
Abstract
The liver is a unique organ with respect to its anatomical location, allowing continuous blood flow from the gastrointestinal tract through the sinusoids, and its cellular composition, comprising metabolically active hepatocytes, nonhepatocytic parenchymal cells, and various immune cell populations. Cytokines are key mediators within the complex interplay of intrahepatic immune cells and hepatocytes, as they can activate effector functions of immune cells, as well as hepatocytic intracellular signaling pathways controlling cellular homeostasis. Kupffer cells and liver-infiltrating monocyte-derived macrophages are primary sources of cytokines such as tumor-necrosis factor-alpha (TNF-alpha) and interleukin-6. The liver is also enriched in natural killer (NK) and NK T cells, which fulfill functions in pathogen defense, T cell recruitment, and modulation of liver injury. TNF-alpha can activate specific intracellular pathways in hepatocytes that influence cell fate in different manners, e.g., proapoptotic signals via the caspase cascade, but also survival pathways, namely the nuclear factor (NF)-kappaB pathway. NF-kappaB regulates important functions in liver physiology and pathology. Recent experiments with genetically modified mice demonstrated important and partly controversial functions of this pathway, e.g., in cytokine-mediated hepatocyte apoptosis or ischemia–reperfusion injury. The exact dissection of the contribution of recruited and resident immune cells, their soluble cytokine and chemokine mediators, and the intracellular hepatocytic response in liver homeostasis and injury could potentially identify novel targets for the treatment of acute and chronic liver disease, liver fibrosis, or cirrhosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
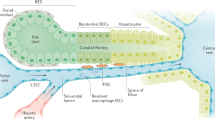
The liver is an exceptional organ in terms of its metabolic, synthetic, and detoxifying function. It has the unique potential to regenerate after tissue loss and, for instance, plays an important role in the regulation of blood glucose or blood lipids. All these and many other functions represent the organ’s ability to execute the proper reaction towards the body’s demands and keeping it in homeostasis. The central function of the liver for homeostasis and inflammatory responses is also underscored by its sole anatomical location, allowing continuous blood supply not only from the arterial system (hepatic arteries) but also from the gastrointestinal tract via the portal vein (Fig. 1). Circulating blood cells, e.g., from the innate or adaptive immune system, are pressed through a network of sinusoids allowing contact to a variety of intrahepatic cell populations, such as parenchymal liver cells (hepatocytes), endothelial cells, liver-resident macrophage (Kupffer cell) or lymphocyte [mainly natural killer T (NKT) cells] populations, hepatic stellate cells, and others (Fig. 1) [1]. Communication between these cell types and the regulation of hepatic functions are primarily achieved by cytokines. Cytokines are small-molecular-weight messengers secreted by one cell to alter the behavior of the cell itself (autocrine messenger), a closely related cell (paracrine messenger), or cells in different organs (endocrine messenger) [2]. This review will address the function of intrahepatic immune cells in homeostasis and inflammation, highlight some of the relevant cytokines and mediators for liver homeostasis, and discuss the manifold consequences of cytokine-driven activation of hepatocellular signaling pathways in liver homeostasis and injury.

Intrahepatic cell populations. The healthy liver comprises about 60–80% hepatocytes; the other intrahepatic cell populations include biliary cells, liver sinusoidal endothelial cells (LSECs) lining the liver sinusoids, Kupffer cells (KC), and hepatic stellate cells (HSC) in the Dissé space between hepatocytes and LSECs. In addition, many immune cells are found in the liver, mainly entering from the circulation via hepatic arteries and portal vein branches, including neutrophils (PMN), monocytes (monos), dendritic cells (DCs), and lymphocytes (T, B, NK, and NKT cells)
Intrahepatic immune cells as producers and targets of cytokines
Besides its various metabolic functions, the liver is also a central “immunological” organ. Blood coming from the gastrointestinal tract is enriched of potential antigens, and immune cells in the liver have the potential to initiate both (a) innate and adaptive immune responses in the case of infections, e.g., in response to lipopolysaccharide (LPS) or bacterial superantigens, or (b) immunological tolerance in the vast majority of harmless antigens during homeostasis.
