
Abstract
Transmissible spongiform encephalopathies (TSEs) are neurodegenerative diseases associated with progressive oligo- and multimerization of the prion protein (PrPC), its conformational conversion, aggregation and precipitation. We recently proposed that PrPC serves as a cell surface scaffold protein for a variety of signaling modules, the effects of which translate into wide-range functional consequences. Here we review evidence for allosteric functions of PrPC, which constitute a common property of scaffold proteins. The available data suggest that allosteric effects among PrPC and its partners are involved in the assembly of multi-component signaling modules at the cell surface, impose upon both physiological and pathological conformational responses of PrPC, and that allosteric dysfunction of PrPC has the potential to entail progressive signal corruption. These properties may be germane both to physiological roles of PrPC, as well as to the pathogenesis of the TSEs and other degenerative/non-communicable diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prion protein (PrPC) was discovered and characterized amid the search for the infectious pathogen involved in the transmissible spongiform encephalopathies (TSEs), a family of severe, still incurable and invariably fatal neurodegenerative diseases [1, 2]. Studies of the pathogenesis of TSEs pointed to the central role of an abnormal conformer of PrPC because of changes in the secondary structure of this protein [1, 3]. The infectious pathogen was designated prion, an acronym for proteinaceous infectious only, after which PrPC was named, and that also led to the alternative term ‘prion diseases’ to denominate TSEs [4].
The cognitive and motor signs and symptoms of the TSEs are attributed to progressive neurodegeneration, associated with oligo- and multimerization, aggregation, occasional amyloid fibrillization, and precipitation of the abnormal protein conformer [5, 6]. In line with present trends in Alzheimer’s disease [7, 8], current thinking favors the hypothesis that TSEs are caused by protein oligomers within a certain range of sizes, on their way to eventually precipitate as the relatively large, compact deposits detectable by light microscopical examination of autopsy specimens [5, 9, 10].
Clinical assays of new treatments for prion diseases have been so far designed on the basis of robust therapeutic effects in prion-infected cell cultures, mostly related to either the prevention or disassembly of aggregates of the abnormal conformer of PrPC. Heretofore the results have been disappointing [11–13]. A significant hurdle is the absence of reliable pathognomonic markers of the TSEs in living patients [14]. Postmortem confirmation of diagnosis is required, through the observation of end-stage spongiosis and cell death in brain tissue, together with immunohistochemical detection of compact, insoluble and protease-resistant aggregates of the abnormal conformer of PrPC [15]. Notably, neuron death, the immunohistopathological marker, and the neurodegenerative disease proper often do not fit together in autopsy specimens [16–18].
An additional, recurrent and frequently underappreciated issue in experimental approaches is the variability of the kinetics of formation of the aggregated, presumably toxic forms of the prion protein in vitro, and the uncertain correlation between those and the actual rates of building up their counterparts in human TSEs, or even in experimental animal models [19–26]. Survival of TSE patients is of the order of several months after diagnosis, varies among distinct forms of TSE [27, 28], and the events required to produce diagnostic symptoms are still unclear. Also, the time courses of experimental approaches may differ from those of the corresponding TSEs by several orders of magnitude. It is, therefore, still difficult to equate human prion diseases with the corresponding animal models, not to mention the responses of cell lines to prions in vitro.
Despite significant advances in the field, fundamental questions remain unanswered [3, 29–33]. Unknown variables include the mechanisms of conformational conversion of PrPC into prions; the events that cause neuronal dysfunction and degeneration; the signal transfer molecules that transduce the pathogenic signals derived from the progressive aggregation of toxic species; the roles of either dysfunction or death of neurons and glia in the diverse symptoms associated with TSEs; the differential topography of brain lesions associated with distinct TSEs; why specific mutations in the prion gene (PRNP) lead to conformational conversion; why specific mutations associate with distinct TSEs; the determinants of the so-called prion strains; the role, if any, of nucleic acids in the composition of prions; and last, but not least, the functional properties of the normal prion protein, together with the role of loss of its function(s) in the pathogenesis of the TSEs.
We have proposed that the prion protein functions as a cell surface scaffold for the assembly of signaling modules, based on which selective interactions with many ligands and transmembrane signaling pathways translate into wide-range consequences upon both physiology and behavior [34]. The assembly of molecular complexes both entails and depends on orchestrated allosteric structural changes propagated in the various components of the complex [35, 36], and scaffold proteins are proposed to act as allosteric effectors on their partners. Thus, they do not only allow the proximity between ligands, but may also impose on the conformation of those binding molecules, thereby affecting their activity [37]. This review attempts to provide a framework for the study of such allosteric properties of PrPC, which may be germane both to the roles of the prion protein in physiological context, as well as to the pathogenesis of the TSEs.
Structure, topology and trafficking of the prion protein
Mammalian prion protein (Fig. 1) is found at the cell surface as a glycosylated, GPI-anchored protein of 208–209 amino acids. PrPC contains an N-terminal flexible, random coil sequence comprising approximately residues 23–124, which is flexible and disordered in solution [38, 39], and a C-terminal globular domain of about 100 amino acids. The latter are arranged in three α-helices corresponding to residues 144–154, 173–194 and 200–228, interspersed with an antiparallel β-pleated sheet formed by β-strands at residues 128–131 and 161–164 in human PrP. A single disulfide bond connects cysteine residues 179 and 214 [39].

The cell surface, GPI-anchored prion protein. Drawing approximately to scale of the prion protein, showing (a) the N-terminal flexible domain; (b) location of the octapeptide repeat domain; (c) the C-terminal globular domain; (d) glycosylation chains, representing a di-glycosylated form; (e) GPI-anchor; (f) plasma membrane. Amino acid sequences are described in the legend to Fig. 4. Globular domain is depicted with Pymol software, from PDB record 1XYX of mouse PrP121-231; GPI anchor and glycosylation residues are from [142]. Modified from [239]
PrPC can be non-, mono- or di-glycosylated with a variety of N-glycans in one residue contained within α-helix 2 and another between α-helices 2 and 3 [40]. Protein glycosylation reportedly affects the recognition of various species of PrPC by monoclonal antibodies [41–43], as well as PrPC trafficking and biophysical features [44–46].
Similar to other GPI-anchored proteins, PrPC molecules attach to low-density, detergent-insoluble membrane domains (DRM) dubbed ‘membrane rafts,’ rich in cholesterol and sphingolipids [47–55]. Membrane attachment through the GPI anchor, as well as other PrPC-membrane interactions, modulate, if only slightly, the structure of the protein [56–59].
A critical issue is that association of PrPC with lipid rafts is highly dynamic [60]. A large fraction of the protein is found at any time in non-raft membrane, on its way to endocytosis [61]. Conversely, cross-linking can induce the recruitment of PrPC from a large pool of non-raft to raft membrane domains [62]. Circulation of PrPC between the cell surface and intracellular compartments is rapid and cyclic [61], and may follow various pathways (reviewed in [34, 60]). Some of the endocytosed PrPC is degraded by lysosomes, but most molecules return to the cell surface for several rounds of recycling. In addition, part of the recycled PrPC may be secreted to the extracellular medium associated with exosomes derived from multi-vesicular bodies [63, 64].
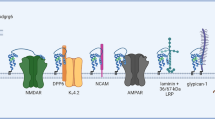
The low-density lipoprotein receptor-related protein (LRP1) is required for clathrin-mediated PrPC endocytosis in individual cells [49, 51, 65], although its role upon the trafficking of PrPC in organized tissue is still unclear (R.J. Morris, personal communication). Other transmembrane proteins, such as the laminin receptor precursor (LRP), may also play a role in the subcellular traffic of PrPC [66]. Endocytosis of PrPC can be regulated by Cu2+ ions [67, 68], by its binding partner hop/STI1 [69], and by sulfated glycans and suramin [70, 71], as well as by antibody cross-linking [72].
Trafficking of the prion protein, particularly along endocytic pathways, allows the encounter of significant variations of local pH. It has been argued that the pH does not significantly affect the three-dimensional structure of PrPC [73]. However, changes in pH were shown to modify the stability of the protein [74, 75], and a folding intermediate was isolated by incubation of PrPC at acidic pH [73–76]. Previous work showed that the conformation of the C-terminal domain (from residue 90) of PrPC is sensitive to pH [56, 74, 76–78]. Acidic pH also imparts changes in antibody binding in the N-terminal flexible domain [78]. Changes in conformation along the progressive acidification of trafficking endocytic vesicles may be relevant for functional properties of PrPC, such as the selection of partners as well as binding affinity.
Minor truncated, transmembrane and cytosolic forms of PrPC were described, usually following abnormal treatments or overexpression in cultured cells. Proteolytic cleavage of PrPC may occur, and clipping usually removes the N-terminal region, leaving a GPI-anchored, truncated C-terminal domain of the protein [79]. PrPC is also released in a soluble form [80, 81], which may be relevant for the modified disease characteristics found associated with anchorless forms of the prion protein in mice [82–84]. Still, the cell surface, GPI-anchored form constitutes almost all of PrPC found during its normal life cycle [49].
The prion protein as a cell surface scaffold protein
Work in various laboratories showed that: (1) the engagement, cross-linking, deletion, overexpression or otherwise change in the content and distribution of PrPC affects proliferation, differentiation, sensitivity to cell death and additional cellular properties not only of neurons, but also of other cells and tissues; (2) multiple signal transduction pathways are involved in such biological responses; (3) several metal ions, proteins, glycoconjugates and nucleic acids bind to PrPC in either its native, normal form or in the anomalous conformation; (4) in some cases, such ligands induce PrPC-dependent cellular responses in physiological context, and their binding sites have been mapped in both the PrPC molecule and the ligand; (5) PrPC undergoes multiple cycles of endocytosis and retrieval to the cell surface, before finally entering a degradation route at a relatively fast turnover rate ([34] for review).
The lack of an overt phenotype in the first reported PrPC-null mice [85] delayed the recognition of physiological properties of the prion protein, and curtailed the hypothesis that the corruption or loss of these functions may hold important clues as to the pathogenesis of TSEs [86]. Further work, nonetheless, eventually disclosed a number of functional consequences of deletion of the prion protein [34, 87].
In particular, evidence is now compelling that the prion protein is involved in signal transduction. Early work showed that engagement of PrPC leads to activation of the soluble tyrosine kinase Fyn, [88], and of the cyclic AMP/protein kinase A and Erk MAP kinase pathways, [89, 90]. The latter work led to the identification of hop/STI1 as a binding partner of PrPC and the mapping of their cognate binding domains [90].
