
Abstract
It has recently been suggested that heat shock protein (Hsp) 70, an intracellular protein, can be released into the extracellular compartment and exert important immunomodulatory functions. Although elevated Hsp70 has been found in synovial fluid from patients with rheumatoid arthritis (RA), its sources and extracellular functions remain unclear. In this study, we explored whether stress response such as heat stress or exposure to tumor necrosis factor-α (TNF-α) could induce Hsp70 release from RA fibroblast-like synoviocytes (FLSs) and whether extracellular Hsp70 would stimulate cytokine production in RA FLSs. Cultured FLSs were obtained from patients with RA. The expression of intracellular Hsp70 was studied by Western blot. Hsp70 release and the production of interleukin (IL)-6, IL-8, and IL-10 by RA FLSs were studied by specific enzyme-linked immunosorbent assays. The levels of Toll-like receptor (TLR) 2 and 4 mRNA and protein in FLSs were analyzed using reverse transcription-polymerase chain reaction and Western blotting. Treatment with sublethal heat shock or TNF-α results in the up-regulation of intracellular Hsp70 in FLSs and Hsp70 release from RA FLSs. In vitro studies show that extracellular Hsp70 can induce anti-inflammatory cytokine IL-10 production in FLSs. The mRNA and protein expression of TLR2 and TLR4 was demonstrated in FLSs, and TLR4 blocking abrogated the up-regulatory effects of Hsp70 on IL-10 production. Thus, these results lend support to the hypothesis that Hsp70 is actively released from FLSs in response to heat shock or TNF-α and Hsp70 may be a major paracrine/autocrine inducer of IL-10 production in FLSs via TLR4.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation of synovial membranes and proliferation of the synovial lining, leading to cartilage damage and ultimately joint destruction (Georganas et al. 2000). Although the pathogenesis of RA remains unclear, a variety of cytokines have been implicated in the development of this disease (Feldmann and Maini 1999). For instance, interleukin (IL)-6 and IL-8, the most abundant cytokines produced mainly by FLSs in the synovium, contribute to joint inflammation in RA (Min et al. 2004; Nakahara and Nishimoto 2006). In contrast to the functions of IL-6 and IL-8, IL-10 is known to down-regulate the production of pro-inflammatory cytokines including TNF-α, IFN-γ, IL-2, and IL-6 (Cho et al. 2002). The production of IL-10 is correlated negatively with arthritis progression and joint destruction and the administration of IL-10 inhibits progression of arthritis in animal model (Isomaki et al. 1996; Tanaka et al. 1996; Walmsley et al. 1996). Consequently, IL-10 is recognized as a major anti-inflammatory molecule and is considered a potentially therapeutic reagent in arthritis and other diseases.
Heat shock proteins (HSPs) are a family of highly conserved proteins found in all eukaryotes and prokaryotes. The HSP70 family is the best characterized of these and include the constitutive HSP70 (Hsc70) and stress-inducible HSP70 (Hsp70). Although HSPs are traditionally regarded as intracellular proteins and their primary functions appear to be as molecular chaperones involved in protein folding and transport (Kiang and Tsokos 1998; Tsan and Gao 2004), accumulating data suggest that HSPs are actively released and have important extracellular functions. It has been shown that Hsp70 is released from a variety of cells in response to cellular stress. Hsp70 release has been demonstrated in cultured glial cells (Guzhova et al. 2001), human peripheral blood mononuclear cells (PBMCs) (Lancaster and Febbraio 2005), human macrophages (Svensson et al. 2006), human epithelial cells (Broquet et al. 2003), and human tumor cells (Mambula and Calderwood 2006). Several studies have shown that Hsp70 is actively released from viable cells via a mechanism involving lipid rafts (Broquet et al. 2003).
In recent years, there has been an intense interest in the extracellular function of HSPs. A growing body of evidence suggests that extracellular HSPs have important immunomodulatory functions (Hauet-Broere et al. 2006; van Eden et al. 2005). Recombinant human Hsp70 stimulates the production of pro-inflammatory cytokines such as TNF-a and IL-6 in human monocytes and macrophages, indicating that extracellular Hsp70, released either as a result of cellular necrosis or via active secretion, may serve as a danger signal to the innate immune system and may also be relevant to the establishment of autoimmune diseases (Asea et al. 2000; Tsan and Gao 2004). While the cell surface receptor for Hsp70 has not yet been completely characterized, some data implicate Toll-like receptors (TLR) 2 and 4 in Hsp70 binding (Asea et al. 2002; Fleshner and Johnson 2005). Autologous release of Hsp70 induces a pro-inflammatory response in innate immune cells potentially mediated via TLR2 and TLR4 (Dybdahl et al. 2002).
