
Abstract
Over the course of time, Hepatitis C has become a universal health menace. Its deleterious effects on human liver encompass a lot of physiological, genetic as well as epigenetic alterations. Fatty liver (Hepatic steatosis) is an inflammation having multifactorial ancestries; one of them is HCV (steatohepatitis). HCV boosts several cellular pathways involving up-regulation of a number of cytokines. Current study reviews the regulation of some selective key cytokines during HCV infection, to help generate an improved understanding of their role. These cytokines, IL-1β, IL-6, TNF-α, and IFN-ϒ, are inflammatory markers of the body. These particular markers along with others help hepatocytes against viral infestation. However, recently, their association has been found in degradation of liver on the trail heading to non-alcoholic steatohepatitis (NASH). Consequently, the disturbance in their equilibrium has been repeatedly reported during HCV infection. Quite a number of findings are affirming their up-regulation. Although these cell markers are stimulated by hepatocytes as their standard protection mechanism, but modern studies have testified the paradoxical nature of this defense line. Nevertheless, direct molecular or epigenetic research is needed to question the actual molecular progressions and directions commanding liver to steatosis, cirrhosis, or eventually HCC (Hepatocellular Carcinoma).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The World Health Organization estimates that every year 3–4 million people get infected with hepatitis C virus (HCV) worldwide. Whereas 130–170 million of chronically infected individuals are at a risk of developing liver cirrhosis, hepatocellular carcinoma (HCC), or both. These infections lead to more than 350 thousand deaths per annum [1], but the manifestation varies widely with geographical deviations. The prevalence is as low as 0.1–1% in northern Europe to 2.5–3.5% in southern Europe. Amongst worst effected countries is Egypt, with 22% of its population infected, then come Pakistan and China, with more than 3% of their population infected with HCV [1].
HCV belongs to genus Hepacivirus [2], resembling the Flaviviridae family of viruses [3]. Its size ranges between 50 and 60 nm in diameter. A lipid bilayer envelope surrounds a capsid. The viral envelope bears E1 and E2 glycoprotein complex that plays a fundamental role in viral and host cell interaction. Viral genome is a single stranded positive sense RNA molecule, comprised of 9600 nucleotides, encoding a 3000 amino acid polyprotein, which is post-translationally modified into three structural proteins and six non-structural proteins. Structural proteins, Core, E1, and E2, make the capsid and the envelope, while non-structural proteins, NS2, NS3, NS4A, NS4B, NS5A, and NS5B, are the building blocks for the particle and play the vital role as genome replicating proteins [3].
Steatohepatitis/hepatic staetosis or more commonly fatty liver frequently causes elevation of serum aminotransferase levels and is commonly prevalent in general population [4]. This hepatic ailment is considered a consequence of metabolic syndrome, whose major underlying factor is insulin resistance, which in turn is a sequela of hyper-triglyceridaemia, obesity, and diabetes mellitus. Alcohol abuse and chronic HCV infection may also lead to hepatic steatosis. Hepatic steatosis without excessive alcohol consumption is termed non-alcoholic fatty liver disease (NAFLD). NAFLD further comprehends a range of diseases from simple steatosis to Non-Alcoholic Steato-Hepatitis (NASH). NASH may lead to development of liver fibrosis and cirrhosis [5]. The only way to cure non-alcoholic steatohepatitis (NASH) is to achieve weight loss and modification of risk factors, such as dyslipidemia, insulin resistance, and hyperglycemia associated with metabolic syndrome [6].
Since ~50% HCV patients develop hepatic steatosis, chance alone cannot be expected to cause this association, thus suggesting some germaneness [7]. Hepatitis B can be another cause of hepatic steatosis, but this association is seen only in ~18% of the cases [8], which is a profoundly smaller percentage. The HCV and hepatic steatosis association is a multifactorial link. While so-called ‘metabolic steatosis’ is the metabolic syndrome, whose features are connected with hepatic steatosis in HCV patients [9]. However, this may occur in the absence of these factors, just through HCV itself, a ‘viral steatosis’. Such steatosis is often linked to HCV genotype 3 infections [7]. HCV genotype 3a has direct association with high intrahepatic viral levels of hepatic steatosis, while other genotypes are involved with metabolic syndrome in NASH patients [10].
Most of the types of liver cells, along with virtually all nucleated cells of the human body produce pleiotropic regulatory peptides, the Cytokines [11, 12]. There are quite a few subfamilies of the cytokine family. These include interleukins, interferons, tumor necrosis factor (TNF) family, IL-6 and its relatives, colony stimulating factors, transforming growth factor, the chemokines, such as IL-8, and others [13]. Production of these autocrine, paracrine, and endocrine effector molecules is absent or minimal during normal body functioning in most of the body tissues, including liver. However, as the cells are activated by pathologic or physiologic stimuli or both, the cytokines production increases and they coordinate the tissue’s response to the stimulus [11, 12].