Engagement of PrPC with hop/STI1 leads to neuroprotection through the cAMP/PKA pathway and neurite outgrowth through the ERK pathway, and endocytosis is required for the latter, but not for the former signaling event [69, 91, 92]. Thus, engagement of PrPC leads to the activation of distinct intracellular signaling pathways with differing biological effects, similar to most membrane receptor-mediated signal transfer systems [93, 94]. A noncommittal interpretation of the functional interaction of hop/STI1 and PrPC, thus, ascribed the role of a neurotrophic factor for the co-chaperone, whereas PrPC may play the role of either a receptor or co-receptor for secreted hop/STI1 [95]. Similar neurotrophic effects follow the binding of PrPC to a defined domain within the laminin gamma chain [96], as well as to vitronectin [97].
Although the physiological relevance of certain cellular responses and ligands has been challenged [98], and in some cases the topology of the presumptive ligands seems inconsistent with interactions in living cells [34], compelling evidence accumulated that PrPC may interact with multiple partners [34, 99, 100], particularly at the cell surface. Indeed, several other transmembrane proteins, such as the neural cell adhesion molecule (N-CAM) and the laminin receptor precursor/laminin receptor (LRP/LR), had been reported both to bind to and to mediate PrPC-dependent signals [34, 66, 101].
An ongoing phage display screening (T.A. Americo, M.H. Magdesian and R. Linden, unpublished) has identified several new candidate PrPC-binding proteins, of which group I metabotropic glutamate receptors (mGluR1 and mGluR5) were validated as mediators of calcium signals triggered by the interaction of laminin gamma chain with the prion protein [102]. Another PrPC phage display hit, the α7 nicotinic acetylcholine receptor (α7nAChR), was also shown to mediate calcium signals that follow the interaction of PrPC with hop/STI1 [103].
The demonstrations of consistent signaling behavior of PrPC in multiple scenarios reiterate the fundamental question as to how a GPI-anchored protein mediates such context- and cell type-specific signal transfer across the plasma membrane. Molecular interactions involved in varied, PrPC-mediated signaling events probably differ among distinct cell types because of the availability of the specific binding partners at the surface and extracellular matrix of the various cells. Importantly, although some overlap has been occasionally shown, spatially segregated binding sites may allow the simultaneous interaction of several such ligands with PrPC [34].
Our concept of the prion protein as a cell surface scaffold protein [34] both follows and extends the current definition of scaffold proteins, which provide physical contact and allosteric interaction of intracellular components of signaling pathways (reviewed in [35, 37]). This hypothesis accounts for both its association with multiple partners, multiple signals and multiple biological responses, as well as with the functional consequences of the presence of PrPC at the cell surface in the immune system and other organs besides the nervous system. It also predicts that the functional properties of PrPC depend on the presence of specific PrPC-binding partners at the cell surface, stoichiometric relationships among PrPC and its partners, and trafficking of PrPC relative to all members of putative signaling complexes (Fig. 2).

Recruitment of a PrPC-mediated signaling complex. The diagram illustrates a model of the recruitment of multicomponent cell surface complexes dependent on the presence of monomeric prion protein. a PrPC is present at the cell surface, together with nearby, non-interacting ligands, which may be either extracellular (ex) molecules (L1) or membrane proteins (L2, L3); b primary ligand L1 binds PrPC, leading to c conformational changes in either PrPC or L1, or both; d membrane partners of either PrPC (L2) or of the primary ligand L1 (L3) bind to their partners and transfer signals into the cell (in). Allosteric interactions are required to allow progression from b to c
Multiple binding partners and allosteric function of the prion protein
High-resolution structural data are available for the prion protein of various species, as determined both by NMR and by protein crystallography, but none in the presence of PrPC ligands. Instead, only limited information is available concerning the latter’s probable binding domains in PrPC. Their distribution along the entire PrPC molecule allows for non-competitive binding mechanisms [34], but steric interaction between ligands cannot be ruled out, which might lead to competition even at quite separate regions along the primary structure of PrPC. Moreover, it is not yet possible to infer the consequences of binding on the conformation of PrPC, which might result in rearrangement of protein domains.
It follows that many of the hypotheses about the mechanisms and consequences of ligand binding remain speculative, because they rest solely upon the currently available, high-resolution structures of monomeric, isolated PrPC. Our aim here is, nevertheless, to stimulate an approach to the problem of how PrPC deals with multiple partners, and the consequences of the dynamic making and breaking of such multicomponent signaling assemblies. The following sections will discuss whether, and to what extent, the concept of the prion protein as a cell surface scaffold protein may fit the hypothesis of allosteric regulation of signal transduction in both physiological and pathophysiological contexts.
Prion protein assembly in the membrane: monomer or homodimer?
The self-assembly of misfolded PrP molecules is a widely accepted model of the infectious scrapie agent [104]. However, the mechanisms of conformational conversion are still poorly defined [105]. Moreover, the supramolecular assembly of the normal prion protein remains elusive.
It has long been assumed that PrPC is monomeric, based on both physicochemical studies [106–112] as well as NMR structures in solution [38, 39, 73, 113–119]. Notwithstanding, the typical conditions for either physicochemical or NMR studies are usually far from physiological, such as the lack of glycosylation, the use of partial constructs instead of the full-length protein, physical and chemical variables, as well as the absence of physiological interactions with putative binding molecules and cells at close range. Furthermore, membrane anchoring restricts diffusion to a two-dimensional space and ultimately results in the decrease of the dissociation constant of membrane-anchored protein complexes [120, 121].
In contrast, dimerization of native, rather than recombinant, prion protein has been suggested [122], and interactome studies support the hypothesis that normal PrPC can undergo self-assemby [99, 123]. PrPC structure has been solved also by protein crystallography, and data are consistent with extensive dimerization interfaces between monomers (Table 1), either from symmetry-related chains or by the swapping of helix 3 between monomers (Fig. 3). These data suggest a possible functional role for such interfaces in the self-assembly of the prion protein into dimers (Table 1), the molecular mechanisms of which await full elucidation. It should be stressed that a dimeric arrangement of the prion protein at the cell surface of normal cells may impart stoichiometric constraints upon signaling complexes scaffolded by PrPC.

Schematic diagram of the interchange between PrPC monomers and dimers. The diagram is based on PrPC crystal structures and shows hypothetical conformational transitions among two monomeric forms (A for 3HEQ.pdb and B for 1I4 M.pdb) and three possible dimers, as derived from their crystal structures. In addition to the interchain contacts stabilizing such dimers, the association between these monomers would result in the exclusion of surface area to the solvent. The α-helix of each PrPC monomer is shown in the dimers by red or cyan colors
Thus, biophysical data support the potential of PrPC to form functional self-oligomers regardless of conformational conversion, and an allosteric function may involve both homo-oligomerization and dissociation equilibria in the extracellular microenvironment of the membrane-anchored PrPC. This may have important physiological consequences, and indeed, evidence for dimerization of the prion protein in trans has been reported, whereby capping of PrPC induced by antibody cross-linking leads to PrPC-mediated homophilic cell adhesion required for the stability of adherens cell junctions during embryogenesis, through a mechanism mediated by reggie/flotillin membrane-associated molecules [124].
Copper binding and structural changes of PrPC
Copper is a well-characterized ligand of the prion protein [125, 126], the functions of which are still unclear, though deemed of major physiological and pathophysiological importance [127–129]. Here we focus on copper-induced modifications of the structure of the prion protein.
Human PrPC can bind Cu2+ at a minimum of five sites, four of which are located in the octarepeat domain at the N-terminus and coordinate one Cu2+ ion each; an additional Cu2+ binding site is composed of His96 and His111, and can bind two copper ions [130, 131]. Interaction with copper reportedly affects the structure and folding stability of the prion protein [132], and Cu2+ binding leads to structuring of the N-terminal domain [133, 134]. It has been reported that the octarepeat segment PrP C61–84 is well structured in solution in the absence of divalent cations [135], with a conformation similar to the Cu2+-bound complex [127, 136]. Further molecular dynamics simulation of the N-terminal domain of human PrP (residues 23–120) loaded with four or five copper ions led to an even more rigid conformation in the octapeptide repeats [134]. This suggests that, depending on the copper/PrPC molar ratio, the dynamics of PrP is changed, which may alter the availability of the N-terminal domain to other ligands.
Recent studies of full-length recombinant PrP (rPrP) showed that copper binding induces both novel interactions between the flexible N-terminal and globular C-terminal of PrPC, as well as a compaction of the prion protein, without signs of aggregation at physiological temperature [137]. These data offer new insights into copper-induced structural transitions of rPrP and suggest that the interaction of PrPC with ligands recognized by either the domain PrP C144–147 in α-helix 1, as well as by the domain PrP C174–185 in α-helix 2, may be allosterically affected by copper binding.
It has also been suggested that copper binding leads to dissociation of PrPC from a lipid raft-resident partner, thus initiating the former’s journey towards non-raft membrane domains, on its way to endocytosis through clathrin-coated vesicles [138]. In addition, glycosaminoglycans (GAGs) bind PrPC, and the heparan sulfate-derived, GPI-anchored GAG glypican-1 (Gpc-1) interacts with copper-loaded PrPC [139, 140]. N2a neuroblastoma cells co-internalize PrPC and Gpc-1 upon induction by copper, indicating that copper loading of PrPC may impose on the endocytosis of a distinct GPI-anchored molecule.
In turn, binding of PrPC to Cu2+ may be influenced by the glycosylation pattern of the prion protein. Full-length, non-glycosylated PrPC from sheep brain bound to IMAC resin loaded with either copper or cobalt ions, with higher efficiency than glycosylated forms [141]. This suggests that the access to the copper-binding sites of PrPC is controlled by the level of glycosylation of the prion protein. This may also affect other PrPC-binding partners, either through analogous allosteric effects or through steric hindrance by the relatively bulky sugar moieties attached to the prion protein [142].
The interaction of recombinant PrP with the constitutive chaperone Hsc70 was investigated in the presence of copper ions at differing pH values, using an ELISA-based assay [143]. Binding of native PrP to Hsc70 was the greatest at low pH, thus disclosing an allosteric effect of protonation upon rPrP. At low pH, pre-incubation with Cu2+ also increased the binding of Hsc70 to rPrP. Two regions within the PrPC globular domain were mapped as the main Hsc70-binding sites at acidic pH, thus highlighting the allosteric effect of both Cu2+ and protonation. The physiological relevance of this interaction is, however, questionable, because measurements were carried out in vitro, using recombinant, non-glycosylated PrPC, and the binding of extracellular PrPC to Hsc70 has not been validated either in vivo or ex vivo. On the other hand, a cytosolyic form of PrPC, which accumulates abnormally upon proteasome inhibition, often forms aggregates that contain Hsc70 [144]. Such molecular chaperones may, in fact, participate in conformational transitions of PrP or protein-protein interactions that lead to TSEs.