Although elevated Hsp70 has been found in synovial fluid from RA patients (Martin et al. 2003), its sources and functions remain unclear. Fibroblast-like synoviocytes (FLSs) in RA have been previously shown to produce large amounts of intracellular Hsp70 in response to heat shock or inflammatory stress (Schett et al. 1998). Thus, the elevated Hsp70 in RA synovial fluid may be derived from fibroblasts that are in close proximity to synovial fluid and play crucial roles in the joint inflammatory and destructive process. It has also been demonstrated that RA FLSs express high levels of TLR2 and TLR4 (Gutierrez-Canas et al. 2006; Kim et al. 2007; Pierer et al. 2004). Therefore, it is of considerable interest to further explore the Hsp70 release from FLSs and its modulation of FLSs function. Accordingly, the aim of this study was to determine whether Hsp70 could be actively released from RA FLSs after stress and whether extracellular Hsp70 would stimulate cytokine production in RA FLSs through the activation of TLRs.
2 Materials and methods
2.1 Cell culture
FLSs were isolated from the synovial tissues of four RA patients undergoing joint replacement surgery as previously described (Aupperle et al. 1999). The diagnoses of RA conformed to the 1987 revised American Council of Rheumatology criteria (Arnett et al. 1988). Informed consent was obtained from each patient in conformity with requirements of the ethics committee at Xiangya School of Medicine, Central South University. Briefly, tissue samples were minced and treated with 1 mg/ml collagenase for 1–2 h at 37°C. After washing, the cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies, Rockville, MD, USA) supplemented with 10% heat-inactivated fetal calf serum (FCS; Life Technologies), 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine in a humidified incubator with 5% CO2 and 95% air. After overnight culture, non-adherent cells were removed and adherent cells were cultivated in DMEM plus 10% FCS. At confluence, cells were trypsinized, split at a 1:3 ratio, and recultured in the same medium. All the experiments described here utilize the FLSs between the fourth and ninth passages. A homogeneous population of FLS was determined by flow cytometry (<1% CD11b, <1% phagocytic, and <1% FcγRII positive).
2.2 Heat shock treatment
FLSs were sealed in screw cap flasks containing an atmosphere of 5% CO2 and 95% air. These flasks were then immersed completely in a thermostatically regulated water bath at indicated temperature for 1 h. Cells were allowed to recover at 37°C, 5% CO2 for different durations for the synthesis and release of Hsp70.
2.3 Hsp70 quantification
Hsp70 levels in supernatants were measured using a commercially available sandwich enzyme-linked immunosorbent assay (ELISA; Stressgen, Victoria, British Columbia, Canada) according to the manufacturer’s instructions. The detection sensitivity is 200 pg/ml. The antibodies (Abs) used in the ELISA are specific for Hsp70 and does not cross-react with Hsc70.
2.4 Cytokine quantification
Following stimulation of human RA FLSs by recombinant human Hsp70 (Stressgen; catalog no. ESP555), supernatants were harvested. IL-6, IL-8, and IL-10 levels were measured using a sandwich ELISA following the manufacturer’s instructions (Jingmei, Shenzhen, China).
2.5 Reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA of cells was extracted with TRIzol reagent (Gibco, Grand Island, NY, USA) as described by the manufacturer’ instructions. Total RNA (1 μg) was then reverse transcribed with avian myeloblastosis virus and oligo (dT) primer (Takara, Dalian, China) and amplified in a final volume of 20 μl containing 1 μl RT mix template, 1 U Taq DNA polymerase (Takara), and 10 pmol of each primer (sense and antisense). For human TLR2 amplification, the primers 5′-GCCAAAGTCTTGATTGATTGG-3′ and 5′-TTGAAGTTCTCCAGCTCCTG-3′ were used. For human TLR4, the primers 5′-TGGATACGTTTCCTTATAAG-3′ and 5′-GAAATGGAGGCACCCCTTC-3′ were used. Amplification parameters were: 30 s of denaturation at 95°C, 30 s of annealing at 61°C, and 30 s of elongation at 72°C for 35 cycles. For glyceraldehyde 3-phosphate dehydrogenase (GAPDH), the primers 5′-CGATGCTGGGCGTGAGTAC-3′ and 5′-CGTTCAGCTCAGGGATGACC-3′ were used. Amplification parameters were: 30 s of denaturation at 95°C, 30 s of annealing at 55°C, and 30 s of elongation at 72°C for 30 cycles.