Innate immune effector cells
The liver is selectively enriched in macrophages (Kupffer cells), natural killer (NK), and NKT cells. Although it has long been known that Kupffer cells principally originate from bone-marrow-derived monocytes [3], Kupffer cells have long been considered a rather sessile, tissue-resident, and (fairly) radio-resistant population [4, 5]. Recent data challenge this concept, as mouse models of bone marrow transplantation and experimental liver transplantation revealed that more than half of the Kupffer cells in a steady state liver originate directly from bone-marrow precursors, indicating a high turnover rate of macrophages in liver homeostasis [6]. In the case of inflammation, blood-derived infiltrating monocytes may become even more dominant for macrophage actions, as suggested by animal models of chronic liver injury and fibrogenesis [5, 7]. Monocytes consist of at least two major subsets with different migratory and functional properties [8–10]; their specific roles in liver homeostasis and inflammation as macrophage-precursors are the subject of intensive ongoing research.
Kupffer cells (resident or monocyte-derived) have the capacity to phagocyte and to release a broad panel of cytokines that critically determine the subsequent reactions of other immune cells and hepatocytes, as well as the degree of organ damage [11]. Kupffer cells can release a variety of proinflammatory cytokines such as tumor-necrosis factor alpha (TNF-alpha), interleukin (IL)-6, IL-1β, or leukotrienes (Fig. 2) [2] but, on the other hand, e.g., in the context of low “physiological” levels of LPS, also anti-inflammatory cytokines like IL-10 [12]. In acute or chronic liver diseases, Kupffer cells and infiltrating monocyte-derived macrophages are well known to promote inflammatory cascades by releasing these proinflammatory mediators with consequences like T cell attraction, induction of hepatocyte apoptosis (Fig. 2), or activation of fibrogenic hepatic stellate cells [11, 13]; however, recent experimental data indicate that macrophages are also required to restrict the inflammatory response and to degrade extracellular matrix proteins for the regression of liver fibrosis [5, 14].

Selected key interactions between innate immune cells and hepatocytes. Kupffer (macrophages), NKT, and NK cells are abundantly found in the liver. Kupffer cells can release IL-6 and TNFα, activating hepatocytic gp130-dependent STAT signaling cascades and TNF-R-dependent activation of apoptotic caspase and/or antiapoptotic (proinflammatory) NF-kappaB (NFkB) pathways. NKT cells exert their actions via release of TNFα, IFNγ, or FasL-mediated apoptosis. NK cells can also release IFNγ and may induce apoptosis via TRAIL in the case of hepatocytic TRAIL-R expression
NK cells represent a population of lymphocytes with potent cytolytic activity against virus-infected or tumor cells. Hepatic NK cells are regulated by Kupffer-cell derived cytokines, e.g., IL-12 and IL-18, as well as NKT cell-driven IL-4; NK cells produce large amounts of (antiviral) interferon gamma (IFNγ) upon activation (Fig. 2) [15]. Hepatic NK cells modulate T cell responses in the liver, promote intracellular changes in endothelial cells and hepatocytes (mainly via IFNγ), and can even directly promote hepatocyte death or cell lysis [16]. In chronic inflammatory conditions, e.g., chronic hepatitis B virus infection, liver injury is closely linked to NK-mediated IL-8 and IFNα synthesis, as well as accumulation and activation of NK cells in the liver expressing the apoptosis-inducing TNF-related apoptosis ligand (TRAIL) [17].
NKT cells are a subpopulation of unconventional T cells, as they express surface markers of T and NK cells, which are found at unusually high frequencies in the liver. From studies of experimental liver injury in mice after administration of the plant-derived lectin Concanavalin A (ConA), NKT have long been identified as critical factors promoting acute liver damage by release of IL-4, IFNγ, and direct induction of Fas-mediated hepatocyte apoptosis (Fig. 2) [18]. However, their physiological function has remained somewhat obscure. Recent studies revealed an important role of NKT cells in the early response against microbial infections, as exogenous and endogenous glycolipid antigens from bacteria were found to activate NKT cells [19, 20]. Furthermore, NKT cells are involved in antiviral defense mechanisms, e.g., during chronic hepatitis B virus infection, as suggested by studies in transgenic mice [21].