Nucleic acids as molecular partners of the prion protein
Nucleic acids (NA) have long been shown to bind to and lead to aggregation of PrPC [145]. The prion protein is, however, prone to transient aggregation upon binding to polyanions [146–149]. Thus, binding to NA was initially thought of as non-specific, such as typically found for NA-binding proteins and non-specific DNAs, or even between interacting polycations and polyanions [150]. However, further studies showed that nucleic acid binding to PrPC is selective [147], and that both aggregation and its time-dependent reversal are tightly regulated, and depend on ligand stoichiometry [151, 152]. In fact, at certain PrPC:NA ratios a soluble complex is formed, in which the prion protein features a conformation similar to pure PrPC [148, 152]. Thus, the binding of membrane-anchored PrPC to NA at the cell surface may not necessarily prompt amyloid formation, and functional properties of the prion protein may be preserved.
Solution-binding studies in equilibrium, achieved after complete reversal of the transient aggregation induced by NA, indicated that small double-stranded DNA binds to the C-terminal domain of PrPC at a 1:1 ratio, accompanied by structural changes in the N-terminal domain [152]. This event is favored by acidification [75], in a condition that produces only minor conformational changes of the C-terminal domain of PrPC [73].
Current data indicate that the binding of NA to either the N- or C-terminals of PrPC results in substantial rearrangement of both protein and nucleic acid [147, 148, 151–153]. Although the NA binding domain in PrPC is still unknown, predictive analysis with dbd-PSSM [154] points to a potential for the PrP C153–174 domain to recognize DNA (A.F. Marques and L.M.T.R. Lima, unpublished results). Interestingly, this domain of PrPC has a high degree of identity with human DNA polymerases 1CLQ.pdb and 1IG9.pdb, differing only in 4 out of 22 amino acids at a region in close proximity to the active site of the latter enzymes.
Attempts made to identify a PrPC-binding consensus sequence, with the use of various DNA and RNA aptamers, reported NA:PrPC binding affinities in the nanomolar concentration range [155], consistent with the typical affinity of regulatory proteins. Moreover, the strict dependence of binding affinity upon both NA sequence and the investigated PrPC domain is also consistent with the typical behavior of classic NA-binding proteins [156–158]. Possible effects of copper, which induces structural gains in the N-terminal domain (see above), upon the interaction of PrPC with NA remain to be investigated.
Interaction of glycosaminoglycans with the prion protein
Glycosaminoglycans have been implicated in the pathogenesis of prion diseases [159–164]. It was shown that GAGs bind the prion protein both when the latter is anchored at the plasma membrane, as well as in a non-glycosylated form in vitro [165, 166], and it was proposed that GAGs in general interact with PrPC through its N-terminal domain [166, 167].
Interaction of the N-terminal of PrPC with pentosan polysulfate (PPS) led to structuring of the octarepeat domain, exposing a hydrophobic surface composed of aligned tryptophan side chains [168]. Similar to the case of nucleic acids, it remains to be investigated whether copper loading of PrP would affect the binding of GAGs, as well as conformational changes [168].
Conflicting results have been reported of the interaction of heparin and heparan sulfate with PrPC (reviewed in [164]). Certain studies suggested that these GAGs prevent PrP aggregation and conformational conversion [169, 170]. Other studies, in contrast, reported that GAGs either induce or accelerate the structural conversion implicated in TSEs [161, 162].
Recently, the interaction of recombinant prion protein with low molecular weight heparin was investigated as a function of pH. Several binding sites for heparin had already been located in the N-terminal domain of PrPC (reviewed in [34]), but, surprisingly, at pH 5.0, PrP presented 2 heparin-binding sites, in contrast with the unique site found at pH 7.4 [148]. This result implies that changes in pH significantly affect the structure of the complex formed by PrPC and GAGs.
As pointed out above, the evidence for pH-dependence of PrPC:heparin interaction may be otherwise relevant for functional properties of PrPC along progressively acidic endocytic compartments [171, 172], such as the selective requirement of endocytosis for the activation of MAP kinases after binding of hop/STI1 to PrPC [91, 92]. It may also be important for putative pH-dependent effects among microdomains with distinct local pH determinants, such as negatively charged oligosaccharides attached to surface glycoproteins, including PrPC itself [173, 174].
Reciprocal remodeling upon binding of PrPC and hop/STI-1
Several spectroscopic techniques provided evidence that interaction of PrPC with the co-chaperone hop/STI1 induces reciprocal conformational changes in both proteins [175]. Thus, the binding of murine, recombinant PrPC to recombinant hop/STI1 entailed a loss in α-helical secondary structure of PrPC, at least part of which was located in α-helix 1 (PrP C143–153 ). This may, for example, affect the interaction of PrPC with ligands such as LPR and N-CAM, which are transmembrane proteins that bind α-helix 1, and transduce PrPC-dependent signals [66, 176] (Fig. 4a–c).

Reciprocal remodeling of binding partners PrPC and hop/STI1. a Diagram of the prion protein, with indicated domains and sequences of amino acids in mouse PrPC (SP, signal peptide 1–22; OR, octapeptide repeats 51–91; CC, charged cluster 95–110; HC, hydrophobic core 112–133; H1, H2, H3, α-helices 144–153, 172–194 and 200–224, respectively; GPIp, GPI-anchoring signal 231–254), glycosylation sites (stars 180 and 196), and location of binding domains (double arrows with amino acid sequences in parentheses) of hop/STI1 (blue), neural cell adhesion molecule (N-CAM, green) and the laminin receptor precursor/laminin receptor (LRP/LR, red); b, c tri-dimensional depiction of the globular domain of PrPC, highlighting the binding domains indicated in a with the same colors; spectrometric techniques unraveled changes in alpha-helix 1 upon binding of hop/STI1230-245 to PrPC. The flexible N-terminal tail was omitted for clarity; d, e low-resolution models (sets of grey spheres) of either hop/STI1 alone (d), or the hop/STI1:PrPC complex (e), generated from SAXS measurements, reveal a compaction of the tertiary structure of hop/STI1 upon binding to PrPC (arrow points to PrPC, where the globular domain is shown in yellow and the flexible N-terminal tail is shown in pink). Modified from [34, 175, 240]
In turn, three-dimensional, low-resolution models generated from small angle X-ray scattering (SAXS) measurements demonstrated a compaction of the C-terminus of hop/STI1 upon binding (Fig. 4d, e). Such tertiary structural changes may either attract or repel ligands of hop/STI1 proper, thus engaging higher order components of the signaling complex in addition to PrPC ligands.
Pertaining to this issue, our data indicated that either the full-length hop/STI1 or a 16 amino acid peptide containing the sequence cognate to PrPC (hop/STI1230–245) induced similar changes in the secondary structure of PrPC [175]. This may allow either the full ligand hop/STI1 or the peptide alone to engage the same PrPC ligand(s) and produce the same intracellular signals (Fig. 5). Indeed, in various cell types, either the full-length hop/STI1 or the hop/STI1230–245 peptide produced similar PrPC-dependent neurotrophic effects [89–91, 177] (Fig. 6a), suggesting that signaling was transferred by a PrPC ligand. As discussed above, one such ligand, detected both by phage display (T.A. Americo et al., unpublished) and validated by biochemical methods may be the α7 nicotinic acetylcholine receptor [103].

PrPC-dependent signaling mediated by similar allosteric effects of either hop/STI1 or the PrPC-binding peptide hop/STI1230–245: the diagrams depict a model of the behavior of PrPC, when its engagement by either the full-length hop/STI1 (a, hop) or only the peptide containing the PrPC-binding domain (b, hoppep) result in the same biological response (c, d, black arrow, see Fig. 6 a). This scenario probably requires the recruitment of a PrPC-binding, transmembrane signal transfer molecule (L2), as a consequence of allosteric effects of hop/STI1 upon PrPC (see Fig. 4b, c). Engagement of a secondary, hop/STI1-binding transmembrane protein (L3) may produce additional signals (dotted arrow) not required for the current biological response

Comparison of PrPC-dependent biological effects of hop/STI1 and hop/STI1230–245. a Neuroprotective effects upon undifferentiated potmitotic cells within the mouse retina. Counts of pyknotic nuclei were done in the ganglion cell layer of retinal explants cultured for 24 h with the inhibitor of protein synthesis anisomycin, either in the absence or the presence of full-length hop/STI1 or the hop/STI1 peptide containing the PrPC-binding domain (adapted from [90]). Notice the protective effects of both the full-length protein and the PrPC-binding peptide in wild-type, but not in PrPC-null nervous tissue. b Mitogenic effects on glioblastoma cell cultures. Incorporation of radioactive nucleotide was measured in cultures of the A172 glioblastoma cell line incubated for 24 h with the indicated full-length, truncated proteins or peptides (adapted from [178] and unpublished data). Notice that only the full-length protein induces cell proliferation, whereas no effect was elicited by the PrPC-binding peptide, by an irrelevant peptide nor by the truncated protein lacking the PrPC-binding domain
Notably, however, whereas full-length hop/STI1 induced proliferation of glioblastoma cells in culture, and deletion of the sequence hop/STI1230–245 prevented this effect [178], the hop/STI1230-245 peptide alone did not induce glioma cell proliferation (S. Kahn et al., unpublished observations) (Fig. 6b). Accordingly, it was recently shown that hop/STI1 enhanced self-renewal of stem/progenitor cells through its interaction with PrPC, and, again, the hop/STI1230–245 peptide by itself had no effect [179]. A likely explanation for these data is that signaling leading to proliferation of either the glioma or the stem/progenitor cells depends on the binding of hop/STI1 to PrPC, but transmembrane signal transfer requires the recruitment of a hop/STI1 ligand (Fig. 7). An ongoing hop/STI1 phage display screening (M. Magdesian, R. Linden et al., unpublished) has identified several potential candidates for such signal transfer, which are currently under scrutiny.

PrPC-dependent signaling mediated by a secondary ligand of hop/STI1: a, b the diagrams depict a model of the events underlying responses induced by hop/STI1, but not by hop/STI1230–245 nor by hop/STI1Δ230–245 lacking the PrPC-binding domain (Fig. 6b). The lack of response to the latter protein indicates that the response (black arrrow) depends on PrPC-hop/STI1 interaction, but the signals produced by the similar allosteric effects of either the full-length or the binding peptide (L2 and dotted arrow) upon PrPC are innefective to produce the biological response. This scenario probably requires engagement of a productive hop/STI1-binding transmembrane signal transfer molecule (L3), as a consequence of allosteric effects of PrPC upon hop/STI1 (see Fig. 4d, e)
It should also be noted that extracellular hop/STI1 may induce biological effects irrespective of the presence of the prion protein. Thus, either full-length hop/STI1 or a recombinant protein lacking the PrPC-binding domain (hop/STI1Δ230–245) equally reduced the proliferation of retinal progenitor cells in either wild-type or PrPC-null retinas in vitro [180], and a similar result was reported for cultures of brain-derived astrocytes [181]. In addition, antibodies against full-length hop/STI1 prevented cell death of distinct retinal cell types induced by differing means, in either wild-type or PrPC-null retinas in vitro, whereas antibodies to the PrPC-binding domain hop/STI1230–245 did not [180]. The simplest interpretation for these findings is that hop/STI1 may, by itself, activate transmembrane signal transfer molecules irrespective of PrPC (Fig. 8a).