2.6 Western blot analysis
Aliquots of total cell lysates were reduced and denatured by boiling in sodium dodecyl sulfate (SDS) sample buffer [2× = 100 mM Tris–HCl (pH 6.8); 4% w/v SDS; 20% glycerol; 200 mM dithiothreitol; 0.1% w/v bromophenol blue], fractionated on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a nitrocellulose membrane (Promega, Madison, WI, USA). Membranes were blocked in blocking buffer (2% bovine serum albumin and 0.2% Tween-20 in Tris-buffered saline) at room temperature for 4 h, incubated 2 h at 25°C with the primary antibody [mouse anti-TLR4 (eBioscience, San Diego, CA, USA; 1:250) and mouse anti-TLR2 monoclonal (eBioscience, 1:400), rabbit anti-Hsp70 (Stressgen, 1:1,000), and mouse anti-GAPDH monoclonal antibody (Upstate, Lake Placid, NY, USA; 1:1,000)], followed by peroxidase-conjugated secondary antibody IgG [anti-mouse (Boster Biotech, Wuhan, China; 1:1,000) and anti-rabbit (Boster Biotech, 1:1,000)] for 1 h at 25°C. The signals were visualized by diaminobenzidine detection (Boster Biotech) following the manufacturer’s instruction and the bands of interest were scanned and counts quantitated with the Band Leader software (Shanghai, China).
2.7 Measurement of lactate dehydrogenase (LDH) release
LDH was measured spectrophotometrically at 340 nm using a commercial kit according to the manufacturer’s instructions (Biosino, Beijing, China). LDH release was expressed as a percentage of the LDH in the medium over the total LDH (lysate).
3 Statistical analysis
Data in the figures and text were expressed as means ± SEM. A significance of differences (p < 0.05) between groups was determined by two-tailed Student’s t test or Fisher’s Least Significant Difference test.
4 Results
4.1 Heat stress or inflammatory stress increases intra- and extracellular Hsp70 in FLSs
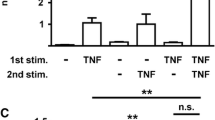
To examine whether FLSs can actively release Hsp70, we tested the effect of heat shock (HS) on the expression of intracellular Hsp70 in FLSs and Hsp70 release from FLSs. As shown in Fig. 1a, exposure of FLSs to HS at 40–44°C resulted in a marked up-regulation of intracellular Hsp70 compared with control conditions (37°C). Exposure of FLSs to HS between 40 and 42°C resulted in a significant increase in Hsp70 release without significant cell death as indicated by LDH release (Fig. 1b and c). Although exposure of FLSs to HS at 43–44°C resulted in a marked increase in Hsp70 release, FLSs already showed significant cell death (Fig. 1b and c). Time course experiments showed the expression of intracellular Hsp70 and Hsp70 release were maximal by 12–24 h post-heat shock (42°C) (Fig. 1d and e). These data show non-lethal HS-induced active release of Hsp70 from FLSs.

Heat shock induces Hsp70 release from FLSs. a–c FLSs were heated at the indicated temperatures for 1 h and allowed to recover to 37°C for 12 h. Hsp70 levels in the cell lysate were determined by Western blot (a) and Hsp70 levels in culture medium were determined by ELISA (b). In parallel, the cell viability was determined by LDH release assay (c). d–e FLSs were heated at 42°C for 1 h and allowed to recover to 37°C for increasing times. Hsp70 levels in the cell lysate were determined by Western blot (d) and Hsp70 levels in culture medium were determined by ELISA (e). Data are expressed as the mean ± SEM of three independent experiments. Asterisks, p < 0.05 versus control
Previous studies showed that inflammatory stress could induce the expression of Hsp70 in FLSs (Schett et al. 1998). We further examined whether inflammatory stress could induce Hsp70 release from FLSs. TNF-α was chosen as an inflammatory stressor for these experiments because TNF-α levels are elevated in patients with RA and it is one of the most biologically active cytokines in synovial fluid of RA patients. Our results showed that treatment of FLSs with TNF-α (1–20 μg/ml) dose- and time-dependently increased the expression of intracellular Hsp70 and the release of Hsp70 into the supernatant, without significant increase in cell death (Fig. 2). Taken together, these data suggest that FLSs can actively release Hsp70 when exposed to non-lethal heat shock or inflammatory stress.