Antigen presenting cells and adaptive immune cells in the liver
For the initiation of adaptive immune responses, antigens need to be processed and professionally presented to T cells, either in the liver itself or in the draining lymph node [1]. Several hepatic cell populations have antigen-presenting properties. In noninflammatory conditions, antigen-presentation by liver sinusoidal endothelial cells appears to be a crucial mechanism for the maintenance of immunological tolerance [22]. Furthermore, Kupffer cells and also bone-marrow-derived dendritic cells, most likely of monocytic origin, can efficiently prime T cells [1, 11]. The nature of the T cell response, e.g., T-cell cytotoxicity in the case of a chronic viral infection or T-cell tolerance in the case of harmless gut-derived antigens or autoantigens, seems to depend on the antigen-presenting cells, the cytokine milieu, and the site of primary T cell activation [23, 24]. More recently, hepatic stellate cells, known for vitamin A storage and collagen synthesis in liver fibrosis, have been identified as cells with antigen-presenting capacity that can activate T cell responses, e.g., after bacterial challenge [25].
Cytokines and cytokine-receptors in the liver
Besides direct effects of immune cells on hepatocytes, inflammatory pathways in the liver are largely regulated by cytokines. Cytokine action is generally mediated by the interaction of cellular receptors, which signal internally to the nucleus, and external factors, which are able to bind these receptors. These networks have evolved early in evolution; pathways with strong homology to human cytokine networks are already found in Drosophila and mollusks [26]. For example, in Drosophila, nuclear factor (NF)-kappaB-like transcription factors are activated in order to combat infections; this represents one major role of cytokine networks in higher organisms like humans. Maintaining the ordered balance between proliferation and controlled cell death (apoptosis) during embryonic development and organogenesis is another important function of cytokines in physiologic conditions. Since these functions are preserved in the adult organism, a disturbance of the critical balance might have deleterious effects [2]. Dysregulated cytokine actions after liver injury can result in excessive apoptosis, a key finding in various acute and chronic liver diseases, e.g., viral and autoimmune hepatitis, cholestatic disease, and alcoholic or drug/toxin-induced liver injury [27]. Among the manifold cytokines relevant for liver homeostasis and injury, we will highlight important findings on TNF-alpha and IL-6, as these represent two extensively studied pathways with exceptional significance in the liver. Studies in patients and animal models have strongly implicated that death receptor ligands such as TNF-alpha or Fas ligand (FasL) are involved in the induction of apoptosis and in triggering destruction of the liver [28]. The IL-6 cytokine family, on the other hand, may have essential functions in protecting the liver during acute or chronic injury [29, 30].
TNF-alpha and its receptors
TNF and FasL belongs to a family of ten ligands (TNF, lymphotoxin-α, TNFβ, FasL, OX40L, CD40L, CD27L, CD30L, 4–1BBL, and lymphotoxin-β) that activate structurally related receptor proteins known as the TNF receptor superfamily. Currently, 12 different death receptors are well established, including TNF receptor 1 (TNF-R1, Fig. 4), TNF-R2, TNF-RP, Fas, OX-40, 4–1BB, CD40, CD30, CD27, pox virus PV-T2, PV-A53R gene products, and the p75 NGFR. In addition, the apoptosis-signaling receptors death receptor (DR) 3, DR4, and DR5; their ligand TRAIL; and a nonsignaling decoy receptor TRID/DcR are recently identified members of these superfamilies [31]. In patients with fulminant hepatic failure, serum levels of TNF-alpha, TNF-R1, and TNF-R2 are markedly increased, and these changes directly correlated with disease activity. In explanted livers of these patients, infiltrating mononuclear cells expressed high amounts of TNF-alpha and hepatocytes overexpressed TNF-R1 [28].
IL-6 and its receptor
IL-6 belongs to a family of cytokines comprising of IL-6, IL-11, leukemia inhibitory factor, oncostatin M, ciliary neurotropic factor, novel neurotrophin-1/B cell stimulating factor-3, and cardiotrophin 1 [32]. IL-6 binds directly to hepatocytes by interacting with an 80-kd membrane glycoprotein (gp80) that complexes with a signal transducing molecule named gp130 (Fig. 3A) [26]. Serum and intrahepatic levels of IL-6 are elevated in patients with acute and chronic liver diseases [29]. The other members of the IL-6 cytokine family have similar receptors and mostly lead to complexes of gp130 with another (related) signaling molecule (Fig. 3B).