Hypothetical balance of PrPC-independent and PrPC-dependent effects of a PrPC-binding protein. The diagrams depict the presumptive behavior of the PrP-binding protein hop/STI1 together with various transmembrane signal transduction molecules, in various scenarios. a hop/STI1 may bear upon various biological functions, such as cell proliferation and cell death (hatched and white arrows), independent of PrPC (see text), and signal transfer may be mediated by either one or more transmembrane molecules; b, c in certain cases, however, hop/STI1 produces conflicting signals (white vs. black arrows) via PrPC-independent and PrPC-dependent mechanisms, and the resulting biological effect may depend on stoichiometric relationships among the various components of the cell surface signaling modules; d alternative events may include intracellular networking of signaling pathways, the net result of which would also depend on the relative amounts of cell surface components
However, in neither of the studies above nor, for that matter, in most studies comparing biological effects between wild-type or PrPC-null cells have kinetic parameters been thoroughly examined. In fact, the conditions in which antibodies to full-length hop/STI1, but not to the PrPC-binding domain, prevented cell death induced by blockade of protein synthesis [180], were the same in which increased levels of either the full-length hop/STI1 or only of the PrPC-binding peptide hop/STI1230-245 produced a PrPC-dependent neuroprotective effect [90]. The results, therefore, suggest that the hop/STI1 protein may have PrPC-dependent, neuroprotective effects, as well as PrPC-independent, neurodegenerative effects on the same cells. It is possible that transmembrane transfer of either cytodegenerative or cytoprotective signals actually involves the same group of molecules, but the relative concentrations of extracellular hop/STI1, PrPC, as well as their ligands dictates the final result (Fig. 8b, c). The conflicting signals may either activate distinct downstream pathways or, alternatively, interact due to intracellular networking of multiple signal transfer molecules, conditioned by allosteric effects at the cell surface (Fig. 8c, d).
The overall data are, thus, consistent with biologically relevant, allosteric interaction of PrPC and hop/STI1, involving ligands of both partners, and fits the concept that PrPC scaffolds multicomponent, cell surface signaling complexes [34]. Further studies, especially of kinetic parameters and stoichiometric requirements of signal transduction, are warranted to critically test this hypothesis.
Allosteric dysfunction of the prion protein and signal corruption
The preceding sections focused mainly on functional properties of the prion protein, as well as on the effects of certain PrPC partners upon protein conformation. Although the possibility of loss-of-function components in prion diseases has long been put forward [86, 182, 183], little attention has been directed at the importance of physiological functions of PrPC in the context of the TSEs. The following discussion will consider hypotheses as to how the scaffold concept, together with allosteric functions of PrPC, may contribute to the understanding of the pathogenesis of prion disease.
The presentation, course, evolution, histo- and molecular pathology of TSEs are highly variable [184, 185]. Notwithstanding, the prevalent idea is that prion diseases are caused by either a single toxin or by oligomeric toxic species of a defined range of sizes [10]. Although this hypothesis cannot be discarded, it fails to account for the coexistence of three ongoing processes in the course of protein conformation diseases: (1) progressive accretion of monomers to growing oligomers and/or fusion of oligomers; (2) progressive conformational conversion; and (3) their progressive compaction. Most importantly, these events likely occur asynchronously both within and among distinct areas of the brain, and their coexistence implies that the set of exposed residues of both PrPC and its anomalous conformer change with time, asynchronously across the affected tissue. This would constantly change the reactivity of the growing aggregates of the prion protein, and their allosteric effects upon the binding and function of physiological or pathophysiological partners in brain tissue.
Notwithstanding the notorious association of anomalous PrP conformers with prion disease, it is still unclear whether or how much conformational conversion is actually required for oligomerization and toxicity. For example, severe overexpression of wild-type PrPC in mice led to neurological dysfunction accompanied by the finding of no more that 10% of insoluble, mildly protease-resistant aggregates among the whole content of the prion protein at advanced stages of the disease [186]. Unfortunately, that study addressed neither the oligomeric state nor the secondary structure of the protein, albeit the authors have acknowledged the hypothesis that ‘…high levels of PrP may be toxic in some other way, for example, by saturating or over-stimulating a normal metabolic or signaling pathway activated by PrPC’ [186]. In another study, cross-linking of PrPC with divalent monoclonal antibodies, but not monovalent Fab fragments, led to neurodegeneration in wild-type mice, with no reported hint of conformational conversion [187]. The latter authors interpreted their findings as a sign that dimerization of PrPC led to the toxic events. However, the evidence for natural dimerization of PrPC (see above) raises the hypothesis that the cross-linking antibodies may have also led to the assembly of higher order oligomers of the prion protein. Finally, recent spectroscopic studies of brain tissue sections have failed to detect anomalous conformers of the prion protein at a stage where both synaptic alterations and behavioral signs are already detectable in a mouse model of infectious prion disease (V.H. Perry, personal communication). It is possible that toxic PrP species that contribute to early pathological events may be too small, diffuse or rare, thus making their detection difficult by the techniques used in the studies mentioned above. Nonetheless, those results are also consistent with an alternative hypothesis that conformational conversion may not be required for PrPC aggregate-dependent toxicity, at least at early stages of the TSEs.
The dynamics of changes among a heterogeneous population of oligo- and multimers, slack and compact aggregates, with variable content of anomalous conformers, is probably relevant for the heterogeneity and the course of the prion diseases. Still, both the time and the asynchrony factors are often ignored in the interpretation of experimental data of TSEs, as well as in other neurodegenerative disorders. In contrast, recent work raised the hypothesis that the pathogenesis of Alzheimer’s disease may actually be linked to the dynamic events of incorporation of β-amyloid monomers into growing protofibrils rather than to a specific class of oligomers [188]. Analogous events involving the prion protein may be relevant to the TSEs as well, and deserve thorough investigation.
It is, thus, questionable that a defined toxic molecular species, at a specific time, is responsible for the complex cellular events that underlie the pathogenesis of TSEs. Misfolded and aggregated anomalous conformers of the prion protein may, by themselves, have toxic gain-of-function properties. Even so, disturbances of the scaffold and allosteric functions of PrPC probably add to the effects of such toxic species, or, alternatively, are responsible for specific pathogenic events. A likely scenario is that, in the course of prion diseases, the brain progresses toward neurodegeneration from a framework of physiological functions of normal PrPC, through either several stages or a continuum of corrupted cellular signals, leading to pathological events, such as the early changes of synapse morphology and function, followed by progressive neuron death [189–191]. It follows that the nature of the molecular interactions of PrPC and their disruption at the earliest stages of the TSEs is pivotal for the understanding of homeostatic corruption associated with prion diseases.
It is particularly noteworthy that the GPI-anchoring of the prion protein to the membrane seems to be required for the full-blown disease in vivo, though not for the accumulation of the pathognomonic compact, protease-resistant aggregates, nor for infectious transmission [82, 83]. These data further highlight the importance of the interactions of PrPC with cell surface and transmembrane molecules, the ensuing allosteric effects, as well as their consequences for signal transduction. The scaffold hypothesis predicts that, in the course of TSEs, the assembly of cell surface signaling complexes organized by the prion protein, together with their downstream effects, change along with progressive oligomerization, conformational conversion and compaction of the aggregates. Such signal corruption may include either the gain or the loss of specific signals associated with PrPC partners [192], as well as effects upon signaling kinetics due to the derangement of multicomponent complexes scaffolded by PrPC [34, 193], both of which are likely to occur upon either the masking or the exposure of specific domains of the prion protein.
Multiple effects can be expected from oligomerization, cross-linking or merely an increased content of the GPI-anchored prion protein. Events may include the disassembly of signaling modules, due to disruption of stoichiometric relationships between PrPC and its partners (Fig. 9a). In fact, the possible interplay of monomers and dimers of PrPC (see above) should also be taken into account to understand the physiological framework of PrPC-mediated signaling, while the accruing of each monomer (or dimer) to a growing oligomer would further disrupt local stoichiometry of cell surface signaling complexes.

Multiple events of signal corruption expected from oligomerization and progressive compaction of aggregates of the prion protein. a Initial events may disrupt stoichiometrical relationships between PrPC and nearby ligands, leading to disassembly of cell surface signaling modules; b further oligo- and multimerization may lead to abnormal oligomerization of PrPC ligands, such as membrane receptors, with ensuing corruption of signal transduction through such receptors; c further compaction of aggregates of the prion protein favors steric hindrance, thus preventing binding of PrPC ligands and the assembly of cell surface signaling modules
This, as well as further accretion of monomers, is expected to exact either physiological or pathological oligomerization of PrPC-binding signal transducers, such as membrane receptors (Fig. 9b). Thus, in cells bearing oligomers of PrPC at their surface and/or internal membranes, abnormal homo- or hetero-oligomerization of receptors may robustly affect ligand affinity, kinetics of signal transduction and receptor trafficking [194–198]. In addition, oligomerization imparts changes in short-term, lateral diffusion at the plasma membrane [199, 200], with likely consequences upon both the kinetics of recycling of the cell surface PrPC, as well as of PrPC-mediated signaling.
The previous events, plus further multimerization and compaction, tend to progressively favor steric hindrance [201–204], eventually blocking the assembly of cell surface signaling modules (Fig. 9c). Notably, several studies suggest that, in various neurodegenerative diseases, the pathognomonic aggregates detectable at light microscopical examination may actually offer neuroprotection [205–210], probably because steric hindrance prevents any productive association of the compact aggregates with molecular partners required for neurotoxic signaling.
It should be noted that separation of these three categories of effects serves only to highlight individual components of a complex, dynamic scenario of signal derangement. Simultaneously, combined with the effects predicted from simple oligomerization, the conformational conversion would add to both the propensity of protein aggregation [211, 212] and to changes in ligand binding, which are expected to follow modification of the secondary structure of the protein (Fig. 10).