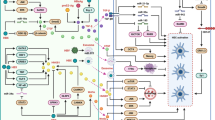
Cytokines in liver mediate its regeneration after any injury [14]. These contribute to upregulation or down-regulation of cytokines production by monocytes/macrophages at the site of inflammation [15]. However, there is increasing evidence that paradoxically these same cytokines mediate inflammation in liver, cell necrosis and apoptosis, fibrosis, and cholestasis [13] This review covers the functioning of four selected cytokines, i.e., Interleukin-1β (IL-1β), Interleukin-6 (IL-6), Tumor Necrosis Factor-α (TNF-α), and Interferon-ϒ (INF-ϒ), during non-inflammatory conditions and alterations in their regulation under the effect of HCV-induced NASH (Fig. 1).

Multifactorial hepatic steatosis leading to hepatocellular carcinoma, HCV being a major factor in development of the disease. HCV infection alone or accompanying some other steatosis inducers lead way to Hepatocellular Carcinoma. Alcohol consumption may play an important role in this development, but non-alcoholism does not prevent the occurring
Selective cytokines operative and HCV-induced regulatory alterations during NASH
IL1β
Almost every tissue and organ system is affected by the polypeptide cytokine interleukin-1 (IL-1α and IL-1β) [16]. IL-1β participates in regulation of immune responses, inflammatory reactions, and hematopoiesis. IL-1β is involved in activation of various pro-inflammatory genes expression that includes autocrine production of IL-1 by binding to IL-1 receptor type І. It itself is produced as a precursor and develops to its mature form by specific cellular proteases. The precursor molecule is cleaved by caspase-1 to its activated mature form [11, 17]. Genetic polymorphism of IL-1β gene is linked to susceptibility of various diseases, including periodontal disease, inflammatory bowel disease, and gastric cancer [18].
IL-1β shows response in viral diseases in both acute and chronic inflammation [19, 20]. It is produced in response of pathogen associated molecular pattern (PAMP) motifs inside macromolecules that are accumulated during microbial infection [21, 22]. Active form of IL-1β is produced by two signals, first signal activates NF-kB (Nuclear Factor-kappaB) and induces IL-1β mRNA expression, and second signal activates NOD like receptor (NLR) for downstream cleavage of caspase-1 and production of biological active cytokine by processing of proIL-1β [23, 24]. During viral infection, production of IL-1β is induced by inflammasome signaling. Flaviviruses that include Hepatitis C Virus, Japanese Encephalitis Virus and West Nile Virus activate production of IL-1β through NLRP3 inflammasome pathway [25]. Its autocrine nature and HCV-induced activation is depicted for the sake of general understanding in Fig. 2a.

Schematic representation of HCV-induced regulatory alterations of IL-1β, IL-6, TNF-α, and IFN-ϒ and their autocrine feedback, leading to production of these cytokines and their deleterious effects in taking liver to Non-Alcoholic Steatohepatitis (NASH) and Hepatocellular Carcinoma (HCC). HCV enhances the normal body production of inflammatory cytokines, through positively influencing different receptors and mediators of our immune system cells. This over expression gives rise to anti-viral responses. At the same time, these cytokines lead liver to degeneration, taking it through Hepatic steatosis, liver fibrosis, cirrhosis and ultimately HCC. a IL-1β Receptor induces the production of IL-1 in macrophages through a series of events, ultimately involving inflammasome and caspase-1 giving rise to IL-1β precursor. This production is further enhanced by HCV. b–d HCV induces production of IL-6, TNF-α, and IFN-ϒ through inducing Toll-Like Receptors, TLR, in different immune cells; stimulating their pro-inflammatory production. Once stimulated, these cytokines keep their production going through autocrine inductions, which involves JAK-STAT pathway in most cases
Although IL-1β is involved to activate expression of pro-inflammatory genes, recruitment of immune cells to the infection site, and modulation of in-filtering cellular immune effector actions, but inside the liver, range of IL-1β responsive genes is not clear yet [26]. IL-1β might have its potential role in HCV pathogenesis by having the ability to promote tissue pathogenesis and to induce pro-fibrogenic mediators production [27, 28]. T cells and macrophages (Kupffer cells) are main sources of IL-1β production and levels of IL-1β increase during chronic hepatitis. HCV-related liver diseases have higher level of IL-1β as compared to other liver diseases, and IL-1β polymorphism has a link to progression of HCC [29]. Serum IL-1β and IL-6 can be used as markers for HCV-infected liver disease progression [30]. During HCV infection elevated levels of c-myc expression, oxidative DNA damage and IL-1β release are associated with development of cirrhosis [31]. IL-1α and IL-1β might have a significant role in conversion of steatosis to steatohepatitis and liver fibrosis [32].
