
Abstract
Cadmium (Cd), a highly toxic heavy metal, is found in soil, environment and contaminated water and food. Moreover, Cd is used in various industrial activities, such as electroplating, batteries production, fertilizers, while an important non-occupational source is represented by cigarette smoking, as Cd deposits in tobacco leaves. Since many years it is clear a strong correlation between Cd body accumulation and incidence of many diseases. Indeed, acute exposure to Cd can cause inflammation and affect many organs such as kidneys and liver. Furthermore, the attention has focused on its activity as environmental pollutant and endocrine disruptor able to interfere with metabolic and energy balance of living beings. Both in vitro and in vivo experiments have demonstrated that the Cd-exposure is related to metabolic diseases such as obesity, diabetes and osteoporosis even if human studies are still controversial. Recent data show that Cd-exposure is associated with atherosclerosis, hypertension and endothelial damage that are responsible for cardiovascular diseases. Due to the large environmental diffusion of Cd, in this review, we summarize the current knowledge concerning the role of Cd in the incidence of metabolic and cardiovascular diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd) is a nonessential heavy metal purified for the first time in 1817 by Friedrich Stromyer[1]. In nature, it is found mainly associated with other ores to form many inorganic salts (i.e., CdS, CdCl2, CdSO4, CdCO3) [2]. The ability to constitute various compounds makes Cd a useful element for industries employed in electroplating (estimated around 83%), alloy production, pigments, production of nickel-Cd batteries and fertilizers. Moreover, Cd is contained in cigarettes, as it accumulates in tobacco leaves, and in soil, leading to contamination of water and food, such as cereals, grain and potatoes [2, 3]. The wide spread presence of this element has brought researchers to evaluate its level of toxicity and, nowadays, Cd (Cas no: 7440-43-9) is classified as a highly toxic metal and, thus, hazardous to human health by the International Agency for Research on Cancer [4] which has included Cd in group I of human carcinogens along with other chemical substances such as formaldehyde, benzene and nickel [5]. Moreover, as already described for other pollutants, it has an endocrine disruptor activity [6] due to its ability to interfere with hormonal homeostasis by binding to the receptors or altering intracellular pathways [7]. Considering the high rate of Cd gathering, it is not surprising that the onset of several serious illnesses is linked with Cd accumulation, since its well-described pro-inflammatory and carcinogenic effect, cell damage induction, that culminate in cell death either by apoptosis or necrosis, with consequent tissue inflammation and fibrosis [8]. Hence, Cd can affect the kidneys, liver, lung, pancreas, testis, placenta and bone (Fig. 1). Moreover, this metal is related with cardiovascular and metabolic diseases (i.e., obesity, diabetes, osteoporosis) and impaired reproduction activity [9,10,11,12].

Schematic representation of the environmental sources of Cd and Cd exposure-related diseases
Due to the large environmental diffusion of Cd, the aim of this review is to potentially clarify the correlation between Cd exposure and pathological risks of some metabolic diseases in humans.
Cadmium tolerability, absorption and excretion
The current tolerable level of Cd exposure appears to be 25 μg/kg body weight per month (62 μg/day for a 70-kg person) [13]. Its toxic effects depend on both length and route of exposure, that could be acute (i.e., single exposure at high doses) or chronic (i.e., repeated exposure at low doses) [14], but it might also differ due to the impairment of the daily system of excretion [15]. Individuals can uptake Cd through inhalation and ingestion [2] and it is then transported from the absorption site through the blood to the body organs. In blood stream, it is found mainly in blood cells but also in plasma where it tightly binds to both high molecular mass proteins such as albumin and low molecular mass molecules such as the metallothionein (MT) [16, 17]. However, Cd has a long half-life (20–40 years), leading to its accumulation in body organs, mainly in the kidneys, for many years [18]. The accumulation, that could be irreversible, depends on the specific organ. In particular, it has been established that the liver (1–3 mg/kg) and, mostly, the kidneys (12–40 mg/kg) are the body districts with highest Cd level concentration since these two organs express large amounts of MT, known to establish tight bindings with this metal, as discussed earlier [2]. The accumulation of Cd is due to constant exposure to it, in particular, it has been reported that smokers have about three times more Cd in their bodies than non-smokers [19, 20].
Cadmium as Endocrine disruptor
The expression Endocrine Disruptors (EDs) indicates a number of chemicals with a specific effect on the endocrine system interfering with the receptor-mediated hormone activity [21]. Thus, EDs may cause the alteration of cellular metabolism leading to long-term and harmful effects. EDs are substances of natural origin or man-made products including over 350 synthetic compounds such as insecticides (i.e., dichlorodiphenyltrichloroethane DDT and metabolites, pyrethroids), herbicides (i.e., atrazine, nitrofen), fungicides (i.e., zineb, ziram), pharmacological agents (i.e., bisphenol A—BPA), chemicals such as the plasticizers polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyl (PCB) [22,23,24,25,26,27], dioxins, dioxin-like compounds, phthalates and heavy metals as lead, mercury and Cd [22]. Due to this peculiarity, there is a rising concern about Cd effect on the endocrine system since it has been demonstrated that this heavy metal might mimic the activity of natural hormones such as estrogens and androgens leading to the activation of specific signaling pathways [7] or blocking the interaction of these hormones with their natural receptors [28, 29].
Cadmium and the thyroid
A thyrotoxicity, leading to either hyper- or hypothyroidism, due to Cd exposure, alone or in combination with other EDs, has been described in both animal and human studies. In particular, this metal might alter serum hormone levels such as thyroxine (T4), triiodothyronine (T3) and thyroid stimulating hormone (TSH) [30]. Moreover, Cd has also been linked to structural damage of thyroid tissue leading to hypertrophy or hyperplasia [31,32,33]. Interestingly, some recent data by Chung and colleagues suggest a sex gender-specific correlation between Cd exposure and thyroid dysfunction, reporting a male-related hypothyroidism in a Korean population [34].
Estrogen activity of Cadmium
Interestingly, Cd is described as a metalloestrogen due to its ability to bind the estrogen receptor (ER), replacing the natural estrogen steroid hormone and affecting the homeostasis of estrogen responsive tissues [35]. A frequent overexpression of ERs and/or overexposure to estrogens may lead to the progression of neoplastic breast epithelium [36]. Considering the estrogen-like activity of Cd, its role in the development of breast cancer has been evaluated in many studies [37, 38]. Our group demonstrated that Cd can bind to and interfere with ER intracellular pathways in MCF7 breast cancer cells contributing to cancer progression by stimulating cell proliferation [7]. Moreover, in the same cell line, low levels of Cd enhanced cancer cells’ adaptability and their malignancy [39]. Another study reported that long-term Cd exposure led to invasion and migration of human breast cancer cells by inducing the TG-interacting factor-matrix metalloproteinase-2 (TGIF-MMP2) signaling pathway [40]. In conclusion, put together these data describe how Cd exposure can contribute to breast cancer incidence.