Diagram of proposed pathogenic events due to dysfunction of the scaffold properties of the prion protein. The figure depicts how the asynchronous accretion of monomeric or dimeric PrPC to growing oligomers, conformational conversion of an unknown proportion of the protein molecules within agregates and progressive compaction of the aggregates may lead to the pathogenesis of TSE and other neurodegenerations. The cycle of accretion of monomers together with conformational conversion is thought to heavily impose upon the properties of PrPC-scaffolded signaling modules, leading to allosteric effects and ensuing signal corruption, which may result in neurodegeneration (arrows). Conformational conversion is, in turn, expected to fuel progressive aggregation and compaction, leading to steric hindrance, which may either cause further signal corruption and neurodegeneration, or, at late stages, contribute to protect the tissue from the recurring cycles of monomer accretion and dynamically changing allosteric effects (dashed arrows). Signal corruption due to accretion of PrPC subunits may also be triggered by non TSE-related aggregation events, such as cross-linking by Aβ oligomers (dotted line)
The progressive, asynchronous occurrence of these dynamic events in any given neuronal population most likely spreads, also asynchronously, along the interconnections of brain areas and nuclei, therefore adding to both the pathogenesis of the diseases, as well as to the production of specific signs and symptoms.
It has recently been demonstrated that the prion protein specifically binds β-amyloid (Aβ) oligomers [213], although the significance of such binding for Alzheimer’s disease is still under debate [214–223]. Nonetheless, it was reported that Aβ disturbs the trafficking of the prion protein, leading to an accumulation of PrPC at the cell surface [224], and that both Aβ and monoclonal antibodies cross-link PrPC, to which synaptic toxicity was attributed [225]. As discussed above, the accumulation and cross-linking of PrPC are both likely relevant for cell signaling (Fig. 9), and the scaffold hypothesis may explain, for example, why an increasing number of differing membrane receptors have been implicated in toxic signaling induced by various β-amyloid aggregates [226–232] (Fig. 10).
Data have also been reported of possible interference of the prion protein in other neurological diseases, such as primary progressive aphasia [233], or in experimental models of amyotrophic lateral sclerosis [234, 235]. It should also be pointed out that changes in the stoichiometry of the components of PrPC-scaffolded signaling modules may be relevant not only for the nervous system, but also for other cell types and tissues, such as muscle and the immune system [34]. An intriguing possibility is that disturbance of the scaffold and allosteric functions of the prion protein may be also involved in the pathogenesis of a variety of diseases other than the TSEs.
Conclusion and perspectives
The preceding sections discussed structural constraints involved in the interaction of the prion protein with various binding molecules. The latter include proteins, such as hop/STI1, nucleic acids and glycosaminoglycans, and extend to the effects of pH. Possible instances of allosteric control were discussed, associated with changes in the interaction of PrPC with a third partner.
Analysis of structural changes due to the interaction of the prion protein with its multiple partners is still precarious, and little data have so far been gathered as to the functional properties of molecular complexes scaffolded by the prion protein. For most putative ligands of PrPC, no structural data relevant to their binding have been published so far. Therefore, this review serves basically to set a framework for future studies. Notwithstanding, the available data higlight the importance of understanding the allosteric function and dysfunction of the prion protein.
One important caveat is that most structural data gathered to date refer to the isolated prion protein in artificial conditions, which may differ substantially from the environment where PrPC resides in living cells. Efforts are warranted to try and reach conditions as close as possible to those prevailing in physiological microenvironments, such as the cell surface, endocytic vesicles involved in the trafficking of PrPC and also the Golgi, where nascent PrPC molecules may interact with either nascent or resident partners. In addition, the events that occur upon the binding of PrPC with multiple partners must be identified to help understand the role of PrPC in cell signaling both in the nervous system as well as in other cell types containing substantial amounts of PrPC.
The scaffold hypothesis further raises key questions regarding the composition of PrPC oligomers. There is pressing need to clarify the components and the kinetics of growth of PrP oligomers in early stages of TSEs, inclusive of the relative contributions of either normal or anomalous conformers, to understand the nature of the cellular signals associated with these deadly diseases.
In conclusion, both the understanding of the pathogenesis of TSEs and the development of their effective treatment, and perhaps the management of other diseases, such as Alzheimer’s disease, where both protein oligomers and the prion protein may play a role, require a change in paradigm, where instead of targeting an elusive toxic species, one should aim at the understanding of the kinetic behavior of rapidly changing oligo- and multimers, together with the allosteric effects associated with the making and breaking of signaling complexes that lead to the dynamic signal corruption at the root of pathogenic events due to dysfunction of the prion protein.
References
Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95(23):13363–13383
Norrby E (2011) Prions and protein-folding diseases. J Intern Med 270(1):1–14
Aguzzi A, Calella AM (2009) Prions: protein aggregation and infectious diseases. Physiol Rev 89(4):1105–1152
Brown K, Mastrianni JA (2010) The prion diseases. J Geriatr Psychiatry Neurol 23(4):277–298
Caughey B, Baron GS, Chesebro B, Jeffrey M (2009) Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem 78:177–204
Wadsworth JD, Collinge J (2007) Update on human prion disease. Biochim Biophys Acta 1772(6):598–609
Klein WL, Krafft GA, Finch CE (2001) Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci 24(4):219–224
Lambert MP, Velasco PT, Viola KL, Klein WL (2009) Targeting generation of antibodies specific to conformational epitopes of amyloid beta-derived neurotoxins. CNS Neurol Disord Drug Targets 8(1):65–81
Simoneau S, Rezaei H, Sales N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier JG, Comte J, Wopfner F, Grosclaude J, Schatzl H, Lasmezas CI (2007) In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog 3(8):e125
Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B (2005) The most infectious prion protein particles. Nature 437(7056):257–261
Zerr I (2009) Therapeutic trials in human transmissible spongiform encephalo-pathies: recent advances and problems to address. Infect Disord Drug Targets 9(1):92–99
Stewart LA, Rydzewska LH, Keogh GF, Knight RS (2008) Systematic review of therapeutic interventions in human prion disease. Neurology 70(15):1272–1281
Brazier MW, Wall VA, Brazier BW, Masters CL, Collins SJ (2009) Therapeutic interventions ameliorating prion disease. Expert Rev Anti Infect Ther 7(1):83–105
Perry DC, Geschwind MD (2011) Prion disease: thorough work-up and new diagnostic criteria needed for CJD. Nat Rev Neurol 7(9):479–480
Kovacs GG, Botond G, Budka H (2010) Protein coding of neurodegenerative dementias: the neuropathological basis of biomarker diagnostics. Acta Neuropathol 119(4):389–408
Dorandeu A, Wingertsmann L, Chretien F, Delisle MB, Vital C, Parchi P, Montagna P, Lugaresi E, Ironside JW, Budka H, Gambetti P, Gray F (1998) Neuronal apoptosis in fatal familial insomnia. Brain Pathol 8(3):531–537
Gray F, Adle-Biassette H, Chretien F, Ereau T, Delisle MB, Vital C (1999) Neuronal apoptosis in human prion diseases. Bull Acad Natl Med 183(2):305–320 discussion 320-301
Gray F, Chretien F, Adle-Biassette H, Dorandeu A, Ereau T, Delisle MB, Kopp N, Ironside JW, Vital C (1999) Neuronal apoptosis in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol 58(4):321–328
Cox DL, Sing RR, Yang S (2006) Prion disease: exponential growth requires membrane binding. Biophys J 90(11):L77–L79
Eghiaian F, Daubenfeld T, Quenet Y, van Audenhaege M, Bouin AP, van der Rest G, Grosclaude J, Rezaei H (2007) Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proc Natl Acad Sci USA 104(18):7414–7419
Jain S, Udgaonkar JB (2008) Evidence for stepwise formation of amyloid fibrils by the mouse prion protein. J Mol Biol 382(5):1228–1241
Almstedt K, Nystrom S, Nilsson KP, Hammarstrom P (2009) Amyloid fibrils of human prion protein are spun and woven from morphologically disordered aggregates. Prion 3(4):224–235
Silva JL, Vieira TC, Gomes MP, Bom AP, Lima LM, Freitas MS, Ishimaru D, Cordeiro Y, Foguel D (2010) Ligand Binding and Hydration in Protein Misfolding: Insights from Studies of Prion and p53 tumor suppressor proteins (dagger). Acc Chem Res 43(2):271–279
Baskakov IV (2007) Branched chain mechanism of polymerization and ultrastructure of prion protein amyloid fibrils. Febs J 274(15):3756–3765
Gonzalez-Montalban N, Makarava N, Ostapchenko VG, Savtchenk R, Alexeeva I, Rohwer RG, Baskakov IV (2011) Highly efficient protein misfolding cyclic amplification. PLoS Pathog 7(2):e1001277
Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326(5959):1533–1537
Boesenberg C, Schulz-Schaeffer WJ, Meissner B, Kallenberg K, Bartl M, Heinemann U, Krasnianski A, Stoeck K, Varges D, Windl O, Kretzschmar HA, Zerr I (2005) Clinical course in young patients with sporadic Creutzfeldt-Jakob disease. Ann Neurol 58(4):533–543
Nagoshi K, Sadakane A, Nakamura Y, Yamada M, Mizusawa H (2011) Duration of Prion Disease is Longer in Japan Than in Other Countries. J Epidemiol 21(4):255–262
Weissmann C (2004) The state of the prion. Nat Rev Microbiol 2(11):861–871
Aguzzi A, Baumann F, Bremer J (2008) The prion’s elusive reason for being. Annu Rev Neurosci 31:439–477
Weissmann C (2009) Thoughts on mammalian prion strains. Folia Neuropathol 47(2):104–113
Watts JC, Westaway D (2007) The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta 1772(6):654–672
Soto C, Estrada LD (2008) Protein misfolding and neurodegeneration. Arch Neurol 65(2):184–189
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiology of the prion protein. Physiol Rev 88(2):673–728
Bray D, Duke T (2004) Conformational spread: the propagation of allosteric states in large multiprotein complexes. Annu Rev Biophys Biomol Struct 33:53–73