TNF-α induces Hsp70 release from FLSs. a–c FLSs were exposed to TNF-α at the indicated concentration for 12 h. Hsp70 levels in the cell lysate were determined by Western blot (a) and Hsp70 levels in culture medium were determined by ELISA (b). In parallel, the cell viability was determined by LDH release assay (c). d–e FLSs were exposed to TNF-α (20 ng/ml) for increasing times. Hsp70 levels in the cell lysate were determined by Western blot (d) and Hsp70 levels in culture medium were determined by ELISA (e). Data are expressed as the mean ± SEM of three independent experiments. Asterisks, p < 0.05 versus control
4.2 Hsp70 induces IL-10 production in RA FLSs
To investigate the ability of human Hsp70 to stimulate cytokine production in FLSs, we treated human FLSs with recombinant human Hsp70 and examined the culture supernatants for IL-6 and IL-8 as a prototypical pro-inflammatory cytokine by FLSs and IL-10 as a prototypical anti-inflammatory cytokine. Our experiments showed that Hsp70 stimulation did not significantly affect IL-6 and IL-8 production in FLSs (Fig. 3a and b) but increased IL-10 secretion in a dose-dependent manner, with the prominent effects at 1–10 μg/ml of Hsp70 (Fig. 3c). Control protein ovalbumin (OVA) did not up-regulate IL-10 production in FLSs (Fig. 3c). Time course experiments showed that the optimal time for production of IL-10 following Hsp70 stimulation was 24 h (Fig. 3d).

Hsp70 induces the production of IL-10 in FLSs. RA FLSs (5 × 104/well) were seeded in 24-well plates in DMEM plus 10% FCS and incubated at 37°C for 24 h. a–c FLSs were stimulated with Hsp70 at the different concentrations (0.1–10 μg/ml) or control protein OVA (10 μg/ml) for 24 h. The supernatants were collected and the concentrations of IL-6 (a), IL-8 (b), and IL-10 (c) were determined by ELISA. Data are expressed as the mean ± SEM of three independent experiments. Asterisks, p < 0.05 versus control. FLSs were stimulated with Hsp70 (1 μg/ml) for increasing times (d). The supernatants were collected and the concentrations of IL-10 were determined by ELISA. Data are expressed as the mean ± SEM of three independent experiments
Recent studies have shown that contamination of Hsp70 with lipopolysaccharide (LPS) might be responsible for its stimulatory activation on macrophages and dendritic cells (Bausinger et al. 2002; Gao and Tsan 2004). To test whether a potential contamination of our Hsp70 preparation with LPS is responsible for the effects of Hsp70 on IL-10 production in FLSs, using a kinetic turbidimetric method, we first observe that recombinant human Hsp70 used in this study contained <0.01 EU/μg protein (1 pg/μg) of bacterial endotoxin. Secondly, the following studies were performed to exclude the possibility that such minute amounts of LPS might affect IL-10 production by using the LPS inhibitor polymyxin B (PMB) and by boiling the Hsp70. Figure 4 shows that boiling abrogated Hsp70 but not LPS-induced effect on IL-10 production and pre-treatment with PMB abrogated the effect of LPS but not Hsp70 on IL-10 production. In addition, to further demonstrate the specificity of Hsp70 direct induction of IL-10 by FLSs, Hsp70 was treated with a specific antibody for Hsp70 for 30 min before its addition to the FLSs cultures. Figure 4 showed that anti-Hsp70 antibody treatment, but not the irrelevant antibody, could abolish the production of IL-10 in FLSs stimulated by Hsp70. These findings suggest that Hsp70 potently induces IL-10 production by FLSs and the effects of Hsp70 on IL-10 production are genuine properties of Hsp70 rather than due to LPS contamination.