Androgen activity of Cadmium
It has been established that androgens are strong promoters of prostate cancer cells proliferation (PC) and there is growing evidence about the role of Cd exposure in PC incidence [41, 42]. This has been observed both in in vitro models, in LNCaP human prostate cells expressing the androgen receptor (AR), thus androgen sensitive, and in animal studies. In particular, it has been demonstrated that Cd might mimic androgens by binding the AR and acting on both cell growth stimulation [43,44,45] and gene expression modulation [46]. Moreover, it might induce an acquisition of apoptotic resistance in malignant transformation in LNCaP cells and in other cell lines such as the primary adenocarcinoma 22Rv1 and CWR-R1 [47] and in the benign prostatic hyperplasia-1 cells (BPH1) [48]. Despite a recent study reporting that animals exposed to low doses of Cd during gestational stage had histopathological damages in adulthood, without significant changes in prostate weight, cell proliferation or alteration in hormone levels [49], experiments on adult castrated animals (mice and rats) have demonstrated contradictory results [50]. Similar data obtained in 2002 by Martin and colleagues demonstrated that animals treated with T propionate, a PC promoter in rodents, or with Cd, increased prostate glands and seminal vesicle complex wet weight of in a similar manner. Moreover, this effect was blocked using an anti-androgen compound, strongly demonstrating that the effect of Cd was mediated by AR [45] and further indicating that Cd is involved in PC progression likely for its androgen-like activity.
Obesogenic effect of Cadmium
Obesity is a multifactorial, metabolic chronic disease characterized by an excess of adipose tissue, and nowadays it represents an important health issue with global diffusion [51, 52]. This pathological condition is marked by several and complex clinical manifestations, leading to a significant increase in the risk of developing chronic metabolic consequences such as type 2 diabetes mellitus (T2DM), cardiovascular diseases (CVD), cancers, alterations of musculoskeletal metabolism [53, 54]. Recently, attention has been focused on the environmental pollutants able to interfere with metabolic and energy balance of living beings, including humans. However, data from human studies on the interaction between Cd exposure and obesity, diabetes mellitus and bone diseases are still controversial [2, 11].
Interestingly, a correlation between Cd exposure and the incidence of obesity has been hypothesized and this heavy metal, along with other EDs (i.e., DDT, heavy metals, pthtalates, PBDEs), is included in the group of factors that might increase the incidence of obesity through various mechanisms such as increasing the number and the size of fat cells, altering endocrine homeostasis, modifying appetite and satiety processes regulation, shifting insulin sensitivity and metabolism rate [11, 55]. The main mechanism triggered by EDs appears to be the peroxisome proliferator-activated receptor γ (PPAR-γ), a regulatory gene of pre-adipocyte proliferation and adipocytes differentiation [56,57,58,59,60]. Moreover, several other intracellular targets and pathways have been described as potential factors [60,61,62] often acting in a gender-specific manner [63]. In experimental animal models, an early exposure to EDs alters pluripotent mesenchymal cells differentiation, favoring the adipocyte lineage [57] and gestational exposure increases the risk of juvenile obesity in the next generation [64].
Even if the mechanism is not completely clear, it appears that Cd might lead to obesity likely through epigenetic modifications, altering adipose tissue physiology and metabolic profile [11, 52].
Clinical studies
Due to the preclinical data, several studies have evaluated a potential correlation between Cd exposure and obesity in humans as well, with contradictory results.
A study demonstrated that indigenous women of Torres Strait Island (Australia) revealed a significant direct correlation between urinary Cd (UCd) levels and increase in waist circumference (WC) [65].
Padilla et al., used the National Health and Nutrition Examination Survey (NHANES) 1999–2002 data to analyze 3816 participants and evaluate the correlation of Cd exposure with either BMI or WC. The analysis showed a direct correlation between BMI, WC and UCd levels. In particular, the regression models reported that the level of Cd was inversely associated with the anthropometric indices of obesity. However, when the analysis was restricted to adolescents (6–18 years of age) or to adults (≥ 19 years), the association in adolescents was not significant [66]. Notably, blood Cd (BCd) levels significantly correlated with BMI in T2DM patients [67].
A cross-sectional SPECT-China study on 5544 adults found that BCd levels were negatively associated with the prevalence of overweight [68]. Equally interesting are the results Skalnaya et al. where they analyzed the correlation between BMI and Cd content in hair. The study examined 1229 people (719 women and 510 men) showing how BMI was positively correlated with high Cd content in women’s hair, while no significant alteration of Cd concentration in males’ hair was observed [69]. Moreover, a study performed on 65 Obese Egyptian Children (11–14 years of age) that followed a diet rich in antioxidants and micronutrients, showed how BMI reduction was associated with a significant reduction in UCd levels, suggesting that a proper diet might reduce the toxic effects of heavy metals in obese children, likely controlling body weight increase [70].
Moreover, NHANES (1999–2011) data were recovered from 6602 American children, adolescents and adults (6–19 years of age). Here, a negative association between Cd [Odds Ratio (OR) 0.46; 95% Confidence Interval (CI) 0.33–0.64; P < 0.001)] and obesity was observed. This association was strongest among the 6–12-year-old children [71].
Interestingly, a recent study established the association between prenatal Cd exposure and obesity in children. First trimester maternal blood samples recorded in the Newborn Epigenetics Study (NEST) were collected, analyzed for the presence of Cd and then cross-analyzed with the weight gain course of children up to 5 years of age. The analysis showed that the presence of Cd in maternal blood during pregnancy was associated with an increased risk of juvenile obesity in the offspring, independently from all other variables, further indicating that Cd could be considered as a potential human obesogenic factor [64].
Another interesting result supporting the role of Cd as potential obesogenic factor is the result reported by Jiang and colleagues who used UCd as biomarker for Cd long-term exposure considering the NHANES data (2007–2012). The authors evaluated the potential correlation of Cd exposure with overweight/obesity as a risk of pre-diabetes among adults (n = 3552; > 20-year-old) demonstrating that overweight/obesity status might significantly increase the Cd-related predisposition to develop-diabetes with higher risk in male adults [72].