Whitty A (2008) Cooperativity and biological complexity. Nat Chem Biol 4(8):435–439
Good MC, Zalatan JG, Lim WA (2011) Scaffold proteins: hubs for controlling the flow of cellular information. Science 332(6030):680–686
Ilc G, Giachin G, Jaremko M, Jaremko L, Benetti F, Plavec J, Zhukov I, Legname G (2010) NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS One 5(7):e11715
Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 97(1):145–150
Haraguchi T, Fisher S, Olofsson S, Endo T, Groth D, Tarentino A, Borchelt DR, Teplow D, Hood L, Burlingame A et al (1989) Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch Biochem Biophys 274(1):1–13
Beringue V, Mallinson G, Kaisar M, Tayebi M, Sattar Z, Jackson G, Anstee D, Collinge J, Hawke S (2003) Regional heterogeneity of cellular prion protein isoforms in the mouse brain. Brain 126(Pt 9):2065–2073
Li R, Liu D, Zanusso G, Liu T, Fayen JD, Huang JH, Petersen RB, Gambetti P, Sy MS (2001) The expression and potential function of cellular prion protein in human lymphocytes. Cell Immunol 207(1):49–58
Monnet C, Gavard J, Mege RM, Sobel A (2004) Clustering of cellular prion protein induces ERK1/2 and stathmin phosphorylation in GT1-7 neuronal cells. FEBS Lett 576(1–2):114–118
Wiseman F, Cancellotti E, Manson J (2005) Glycosylation and misfolding of PrP. Biochem Soc Trans 33(Pt 5):1094–1095
Cancellotti E, Barron RM, Bishop MT, Hart P, Wiseman F, Manson JC (2007) The role of host PrP in Transmissible Spongiform Encephalopathies. Biochim Biophys Acta 1772(6):673–680
Cancellotti E, Wiseman F, Tuzi NL, Baybutt H, Monaghan P, Aitchison L, Simpson J, Manson JC (2005) Altered glycosylated PrP proteins can have different neuronal trafficking in brain but do not acquire scrapie-like properties. J Biol Chem 280(52):42909–42918
Stahl N, Borchelt DR, Hsiao K, Prusiner SB (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51(2):229–240
Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A (1997) Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem 272(10):6324–6331
Taylor DR, Hooper NM (2006) The prion protein and lipid rafts. Mol Membr Biol 23(1):89–99
Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R (1999) Functionally different GPI proteins are organized in different domains on the neuronal surface. Embo J 18(24):6917–6926
Morris RJ, Parkyn CJ, Jen A (2006) Traffic of prion protein between different compartments on the neuronal surface, and the propagation of prion disease. FEBS Lett 580(23):5565–5571
Pike LJ (2004) Lipid rafts: heterogeneity on the high seas. Biochem J 378(Pt 2):281–292
Brugger B, Graham C, Leibrecht I, Mombelli E, Jen A, Wieland F, Morris R (2004) The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J Biol Chem 279(9):7530–7536
Botto L, Masserini M, Cassetti A, Palestini P (2004) Immunoseparation of Prion protein-enriched domains from other detergent-resistant membrane fractions, isolated from neuronal cells. FEBS Lett 557(1–3):143–147
Loberto N, Prioni S, Bettiga A, Chigorno V, Prinetti A, Sonnino S (2005) The membrane environment of endogenous cellular prion protein in primary rat cerebellar neurons. J Neurochem 95(3):771–783
DeMarco ML, Daggett V (2005) Local environmental effects on the structure of the prion protein. C R Biol 328(10–11):847–862
Eberl H, Tittmann P, Glockshuber R (2004) Characterization of recombinant, membrane-attached full-length prion protein. J Biol Chem 279(24):25058–25065
Hicks MR, Gill AC, Bath IK, Rullay AK, Sylvester ID, Crout DH, Pinheiro TJ (2006) Synthesis and structural characterization of a mimetic membrane-anchored prion protein. Febs J 273(6):1285–1299
Morillas M, Swietnicki W, Gambetti P, Surewicz WK (1999) Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem 274(52):36859–36865
Prado MA, Alves-Silva J, Magalhaes AC, Prado VF, Linden R, Martins VR, Brentani RR (2004) PrPc on the road: trafficking of the cellular prion protein. J Neurochem 88(4):769–781
Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R (2003) The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. Embo J 22(14):3591–3601
Hugel B, Martinez MC, Kunzelmann C, Blattler T, Aguzzi A, Freyssinet JM (2004) Modulation of signal transduction through the cellular prion protein is linked to its incorporation in lipid rafts. Cell Mol Life Sci 61(23):2998–3007
Porto-Carreiro I, Fevrier B, Paquet S, Vilette D, Raposo G (2005) Prions and exosomes: from PrPc trafficking to PrPsc propagation. Blood Cells Mol Dis 35(2):143–148
Robertson C, Booth SA, Beniac DR, Coulthart MB, Booth TF, McNicol A (2006) Cellular prion protein is released on exosomes from activated platelets. Blood 107(10):3907–3911
Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, Morris RJ (2008) LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci 121(Pt 6):773–783
Gauczynski S, Peyrin JM, Haik S, Leucht C, Hundt C, Rieger R, Krasemann S, Deslys JP, Dormont D, Lasmezas CI, Weiss S (2001) The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. Embo J 20(21):5863–5875
Pauly PC, Harris DA (1998) Copper stimulates endocytosis of the prion protein. J Biol Chem 273(50):33107–33110
Perera WS, Hooper NM (2001) Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol 11(7):519–523
Caetano FA, Lopes MH, Hajj GN, Machado CF, Pinto Arantes C, Magalhaes AC, Vieira Mde P, Americo TA, Massensini AR, Priola SA, Vorberg I, Gomez MV, Linden R, Prado VF, Martins VR, Prado MA (2008) Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J Neurosci 28(26):6691–6702
Shyng SL, Lehmann S, Moulder KL, Harris DA (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J Biol Chem 270(50):30221–30229
Kiachopoulos S, Heske J, Tatzelt J, Winklhofer KF (2004) Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic 5(6):426–436
Stuermer CA, Langhorst MF, Wiechers MF, Legler DF, Von Hanwehr SH, Guse AH, Plattner H (2004) PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. Faseb J 18(14):1731–1733
Calzolai L, Zahn R (2003) Influence of pH on NMR structure and stability of the human prion protein globular domain. J Biol Chem 278(37):35592–35596
Swietnicki W, Petersen R, Gambetti P, Surewicz WK (1997) pH-dependent stability and conformation of the recombinant human prion protein PrP(90–231). J Biol Chem 272(44):27517–27520
Marques AF, Cordeiro Y, Silva JL, Lima LM (2009) Enhanced prion protein stability coupled to DNA recognition and milieu acidification. Biophys Chem 141(2–3):135–139
Hornemann S, Glockshuber R (1998) A scrapie-like unfolding intermediate of the prion protein domain PrP(121–231) induced by acidic pH. Proc Natl Acad Sci USA 95(11):6010–6014
Alonso DO, DeArmond SJ, Cohen FE, Daggett V (2001) Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc Natl Acad Sci USA 98(6):2985–2989
Matsunaga Y, Peretz D, Williamson A, Burton D, Mehlhorn I, Groth D, Cohen FE, Prusiner SB, Baldwin MA (2001) Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: dependence of conformation on pH. Proteins 44(2):110–118
Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R (1993) Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 32(4):1009–1016
Starke R, Harrison P, Drummond O, Macgregor I, Mackie I, Machin S (2003) The majority of cellular prion protein released from endothelial cells is soluble. Transfusion 43 (5):677–678; author reply 678
Roberts TK, Eugenin EA, Morgello S, Clements JE, Zink MC, Berman JW (2010) PrPC, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am J Pathol 177(4):1848–1860
Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308(5727):1435–1439
Klingeborn M, Race B, Meade-White KD, Rosenke R, Striebel JF, Chesebro B (2011) Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol 85(4):1484–1494
Campana V, Caputo A, Sarnataro D, Paladino S, Tivodar S, Zurzolo C (2007) Characterization of the properties and trafficking of an anchorless form of the prion protein. J Biol Chem 282(31):22747–22756
Weissmann C, Bueler H, Fischer M, Sauer A, Aguet M (1994) Susceptibility to scrapie in mice is dependent on PrPC. Philos Trans R Soc Lond B Biol Sci 343(1306):431–433
Samaia HB, Brentani RR (1998) Can loss-of-function prion-related diseases exist? Mol Psychiatry 3(3):196–197
Weissmann C, Flechsig E (2003) PrP knock-out and PrP transgenic mice in prion research. Br Med Bull 66:43–60
Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O (2000) Signal transduction through prion protein. Science 289(5486):1925–1928
Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R (2002) Cellular prion protein transduces neuroprotective signals. Embo J 21(13):3317–3326
Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. Embo J 21(13):3307–3316
Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R, Martins VR (2005) Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J Neurosci 25(49):11330–11339
Americo TA, Chiarini LB, Linden R (2007) Signaling induced by hop/STI-1 depends on endocytosis. Biochem Biophys Res Commun 358(2):620–625
Siegel GJ (2006) Basic neurochemistry: molecular, cellular, and medical aspects, 7th edn. Elsevier, Amsterdam
Feng XH, Derynck R (2005) Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 21:659–693
Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MM, Linden R (2010) Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol 12(2):63–86
Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR (2000) Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res 76(1):85–92
Hajj GN, Lopes MH, Mercadante AF, Veiga SS, da Silveira RB, Santos TG, Ribeiro KC, Juliano MA, Jacchieri SG, Zanata SM, Martins VR (2007) Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J Cell Sci 120(Pt 11):1915–1926
Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, Daude N, Lau A, Young R, Xu L, Carlson GA, Williams D, Westaway D, Schmitt-Ulms G (2009) Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 5(10):e1000608
Rutishauser D, Mertz KD, Moos R, Brunner E, Rulicke T, Calella AM, Aguzzi A (2009) The comprehensive native interactome of a fully functional tagged prion protein. PLoS One 4(2):e4446
Sorgato MC, Peggion C, Bertoli A (2009) Is, indeed, the prion protein a Harlequin servant of “many” masters? Prion 3(4):202–205
Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB (2001) Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 314(5):1209–1225
Beraldo FH, Arantes CP, Santos TG, Machado CF, Roffe M, Hajj GN, Lee KS, Magalhaes AC, Caetano FA, Mancini GL, Lopes MH, Americo TA, Magdesian MH, Ferguson SS, Linden R, Prado MA, Martins VR (2011) Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. Faseb J 25(1):265–279
Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA, Martins VR (2010) Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem 285(47):36542–36550
Aguzzi A, O’Connor T (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov 9(3):237–248
Cobb NJ, Surewicz WK (2009) Prion diseases and their biochemical mechanisms. Biochemistry 48(12):2574–2585