Cytokines of the interleukin 6 family and their receptors. A Interleukin 6 (IL-6) binds to the IL-6R/gp80, e.g., on hepatocytes. IL-6-gp80 then complexes with the signal transducing molecule gp130. Dimerization of two gp130 molecules activates intracellular signaling cascades. B Members of the IL-6 cytokine family and their receptors (schematic). LIF, leukemia inhibitory factor; OSM, oncostatin M; CNTF, ciliary neurotropic factor; CT, cardiotrophin; NP, novel neurotrophin-1; R, receptor
Intracellular pathways controlling liver homeostasis and inflammation
TNF-alpha signaling in the liver
In many instances, hepatic failure might result from an imbalance between damaging and protective signals that are very tightly regulated under physiologic conditions. As mentioned above, TNF-alpha and related cytokines are key players in liver homeostasis, as they can activate both proapoptotic (mainly caspases) and antiapoptotic (mainly NF-kappaB) pathways in hepatocytes (Figs. 2 and 4).

TNF-alpha signaling in the liver. TNF-alpha binds to its receptors, e.g., TNF-R1, and can thereby activate the proapoptotic caspase cascades (via TRADD, FADD, and cleavage of procaspase 8) or the antiapoptotic NF-kappaB pathway (via activation of the IKK complex resulting in phosphorylation of IkBα, subsequent translocation of NF-kappaB to the nucleus, and expression of NF-kappaB responsive genes). For details, please see main text
Activation of proapoptotic signaling cascades
FasL and TNF-alpha facilitate programmed cell death in a similar manner by activation of caspases. The most important target of both pathways orchestrating cellular death is the aspartate-specific cysteine protease- or caspase-cascade, consisting of initiator caspases such as caspases 8 and 9 and executioner caspases, e.g., caspase 3, 6, and 7. Since proteolytic cleavage generates the mature caspases, one way in which these enzymes are activated is via the action of proteases, including other caspases [33]. TNF-alpha signals through two distinct cell surface receptors, TNF-R1 and TNF-R2, of which TNF-R1 initiates the majority of TNF-alpha’s biological activities. Binding of TNF-alpha to its receptor leads to the release of the inhibitory protein silencer of death domains from TNF-R1’s intracellular domain. This results in the recognition of the intracellular TNF-R1 domain by the adaptor protein TNF receptor-associated death domain (TRADD), which in turn recruits Fas-associated death domain (FADD). FADD recruits caspase-8 to the TNF-R1 complex, where it becomes activated and initiates the protease cascade leading to activation of executioner caspases and apoptosis (Fig. 4). In contrast to TNF-dependent signaling, FasL can interact directly with the death domain of FADD without recruiting TRADD [34, 35]. In several studies, involvement of mitochondria and the release of cytochrome c in the apoptotic process has been demonstrated, and this was also shown for FasL and TNF-alpha mediated apoptosis in the liver [36, 37].
Activation of the NF-kappaB pathway
Next to the activation of caspases, binding of TNF-alpha to its receptor also leads to the activation of the NF-kappaB pathway (Fig. 4). NF-kappaB is a dimer of members of the Rel family of DNA-binding proteins. The mammalian NF-kappaB family includes five cellular DNA-binding subunit proteins: p50 (NF-κB1), p52 (NF-κB2), c-Rel (Rel), p65 (RelA), and RelB [38]. The NF-kappaB DNA-binding subunits share an N-terminal Rel homology domain, which is responsible for DNA-binding, dimerization, nuclear translocation, and interaction with the inhibitory IκB proteins [39].
TNF-induced activation of NF-kappaB relies on the phosphorylation of two conserved serines (S32 and S36 in human IκBα) in the N-terminal regulatory domain of IκBs. After phosphorylation, the IκBs undergo a second posttranslational modification: polyubiquitination by a cascade of enzymatic reactions, followed by the degradation of IκB proteins by the proteasome, thus releasing NF-kappaB from its inhibitory IκB-binding partner so it can translocate to the nucleus and activate transcription of NF-kappaB-dependent target genes (Fig. 4) [40, 41]. Since the enzymes that catalyze the ubiquitination of I-κB are constitutively active, the only regulated step in NF-kappaB activation appears to be, in most cases, the phosphorylation of I-κB molecules.
A high-molecular-weight complex, called “I-κB kinase (IKK)-complex,” that mediates the phosphorylation of I-κB has been purified and characterized (Fig. 4). This complex consists of three tightly associated IKK polypeptides: IKK1 (also called IKKα) and IKK2 (IKKβ), the catalytic subunits of the kinase complex [42–44], and a regulatory subunit called NF-κB essential modulator, IKKγ (NEMO) [42, 45]. In vitro, IKK1 and IKK2 can form homo- and heterodimers [46]. Both IKK1 and IKK2 are able to phosphorylate I-κB in vitro, but IKK2 has a higher kinase activity in vitro compared with IKK1 [42, 47–50].