Noor et al., obtained data from NHANES (2001–2014; n = 3982; 20– < 80 years old) about UCd levels adjusted for creatinine using spot urine samples and evaluated them in quintiles (Q). In the general population, higher UCd levels were associated with a reduction in the odds of additional obesity (adj. OR for Q5 versus Q1: 0.5; 95% CI: 0.3–0.7). When stratified by gender, both men and women looked at similar reduced odds of additional obesity [men (adj. OR for Q5: 0.4; 95% CI: 0.2–0.7) and women (adj. OR for Q5: 0.5; 95% CI: 0.3–0.8)] [73]. Another stratified analysis by smoking status, found higher Cd concentrations in smokers compared to the overall study population. Moreover, among the non-smokers group, the researchers found significant inverse associations between UCd and central obesity (adj. OR for Q5 versus Q1: 0.4; 95% CI: 0.3–0.7) [73].
However, other studies have failed to identify any significant association between BCd levels and excessive body weight [74,75,76,77]. Correspondingly, a study based on data from the 2008–2010 Korean National Health and Nutritional Examination Survey (n = 4522, aged ≥ 20 years), showed no significant relationship between BCd and body fat [78]. In addition, no effect of obesity on hair Cd levels was found in a healthy population of the Canary Islands[79] and in the Korea National Health and Nutrition Examination Survey 2010–2013 [80]. Equally, a cross-sectional analysis of adult males in Poland (250 overweight/obese and 61 normal weight) with and without Metabolic Syndrome (MS) identified no association between BCd and obesity [81].
As a result, all data suggest that between Cd levels and the overweight/obesity condition, both negative and positive correlations may exist, or Cd may have no significant impact on weight gain. According to Tinkov et al., the contradiction among the results of these studies could be due to differences in Cd exposure levels in different parts of human body (urine, hair, nails) [11].
In vivo studies
The first study performed in an experimental animal model showed that oral Cd administration (9.7 mg/L) for 6 weeks did not lead to an increase in BMI and adipocytes size, but significantly increased serum glucose and insulinemia, due to a reduction (~ 50%) of both number and density of insulin receptors in target cells [82].
In a consequent study, healthy female rats were randomly divided into three groups with a daily Cd oral dose (1–0.25 mg/kg weight), 5 days a week, for 6 weeks showing that the high-dose group had a lower body weight than the control group [83] and similar data were obtained more recently by Singh et al. [84]. Likewise, Treviño demonstrated that Cd increased insulin release, and altered both glycemic and lipid metabolism, without influencing body weight [85].
MT is known to have protective effects against the toxicity of heavy metals such as Cd. One of the first studies by Kawakami et al., showed that the administration of Cd (0–0.75 mg/kg weight per day) for 7 days, reduced the size of adipocytes in MT-null mice and it modulated the expression of some adipokines, such as adiponectin, leptin and resistin [86]. Furthermore, after Cd exposure, the gene expression of the monocyte chemoattractant protein-1 (MCP-1), which has a role in the recruitment of macrophages into adipose tissue, increased in the white adipose tissue (WAT). When mice were not exposed to Cd, the adipocyte recovered its size in 6 weeks, but the expression of adiponectin and leptin remained at low levels. In addition, it was suggested that the reduction of the size of the adipocytes by Cd could derive from an imbalance between lipid synthesis and lipolysis [86].
Another study evaluated the effects of subchronic exposure (10 weeks) in mice with low doses of Cd (10 mg/L) on energy metabolism and intestinal microbiome [87]. Exposure to Cd caused a significant increase in liver GluT2, glucokinase, carbohydrate responsive element binding protein (Chrebp) and pyruvate kinase mRNA. There was also an increase in hepatic triacylglycerols (TG), serum free fatty acid (FFA) and TG levels. Moreover, the alteration of the intestinal microbiome led to an increase in serum lipopolysaccharide (LPS). LPS caused liver inflammation through the elevation of interleukin-1β (IL-1β), tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) mRNA. Therefore, this study indicated that sub-chronic Cd exposure caused the deregulation of energy metabolism and altered the gut microbiome composition in mice [87].
More recently, Green et al., confirmed also in zebrafish that Cd increases lipid accumulation, leading to obesity. The zebrafish prenatal exposure to Cd showed a significantly higher lipid accumulation compared to the unexposed controls. Zebrafish that were followed until sexual maturity exhibited reduced lipid accumulation [64].
In vitro studies
Early in vitro studies showed that the adipocytes exposed to Cd were characterized by an increase in the formation of carbon dioxide (CO2) and in the rate of lipogenesis from glucose [88]. Subsequently, it was demonstrated that the exposure of 3T3-L1 fibroblasts to 10 and 25 μM of CdCl2 for 12 h led to a 5–6 times increase of the absorption of 2-deoxyglucose or 3-O-methylglucose. Furthermore, the authors demonstrated that Cd-induced glucose uptake in adipocytes is related to impaired Ca2+ signaling in place of insulin signaling [89]. Another study showed how exposure of 3T3-L1 fibroblasts to Cd (5–10 μM) increases glucose uptake mediated by the modulation of GluT1 activity [90]. This result is sustained by another study that reported an increase in GluT4 activity [91, 92]. A subsequent study showed that adipocytes extracted from rats exposed subcutaneously to CdCl2 (2 mg/kg weight—4 days) were characterized by a significant dose-dependent reduction in GluT4 both in terms of proteins and mRNA, while neither glucose transporter type 1 (GluT1) nor glucose transporter type 2 (GluT2) were affected [93].
The viability of 3T3-L1 adipocytes treated with Cd (30 μM for 24 h) was significantly decreased (28.3%) in a concentration-dependent manner. Hence, Cd disrupted multiple metabolic pathways [94]. Accordingly, another in vitro study in 3T3-L1 adipocytes showed that fat cells exposed to Cd presented a significant reduction in cell viability, in a dose-dependent manner, and a decrease of adiponectin and resistin expression. This demonstrated the toxicity of Cd [95].
Lee et al., demonstrated, for the first time, the capacity of Cd to inhibit preadipocyte 3T3-L1 differentiation. This effect was mediated by the downregulation of the expression of CCAAT-enhancer-binding protein alpha (C/EBPα) and PPAR-γ, two main adipogenic transcriptional activators. In addition, this study reported that exposure to 0.3–3 μM CdCl2 resulted in a significant dose-dependent decrease in lipid accumulation in differentiating 3T3-L1 cells on the stage of preadipocyte differentiation. It was also specified that Cd exposure significantly altered the expression of adipogenesis activators [96].
Levy et al., showed how exposure to Cd (0.01–0.1–1–10 mM) induced a dose-dependent reduction of leptin levels [97]. Accordingly, Kawakami described how the in vitro exposure of the adipocytes to Cd (0–100 M for 6–48 h) induced a significant reduction in the expression of leptin, adiponectin and resistin, being associated with a reduced synthesis of fatty acids and lipid degradation mediated by perilipine [86].