The effects of human Hsp70 on IL-10 production in FLSs are not due to the contamination of LPS. RA FLSs (5 × 104/well) were seeded in 24-well plates in DMEM plus 10% FCS and incubated at 37°C for 24 h. FLSs were treated with human Hsp70 (1 μg/ml) or LPS (100 ng/ml) for 24 h. As indicated, Hsp70 and LPS were pre-treated with PMB (10 μg/ml, 30 min), anti-Hsp70 mAb (20 μg/ml, 30 min), or isotype-matched mAb (20 μg/ml, 30 min) or boiled (100°C, 30 min) before addition to FLSs. The IL-10 concentrations of supernatants were determined with ELISA. Data are shown as means ± SEM of three independent experiments. Asterisks, p < 0.05 versus Hsp70 alone. Double asterisk, p < 0.05 versus LPS alone
4.3 The effects of Hsp70 on IL-10 production in FLSs are involved in TLR4
Hsp70 has been found to activate macrophages and other cells via TLR4 or TLR2 signaling (Asea et al. 2002; Fleshner and Johnson 2005). By RT-PCR and Western blotting, we demonstrated that, like macrophages, human RA FLSs also expressed TLR2 and TLR4 (Fig. 5a and b), in accordance with previous reports (Kim et al. 2007; Radstake et al. 2004). To test whether TLR2 or TLR4 might be involved functionally in the effects of Hsp70 on the IL-10 production in FLSs, human RA FLSs were pre-incubated with antibodies to TLR2 or TLR4 proteins and then stimulated by Hsp70, and IL-10 production was assessed. Figure 5c shows that the augmentation of IL-10 production by Hsp70 was blocked by the antibody to TLR4 but not by the antibody to TLR2. In addition, to test the effectiveness of anti-TLR2 and TLR4 antibodies used in our experiments, FLSs were stimulated by peptidoglycan (PGN, agonists for TLR2) and LPS (agonist for TLR4) respectively, in the presence or absence of the antibody, and IL-10 production was assessed. As shown in Fig. 5d, PGN and LPS also induced IL-10 production but their effects were blocked by anti-TLR2 or anti-TLR4, respectively. Thus, the effect of human Hsp70 on IL-10 production in RA FLSs was mediated by TLR4, not by TLR2.

TLR4 is involved in Hsp70-induced IL-10 production in FLSs. a Total RNA from RA FLSs or RAW264.7 macrophages was extracted and analyzed by RT-PCR for TLR2 and TLR4. GAPDH was used as a loading control. b RA FLSs or RAW264.7 macrophages were lysed and the protein levels of TLR2 and TLR4 expression in the cells were determined by Western blotting using specific mAb against TLR2 and TLR4. GAPDH was used as a loading control. c RA FLSs were pre-treated with anti-TLR2, anti-TLR4, or isotype-matched mAb (10 μg/ml) for 30 min. Then, the cells were incubated with Hsp70 (1 μg/ml) for 24 h and IL-10 concentrations were determined with ELISA. Data are shown as means ± SEM of three independent experiments. Asterisk, p < 0.05 versus Hsp70 alone group. d RA FLSs were pre-treated with monoclonal anti-TLR2 and anti-TLR4 (10 μg/ml) for 30 min. Then, the cells were incubated with PGN or LPS (100 ng/ml) for 24 h and IL-10 concentrations were determined with ELISA. Data are shown as means ± SEM of three independent experiments. Asterisk, p < 0.05 versus LPS alone group. Double asterisk, p < 0.05 versus PGN alone group
5 Discussion
In this study, we show that Hsp70 can be actively released from RA FLSs in response to heat shock or TNF-α. The in vitro studies show that extracellular Hsp70 can induce anti-inflammatory cytokine IL-10 production in FLSs through TLR4-mediated interaction. Our results suggest that in the inflamed RA joint, stress conditions, especially inflammatory stress, can induce Hsp70 release from FLSs and that the extracellular Hsp70 may induce anti-inflammatory cytokine IL-10 production in FLSs through an autocrine or paracrine stimulation of TLR4.
Although HSPs were once regarded as intracellular proteins, there is growing evidence suggesting that HSPs can also be expressed on cellular membranes and even released into the extracellular environment after stress. HSPs have been found in the circulation of both healthy individuals and those suffering from autoimmune disease and inflammatory conditions (Dybdahl et al. 2005; Dybdahl et al. 2002; Satoh et al. 2006). Elevated Hsp70 levels have also been found in synovial fluid from RA patients (Martin et al. 2003). However, how Hsp70 is released into the synovial fluid is currently unknown. In earlier studies, it was suggested that cellular Hsp70 release may be the result of cell lysis, but it is now recognized that elevated Hsp70 may be found in the absence of necrosis. In fact, Hsp70 could be actively released from glial cells (Guzhova et al. 2001), epithelial cells (Broquet et al. 2003), tumor cells (Mambula and Calderwood 2006), and PBMC (Lancaster and Febbraio 2005) in the absence of detectable cell death. It was previously shown that heat shock and pro-inflammatory cytokines induced the expression of intracellular Hsp70 in RA FLSs (Schett et al. 1998). Here we extended these observations showing that Hsp70 is also released from RA FLSs in response to heat shock or TNF-α without significant cell death. This indicates that, in the inflamed synovial tissue of RA patients, a variety of different stressors including pro-inflammatory cytokine TNF-α may induce Hsp70 expression and active release from FLSs, which contributes to the elevated Hsp70 levels in synovial fluid.