In human hepatoma cells, IL-1 has direct antiviral activity through activation of Extracellular signal-regulated kinases- mitogen-activated protein kinases, ERK MAP kinase pathway. Potential antiviral activity of IL-1 could be administered, as anti-viral treatment to chronic HCV patients having IFN resistance [33]. Major target for IL-1-mediated inflammation is endothelium. Systemic IL-1 triggers its receptors and activates production of prostaglandin E, neutrophil release from bone marrow and production of IL-6. IL-6 induction by IL-1 accounts for thrombocytosis and hepatic acute-phase protein (APP) synthesis [34]. High levels of IL-1β in serum and liver inhibit IFN-α/β-activated signals and antiviral activity results in reduction of therapeutic responses to IFN-α in chronic HCV infected patients by suppressing Signal Transducer and Activator of Transcription 1 (STAT1) activity in a proteosome-dependant approach [35]. Studies have shown that TNFα, IL6, and IL1b are upregulated in NASH patients, which are involved in hepatocyte apoptosis, Malloray–Denk bodies, and hepatic insulin resistance [36].
IL-6
IL-6 plays a central role as a distress cytokine during inflammation in body. It is the key cytokine in liver which makes dramatic amendments to the synthesis of a number of APP proteins [37–39]. Being a multifactorial cytokine, it is not only involved in inflammation responses of body but in a varied range of cell functions, such as cell survival, apoptosis, and proliferation etc [40]. During Hepatitis C infection, the production of IL-6 along with interferon beta, from B cells is immensely increased. These events follow a strong activation of TLRs (Toll-like Receptors). TLRs are actually trans-membrane proteins which detect and respond to microbial ligands, acting as pattern recognition receptors. TLR4 is one member of the family, triggering IL-6 production [41]. TLRs activate NF-κB [42], which in turn triggers IL-6 production by binding the promotor region of IL-6 gene [43]. Being autocrine in nature, IL-6 can help self-production through binding to a heterodimeric complex, IL-6R/gp130 complex, present on surface of active B cells, as well as number of other cells [44]. This heterodimer triggers an activation process of different cell modulators, such as Janus Kinases (JAK), and the effectors downstream, such as Phosphatidylinositol-3-Kinase and Protein Kinase B (PI3K/Akt), STAT3, and SH2-domain-containing protein tyrosine phosphatase-2/Ras (SHP-2/Ras) [40]. This JAK-STAT3 pathway keeps the production cycle going [45], as evident in Fig. 2b.
It was implicated earlier that IL-6 along with one of its major effectors, STAT3, are pro-tumorigenic agents in a variety of cancers [46]. It is a key cytokine to regenerate liver after liver injury [14, 47–49]. However, if its signaling goes uncontrolled, several chronic diseases, such as liver fibrosis, are triggered and maintained [50], thus emphasizing a need of effective signal termination mechanisms along with signal initiation processes [51]. The signal termination is acquired through several counter-balancing mechanisms. These include the protein inhibitor of activated STAT3 (PIAS3) and the protein tyrosine phosphatase SHP2, but the critical negative signaling component is Suppressor of Cytokine Signaling 3 (SOCS3), induced by IL-6 itself as a feedback inhibitor. Transitory IL-6 signal transduction termination is achieved through rapid and short lived induction of SOCS3. SOCS3 binds to phosphotyrosine-759 of activated gp130 in IL-6R/gp130 complex and inhibits JAK activity. This inhibition further prevents the activation of above-mentioned signaling components through terminating their phosphorylation [52].
IL-6 role in insulin resistance has not been much investigated, though it is secreted by adipocytes and adipose stromal cells also, along with many others [53, 54]. This production increases notably after meals [55]. IL-6 along with TNF inhibits Lipoprotein lipase (LPL) expression but does not stimulate lipolysis, which TNF does [56, 57]. While studies rendering IL-6 and insulin resistance linkage have shown an increase in adipocytes secretion of IL-6 in obese [58] and diabetic objects [59].
HCV has been reported to induce inflammation through a significant raise in serum IL-6 levels in chronically infected HCV patients [60]. These increased serum levels were also observed during alcoholic hepatitis as well as chronic hepatitis B and C that lead to further development of HCC. IL-6 itself signals promotion of HCC [61]. This role of inflammatory pathways in developing HCC was also observed by Park et al. They established that obesity promoted HCC has its roots in IL-6 and TNF signaling [62]. Similar results were found while working with immortalized human hepatocytes (Human hepatocarcinoma cell line-Huh 7). Significant upregulation of IL-6 and STAT3 was observed after induction of Huh 7 cultures with HCV core protein [63].
IL-6 expression level elevation in objects during NAFLD and NASH has been recurrently reported in different studies [64–67]; however, Lima-Cabello et al. have reported that level of upregulation of IL-6 and TNF-α is higher in steatotic objects as compared to objects having hepatic steatosis [64]. Similar findings of increased IL-6 expression during NASH have been reported in different studies conducted on humans as well as transgenic animal models [65, 68, 69].