According to Planchart, laboratory studies clearly demonstrated the correlation between Cd and adipose tissue. These data showed the anti-obesogenic activity of Cd in adult animals by promoting the release of lipids from the liver and adipose tissue, causing dyslipidemia. Conversely, prenatal exposure to Cd could increase the risk of lipid accumulation [98].
Cadmium and diabetes
T2DM is a metabolic chronic disorder with an increasing incidence worldwide presenting, to date, an important issue of global diffusion [99].
The factors involved in this dramatic increase have been extensively studied to further understand and characterize possible prevention strategies. Taking into account the potential mechanisms that contribute to the development of the disease, recent studies have hypothesized a role of EDs, including Cd. It is in fact known that the pancreas, along with the kidneys and liver, is one of the organs with prominent Cd build-up. Thus, several studies have evaluated the correlation between EDs and glucose metabolism alterations [100,101,102].
Clinical studies
Following preclinical results, clinical cross-sectional studies were performed showing a strong relationship between Cd, as measured with UCd and T2DM [2, 11].
These interesting data corroborated previous results by Lei et al., demonstrating that a long exposure to Cd could induce a reduction of serum levels of insulin and amylase, biomarkers of both endocrine and exocrine toxic effects [2, 103].
Moreover, Nie et al., through multinomial logistic regression analysis demonstrated a positive correlation between pre-diabetes and BCd [68].
Tinkov et al., completed a meta-analysis regarding the risk of prevalence and incidence of diabetes and pre-diabetes, remarking the similarities and differences between the highest Cd exposure categories and the lowest intake categories. They used random-effect models to account for heterogeneity in specific study results, and they performed stratified analyses by diabetes or prevalence and incidence of pre-diabetes, type of sample (blood or urine), sex of the participants. Not all studies were adjusted for gender, age and smoke, but they performed stratified analyses for smokers and non-smokers [11]. Two studies were accomplished in Europe (Sweden), three in America (United States and Mexico) and six in Asia. ORs for prevalence of pre-diabetes and diabetes were 1.60 (95% CI 1.25–2.06) and 1.04 (95% CI 0.99–1, 10), respectively, and the risk of diabetes incidence was 1.38 (95% CI 1.12–1.71) [11]. These analyses showed that the highest OR was observed in studies focused on UCd in both pre-diabetes prevalence and diabetes incidence, but not in diabetes prevalence. Higher risk of prevalent diabetes was found in men respect to females, while for diabetes incidence studies women showed a higher risk of Cd exposure, however only one study was conducted in men [11].
A recent case–control study by Lei et al., (166 cases and 427 controls), demonstrated the association between UCd and T2DM. Regression analysis data showed how UCd appeared to be a risk factor for T2DM (OR = 1.61, 95% CI: 1.08–2.41) [104].
Guo et al., in a recent meta-analysis, despite the presence of heterogeneity among studies, showed an association between Cd exposure and T2DM. The results showed a positive association between individual Cd levels and T2DM (OR = 1.27; 95% CI, 1.07–1.52) [105].
Another recent study was carried out on 2749 middle-aged adults from the cross-sectional ELISABET survey to analyze the relationship between the levels of BCd and Hemoglobin A1c (HbA1c) separately in never-, previous- and current smokers. Even though the effects observed in the non-smoking population with low Cd exposure suggested that the risk attributable to this metal is not high, the impact of exposure to high Cd levels (such as occupational exposure) on diabetes risk might be of concern [106].
Another recent study, involving 3140 adults from the Wuhan-Zhuhai cohort, explored a potential relationships between UCd, plasma C-reactive protein (CRP) and T2DM using multivariate logistic regression demonstrating that individuals with high levels of UCd and plasma CRP potentially have a significant higher risk of T2DM [107].
In vivo studies
Studies performed on experimental animal models indicated that an acute and sub chronic Cd exposure induces a diabetogenic effect. Initial studies in early 1990, demonstrated that an acute exposure to Cd (intraperitoneal injection of Cd 0.84 mg/Kg weight) induced an increase in plasma glucose levels in non-fasted rats [108]. Moreover, a Cd chronic exposure (oral gavage for 45 days) exhibited a significant elevation of fasting blood glucose levels [109] as well as in other studies which showed that a subcutaneous injection of Cd led to a compelling increase of blood glucose levels [110, 111].
Moreover, Lei et al., showed that an oral Cd administration (0, 50, 100, 200 mg/L) with water (30–60–90 days) promoted a reduction of insulin levels both in mice treated with 100–200 mg/L for 30 days and with 100 mg/L for 60 days. Also, Cd could be accumulated in the pancreas, inducing the alteration of genes and proteins, hence; this influenced the endocrine and exocrine functions [112, 113].
Edwards et al., using a model of chronic 8 weeks Cd exposure, demonstrated a notable increase in fasting blood glucose levels following, a reduction of serum insulin and an accumulation of Cd in the pancreas [114]. The latter was in agreement with Lei et al., who detected that Cd accumulation in the pancreas caused changes in insulin genes expression. Therefore, it appears that Cd can influence insulin biosynthesis, but not its secretion [111], suggesting a direct toxic effect of Cd on the pancreas [114] and an impairment of glucose homeostasis as recently suggested [115].
In vitro studies
Interestingly, old studies showed that pancreatic β-cells are targets of Cd toxicity. Yau and Mennear demonstrated that Cd significantly reduced insulin secretion and increased MT pancreatic concentration after 6 h of exposure [116]. Moreover, pancreatic β-cells isolated by mice after Cd-exposure, showed a reduction of insulin secretion in the presence of high glucose levels [109].
Muayed et al., showed that pancreatic β cell line MIN6 accumulated Cd in a dose-dependent manner (0.1–1.0 μmol/L) and over time (over 72 h). Cd uptake led to a selective activation of MT and inhibition of glucose-stimulated insulin secretion. Hence, Cd accumulation caused a functional impairment of β-cell function, but did not affect either cell viability or gene expression and did not induce oxidative stress [117].
Moreover, Cd-induced phophosphorylation of several kinases such as c-jun N-terminal kinases (JNK), extracellular signal-regulated kinases (ERK) 1/2, and p38-mitogen-activated protein kinase (MAPK), leading to pancreatic β-cell death by oxidative stress downstream-mediated by JNK activation triggering mitochondria-regulated apoptotic pathway [118].
The importance of oxidative stress was further confirmed by the demonstration that this pollutant decreased islets viability along with an increase in the formation of both ROS and apoptosis markers. Interestingly, β-estradiol appears to have a protective role against Cd toxicity, in particular suggesting a protective role for the Cd-induced β-cells damages [119].