Activation of the IKK complex upon TNF-alpha stimulation involves IKK recruitment to the TNF-R1 [51–53]. Besides TNF-R1, this process involves TNF-receptor-associated-factor 2 (TRAF2) and the death-domain kinase receptor-interacting protein (RIP). In response to TNF-alpha treatment, TRAF2 recruits the IKK complex to TNF-R1 via the interaction of the RING-finger motifs of TRAF2 with the leucin-zipper motif of both IKK1 and IKK2 [51, 53]. RIP can directly interact with NEMO and mediate IKK activation, although the enzymatic activity of RIP is not required for this process [52]. The mechanism by which recruitment of the IKK complex to the TNF receptor leads to IKK activation is not clear, but it might involve NEMO-induced autophosphorylation of the IKK complex. Moreover, ubiquitination of multiple factors that regulate the IKK complex, like TRAF6/TAK1 or c-IAP1, an inhibitor of apoptosis that is also part of the TNF receptor complex, modulates the activity of the NF-kappaB pathway [41].
Consequences of NF-kappaB activation in the liver
Numerous studies have shown that NF-kappaB provides survival signals in the context of death-receptor-induced apoptosis in the liver. This process is assumed to involve the transcriptional induction of various apoptotic suppressors [54]. Evidence that NF-kappaB governs critical antiapoptotic proteins comes from well-described animal models. Injection of TNF-alpha into mice and addition of TNF-alpha to hepatic cells resulted in activation of nuclear translocation and of DNA binding of NF-kappaB [55], and hepatocytes are resistant to apoptosis induced by TNF-alpha or LPS, a potent inductor for endogenous TNF-alpha in the liver, unless they are treated with inhibitors of transcription of (antiapoptotic) proteins like cycloheximide or actinomycin D [56–58]. Knockout mice lacking the p65 subunit of NF-kappaB die between days E15 and E16 post coitum as a result of fetal hepatocyte apoptosis [59]. This is caused by increased sensitivity towards TNF-alpha since TNF/p65 double-deficient mice are rescued from embryonic lethality [60].
Genetic experiments have also highlighted the differential functions of the IKK subunits in TNF-alpha-mediated liver apoptosis. Mice lacking IKK1 die shortly after birth and display a phenotype marked by thickening of skin and limb, as well as skeletal defects [61, 62]. IKK2-deficient mice die in utero approximately at embryonic day12.5 as a result of massive apoptosis in the liver, and fibroblasts from these mice show no activation of NF-kappaB in response to TNF-alpha [63–65]. A similar phenotype was noted in mice lacking the regulatory subunit NEMO, which also die from massive apoptosis in the liver and show a defect in NF-kappaB activation upon TNF-alpha stimulation in their primary murine embryonic fibroblasts [66]. Therefore, at least during embryogenesis, IKK2 and NEMO appear to be the critical subunits for NF-kappaB activation and protection of liver cells from proinflammatory cytokines like TNF-alpha.
The role of the IKK subunits in the adult animal is less well understood. Conditional knockout mice based on the cre/loxP system have emerged as new powerful tools to study gene functions in the adult animal in vivo [67, 68]. In a recent study, we showed that hepatocyte-specific ablation of IKK2 does not lead to a strongly impaired activation of NF-kappaB or increased apoptosis after TNF-alpha stimulation, probably because IKK1 homodimers can take over this function in the absence of IKK2 in the adult mouse [69]. In contrast, conditional hepatocyte-specific knockout of NEMO resulted in complete block of NF-kappaB activation and massive hepatocyte apoptosis, underlining that NEMO is the only irreplaceable IKK subunit for prevention of TNF-alpha-mediated liver apoptosis [70].
Besides its antiapoptotic function in TNF-alpha-mediated liver apoptosis, NF-kappaB appears to be also critically involved in various models of experimental liver injury. For instance, NF-kappaB DNA-binding occurs quickly upon hepatic ischemia–reperfusion (I/R) injury [71], but it has also long been unclear whether NF-kappaB-dependent signaling withholds a protective or damaging role in I/R injury. We showed that hepatocyte-specific conditional knockout mice for IKK2 show a defect in NF-kappaB activation after I/R [69]. Inhibition of NF-kappaB activation in conditional IKK2-deficient mice protected them from liver injury due to I/R [69], thus underlining that, depending on the experimental model, the NF-kappaB pathway does not serve as a survival pathway, but instead can aggravate hepatocyte death and liver damage. However, complete abolishment of NF-kappaB activation in conditional NEMO-knockout mice resulted in massive hepatic inflammation and apoptosis after I/R injury [70].