TNF-α
During acute phase inflammatory response, TNF-α along with other cytokines is produced to activate endothelial cells and leukocytes. In many inflammatory diseases, such as osteoporosis, cachexia in cancer patients, chronic rheumatoid arthritis, and viral hepatitis, dysregulation of TNF-α levels is observed [70, 71]. TNF-α might be key player in both cell death and cell proliferation. Studies have shown that TNF-α promotes hepatocyte proliferation rather than their death when it is administrated to animals or incubated with hepatocytes in vitro [72]. Antibody neutralization studies and experiments with TNF type І receptors deficient mice have revealed that TNF has a role in initiation of regenerating liver tissues by activation of type І TNF receptors, followed by partial hepatectomy, an offend of liver tissues that causes increased production of TNF-α [14, 73]. TNF-α, cytokines that regulate TNF and cytokines involve in altering synthesis and biological events of TNF mediate a variety of aspects of liver damage and repair. Therefore, cell death is not a usual result when hepatocytes experience to TNF-α and it indicates that during liver injury or damage, normal hepatocytes response to TNF-α in a destabilized manner. TNF-α has not only role in initial stages of fatty liver disease, but it is also involved in evolution of advanced stages of liver injury [13].
TNF is type ІІ protein which upon proteolytic cleavage produces a soluble homotrimer of 17 kDa. TNF yields its function by interacting with one of its receptors: type І (p55) and type ІІ (p75), which are present on various cell types, including hepatocytes [71]. TNF–α signal is transduced by activation of transcription factor NF-kB that results in activation of various genes involved in cell proliferation and death, inflammation, and cancer [74]. Macrophages and monocytes primarily produce TNF as well as lymphocytes; endothelial cells, fibroblasts, and neurons also produce TNF-α. In liver, macrophages and Kupffer cells are its main source [75]. Although hepatocytes comprise major part of liver (~60–80%), they only produce it during chronic Hepatitis C virus infection [76].
Several gene expression mechanisms regulate the levels of TNF-α. In response to stimuli, TNF-α mRNA induction takes place, and then, it is rapidly degraded. First, induction of TNF-α mRNA is regulated by transcription of NF-kB. Second, cis control elements: a cluster of AU-rich elements (AREs) located in the 32-untranslated region (32’UTR) stabilize and regulate mRNA [77]. ARE motifs are frequently found in 32′UTR of inducible genes, such as chemokines and inflammatory cytokines [78]. Third, TNF-α mRNA stability is regulated by RNA binding proteins by binding to the ARE in 32′UTR [79]. In an autocrine response, TNF receptors, TNFR1 and TNFR2 present on macrophages receive TNF-α [80] which keeps the cycle going for itself through activating NF-kB, [81] until negative regulation. This attribute is portrayed briefly in Fig. 2c.
Some experimental studies have shown that endotoxin-mediated release of cytokine has a role in development of liver damage in NASH; Yang et al. demonstrated that during severe steatosis, obese mice showed more sensitivity to endotoxin than lean ones. They also showed that TNF-α and interferon gamma might have role in liver injury [82]. Studies have reported that TNF receptor deficient mice showed complete protection from alcoholic induced steatohepatitis, which reveal the significance of TNF-α in inflammatory stages of fatty liver disease [83]. In obese leptin deficient mice, metformin plays a role in reversion of fatty liver disease by adopting a mechanism that inhibits expression of TNF-α and TNF-inducible factors that are involved in adenosine triphosphate depletion and hepatic lipid accumulation [84]. NASH patients with significant fibrosis are associated with elevated level of TNF-α mRNA expression. Over expression of TNF-α mRNA in liver and adipose tissues, and increased level of p55- mRNA in NASH patients demonstrate that TNF-α system may be involved in pathogenesis of NASH [85].
INF-ϒ
The interferons, α, β, and ϒ, are the extracellular signaling proteins (ESPs) that bind with cellular receptors at cell surface, to play role in activating previously quiescent genes at transcriptional levels-the epigenetic interaction. Some structurally unrelated ESPs remain successful in rapidly activating some of the genes [86–92], while some other activator specific genes require specific ESPs for their rapid activation [89, 93]. Intra cellular pathogens resistance and mediator activation are mediated principally by INF-ϒ, the lymphokine [94].
Certain T-cell-derived cytokines are further classified as T-helper type-1 (Th1) and T-helper type-2 (Th2), on the basis of their production pattern. IFN-ϒ and IL-2 are Th1, while IL-4 and IL-10 are Th2 [95]. IFN-ϒ along with others performs a wide range of regulatory functions, signaling for activating and promoting growth and differentiation of T-lymphocytes, granulocytes, macrophages, and Natural Killer Cells (NK) are few of them [96–98].