Baskakov IV, Legname G, Gryczynski Z, Prusiner SB (2004) The peculiar nature of unfolding of the human prion protein. Protein Sci 13(3):586–595
Baskakov IV, Legname G, Prusiner SB, Cohen FE (2001) Folding of prion protein to its native alpha-helical conformation is under kinetic control. J Biol Chem 276(23):19687–19690
Hart T, Hosszu LL, Trevitt CR, Jackson GS, Waltho JP, Collinge J, Clarke AR (2009) Folding kinetics of the human prion protein probed by temperature jump. Proc Natl Acad Sci USA 106(14):5651–5656
Apetri AC, Surewicz K, Surewicz WK (2004) The effect of disease-associated mutations on the folding pathway of human prion protein. J Biol Chem 279(17):18008–18014
Apetri AC, Maki K, Roder H, Surewicz WK (2006) Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J Am Chem Soc 128(35):11673–11678
Apetri AC, Surewicz WK (2002) Kinetic intermediate in the folding of human prion protein. J Biol Chem 277(47):44589–44592
Jackson GS, Hosszu LL, Power A, Hill AF, Kenney J, Saibil H, Craven CJ, Waltho JP, Clarke AR, Collinge J (1999) Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 283(5409):1935–1937
Perez DR, Damberger FF, Wuthrich K (2010) Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol 400(2):121–128
Perez DR, Damberger FF, Wuthrich K (2010) Erratum to “Horse Prion Protein NMR Structure and Comparisons with Related Variants of the Mouse Prion Protein” [J Mol Biol 400/2 (2010) 121–128]. J Mol Biol
Wen Y, Li J, Yao W, Xiong M, Hong J, Peng Y, Xiao G, Lin D (2010) Unique structural characteristics of the rabbit prion protein. J Biol Chem 285(41):31682–31693
Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K (2005) Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci USA 102(3):646–650
Calzolai L, Lysek DA, Perez DR, Guntert P, Wuthrich K (2005) Prion protein NMR structures of chickens, turtles, and frogs. Proc Natl Acad Sci USA 102(3):651–655
Lysek DA, Schorn C, Nivon LG, Esteve-Moya V, Christen B, Calzolai L, von Schroetter C, Fiorito F, Herrmann T, Guntert P, Wuthrich K (2005) Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci USA 102(3):640–645
Hornemann S, Schorn C, Wuthrich K (2004) NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep 5(12):1159–1164
Grasberger B, Minton AP, DeLisi C, Metzger H (1986) Interaction between proteins localized in membranes. Proc Natl Acad Sci USA 83(17):6258–6262
Fan QR, Hendrickson WA (2005) Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433(7023):269–277
Meyer RK, Lustig A, Oesch B, Fatzer R, Zurbriggen A, Vandevelde M (2000) A monomer-dimer equilibrium of a cellular prion protein (PrPC) not observed with recombinant PrP. J Biol Chem 275(48):38081–38087
Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA (2004) Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol 22(6):724–731
Malaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA (2009) Regulation of embryonic cell adhesion by the prion protein. PLoS Biol 7(3):e55
Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H (1997) The cellular prion protein binds copper in vivo. Nature 390(6661):684–687
Hornshaw MP, McDermott JR, Candy JM (1995) Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem Biophys Res Commun 207(2):621–629
Millhauser GL (2007) Copper and the prion protein: methods, structures, function, and disease. Annu Rev Phys Chem 58:299–320
Davies P, Brown DR (2008) The chemistry of copper binding to PrP: is there sufficient evidence to elucidate a role for copper in protein function? Biochem J 410(2):237–244
Haigh CL, Marom SY, Collins SJ (2010) Copper, endoproteolytic processing of the prion protein and cell signalling. Front Biosci 15:1086–1104
Walter ED, Stevens DJ, Spevacek AR, Visconte MP, Dei Rossi A, Millhauser GL (2009) Copper binding extrinsic to the octarepeat region in the prion protein. Curr Protein Pept Sci 10(5):529–535
Viles JH, Klewpatinond M, Nadal RC (2008) Copper and the structural biology of the prion protein. Biochem Soc Trans 36(Pt 6):1288–1292
Younan ND, Klewpatinond M, Davies P, Ruban AV, Brown DR, Viles JH (2011) Copper(II)-induced secondary structure changes and reduced folding stability of the prion protein. J Mol Biol 410(3):369–382
Riihimaki ES, Martinez JM, Kloo L (2008) Structural effects of Cu(II)-coordination in the octapeptide region of the human prion protein. Phys Chem Chem Phys 10(18):2488–2495
Valensin G, Molteni E, Valensin D, Taraszkiewicz M, Kozlowski H (2009) Molecular dynamics study of the Cu2 + binding-induced “structuring” of the N-terminal domain of human prion protein. J Phys Chem B 113(11):3277–3279
Zahn R (2003) The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J Mol Biol 334(3):477–488
Millhauser GL (2004) Copper binding in the prion protein. Acc Chem Res 37(2):79–85
Thakur AK, Srivastava AK, Srinivas V, Chary KV, Rao CM (2011) Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J Biol Chem. doi:10.1074/jbc.M111.265645
Taylor DR, Watt NT, Perera WS, Hooper NM (2005) Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci 118(Pt 21):5141–5153
Mani K, Cheng F, Havsmark B, Jonsson M, Belting M, Fransson LA (2003) Prion, amyloid beta-derived Cu(II) ions, or free Zn(II) ions support S-nitroso-dependent autocleavage of glypican-1 heparan sulfate. J Biol Chem 278(40):38956–38965
Cheng F, Lindqvist J, Haigh CL, Brown DR, Mani K (2006) Copper-dependent co-internalization of the prion protein and glypican-1. J Neurochem 98(5):1445–1457
Moudjou M, Bernard J, Sabuncu E, Langevin C, Laude H (2007) Glycan chains modulate prion protein binding to immobilized metal ions. Neurochem Int 50(5):689–695
Rudd PM, Wormald MR, Wing DR, Prusiner SB, Dwek RA (2001) Prion glycoprotein: structure, dynamics, and roles for the sugars. Biochemistry 40(13):3759–3766
Wilkins S, Choglay AA, Chapple JP, van der Spuy J, Rhie A, Birkett CR, Cheetham ME (2010) The binding of the molecular chaperone Hsc70 to the prion protein PrP is modulated by pH and copper. Int J Biochem Cell Biol 42(7):1226–1232
Ma J, Lindquist S (2001) Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A 98(26):14955–14960
Nandi PK, Leclerc E (1999) Polymerization of murine recombinant prion protein in nucleic acid solution. Arch Virol 144(9):1751–1763
Nandi PK (1997) Interaction of prion peptide HuPrP106–126 with nucleic acid. Arch Virol 142(12):2537–2545
Cordeiro Y, Machado F, Juliano L, Juliano MA, Brentani RR, Foguel D, Silva JL (2001) DNA converts cellular prion protein into the beta-sheet conformation and inhibits prion peptide aggregation. J Biol Chem 276(52):49400–49409
Vieira TC, Reynaldo DP, Gomes MP, Almeida MS, Cordeiro Y, Silva JL (2011) Heparin binding by murine recombinant prion protein leads to transient aggregation and formation of rna-resistant species. J Am Chem Soc 133(2):334–344
Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425(6959):717–720
Record MT Jr, Lohman ML, De Haseth P (1976) Ion effects on ligand-nucleic acid interactions. J Mol Biol 107(2):145–158
Gomes MP, Millen TA, Ferreira PS, e Silva NL, Vieira TC, Almeida MS, Silva JL, Cordeiro Y (2008) Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J Biol Chem 283(28):19616–19625
Lima LM, Cordeiro Y, Tinoco LW, Marques AF, Oliveira CL, Sampath S, Kodali R, Choi G, Foguel D, Torriani I, Caughey B, Silva JL (2006) Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 45(30):9180–9187
Bera A, Roche AC, Nandi PK (2007) Bending and unwinding of nucleic acid by prion protein. Biochemistry 46(5):1320–1328
Ahmad S, Sarai A (2005) PSSM-based prediction of DNA binding sites in proteins. BMC Bioinformatics 6:33
Silva JL, Lima LM, Foguel D, Cordeiro Y (2008) Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem Sci 33(3):132–140
King DJ, Safar JG, Legname G, Prusiner SB (2007) Thioaptamer interactions with prion proteins: sequence-specific and non-specific binding sites. J Mol Biol 369(4):1001–1014
Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D (2005) Search for a prion-specific nucleic acid. J Virol 79(16):10796–10806
Takemura K, Wang P, Vorberg I, Surewicz W, Priola SA, Kanthasamy A, Pottathil R, Chen SG, Sreevatsan S (2006) DNA aptamers that bind to PrP(C) and not PrP(Sc) show sequence and structure specificity. Exp Biol Med (Maywood) 231(2):204–214
Snow AD, Wight TN, Nochlin D, Koike Y, Kimata K, DeArmond SJ, Prusiner SB (1990) Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab Invest 63(5):601–611
Ladogana A, Casaccia P, Ingrosso L, Cibati M, Salvatore M, Xi YG, Masullo C, Pocchiari M (1992) Sulphate polyanions prolong the incubation period of scrapie-infected hamsters. J Gen Virol 73(Pt 3):661–665
Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, Caughey B (2001) Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. Embo J 20(3):377–386
Ben-Zaken O, Tzaban S, Tal Y, Horonchik L, Esko JD, Vlodavsky I, Taraboulos A (2003) Cellular heparan sulfate participates in the metabolism of prions. J Biol Chem 278(41):40041–40049
Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S (2005) Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem 280(29):26873–26879
Silva JL, Gomes MP, Vieira TC, Cordeiro Y (2010) PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front Biosci 15:132–150
Caughey B, Race RE (1994) Scrapie-associated PrP accumulation and its inhibition: revisiting the amyloid-glycosaminoglycan connection. Ann N Y Acad Sci 724:290–295
Warner RG, Hundt C, Weiss S, Turnbull JE (2002) Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem 277(21):18421–18430
Pan T, Wong BS, Liu T, Li R, Petersen RB, Sy MS (2002) Cell-surface prion protein interacts with glycosaminoglycans. Biochem J 368(Pt 1):81–90
Taubner LM, Bienkiewicz EA, Copie V, Caughey B (2010) Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J Mol Biol 395(3):475–490
Caughey B, Raymond GJ (1993) Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol 67(2):643–650
Gabizon R, Meiner Z, Halimi M, Ben-Sasson SA (1993) Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J Cell Physiol 157(2):319–325
Brenot F, Aubry L, Martin JB, Satre M, Klein G (1992) Kinetics of endosomal acidification in Dictyostelium discoideum amoebae. 31P-NMR evidence for a very acidic early endosomal compartment. Biochimie 74(9–10):883–895
Weisz OA (2003) Acidification and protein traffic. Int Rev Cytol 226:259–319
Freire E, Coelho-Sampaio T (2000) Self-assembly of laminin induced by acidic pH. J Biol Chem 275(2):817–822
Mitra RC, Zhang Z, Alexov E (2011) In silico modeling of pH-optimum of protein-protein binding. Proteins 79(3):925–936
Romano SA, Cordeiro Y, Lima LM, Lopes MH, Silva JL, Foguel D, Linden R (2009) Reciprocal remodeling upon binding of the prion protein to its signaling partner hop/STI1. Faseb J 23(12):4308–4316
Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M (2005) Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 169(2):341–354