Cadmium and bone and mineral metabolism
It is a known that too much exposure to Cd might affect bone mineral metabolism leading to skeletal alterations. In particular, osteoporosis is a skeleton metabolic disease characterized by a reduction of bone strength leading to increased risk of traumatic and/or spontaneous fracture. The decrease in strength of bone tissue is caused by changes in its quantity, in terms of reduction of bone mineral density, and its quality, in terms of alterations of the micro- and macro-architecture of the bone [120].
Clinical studies
Interestingly, it has been established that prolonged exposure to Cd can induce skeleton fragility with higher fracture risk and high incidence of osteoporosis for both direct bone demineralization and renal dysfunction, even if critical exposure levels and underlying mechanisms are still unknown [2, 12, 121, 122].
Likely, high Cd levels might induce greater urinary calcium excretion and lack of vitamin D activation in the kidneys [123,124,125]. Furthermore, it is interesting to note that some recent published results also demonstrated that Cd exposure might alter parathyroid hormone (PTH) secretion likely linked to kidneys dysfunction [126,127,128]. Interestingly, a recent study indicated that exposed workers had higher Cd concentration in serum and urine than controls subjects. Further, Cd exposed group had PTH, serum phosphorus and magnesium levels significantly lower vs control and, also, experienced musculoskeletal complaints, bone ache, joint pain and muscle spasm as compared to controls [126]. In addition, it also appears that, as demonstrated by some Authors, a potential gender-specific negative effect of Cd on PTH and skeletal metabolism [127, 128].
Moreover, several studies also identified a direct action of this ED on bone cells [125, 129, 130]. Akesson et al., demonstrated the negative effects of low-level Cd exposure on the bone; these effects could be caused by an increase in bone resorption, which intensified after menopause [130].
A recent study evaluated the association between cumulative Cd intake and osteoporosis and fracture risk in a Chinese population. The results indicated that a high level of cumulative Cd intake was associated with an increase in the rate of osteoporosis and fracture among women [131]. Furthermore, another study showed that Cd chronic exposure during early childhood could affect bone remodeling and prepubertal growth [132].
In contrast, Li et al., found no positive association of prevalence of osteoporosis with Cd exposure. However, positive UCd interactions on the prevalence of osteoporosis for women and non-smoking women and the same interactions with BCd for men have been found [133]. In conclusion, according to Nordberg, there was an association between Cd exposure and the onset of osteoporosis. A correlation between UCd, BCd and osteoporosis was noticed. Moreover, it would seem that Cd accelerated resorption by promoting osteoclastogenesis and inhibiting osteogenesis.
In vivo studies
Female Wistar rats exposed to chronic Cd (1 mg Cd/l in drinking water for 24 months), showed a decreased mineralization and determined an alteration in bone formation leading to deformity and fragility fractures [134]. Cd induced a decreased expression of Runt-related transcription factor 2 (Runx2) and matrix proteins such as osteocalcin (OCN), type I collagenase(COL1a2), alkaline phosphatase [ALP (enzyme involved in the mineralization process)] [135] and another study demonstrated how Cd chronic oral administration (50 mg Cd/L for 3 months) produced marked abnormalities in bone biomarkers (OCN, ALP) and increasing risk of fracture [136].
In addition, an increase in fat has been found in the bone marrow, suggesting that Cd might also affect mesenchymal stem cells (MSCs) differentiation by stimulating adipogenesis at the expense of osteoblastogenesis [137].
A recent study showed that chronic Cd exposure directly acts on MSCs through receptor activator of nuclear factor (NF)-kB-ligand/osteoprotegerin (RANKL/OPG) pathway and down-regulates genes involved in osteogenic differentiation of MSCs (COL1a2, Osteopontin, ALP, Osterix, and RUNX2) [138]. In accordance, He et al., demonstrated how Cd increases RANKL expression, but has a lower effect on OPG expression in bone marrow cells and bone tissue. Therefore, Cd favors the formation of osteoclasts in the bone tissue and accelerates the bone resorption. Furthermore, Cd has no significant effects on serum ALP activity [139].
In vitro studies
The observations in vivo, were supported by in vitro studies that showed how Cd increased RANKL expression and it might stimulate osteoclastogenesis [140, 141]. In addition, our group published results demonstrating that Cd-induced cell apoptosis and homeostasis by cytoskeletal disruptionand by alteration of Wnt/β-catenin pathway, activation of caspases [142]. Further studies also suggested an increase in ROS [125, 143, 144].
Moreover, a recent study shows that Cd suppresses the osteogenesis from MSCs by inhibiting the Wnt/β-catenin pathway [145].
In conclusion, according to Nordberg, there was an association between Cd exposure and the onset of osteoporosis. A correlation between UCd, BCd and osteoporosis was noticed. Moreover, it would seem that Cd accelerated resorption by promoting osteoclastogenesis and inhibiting osteogenesis.
Cadmium and cardiovascular diseases (CVD)
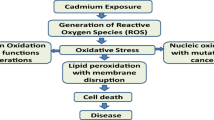
Cardiovascular diseases (CVD) are a group of disorders that affect blood vessels and heart, and they represent one of the major causes of morbidity and mortality in the world. The risk factors for CVD include sedentary, unhealthy diet, frequent consumption of alcohol, smoke and pollution [146]. The environmental distribution of Cd, its strong concentration in cigarettes, food and water and its effects even at vascular level indicate that this metal correlates with CVD [11, 147, 148]. The molecular mechanisms by which Cd exerts toxic effects in the cardiovascular tissues are associated with the induction of oxidative stress, indeed this metal can cause alteration of the endogenous antioxidant defense such as glutathione peroxidase (GPx), catalase (CAT) and superoxide dismutase (SOD). Furthermore, Cd can indirectly induce ROS generation [149]. In particular, it can supply other metals, such as iron, in many proteins (i.e., Ferritin), by freeing unbound ions that may generate oxidative stress through Fenton reactions. Moreover, Cd can impair the mitochondrial electron chain transport and it can diminish the antioxidant scavengers such as Glutathione (GSH), thus unbalancing the cellular redox state and, consequently, causing the production of ROS [150,151,152].