IFN-ϒ is produced by a number of different cells, and these include T cells-CD4 or CD8+, NK, and NKT cells. Its receptor is a heterodimer of IFN-ϒR1 and IFN-ϒR2. The control of microbial infection is achieved through mutation in the receptor or the cytokine itself. Several viruses interfere with its receptor signaling by encoding proteins specific for this task, thus indicating its importance in viral infection cycles. Its role in adaptive immune response is as vital. CD8 + T cell activity is highly affected by IFN-ϒ, as it induces immunoproteasome, TAP (Transporter associated with antigen processing) and MHC (Major histocompatibility complex) class I expression. These inductions lead to increased visibility of intracellular pathogens to the adaptive immune response. These “positive looking” effects though actually suppress the CD8+T-cell activity. IFN-ϒ-treated T cells show increased apoptosis and reduced proliferation. It directly acts on these cells to control their abundance [99]. Viral induction of TLR leads to activation of NFκB and AP1, which along with a lot many other cytokines stimulate IFN-ϒ production [100]. Once initiated, the supply goes on by autocrine self-production. This is achieved through binding with IFN-ϒR1, IFN-ϒR2 complex, involving JAK1, JAK2, and finally activating STAT1 homodimer to elicit transcriptional response [101]. A simplified illustration can be seen here in Fig. 2d.
Kusters et al. have studied that IFN-ϒ plays its role in mediating liver injury along with other cytokines, such as TNF [102]. It is also an important factor in other immune responses and also in inflammatory diseases, such as chronic HCV [103]. IFN-ϒ along with other immunoregulatory cytokines, IL-2, IL-4, and IL-10, are particular modulators in HCV infection and host immune responses [95]. IFN-ϒ producing cells has been found localized in liver of chronic hepatitis B patients as well [104] quite a number of other studies have got similar findings [105, 106], leading to its widely known establishment as an anti-viral agent. These finding are not different than studies conducted on HCV in this regard. Cacciarelli et al. have found significant raise of IFN-ϒ in serum of HCV patients as compared with normal controls [107]. Similar findings by Tilg et al. regarding IFN-ϒ have suggested its role in viral-induced liver inflammation [70]. In vitro studies conducted on HCV genomic and sub-genomic replicons also supported these findings. The protein synthesis as well as RNA replication of these particles is prohibited by IFN-ϒ. This study has shown that IFN-ϒ produced by cytotoxic T cells and NK cells is an extra defense mechanism to enhance the intracellular viral replication inhibition along with their cell killing accomplishments [108]. Although this NK cells mediated, IFN-ϒ production raise is not the case with all viral infections [109].
Different studies conducted in vitro [110] and clinical [111] demonstrate an increase in NK cells fashioned Th1 cytokines production in liver during obesity. This leads to hepatic steatosis [112], NASH and eventually HCC [113]. Bertola et al. have reported an antagonistic relation between pro-inflammatory IFN-ϒ and Th2 cytokine IL-10 genes regulation during NASH, with IFN-ϒ upregulated remarkably [114]. Increased IFN-ϒ production during steatosis and NASH has been reported in a number of studies illustrating a shift of NK cells towards Th1 cytokine production [111, 115, 116].
Conclusions and future perspectives
Our understanding of viral detection through innate immune system and corresponding antiviral responses has immensely increased during the past decade. We have gained better discernments in the functioning and effects of cytokines as host defense mediators against inflammation. A number of overlapping and intersected pathways have been generated by the host in order to defy viral attack. These pathways comprise a large number of cytokines working along with each other to produce an anti-viral effect. IL-1β, IL-6, TNF-α, and IFN-ϒ are few among them. Hepatocytes like the rest of body cells deviate from routine signaling towards preferment of these particular cytokines to protect themselves from invading viruses. These triggered mechanisms are a consequence of the changes that viral nucleic acid persuades in intracellular environment. IL-1B, IL-6, TNF-α, and IFN-ϒ play a pivotal role in cell defense, thus are subsequently up-regulated during HCV induced NASH.
What remains unrequited is the direct evidence to their connection with HCV-induced NASH in particular at a molecular level as compared to the incidental inferences obtained through clinical and experimental studies. By scheming laboratory studies regarding the unswerving epigenetic effects of viral nucleic acid on the regulation of cytokines can help generate more clear and strong evidences for further development of more useful drugs in future and may be ways to even revoke the damage already done to liver in the course of NASH, which ultimately leads to HCC [113].
References
Durier N, Nguyen C, White LJ. Treatment of hepatitis C as prevention: a modeling case study in Vietnam. PLoS One. 2012;7(4):e34548.
Robertson B, Myers G, Howard C, et al. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch Virol. 1998;143(12):2493–503.
Timpe J, McKeating J. Hepatitis C virus entry: possible targets for therapy. Gut. 2008;57(12):1728–37.
Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346(16):1221–31.
Patel JH, Cobbold JFL, Thomas HC, Taylor-Robinson SD. Hepatitis C and hepatic steatosis. QJM. 2010;103(5):293–303.
van der Poorten D, George J. Current and novel therapies for the treatment of nonalcoholic steatohepatitis. Hepatol Int. 2007;1(3):343–54.
Hwang SJ, Luo JC, Chu CW, et al. Hepatic steatosis in chronic hepatitis C virus infection: prevalence and clinical correlation. J Gastroenterol Hepatol. 2001;16(2):190–5.
Thomopoulos KC, Arvaniti V, Tsamantas AC, et al. Prevalence of liver steatosis in patients with chronic hepatitis B: a study of associated factors and of relationship with fibrosis. Eur J Gastroenterol Hepatol. 2006;18(3):233–7.
Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365(9468):1415–28.
Shrivastava S, Meissner EG, Funk E, et al. Elevated hepatic lipid and interferon stimulated gene expression in HCV GT3 patients relative to non-alcoholic steatohepatitis. Hepatol Int. 2016;10(6):937–946.
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87(6):2095–147.
Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annu Rev Cell Biol. 1993;9(1):317–43.
Epstein FH, Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343(20):1467–76.
Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci. 1997;94(4):1441–6.
Danis V, Millington M, Hyland V, Grennan D. Cytokine production by normal human monocytes: inter-subject variation and relationship to an IL-1 receptor antagonist (IL-1Ra) gene polymorphism. Clin Exp Immunol. 1995;99(2):303.
Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77(8):1627–52.
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
Donaldson P, Agarwal K, Craggs A, Craig W, James O, Jones D. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut. 2001;48(3):397–402.
Allen IC, Scull MA, Moore CB, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30(4):556–65.
Dinarello CA. Interleukin-1 and the pathogenesis of the acute-phase response. N Engl J Med. 1984;311(22):1413–8.
Kanneganti T-D, Body-Malapel M, Amer A, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006;281(48):36560–8.
Vance RE, Isberg RR, Portnoy DA. Patterns of pathogenesis: discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell host microbe. 2009;6(1):10–21.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10(2):417–26.
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–7.
Ramos HJ, Lanteri MC, Blahnik G, et al. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012;8(11):e1003039.
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87.
Dombrowski Y, Peric M, Koglin S, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. 2011;3(82):82ra38–8.
Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res. 2010;88(8):1615–31.
Bahr MJ, El Menuawy M, Boeker KH, Musholt PB, Manns MP, Lichtinghagen R. Cytokine gene polymorphisms and the susceptibility to liver cirrhosis in patients with chronic hepatitis C. Liver Int. 2003;23(6):420–5.
Huang Y, Hwang S, Chan C, et al. Serum levels of cytokines in hepatitis C-related liver disease: a longitudinal study. Zhonghua yi xue za zhi =. Chinese medical journal; Free China ed. 1999;62(6):327–33.
Farinati F, Cardin R, Bortolami M, Guido M, Rugge M. Oxidative damage, pro-inflammatory cytokines, TGF-alpha and c-myc in chronic HCV-related hepatitis and cirrhosis. World J Gastroenterol. 2006;12(13):2065–9.
Kamari Y, Shaish A, Vax E, et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. 2011;55(5):1086–94.
Zhu H, Liu C. Interleukin-1 inhibits hepatitis C virus subgenomic RNA replication by activation of extracellular regulated kinase pathway. J Virol. 2003;77(9):5493–8.
Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006;83(2):447–55.
Tian Z, Shen X, Feng H, Gao B. IL-1β attenuates IFN-αβ-induced antiviral activity and STAT1 activation in the liver: involvement of proteasome-dependent pathway. J Immunol. 2000;165(7):3959–65.
Cortez-Pinto H, de Moura MC, Day CP. Non-alcoholic steatohepatitis: from cell biology to clinical practice. J Hepatol. 2006;44(1):197–208.
Castell JV, Gómez-Lechón MJ, David M, et al. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett. 1989;242(2):237–9.
Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265(3):621.
Geiger T, Andus T, Klapproth J, Hirano T, Kishimoto T, Heinrich PC. Induction of rat acute-phase proteins by interleukin 6 in vivo. Eur J Immunol. 1988;18(5):717–21.
Kishimoto T. Interleukin-6: from basic science to medicine-40 years in immunology. Annu Rev Immunol. 2005;23:1–21.
Machida K, Cheng KT, Sung VM-H, Levine AM, Foung S, Lai MM. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006;80(2):866–74.
Yamamoto M, Uematsu S, Okamoto T, et al. Enhanced TLR-mediated NF-IL6–dependent gene expression by Trib1 deficiency. J Exp Med. 2007;204(9):2233–9.
Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–34.
Kitani A, Hara M, Hirose T, et al. Autostimulatory effects of IL-6 on excessive B cell differentiation in patients with systemic lupus erythematosus: analysis of IL-6 production and IL-6R expression. Clin Exp Immunol. 1992;88(1):75.
Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer cell. 2008;13(1):7–9.
Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41(16):2502–12.
Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6–deficient mice. Hepatology. 2000;31(1):149–59.
Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274(5291):1379–83.
Yamada Y, Fausto N. Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am J Pathol. 1998;152(6):1577.
Yoshimura A, Mori H, Ohishi M, Aki D, Hanada T. Negative regulation of cytokine signaling influences inflammation. Curr Opin Immunol. 2003;15(6):704–8.
Yang X-P, Schaper F, Teubner A, et al. Interleukin-6 plays a crucial role in the hepatic expression of SOCS3 during acute inflammatory processes in vivo. J Hepatol. 2005;43(4):704–10.