The release of Hsp70 from FLSs may be of functional importance in RA. Extracellular HSPs have previously been thought of as initiators of a pro-inflammatory response within the innate immune system. It has been shown that extracellular Hsp70 and Hsp60 can induce the production of pro-inflammatory cytokines including TNF-α, IL-1, and IL-6 in monocyte and macrophage (Asea et al. 2000; Quintana and Cohen 2005). Recently, there is evidence that, in contrast to the pro-inflammatory response, some self extracellular HSPs including human BiP (Corrigall et al. 2004), human Hsp60 (Zanin-Zhorov et al. 2005), human Hsp27 (De et al. 2000), and human Hsp10 (Johnson et al. 2005) induce a strong anti-inflammatory response with sustained production of IL-10 in vitro and in vivo. For example, BiP stimulation of human PBMCs in vitro was found to trigger the production of large amounts of IL-10 while the production of pro-inflammatory cytokines such as TNF-α is minimal (Corrigall et al. 2004; Panayi and Corrigall 2006). Human Hsp60 treatment of T cells in vitro was found to inhibit the production of pro-inflammatory cytokine TNF-α and IFN-γ and trigger the production of anti-inflammatory cytokines IL-10 (Zanin-Zhorov et al. 2005). These findings suggest that, rather than being pro-inflammatory, self HSP reactivity might be a physiological mechanism for regulation of pro-inflammatory responses and inflammatory disease. It should therefore not be surprising if self Hsp70 is found to have an anti-inflammatory effect in RA. Indeed, our studies showed that human Hsp70 significantly induced the production of anti-inflammatory cytokine IL-10 but failed to induce the production of pro-inflammatory cytokines including IL-6 and IL-8 in FLSs (Fig. 3), suggesting that extracellular Hsp70 have an anti-inflammatory property in RA. Thus, it is conceivable that in the inflamed RA joint, inflammatory stress contributes to the expression and release of Hsp70 and the extracellular Hsp70 may act as natural dimmers of inflammation by specially inducing FLSs to produce IL-10, which is a part of a normal mechanism to down-regulate an inflammatory response.
The TLRs are a family of pattern recognition receptors, by means of which cells of the innate immune system, including macrophages and dendritic cells, recognize microbial pathogens (Medzhitov 2001). Ten different proteins have so far been defined as members of human TLR family. The motif pathogen-associated molecular patterns such as LPS are recognized by TLR4 and those such as lipoteichoic acid and PGN are recognized mainly by TLR2. TLRs have been shown to mediate activation of NF-κB and mitogen-activated protein kinase signaling pathways, resulting in the production of pro-inflammatory mediators such as IL-1, IL-6, IL-8, or TNF-α from the innate immune cells (Medzhitov 2001; Muzio et al. 1998; Seibl et al. 2003). There is considerable evidence that activation of the TLRs can induce or exacerbate inflammatory diseases including RA (Sacre et al. 2007). Injection of bacterial products such as the TLR2 ligand PGN, TLR4 ligand LPS, or the LTR-9 ligand CpG DNA into the joints of mice results in arthritis. It has recently been shown that TLR2, TLR4, or both are implicated in the recognition of Hsp70 (Asea et al. 2002). TLR2 and TLR4 are abundantly expressed on innate immune cells such as macrophages and dendritic cells (Akira et al. 2001). Although several studies have demonstrated that TLR2 and TLR4 are also expressed in a variety of non-immune cells, their function is less well understood. Our results and other’s have demonstrated that TLR2 and TLR4 are also expressed in RA FLSs (Fig. 5a and b) (Gutierrez-Canas et al. 2006; Kim et al. 2007; Pierer et al. 2004), suggesting that human Hsp70 might affect RA FLSs by its binding to TLR2 or TLR4. To test whether TLR2 or TLR4 might be functionally involved in the regulation of FLSs by Hsp70, we assayed the effects of blocking antibodies to the TLR2 or TLR4 proteins on FLSs. As we have shown here, a specific monoclonal antibody (mAb) to TLR4 but not anti-TLR2 abrogated increase of IL-10 in RA FLSs, indicating that the effect of human Hsp70 on FLSs would be TLR4-dependent and TLR4 might be involved in the IL-10 production in RA FLSs. In addition, whether TLR4 serves as a primary receptor for extracellular Hsp70 or whether TLR4 is a necessary participant in the response of other receptors to the Hsp70 molecule remains unclear. The exact mechanism of interaction between human Hsp70 and the TLR4 on RA FLSs remains to be elucidated.