Clinical studies
Cd has been identified as a pro-atherogenic factor since it largely accumulates in carotid plaques with an increase of 50 times in vulnerable plaques compared to blood levels or plaques where the rupture does not occur [9, 153, 154]. Moreover, epidemiological studies have shown that BCd level is associated with CVD mortality and carotid plaques prevalence in a Swedish population and with CVD risk in Korean men [155, 156]. Both longitudinal analyses and cross-sectional studies in a group of 64-year-old women have demonstrated a correlation between high concentration of UCd and BCd and plaques formation [157]. The sex gender specificity of Cd is currently unclear. Other studies reported that Cd accumulation is linked with an increase in macrophages content that is a hallmark of symptomatic and vulnerable carotid plaques [158, 159]. Moreover, a recent paper demonstrates an association between pollutants and carotid intima-media thickness (CIMT)in a Canadian population sample [160]. However, BCd did not result as a predictor of juvenile CIMT in Indonesian young adults [161]. Bornè and colleagues in 2017 demonstrated that Cd accumulation was associated with incidence of ischemic stroke, supporting that this element promotes the vulnerability of carotid plaques, by increasing the possibility of rupture and ischemic stroke [9]. Cigarette smoke is an important risk factor for CVD, and it is one of the main Cd sources and many studies tried to explain the Cd-related CVD incidence [162,163,164]. Another study showed that both cigarette smoke and Cd produce vascular damage such as vascular plaque inflammation and vasomotor dysfunction[165]. More recently, the NHANES realized 5 cross-sectional studies including a random sample of the US population, demonstrating that individuals with higher levels of BCd or UCd showed increased risks of, among the other disturbs, hypertension, stroke, heart failure, myocardial infarction, and peripheral artery disease [166,167,168,169]. A follow-up study for 16–19 years realized on a Swedish population-based cohort of 4304 middle-aged men and women, analyzed BCd levels to investigate the effect of smoking on CVD, demonstrating that Cd may play a significant role in smoking-induced CVDs [170].
In vitro studies
In vitro studies have identified Cd as a pro-atherogenic factor with a cytotoxic effect in macrophages [3]. Our group has recently demonstrated that the in vitro exposure of endothelial cells (HUVECs) to Cd modifies AR levels and affects pro-inflammatory signaling suggesting a role for this heavy metal in cell injury related to cardiovascular diseases, and this might partially explain the risk of prostate cancer due to this ED [6]. Another process caused by Cd is the endothelial dysfunction, indeed it has been demonstrated that Cd increases vascular endothelial permeability, reduces nitric oxide (NO) production, inhibits endothelial cell proliferation, it can lead to the upregulation of Vascular Cell Adhesion protein 1 (VCAM-1) expression level and induce apoptosis in endothelial cells [171].
In summary, these findings demonstrate a correlation between Cd exposure and CVD incidence, however further studies are needed to define the dose–response relationship and the possible existence of a specificity dependent on sex.
Future directions and conclusions
Cd is a heavy metal considered harmful for plants, and animals. Moreover, it is implicated in serious human illnesses such as metabolic diseases, osteoporosis, renal dysfunctions and CVDs. Equally important are the pro-inflammatory activity and the carcinogenic effect of this metal. The presence of Cd in soil, foods and human sources shows a great variability in the geographical distribution as demonstrated by many epidemiological studies [2]. It has been calculated that, to maintain the strong demand for Cd worldwide, the amount of the anthropic emission released into the atmosphere each year is approximately 30,000 t [172] and for example, every year the utilization of Cd pigments exceeds 2500 t [173]. Hence, considering the dangerous effects of Cd exposition and its high diffusion for many years, there is a necessity to identify methods to mitigate Cd toxicity. In recent years, several regulations applied on Cd emissions have allowed to observe a decreasing trend on Cd release. In particular, European countries have recorded that in 2017 Cd level represented only the 35% of the same amount detected in 1990 [174]. To achieve this aim, a global strategy could be the mitigation of Cd in foods and it includes many steps. The first one concerns the crops, in particular it should be recommended to perform preventive measures such as avoiding phosphate fertilizers, recording Cd uptake in the crop, selecting, prudentially, plant varieties with low Cd levels or doing tests on irrigation water. The second step involves industries; indeed, it should be borne by manufacturers who produce food equipment, and they should only use material with low Cd levels. The last step is in charge of consumers who should commit themselves to follow a healthy diet, for instance, by consuming foods that can protect against Cd toxicity [175]. Other strategies could be the employment of plants with a strong resistance to high concentration of heavy metals and able to realize a phytoremediation, known as hyper accumulators, or the use of compounds known to induce protective mechanisms in plants [176]. Indeed, despite plants having a strong antioxidant defence, this is effective only at low Cd concentration. In 2019, Mostofa and colleagues showed that treating rice seedlings with salicylic acid (SA) and sodium nitroprusside (SNP), that is a source of nitric oxide (NO), could support the defence against Cd toxicity. In particular, they reported that this treatment could restore plant growth and biomass, revive the colour, reduce leaf rolling and ameliorate the phenotypic appearance [177].
Since many years the potential role of chemical compounds, both synthetic and natural, to counteract the detrimental Cd effects has been clear. Among synthetic molecules infliximab, a chimeric immunoglobulin 1 (IgG1) monoclonal antibody that targets TNF-a, has been shown to protect the testicular tissue of rats from the harmful effects of Cd, as it has antioxidant, anti-inflammatory and anti-apoptotic activity [178]. Among natural compounds, the antioxidants curcumin and tetrahydrocurcumin protect vascular endothelium by increasing NO bioavailability and improving vascular function in chronic Cd exposure [149].
Despite these findings, there is an urgency to develop new studies and research focused on reducing Cd amount to the minimum level to ameliorate health-related quality of life.