Heinrich P, Behrmann I, Haan S, Hermanns H, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem j. 2003;374:1–20.
Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid 1. J Clin Endocrinol Metab. 1998;83(3):847–50.
Crichton MB, Nichols JE, Zhao Y, Bulun SE, Simpson ER. Expression of transcripts of interleukin-6 and related cytokines by human breast tumors, breast cancer cells, and adipose stromal cells. Mol Cell Endocrinol. 1996;118(1):215–20.
Orban Z, Remaley AT, Sampson M, Trajanoski Z, Chrousos GP. The differential effect of food intake and β-adrenergic stimulation on adipose-derived hormones and cytokines in man. J Clin Endocrinol Metab. 1999;84(6):2126–33.
Greenberg AS, Nordan RP, McIntosh J, Calvo JC, Scow RO, Jablons D. Interleukin 6 reduces lipoprotein lipase activity in adipose tissue of mice in vivo and in 3T3-L1 adipocytes: a possible role for interleukin 6 in cancer cachexia. Cancer Res. 1992;52(15):4113–6.
Feingold KR, Doerrler W, Dinarello CA, Fiers W, Grunfeld C. Stimulation of lipolysis in cultured fat cells by tumor necrosis factor, interleukin-1, and the interferons is blocked by inhibition of prostaglandin synthesis. Endocrinology. 1992;130(1):10–6.
Mohamed-Ali V, Goodrick S, Rawesh A, et al. Subcutaneous Adipose Tissue Releases Interleukin-6, But Not Tumor Necrosis Factor-α, in Vivo 1. J Clin Endocrinol Metab. 1997;82(12):4196–200.
Bastard J-P, Jardel C, Bruckert E, et al. Elevated Levels of Interleukin 6 Are Reduced in Serum and Subcutaneous Adipose Tissue of Obese Women after Weight Loss 1. J Clin Endocrinol Metab. 2000;85(9):3338–42.
Malaguarnera M, Di Fazio I, Romeo MA, Restuccia S, Laurino A, Trovato BA. Elevation of interleukin 6 levels in patients with chronic hepatitis due to hepatitis C virus. J Gastroenterol. 1997;32(2):211–5.
Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–4.
Park EJ, Lee JH, Yu G-Y, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208.
Basu A, Meyer K, Lai KK, et al. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology. 2006;349(2):347–58.
Lima-Cabello E, Garcia-Mediavilla M, Miquilena-Colina M, et al. Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clin Sci. 2011;120:239–50.
Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103(6):1372–9.
Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38(2):413–9.
Haukeland JW, Damås JK, Konopski Z, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44(6):1167–74.
Mas E, Danjoux M, Garcia V, Carpentier S, Ségui B, Levade T. IL-6 deficiency attenuates murine diet-induced non-alcoholic steatohepatitis. PLoS One. 2009;4(11):e7929.
Yamaguchi K, Itoh Y, Yokomizo C, et al. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Laboratory Investig. 2010;90(8):1169–78.
Tilg H, Wilmer A, Vogel W, et al. Serum levels of cytokines in chronic liver diseases. Gastroenterology. 1992;103(1):264–74.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65.
Diehl A, Yin M, Fleckenstein J, et al. Tumor necrosis factor-alpha induces c-jun during the regenerative response to liver injury. Am J Physiol Gastrointest Liver Physiol. 1994;267(4):G552–G61.
Akerman P, Cote P, Yang SQ, et al. Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 1992;263(4):G579–G85.
Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harbor Perspect Biol. 2009;1(5):a000141.
DECKER K. Biologically active products of stimulated liver macrophages (Kupffer cells). Eur J Biochem. 1990;192(2):245–61.
Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36(1):4–12.
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–98.
Yoshigai E, Hara T, Okuyama T, et al. Characterization of natural antisense transcripts expressed from interleukin 1β-inducible genes in rat hepatocytes. HOAJ Biol. 2012;1(1):10.
Matsui K, Nishizawa M, Ozaki T, et al. Natural antisense transcript stabilizes inducible nitric oxide synthase messenger RNA in rat hepatocytes. Hepatology. 2008;47(2):686–97.
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481.
Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci. 1989;86(7):2336–40.
Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci. 1997;94(6):2557–62.
Yin M, Wheeler MD, Kono H, et al. Essential role of tumor necrosis factor α in alcohol-induced liver injury in mice. Gastroenterology. 1999;117(4):942–52.
Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6(9):998–1003.
Crespo J, Fern P, Hern M, Mayorga M, Pons-Romero F. Gene expression of tumor necrosis factor [alpha] and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34(6):1158–63.
Almendral JM, Sommer D, Macdonald-Bravo H, Burckhardt J, Perera J, Bravo R. Complexity of the early genetic response to growth factors in mouse fibroblasts. Mol Cell Biol. 1988;8(5):2140–8.
Greenberg ME, Greene L, Ziff E. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem. 1985;260(26):14101–10.
Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1983;311(5985):433–8.
Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell. 1984;38(3):745–55.