IL-6 signaling in the liver
IL-6 binds to the gp80/IL-6 receptor on hepatocytes that then complex with the signal transducer gp130 (Fig. 3). Binding of gp130 leads to dimerization of the intracellular domains of two gp130 molecules, which promotes association with receptor-associated Janus kinases (JAKs; JAK1, JAK2, and TYK). The JAKs become activated and phosphorylate different tyrosine residues on the gp130 molecule. Depending on the location of the phosphorylated tyrosines, signal transducers and activators of transcription (STAT) proteins (mainly STAT-3) and also the Ras/mitogen-activated protein kinase pathway become activated and trigger numerous downstream effects mediated by the signaling of IL-6 and related cytokines (Fig. 5) [32].

IL-6 signaling in the liver. The complex of IL-6, gp80 (IL-6R), and two gp130 molecules mediates IL-6 signaling via phosphorylation of tyrosine (Y) residues of the intracellular gp130 molecule. Depending on the location of the phosphorylated tyrosines, signal transducers, and activators of transcription (STAT) proteins (mainly STAT-3), and also the Ras/mitogen-activated protein (MAP) kinase pathway become activated and trigger the downstream effects
An important role of IL-6-dependent signaling in the liver is the induction of the acute phase response [72, 73], and STAT3 participates in its transcriptional activation. In patients with fulminant hepatic failure or with chronic liver diseases, IL-6 expression in serum and liver tissue correlates with disease progression [29, 30]. In experimental models of liver injury, mice deficient for the gp130 receptor in hepatocytes show an abolished acute phase response and an increased susceptibility to LPS-induced liver failure or to bacterial infections [30, 74]. In the model of ConA-induced hepatitis, pretreatment with IL-6 can protect mice from liver injury. This protection from ConA-induced liver damage requires gp130 signaling in hepatocytes and is mediated via the gp130/STAT3 signaling cascade, resulting in the upregulation of other cytokines, such as the IL8 ortholog KC (CXCL1) and serum amyloid A-2 (SAA-2) [75]. Gp130-signaling in nonparenchymal cells is, on the other hand, essential for mediating protective IL-6/gp130 effects during experimental chronic liver diseases and liver fibrogenesis [29]. Studies are ongoing to translate findings of hepatoprotective effects of IL-6/gp130 into novel therapeutic approaches in acute and chronic liver diseases [26].
Conclusions
A growing number of studies have implicated immune cells, cytokines, and cytokine-dependent pathways in the development of liver failure, chronic liver disease, hepatic inflammation, and liver fibrogenesis. Resident and infiltrating immune cells have been linked to progression, but also to restriction and regression of liver injury. Parenchymal and nonparenchymal survival pathways like NF-kappaB or IL-6 withhold a protective function in many experimental liver disease models and thus are an attractive target for a pharmacological intervention [76]. However, many experimental models highlighted dual functions of these key players, namely beneficial or adverse effects. For instance, blockage of monocyte infiltration into the liver may limit disease progression but would negatively affect the resolution of liver fibrosis [5]. Thus, an inflammatory immune cell is not necessarily detrimental in any context. Also, an inhibition of NF-κB in the liver can have different outcomes depending on the experimental model applied: protecting from apoptosis in a model of TNF-dependent cell death vs aggravating cellular necrosis in a model of I/R injury [67]. Thus, a survival pathway is not necessarily protective in any context. Dissecting the cellular and molecular inflammatory pathways during liver homeostasis and during liver injury will hopefully provide the basis for novel specific therapeutic approaches in the near future.