References
Tarakina NV, Verberck B (2017) A portrait of cadmium. Nat Chem 9(1):96. https://doi.org/10.1038/nchem.2699
Nordberg GF, Bernard A, Diamond GL, Duffus JH, Illing P, Nordberg M, Bergdahl IA, Jin T, Skerfving S (2018) Risk assessment of effects of cadmium on human health (IUPAC Technical Report). Pure Appl Chem 90(4):755–808. https://doi.org/10.1515/pac-2016-0910
Zhang D, Zhang T, Liu J, Chen J, Li Y, Ning G, Huo N, Tian W, Ma H (2019) Zn supplement-antagonized cadmium-induced cytotoxicity in macrophages in vitro: involvement of cadmium bioaccumulation and metallothioneins regulation. J Agric Food Chem 67(16):4611–4622. https://doi.org/10.1021/acs.jafc.9b00232
Vainio H, Heseltine E, Partensky C, Wilbourn J (1993) Meeting of the IARC working group on beryllium, cadmium, mercury and exposures in the glass manufacturing industry. Scand J Work Environ Health. https://doi.org/10.5271/sjweh.1461
Chowdhury R (2019) Kontamination mit Schwermetallen erhöht kardiovaskuläres Risiko. Dtsch Med Wochenschr. https://doi.org/10.1136/bmj.k3310
Fittipaldi S, Bimonte V, Soricelli A, Aversa A, Lenzi A, Greco E, Migliaccio S (2019) Cadmium exposure alters steroid receptors and proinflammatory cytokine levels in endothelial cells in vitro: a potential mechanism of endocrine disruptor atherogenic effect. J Endocrinol Invest 42(6):727–739. https://doi.org/10.1007/s40618-018-0982-1
Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, Mariani S, Cherubini S, d’Abusco AS, Scandurra R, Migliaccio S (2007) Cadmium induces mitogenic signaling in breast cancer cell by an ERα-dependent mechanism. Mol Cell Endocrinol 264(1–2):102–108. https://doi.org/10.1016/j.mce.2006.10.013
Thijssen S, Lambrichts I, Maringwa J, Van Kerkhove E (2007) Changes in expression of fibrotic markers and histopathological alterations in kidneys of mice chronically exposed to low and high Cd doses. Toxicology 238(2–3):200–210. https://doi.org/10.1016/j.tox.2007.06.087
Borné Y, Fagerberg B, Persson M, Östling G, Söderholm M, Hedblad B, Sallsten G, Barregard L, Engström G (2017) Cadmium, carotid atherosclerosis, and incidence of ischemic stroke. J Am Heart Assoc 6(12):e006415. https://doi.org/10.1161/JAHA.117.006415
de Angelis C, Galdiero M, Pivonello C, Salzano C, Gianfrilli D, Piscitelli P, Lenzi A, Colao A, Pivonello R (2017) The environment and male reproduction: the effect of cadmium exposure on reproductive function and its implication in fertility. Reprod Toxicol 73:105–127. https://doi.org/10.1016/j.reprotox.2017.07.021
Tinkov AA, Filippini T, Ajsuvakova OP, Aaseth J, Gluhcheva YG, Ivanova JM, Bjørklund G, Skalnaya MG, Gatiatulina ER, Popova EV (2017) The role of cadmium in obesity and diabetes. Sci Total Environ 601:741–755. https://doi.org/10.1016/j.scitotenv.2017.05.224
Buha A, Jugdaohsingh R, Matovic V, Bulat Z, Antonijevic B, Kerns JG, Goodship A, Hart A, Powell JJ (2019) Bone mineral health is sensitively related to environmental cadmium exposure-experimental and human data. Environ Res 176:108539. https://doi.org/10.1016/j.envres.2019.108539
FAO/WHO (2013). https://apps.who.int/food-additives-contaminants-jecfa-database/chemical.aspx?chemID=1376
Faroon O, Ashizawa A, Wright S, Tucker P, Jenkins K, Ingerman L (2012) Toxicological profile for Cadmium. Atlanta (GA): Agency for Toxic Substances and Disease Registry (US) Public Health Statement Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK158840/
Vacchi-Suzzi C, Kruse D, Harrington J, Levine K, Meliker JR (2016) Erratum to: is urinary cadmium a biomarker of long-term exposure in humans? A review. Curr Environ Health Rep 3(4):493–494. https://doi.org/10.1007/s40572-016-0107-y
Fels J, Scharner B, Zarbock R, Zavala Guevara IP, Lee W-K, Barbier OC, Thévenod F (2019) Cadmium complexed with β2-microglubulin, albumin and lipocalin-2 rather than metallothionein cause megalin: cubilin dependent toxicity of the renal proximal tubule. Int J Mol Sci 20(10):2379. https://doi.org/10.3390/ijms20102379
Li Y, Huang Y-s, He B, Liu R, Qu G, Yin Y, Shi J, Hu L, Jiang G (2020) Cadmium-binding proteins in human blood plasma. Ecotoxicol Environ Saf 188:109896
Akerstrom M, Barregard L, Lundh T, Sallsten G (2014) Variability of urinary cadmium excretion in spot urine samples, first morning voids, and 24 h urine in a healthy non-smoking population: implications for study design. J Expo Sci Environ Epidemiol 24(2):171–179. https://doi.org/10.1038/jes.2013.58
Becker K, Kaus S, Krause C, Lepom P, Schulz C, Seiwert M, Seifert B (2002) German Environmental Survey 1998 (GerES III): environmental pollutants in blood of the German population. Int J Hyg Environ Health 205(4):297–308. https://doi.org/10.1078/1438-4639-00188
Ganguly K, Levänen B, Palmberg L, Åkesson A, Lindén A (2018) Cadmium in tobacco smokers: a neglected link to lung disease? Eur Respir Rev. https://doi.org/10.1183/16000617.0122-2017
Pickering AD, Sumpter JP (2003) Peer reviewed: comprehending endocrine disruptors in aquatic environments. ACS Pubs. https://doi.org/10.1021/es032570f
Colborn T, Vom Saal FS, Soto AM (1993) Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ Health Perspect 101(5):378–384
Rudel RA, Perovich LJ (2009) Endocrine disrupting chemicals in indoor and outdoor air. Atmos Environ 43(1):170–181. https://doi.org/10.1016/j.atmosenv.2008.09.025
Brander SM, Gabler MK, Fowler NL, Connon RE, Schlenk D (2016) Pyrethroid pesticides as endocrine disruptors: molecular mechanisms in vertebrates with a focus on fishes. Environ Sci Technol 50(17):8977–8992. https://doi.org/10.1021/acs.est.6b02253
Combarnous Y (2017) Endocrine disruptor compounds (EDCs) and agriculture: the case of pesticides. CR Biol 340(9–10):406–409. https://doi.org/10.1016/j.crvi.2017.07.009
Rochefort H (2017) Endocrine disruptors (EDs) and hormone-dependent cancers: correlation or causal relationship? CR Biol 340(9–10):439–445. https://doi.org/10.1016/j.crvi.2017.07.007
Beausoleil C, Emond C, Cravedi J-P, Antignac J-P, Applanat M, Appenzeller BR, Beaudouin R, Belzunces LP, Canivenc-Lavier M-C, Chevalier N (2018) Regulatory identification of BPA as an endocrine disruptor: context and methodology. Mol Cell Endocrinol 475:4–9. https://doi.org/10.1016/j.mce.2018.02.001