Larner A, Jonak G, Cheng Y, Korant B, Knight E, Darnell J. Transcriptional induction of two genes in human cells by beta interferon. Proc Natl Acad Sci. 1984;81(21):6733–7.
Larner A, Chaudhuri A, Darnell J. Transcriptional induction by interferon. New protein (s) determine the extent and length of the induction. J Biol Chem. 1986;261(1):453–9.
Lee T, Lee G, Ziff E, Vilcek J. Isolation and characterization of eight tumor necrosis factor-induced gene sequences from human fibroblasts. Mol Cell Biol. 1990;10(5):1982–8.
Beadling C, Johnson KW, Smith KA. Isolation of interleukin 2-induced immediate-early genes. Proc Natl Acad Sci. 1993;90(7):2719–23.
Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178(6):2249–54.
Mosmann T, Coffman R. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7(1):145–73.
Aoki T, Kikuchi H, Miyatake S, et al. Interleukin 5 enhances interleukin 2-mediated lymphokine-activated killer activity. J Exp Med. 1989;170(2):583–8.
Balkwill F, Burke F. The cytokine network. Immunology today. 1989;10(9):299–304.
Spits H, Yssel H, Paliard X, Kastelein R, Figdor C, De Vries J. IL-4 inhibits IL-2-mediated induction of human lymphokine-activated killer cells, but not the generation of antigen-specific cytotoxic T lymphocytes in mixed leukocyte cultures. J Immunol. 1988;141(1):29–36.
Whitmire JK, Tan JT, Whitton JL. Interferon-γ acts directly on CD8 + T cells to increase their abundance during virus infection. J Exp Med. 2005;201(7):1053–9.
Ye J, Ortaldo JR, Conlon K, Winkler-Pickett R, Young HA. Cellular and molecular mechanisms of IFN-gamma production induced by IL-2 and IL-12 in a human NK cell line. J Leukoc Biol. 1995;58(2):225–33.
Darnell J, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21.
Kusters S, Gantner F, Kunstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 1996;111(2):462–71.
Toyonaga T, Hino O, Sugai S, et al. Chronic active hepatitis in transgenic mice expressing interferon-gamma in the liver. Proc Natl Acad Sci. 1994;91(2):614–8.
Dienes HP, Hess G, Wöorsdörfer M, et al. Ultrastructural localization of interferon-producing cells in the livers of patients with chronic hepatitis B. Hepatology. 1991;13(2):321–6.
Daniels H, Eddleston A, Alexander G, Williams R, Meager A. Spontaneous production of tumour necrosis factor α and interleukin-1β during interferon-α treatment of chronic HBV infection. Lancet. 1990;335(8694):875–7.
Kakumu S, Fuji A, Yoshioka K, Tahara H. Serum levels of alpha-interferon and gamma-interferon in patients with acute and chronic viral hepatitis. Hepatogastroenterology. 1989;36(2):97–102.
Cacciarelli TV, Martinez OM, Gish RG, Villanueva JC, Krams SM. Immunoregulatory cytokines in chronic hepatitis C virus infection: Pre-and posttreatment with interferon alfa. Hepatology. 1996;24(1):6–9.
Frese M, Schwärzle V, Barth K, et al. Interferon-γ inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology. 2002;35(3):694–703.
Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17(1):189–220.
Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4 A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102(8):2992.
Pacifico L, Di Renzo L, Anania C, et al. Increased T-helper interferon-γ-secreting cells in obese children. Eur J Endocrinol. 2006;154(5):691–7.
Caldwell SH, Crespo DM, Kang HS, Al-Osaimi AM. Obesity and hepatocellular carcinoma. Gastroenterology. 2004;127(5):S97–S103.
Stickel F, Hellerbrand C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: mechanisms and implications. Gut. 2010:gut. 2009.199661.
Bertola A, Bonnafous S, Anty R, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One. 2010;5(10):e13577.
Guebre-Xabier M, Yang S, Lin HZ, Schwenk R, Krzych U, Diehl AM. Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: Potential mechanism for sensitization to liver damage. Hepatology. 2000;31(3):633–40.
Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005;42(4):880–5.
Author contributions
The work is a product of the intellectual environment of the whole team. Both RN and SZ have contributed equally to the exploration of data, its compilation, acquisition, and writing of the manuscript. AA has helped in data compilation and manuscript writing. MS and IA have helped in acquisition of the data of the manuscript. MI, SR, and SA have critically gone through the whole work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was obtained for this study.
Conflict of interest
Authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Responsible Editor: John Di Battista.
Rabia Nawaz and Sadia Zahid contributed equally to this work.
Rights and permissions
About this article

Cite this article
Nawaz, R., Zahid, S., Idrees, M. et al. HCV-induced regulatory alterations of IL-1β, IL-6, TNF-α, and IFN-ϒ operative, leading liver en-route to non-alcoholic steatohepatitis. Inflamm. Res. 66, 477–486 (2017). https://doi.org/10.1007/s00011-017-1029-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-017-1029-3