References
Racanelli V, Rehermann B (2006) The liver as an immunological organ. Hepatology 43:S54–S62
Luedde T, Liedtke C, Manns MP, Trautwein C (2002) Losing balance: cytokine signaling and cell death in the context of hepatocyte injury and hepatic failure. Eur Cytokine Netw 13:377–383
Gale RP, Sparkes RS, Golde DW (1978) Bone-marrow origin of hepatic macrophages (Kupffer cells) in humans. Science 201:937–938
Naito M, Hasegawa G, Takahashi K (1997) Development, differentiation, and maturation of Kupffer cells. Microsc Res Tech 39:350–364
Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu SJ, Lang R, Iredale JP (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65
Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, Crispe IN (2007) Kupffer cell heterogeneity: functional properties of bone marrow-derived and sessile hepatic macrophages. Blood 110:4077–4085
Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H (2005) Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology 128:138–146
Tacke F, Randolph GJ (2006) Migratory fate and differentiation of blood monocyte subsets. Immunobiol 211:609–618
Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117:185–194
Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M, Randolph GJ (2006) Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J Exp Med 203:583–597
Bilzer M, Roggel F, Gerbes AL (2006) Role of Kupffer cells in host defense and liver disease. Liver Int 26:1175–1186
Knolle PA (2006) Involvement of the liver in the induction of CD8 T cell tolerance towards oral antigen. Z Gastroenterol 44:51–56
Schumann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N, Tiegs G (2000) Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol 157:1671–1683
Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP (2007) Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 178:5288–5295
Ajuebor MN, Wondimu Z, Hogaboam CM, Le T, Proudfoot AE, Swain MG (2007) CCR5 Deficiency Drives Enhanced Natural Killer Cell Trafficking to and Activation within the Liver in Murine T Cell-Mediated Hepatitis. Am J Pathol 170:1975–1988
Dong ZJ, Wei HM, Sun R, Tian ZG (2007) The roles of innate immune cells in liver injury and regeneration. Cell Mol Immunol 4:241–252
Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, Das A, Lopes AR, Borrow P, Williams K, Humphreys E, Afford S, Adams DH, Bertoletti A, Maini MK (2007) Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med 204:667–680
Tiegs G (2007) Cellular and cytokine-mediated mechanisms of inflammation and its modulation in immune-mediated liver injury. Z Gastroenterol 45:63–70
Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, Tsuji M, Kawahara K, Wong CH, Kronenberg M (2005) Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 434:520–525
Mattner J, Debord KL, Ismail N, Goff RD, Cantu C III, Zhou D, Saint-Mezard P, Wang V, Gao Y, Yin N, Hoebe K, Schneewind O, Walker D, Beutler B, Teyton L, Savage PB, Bendelac A (2005) Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 434:525–529
Kakimi K, Guidotti LG, Koezuka Y, Chisari FV (2000) Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med 192:921–930
Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, Momburg F, Arnold B, Knolle PA (2000) Efficient presentation of exogenous antigen by liver endothelial cells to CD8 + T cells results in antigen-specific T-cell tolerance. Nat Med 6:1348–1354
Bowen DG, Zen M, Holz L, Davis T, McCaughan GW, Bertolino P (2004) The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J Clin Invest 114:701–712
Crispe IN, Giannandrea M, Klein I, John B, Sampson B, Wuensch S (2006) Cellular and molecular mechanisms of liver tolerance. Immunol Rev 213:101–118
Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, Modlin RL, Liblau RS, Gressner AM, Kaufmann SH (2007) Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 26:117–129
Luedde T, Trautwein C (2006) Intracellular survival pathways in the liver. Liver Int 26:1163–1174
Neuman MG (2001) Apoptosis in diseases of the liver. Crit Rev Clin Lab Sci 38:109–166
Streetz K, Leifeld L, Grundmann D, Ramakers J, Eckert K, Spengler U, Brenner D, Manns M, Trautwein C (2000) Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology 119:446–460
Streetz KL, Tacke F, Leifeld L, Wustefeld T, Graw A, Klein C, Kamino K, Spengler U, Kreipe H, Kubicka S, Muller W, Manns MP, Trautwein C (2003) Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology 38:218–229
Streetz KL, Wustefeld T, Klein C, Kallen KJ, Tronche F, Betz UA, Schutz G, Manns MP, Muller W, Trautwein C (2003) Lack of gp130 expression in hepatocytes promotes liver injury. Gastroenterology 125:532–543
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME (1998) Apoptosis signaling by death receptors. Eur J Biochem 254:439–459
Streetz KL, Luedde T, Manns MP, Trautwein C (2000) Interleukin 6 and liver regeneration. Gut 47:309–312
Martin SJ, Green DR (1995) Protease activation during apoptosis: death by a thousand cuts? Cell 82:349–352
Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281:1305–1308
Chen G, Goeddel DV (2002) TNF-R1 signaling: a beautiful pathway. Science 296:1634–1635
Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ (1998) The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Mol Cell Biol 18:6353–6364
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400:886–891
Ghosh S, May MJ, Kopp EB (1998) NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16:225–260
Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109:S81–S96, (Suppl)
Karin M (1999) How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 18:6867–6874
Yamamoto Y, Gaynor RB (2004) IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci 29:72–79
Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, Pascual G, Motiwala A, Zhu H, Mann M, Manning AM (1999) IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol 19:1526–1538
Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M (1997) Identification and characterization of an IkappaB kinase. Cell 90:373–383
DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M (1997) A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388:548–554
Rothwarf DM, Zandi E, Natoli G, Karin M (1998) IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395:297–300
Zandi E, Chen Y, Karin M (1998) Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science 281:1360–1363
Delhase M, Hayakawa M, Chen Y, Karin M (1999) Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284:309–313
Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A (1997) IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278:860–866
Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV (1997) IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science 278:866–869
Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M (1997) The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91:243–252
Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z (2000) The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity 12:419–429
Zhang SQ, Kovalenko A, Cantarella G, Wallach D (2000) Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity 12:301–311
Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Z (2001) The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol Cell Biol 21:3986–3994
Liu ZG, Hsu H, Goeddel DV, Karin M (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87:565–576
FitzGerald MJ, Webber EM, Donovan JR, Fausto N (1995) Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ 6:417–427
Lehmann V, Freudenberg MA, Galanos C (1987) Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med 165:657–663
Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A (1994) Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol 153:1778–1788
Leist M, Gantner F, Naumann H, Bluethmann H, Vogt K, Brigelius-Flohe R, Nicotera P, Volk HD, Wendel A (1997) Tumor necrosis factor-induced apoptosis during the poisoning of mice with hepatotoxins. Gastroenterology 112:923–934
Beg AA, Baltimore D (1996) An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274:782–784
Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y (1999) Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci USA 96:2994–2999
Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M (1999) Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science 284:316–320
Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S (1999) Limb and skin abnormalities in mice lacking IKKalpha. Science 284:313–316
Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM (1999) Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 284:321–325
Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M (1999) The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med 189:1839–1845
Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV (1999) Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity 10:421–429
Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW (2000) Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev 14:854–862
Luedde T, Beraza N, Trautwein C (2006) Evaluation of the role of nuclear factor-kappaB signaling in liver injury using genetic animal models. J Gastroenterol Hepatol 21(Suppl 3):S43–S46
Pasparakis M, Luedde T, Schmidt-Supprian M (2006) Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ 13:861–872
Luedde T, Assmus U, Wustefeld T, Meyer ZV, Roskams T, Schmidt-Supprian M, Rajewsky K, Brenner DA, Manns MP, Pasparakis M, Trautwein C (2005) Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J Clin Invest 115:849–859
Beraza N, Ludde T, Assmus U, Roskams T, Vander BS, Trautwein C (2007) Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology 132:2504–2517
Zwacka RM, Zhang Y, Zhou W, Halldorson J, Engelhardt JF (1998) Ischemia/reperfusion injury in the liver of BALB/c mice activates AP-1 and nuclear factor kappaB independently of IkappaB degradation. Hepatology 28:1022–1030
Trautwein C, Boker K, Manns MP (1994) Hepatocyte and immune system: acute phase reaction as a contribution to early defence mechanisms. Gut 35:1163–1166
Zhang D, Sun M, Samols D, Kushner I (1996) STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem 271:9503–9509
Wuestefeld T, Klein C, Streetz KL, Beraza N, Scholmerich J, Burgart LJ, Zender L, Kubicka S, Baskin-Bey E, Gores GJ, Manns MP, Trautwein C (2005) Lack of gp130 expression results in more bacterial infection and higher mortality during chronic cholestasis in mice. Hepatology 42:1082–1090
Klein C, Wustefeld T, Assmus U, Roskams T, Rose-John S, Muller M, Manns MP, Ernst M, Trautwein C (2005) The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J Clin Invest 115:860–869
Karin M, Cao Y, Greten FR, Li ZW (2002) NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2:301–310
Acknowledgments
This work was supported by the START program (to FT and TL) and the Interdisciplinary Centre for Clinical Research “BIOMAT.” (to FT) within the faculty of Medicine at the Rheinisch Westfälische Technische Hochschule Aachen University and by the German Research Foundation (DFG Ta 434/2–1 to FT and SFB 542 to CT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tacke, F., Luedde, T. & Trautwein, C. Inflammatory Pathways in Liver Homeostasis and Liver Injury. Clinic Rev Allerg Immunol 36, 4–12 (2009). https://doi.org/10.1007/s12016-008-8091-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-008-8091-0