1007/s00018-011-0847-7
Acknowledgments
We are very grateful to Dr. Wes Sundquist for the donation of VPS4A plasmids. We thank Leica Microsystems and Ms. Lianne Dale for help in experiments done in a demo Leica GSD unit. This investigation was supported by grants from FAPESP to VRM (2009/14027-2) and GNMH (2012/04370-4), PrioNet-Canada, Canadian Institutes of Health Research (CIHR), Canadian Foundation for Innovation and Ontario Research Fund to MAMP, CNPq and FAPERJ to RL, FAPESP fellowships to CA, MVSD, MR and BS, and a postdoctoral CNPq fellowship to IPC are gratefully acknowledged.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
G. N. M. Hajj and C. P. Arantes contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2013_1328_MOESM1_ESM.tif
Supplementary Fig. 1 CM collected either from serum free (0 %) astrocytes or from astrocytes cultured in 10 % serum were applied to gel filtration chromatography columns and fractions 1, 2, 5, 6, 11, and 12 were subjected to immunoblotting for STI1 (TIFF 121 kb)
18_2013_1328_MOESM2_ESM.tif
Supplementary Fig. 2 a Astrocyte cultures were transfected with GFP and CM was ultracentrifuged for 2 h. Pellets and supernatant were added to primary hippocampal neurons SN-56 cells. Scale bar 10 μm (TIFF 2555 kb)
18_2013_1328_MOESM3_ESM.mov
Supplementary movie 1 STI1-GFP was co-transfected in astrocytes with mCherry Rab7 and live imaged in a TIRF microscope. Total time 3 min (MOV 8907 kb)
18_2013_1328_MOESM4_ESM.avi
Supplementary movie 2 STI1-GFP was co-transfected in astrocytes with mCherry Rab5 and live imaged in a TIRF microscope. Total time 5 min (AVI 4065 kb)
Rights and permissions
About this article
Cite this article
Hajj, G.N.M., Arantes, C.P., Dias, M.V.S. et al. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci. 70, 3211–3227 (2013). https://doi.org/10.1007/s00018-013-1328-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1328-y