In summary, we show that Hsp70 is actively released from FLSs in response to HS or TNF-α and that Hsp70 may be a major paracrine/autocrine inducer of IL-10 production in FLSs via TLR4. The results of this study indicate that human Hsp70, which is up-regulated and released by RA FLSs exposed to stress and inflammation, may have immunomodulatory and anti-inflammatory effects on RA inflammation.
References
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2(8):675–680
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS et al (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31(3):315–324
Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK (2000) HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 6(4):435–442
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK (2002) Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 277(17):15028–15034
Aupperle KR, Bennett BL, Boyle DL, Tak PP, Manning AM, Firestein GS (1999) NF-kappa B regulation by I kappa B kinase in primary fibroblast-like synoviocytes. J Immunol 163(1):427–433
Bausinger H, Lipsker D, Ziylan U, Manie S, Briand JP, Cazenave JP, Muller S, Haeuw JF, Ravanat C, de la Salle H et al (2002) Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol 32(12):3708–3713
Broquet AH, Thomas G, Masliah J, Trugnan G, Bachelet M (2003) Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J Biol Chem 278(24):21601–21606
Cho ML, Kim WU, Min SY, Min DJ, Min JK, Lee SH, Park SH, Cho CS, Kim HY (2002) Cyclosporine differentially regulates interleukin-10, interleukin-15, and tumor necrosis factor α production by rheumatoid synoviocytes. Arthritis Rheum 46(1):42–51
Corrigall VM, Bodman-Smith MD, Brunst M, Cornell H, Panayi GS (2004) Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: relevance to the treatment of inflammatory arthritis. Arthritis Rheum 50(4):1164–1171
De AK, Kodys KM, Yeh BS, Miller-Graziano C (2000) Exaggerated human monocyte IL-10 concomitant to minimal TNF-alpha induction by heat-shock protein 27 (Hsp27) suggests Hsp27 is primarily an antiinflammatory stimulus. J Immunol 165(7):3951–3958
Dybdahl B, Wahba A, Lien E, Flo TH, Waage A, Qureshi N, Sellevold OF, Espevik T, Sundan A (2002) Inflammatory response after open heart surgery: release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 105(6):685–690
Dybdahl B, Slordahl SA, Waage A, Kierulf P, Espevik T, Sundan A (2005) Myocardial ischaemia and the inflammatory response: release of heat shock protein 70 after myocardial infarction. Heart 91(3):299–304
Feldmann M, Maini RN (1999) The role of cytokines in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 38(Suppl 2):3–7
Fleshner M, Johnson JD (2005) Endogenous extra-cellular heat shock protein 72: releasing signal(s) and function. Int J Hyperthermia 21(5):457–471
Gao B, Tsan MF (2004) Induction of cytokines by heat shock proteins and endotoxin in murine macrophages. Biochem Biophys Res Commun 317(4):1149–1154
Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope RM (2000) Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol 165(12):7199–7206
Gutierrez-Canas I, Juarranz Y, Santiago B, Arranz A, Martinez C, Galindo M, Paya M, Gomariz RP, Pablos JL (2006) VIP down-regulates TLR4 expression and TLR4-mediated chemokine production in human rheumatoid synovial fibroblasts. Rheumatology (Oxford) 45(5):527–532
Guzhova I, Kislyakova K, Moskaliova O, Fridlanskaya I, Tytell M, Cheetham M, Margulis B (2001) In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res 914(1–2):66–73
Hauet-Broere F, Wieten L, Guichelaar T, Berlo S, van der Zee R, Van Eden W (2006) Heat shock proteins induce T cell regulation of chronic inflammation. Ann Rheum Dis 65(Suppl 3):iii65–iii68
Isomaki P, Luukkainen R, Saario R, Toivanen P, Punnonen J (1996) Interleukin-10 functions as an antiinflammatory cytokine in rheumatoid synovium. Arthritis Rheum 39(3):386–395
Johnson BJ, Le TT, Dobbin CA, Banovic T, Howard CB, Flores Fde M, Vanags D, Naylor DJ, Hill GR, Suhrbier A (2005) Heat shock protein 10 inhibits lipopolysaccharide-induced inflammatory mediator production. J Biol Chem 280(6):4037–4047
Kiang JG, Tsokos GC (1998) Heat shock protein 70 kDa: molecular biology, biochemistry, and physiology. Pharmacol Ther 80(2):183–201
Kim KW, Cho ML, Lee SH, Oh HJ, Kang CM, Ju JH, Min SY, Cho YG, Park SH, Kim HY (2007) Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett 110(1):54–64