Roffe M, Beraldo FH, Bester R, Nunziante M, Bach C, Mancini G, Gilch S, Vorberg I, Castilho BA, Martins VR, Hajj GN (2010) Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc Natl Acad Sci USA 107(29):13147–13152
Erlich RB, Kahn SA, Lima FR, Muras AG, Martins RA, Linden R, Chiarini LB, Martins VR, Moura Neto V (2007) STI1 promotes glioma proliferation through MAPK and PI3 K pathways. Glia 55(16):1690–1698
Santos TG, Silva IR, Costa-Silva B, Lepique AP, Martins VR, Lopes MH (2011) Enhanced Neural Progenitor/Stem Cells Self-Renewal via the Interaction of Stress Inducible Protein 1 with the Prion Protein. Stem Cells 29(7):1126–1136
Arruda-Carvalho M, Njaine B, Silveira MS, Linden R, Chiarini LB (2007) Hop/STI1 modulates retinal proliferation and cell death independent of PrPC. Biochem Biophys Res Commun 361(2):474–480
Arantes C, Nomizo R, Lopes MH, Hajj GN, Lima FR, Martins VR (2009) Prion protein and its ligand stress inducible protein 1 regulate astrocyte development. Glia 57(13):1439–1449
Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG (1994) Prion protein is necessary for normal synaptic function. Nature 370(6487):295–297
Borchelt DR, Sisodia SS (1996) Loss of functional prion protein: a role in prion disorders? Chem Biol 3(8):619–621
Parchi P, Strammiello R, Giese A, Kretzschmar H (2011) Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol 121(1):91–112
Wadsworth JD, Collinge J (2011) Molecular pathology of human prion disease. Acta Neuropathol 121(1):69–77
Chiesa R, Piccardo P, Biasini E, Ghetti B, Harris DA (2008) Aggregated, wild-type prion protein causes neurological dysfunction and synaptic abnormalities. J Neurosci 28(49):13258–13267
Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA (2004) Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303(5663):1514–1516
Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, Muhs A, Lashuel HA (2011) Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J Biol Chem 286(10):8585–8596
Siskova Z, Page A, O’Connor V, Perry VH (2009) Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping. Am J Pathol 175(4):1610–1621
Gray BC, Siskova Z, Perry VH, O’Connor V (2009) Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology. Neurobiol Dis 35(1):63–74
Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP, Scott H, Rawlins JN, Perry VH (2003) Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci 17(10):2147–2155
Caughey B, Baron GS (2006) Prions and their partners in crime. Nature 443(7113):803–810
Martins VR, Linden R, Prado MA, Walz R, Sakamoto AC, Izquierdo I, Brentani RR (2002) Cellular prion protein: on the road for functions. FEBS Lett 512(1–3):25–28
Ehrlich M, Horbelt D, Marom B, Knaus P, Henis YI (2011) Homomeric and heteromeric complexes among TGF-beta and BMP receptors and their roles in signaling. Cell Signal 23(9):1424–1432
Ciruela F, Gomez-Soler M, Guidolin D, Borroto-Escuela DO, Agnati LF, Fuxe K, Fernandez-Duenas V (2011) Adenosine receptor containing oligomers: their role in the control of dopamine and glutamate neurotransmission in the brain. Biochim Biophys Acta 1808(5):1245–1255
Pin JP, Comps-Agrar L, Maurel D, Monnier C, Rives ML, Trinquet E, Kniazeff J, Rondard P, Prezeau L (2009) G-protein-coupled receptor oligomers: two or more for what? Lessons from mGlu and GABAB receptors. J Physiol 587(Pt 22):5337–5344
Kramp BK, Sarabi A, Koenen RR, Weber C (2011) Heterophilic chemokine receptor interactions in chemokine signaling and biology. Exp Cell Res 317(5):655–663
Nakata H, Yoshioka K, Kamiya T (2004) Purinergic-receptor oligomerization: implications for neural functions in the central nervous system. Neurotox Res 6(4):291–297
Schmidt U, Weiss M (2011) Anomalous diffusion of oligomerized transmembrane proteins. J Chem Phys 134(16):165101
Guigas G, Weiss M (2006) Size-dependent diffusion of membrane inclusions. Biophys J 91(7):2393–2398
Xu W, Kimelman D (2007) Mechanistic insights from structural studies of beta-catenin and its binding partners. J Cell Sci 120(Pt 19):3337–3344
Enghild JJ, Salvesen G, Thogersen IB, Pizzo SV (1989) Proteinase binding and inhibition by the monomeric alpha-macroglobulin rat alpha 1-inhibitor-3. J Biol Chem 264(19):11428–11435
Rebowski G, Namgoong S, Boczkowska M, Leavis PC, Navaza J, Dominguez R (2010) Structure of a longitudinal actin dimer assembled by tandem w domains: implications for actin filament nucleation. J Mol Biol 403(1):11–23
Kiermayr S, Stiasny K, Heinz FX (2009) Impact of quaternary organization on the antigenic structure of the tick-borne encephalitis virus envelope glycoprotein E. J Virol 83(17):8482–8491
Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM (2004) Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem 279(6):4625–4631
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95(1):55–66
Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M (1999) Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci 19(3):964–973
Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT (1998) Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95(1):41–53
Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH (2003) Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 12(7):749–757
Marx FP, Holzmann C, Strauss KM, Li L, Eberhardt O, Gerhardt E, Cookson MR, Hernandez D, Farrer MJ, Kachergus J, Engelender S, Ross CA, Berger K, Schols L, Schulz JB, Riess O, Kruger R (2003) Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Hum Mol Genet 12(11):1223–1231
Sanghera N, Swann MJ, Ronan G, Pinheiro TJ (2009) Insight into early events in the aggregation of the prion protein on lipid membranes. Biochim Biophys Acta 1788(10):2245–2251
Baskakov IV, Breydo L (2007) Converting the prion protein: what makes the protein infectious. Biochim Biophys Acta 1772(6):692–703
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457(7233):1128–1132
Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L (2011) Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci 31(29):10427–10431
Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J (2011) Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2:336
Saijo E, Scheff SW, Telling GC (2011) Unaltered prion protein expression in Alzheimer disease patients. Prion 5(2):109–116
Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ (2011) Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 31(20):7259–7263
Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, Zhu X, McGeer PL, McGeehan J, Kneale G, Rincon-Limas DE, Fernandez-Funez P, Lee HG, Smith MA, Petersen RB, Guo JP (2011) Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem 286(17):15095–15105
Forloni G, Balducci C (2011) beta-amyloid oligomers and prion protein: Fatal attraction? Prion 5(1):10–15
Kessels HW, Nguyen LN, Nabavi S, Malinow R (2010) The prion protein as a receptor for amyloid-beta. Nature 466(7308):E3–E4 discussion E4-5
Benilova I, De Strooper B (2010) Prion protein in Alzheimer’s pathogenesis: a hot and controversial issue. EMBO Mol Med 2(8):289–290
Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A (2010) Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med 2(8):306–314
Chen S, Yadav SP, Surewicz WK (2010) Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem 285(34):26377–26383
Caetano FA, Beraldo FH, Hajj GN, Guimaraes AL, Jurgensen S, Wasilewska-Sampaio AP, Hirata PH, Souza I, Machado CF, Wong DY, De Felice FG, Ferreira ST, Prado VF, Rylett RJ, Martins VR, Prado MA (2011) Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem 117(3):538–553
Bate C, Williams A (2011) Amyloid-{beta}-induced synapse damage is mediated via cross-linkage of the cellular prion protein. J Biol Chem. doi:10.1074/jbc.M111.248724
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31(18):6627–6638
Wang D, Govindaiah G, Liu R, De Arcangelis V, Cox CL, Xiang YK (2010) Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. Faseb J 24(9):3511–3521
Uhasz GJ, Barkoczi B, Vass G, Datki Z, Hunya A, Fulop L, Budai D, Penke B, Szegedi V (2010) Fibrillar Abeta (1–42) enhances NMDA receptor sensitivity via the integrin signaling pathway. J Alzheimers Dis 19(3):1055–1067
Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL (2008) Amyloid beta oligomers induce impairment of neuronal insulin receptors. Faseb J 22(1):246–260
Abbott JJ, Howlett DR, Francis PT, Williams RJ (2008) Abeta(1–42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol Aging 29(7):992–1001
Parameshwaran K, Sims C, Kanju P, Vaithianathan T, Shonesy BC, Dhanasekaran M, Bahr BA, Suppiramaniam V (2007) Amyloid beta-peptide Abeta(1–42) but not Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse 61(6):367–374
Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA (2010) Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci 30(27):9166–9171
Li X, Rowland LP, Mitsumoto H, Przedborski S, Bird TD, Schellenberg GD, Peskind E, Johnson N, Siddique T, Mesulam MM, Weintraub S, Mastrianni JA (2005) Prion protein codon 129 genotype prevalence is altered in primary progressive aphasia. Ann Neurol 58(6):858–864
Dupuis L, Mbebi C, Gonzalez de Aguilar JL, Rene F, Muller A, de Tapia M, Loeffler JP (2002) Loss of prion protein in a transgenic model of amyotrophic lateral sclerosis. Mol Cell Neurosci 19(2):216–224
Steinacker P, Hawlik A, Lehnert S, Jahn O, Meier S, Gorz E, Braunstein KE, Krzovska M, Schwalenstocker B, Jesse S, Propper C, Bockers T, Ludolph A, Otto M (2010) Neuroprotective Function of Cellular Prion Protein in a Mouse Model of Amyotrophic Lateral Sclerosis. Am J Pathol 176(3):1409–1420
Lee S, Antony L, Hartmann R, Knaus KJ, Surewicz K, Surewicz WK, Yee VC (2010) Conformational diversity in prion protein variants influences intermolecular beta-sheet formation. Embo J 29(1):251–262
Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC (2001) Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 8(9):770–774
Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, Pai EF, Chakrabartty A (2010) Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci USA 107(46):19808–19813
Linden R, Martins VR, Prado MA (2012) Prion protein. In: Choi S (ed) Encyclopedia of signaling molecules. Springer, p in press
Romano SA, Cordeiro Y, Lima LM, Lopes MH, Silva JL, Foguel D, Linden R (2009) Uncovering molecular structural mechanisms of signaling mediated by the prion protein. LNLS activity report-science highlights:30–36
Acknowledgments
The authors' work is supported by grants from CNPq, FAPERJ, and the National Institute of Structural Biology and Bioimaging (INBEB-MCT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Linden, R., Cordeiro, Y. & Lima, L.M.T.R. Allosteric function and dysfunction of the prion protein. Cell. Mol. Life Sci. 69, 1105–1124 (2012). https://doi.org/10.1007/s00018-011-0847-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0847-7