Lancaster GI, Febbraio MA (2005) Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 280(24):23349–23355
Mambula SS, Calderwood SK (2006) Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol 177(11):7849–7857
Martin CA, Carsons SE, Kowalewski R, Bernstein D, Valentino M, Santiago-Schwarz F (2003) Aberrant extracellular and dendritic cell (DC) surface expression of heat shock protein (hsp) 70 in the rheumatoid joint: possible mechanisms of hsp/DC-mediated cross-priming. J Immunol 171(11):5736–5742
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1(2):135–145
Min DJ, Cho ML, Lee SH, Min SY, Kim WU, Min JK, Park SH, Cho CS, Kim HY (2004) Augmented production of chemokines by the interaction of type II collagen-reactive T cells with rheumatoid synovial fibroblasts. Arthritis Rheum 50(4):1146–1155
Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A (1998) The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J Exp Med 187(12):2097–2101
Nakahara H, Nishimoto N (2006) Anti-interleukin-6 receptor antibody therapy in rheumatic diseases. Endocr Metab Immune Disord Drug Targets 6(4):373–381
Panayi GS, Corrigall VM (2006) BiP regulates autoimmune inflammation and tissue damage. Autoimmun Rev 5(2):140–142
Pierer M, Rethage J, Seibl R, Lauener R, Brentano F, Wagner U, Hantzschel H, Michel BA, Gay RE, Gay S et al (2004) Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J Immunol 172(2):1256–1265
Quintana FJ, Cohen IR (2005) Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol 175(5):2777–2782
Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PL, Barrera P, Joosten LA, van den Berg WB (2004) Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum 50(12):3856–3865
Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, Feldmann M, Brennan F, Foxwell BM (2007) The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol 170(2):518–525
Satoh M, Shimoda Y, Akatsu T, Ishikawa Y, Minami Y, Nakamura M (2006) Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte Toll signal in patients with heart failure after acute myocardial infarction. Eur J Heart Fail 8(8):810–815
Schett G, Redlich K, Xu Q, Bizan P, Groger M, Tohidast-Akrad M, Kiener H, Smolen J, Steiner G (1998) Enhanced expression of heat shock protein 70 (hsp70) and heat shock factor 1 (HSF1) activation in rheumatoid arthritis synovial tissue. Differential regulation of hsp70 expression and hsf1 activation in synovial fibroblasts by proinflammatory cytokines, shear stress, and antiinflammatory drugs. J Clin Invest 102(2):302–311
Seibl R, Birchler T, Loeliger S, Hossle JP, Gay RE, Saurenmann T, Michel BA, Seger RA, Gay S, Lauener RP (2003) Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol 162(4):1221–1227
Svensson PA, Asea A, Englund MC, Bausero MA, Jernas M, Wiklund O, Ohlsson BG, Carlsson LM, Carlsson B (2006) Major role of HSP70 as a paracrine inducer of cytokine production in human oxidized LDL treated macrophages. Atherosclerosis 185(1):32–38
Tanaka Y, Otsuka T, Hotokebuchi T, Miyahara H, Nakashima H, Kuga S, Nemoto Y, Niiro H, Niho Y (1996) Effect of IL-10 on collagen-induced arthritis in mice. Inflamm Res 45(6):283–288
Tsan MF, Gao B (2004) Cytokine function of heat shock proteins. Am J Physiol Cell Physiol 286(4):C739–C744
van Eden W, van der Zee R, Prakken B (2005) Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol 5(4):318–330
Walmsley M, Katsikis PD, Abney E, Parry S, Williams RO, Maini RN, Feldmann M (1996) Interleukin-10 inhibition of the progression of established collagen-induced arthritis. Arthritis Rheum 39(3):495–503
Zanin-Zhorov A, Bruck R, Tal G, Oren S, Aeed H, Hershkoviz R, Cohen IR, Lider O (2005) Heat shock protein 60 inhibits Th1-mediated hepatitis model via innate regulation of Th1/Th2 transcription factors and cytokines. J Immunol 174(6):3227–3236
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (30330280, 30671947), the National Basic Research Program of China (2007CB512007), and the Specialized Research Fund for the Doctoral Program of Higher Education of China (20060533009).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Luo, X., Zuo, X., Zhang, B. et al. Release of heat shock protein 70 and the effects of extracellular heat shock protein 70 on the production of IL-10 in fibroblast-like synoviocytes. Cell Stress and Chaperones 13, 365–373 (2008). https://doi.org/10.1007/s12192-008-0036-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-008-0036-2