Diamanti-Kandarakis E, Bourguignon J-P, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC (2009) Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 30(4):293–342. https://doi.org/10.1210/er.2009-0002
Schug TT, Janesick A, Blumberg B, Heindel JJ (2011) Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 127(3–5):204–215
Buha A, Matovic V, Antonijevic B, Bulat Z, Curcic M, Renieri EA, Tsatsakis AM, Schweitzer A, Wallace D (2018) Overview of cadmium thyroid disrupting effects and mechanisms. Int J Mol Sci 19(5):1501. https://doi.org/10.3390/ijms19051501
Nie X, Chen Y, Chen Y, Chen C, Han B, Li Q, Zhu C, Xia F, Zhai H, Wang N (2017) Lead and cadmium exposure, higher thyroid antibodies and thyroid dysfunction in Chinese women. Environ Pollut 230:320–328. https://doi.org/10.1016/j.envpol.2017.06.052
Yu Y, Ma R, Yu L, Cai Z, Li H, Zuo Y, Wang Z, Li H (2018) Combined effects of cadmium and tetrabromobisphenol a (TBBPA) on development, antioxidant enzymes activity and thyroid hormones in female rats. Chem Biol Interact 289:23–31. https://doi.org/10.1016/j.cbi.2018.04.024
Rezaei M, Javadmoosavi SY, Mansouri B, Azadi NA, Mehrpour O, Nakhaee S (2019) Thyroid dysfunction: how concentration of toxic and essential elements contribute to risk of hypothyroidism, hyperthyroidism, and thyroid cancer. Environ Sci Pollut Res 26(35):35787–35796. https://doi.org/10.1007/s11356-019-06632-7
Chung SM, Moon JS, Yoon JS, Won KC, Lee HW (2019) Sex-specific effects of blood cadmium on thyroid hormones and thyroid function status: Korean nationwide cross-sectional study. J Trace Elem Med Biol 53:55–61. https://doi.org/10.1016/j.jtemb.2019.02.003
Byrne C, Divekar SD, Storchan GB, Parodi DA, Martin MB (2009) Cadmium—a metallohormone? Toxicol Appl Pharmacol 238(3):266–271. https://doi.org/10.1016/j.taap.2009.03.025
Aquino NB, Sevigny MB, Sabangan J, Louie MC (2012) The role of cadmium and nickel in estrogen receptor signaling and breast cancer: metalloestrogens or not? J Environ Sci Health C 30(3):189–224. https://doi.org/10.1080/10590501.2012.705159
Kresovich JK, Erdal S, Chen HY, Gann PH, Argos M, Rauscher GH (2019) Metallic air pollutants and breast cancer heterogeneity. Environ Res 177:108639. https://doi.org/10.1016/j.envres.2019.108639
Strumylaite L, Kregzdyte R, Bogusevicius A, Poskiene L, Baranauskiene D, Pranys D (2019) Cadmium exposure and risk of breast cancer by histological and tumor receptor subtype in white caucasian women: a hospital-based case-control study. Int J Mol Sci 20(12):3029. https://doi.org/10.3390/ijms20123029
Bloomfield M, Louie MC (2019) Chronic cadmium exposure decreases the dependency of MCF7 breast cancer cells on ERα. Sci Rep 9(1):1–11
Wang Y, Shi L, Li J, Li L, Wang H, Yang H (2019) Long-term cadmium exposure promoted breast cancer cell migration and invasion by up-regulating TGIF. Ecotoxicol Environ Saf 175:110–117. https://doi.org/10.1016/j.ecoenv.2019.03.046
Waalkes MP, Rehm S (1994) Cadmium and prostate cancer. J Toxicol Environ Health Part A 43(3):251–269. https://doi.org/10.1080/15287399409531920
Dai C, Heemers H, Sharifi N (2017) Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med 7(9):a030452. https://doi.org/10.1101/cshperspect.a030452
Webber MM (1985) Selenium prevents the growth stimulatory effects of cadmium on human prostatic epithelium. Biochem Biophys Res Commun 127(3):871–877
Voeller HJ, Wilding G, Gelmann EP (1991) V-ras H expression confers hormone-independent in vitro growth to LNCaP prostate carcinoma cells. Mol Endocrinol 5(2):209–216. https://doi.org/10.1210/mend-5-2-209
Martin MB, Voeller HJ, Gelmann EP, Lu J, Stoica E-G, Hebert EJ, Reiter R, Singh B, Danielsen M, Pentecost E (2002) Role of cadmium in the regulation of AR gene expression and activity. Endocrinology 143(1):263–275. https://doi.org/10.1210/endo.143.1.8581
Neslund-Dudas CM, McBride RB, Kandegedara A, Rybicki BA, Kryvenko ON, Chitale D, Gupta N, Williamson SR, Rogers CG, Cordon-Cardo C (2018) Association between cadmium and androgen receptor protein expression differs in prostate tumors of African American and European American men. J Trace Elem Med Biol 48:233–238. https://doi.org/10.1016/j.jtemb.2018.04.006
Aimola P, Carmignani M, Volpe AR, Di Benedetto A, Claudio L, Waalkes MP, Van Bokhoven A, Tokar EJ, Claudio PP (2012) Cadmium induces p53-dependent apoptosis in human prostate epithelial cells. PLoS ONE 7(3):e33647. https://doi.org/10.1371/journal.pone.0033647
Chandrasekaran B, Dahiya NR, Tyagi A, Kolluru V, Saran U, Baby BV, States JC, Haddad AQ, Ankem MK, Damodaran C (2020) Chronic exposure to cadmium induces a malignant transformation of benign prostate epithelial cells. Oncogenesis 9(2):1–10. https://doi.org/10.1038/s41389-020-0202-7
Santana VP, Salles ÉS, Correa DE, Gonçalves BF, Campos SG, Justulin LA, Godinho AF, Scarano WR (2016) Long-term effects of perinatal exposure to low doses of cadmium on the prostate of adult male rats. Int J Exp Pathol 97(4):310–316
Visser A, Deklerk J (1978) The effect of dietary cadmium on prostate growth. Trans Am Assoc Genitourin Surg 70:66–68
Lubrano C, Genovesi G, Specchia P, Costantini D, Mariani S, Petrangeli E, Lenzi A, Gnessi L (2013) Obesity and metabolic comorbidities: environmental diseases? Oxi Med Cell Longev. https://doi.org/10.1155/2013/640673
Park SS, Skaar DA, Jirtle RL, Hoyo C (2017) Epigenetics, obesity and early-life cadmium or lead exposure. Epigenomics 9(1):57–75. https://doi.org/10.2217/epi-2016-0047
Baillie-Hamilton PF (2002) Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med 8(2):185–192. https://doi.org/10.1089/107555302317371479
Papa V, Wannenes F, Crescioli C, Caporossi D, Lenzi A, Migliaccio S, Di Luigi L (2014) The environmental pollutant cadmium induces homeostasis alteration in muscle cells in vitro. J Endocrinol Invest 37(11):1073–1080
Darbre PD (2017) Endocrine disruptors and obesity. Current Obes Rep 6(1):18–27. https://doi.org/10.1007/s13679-017-0240-4
González-Casanova JE, Pertuz-Cruz SL, Caicedo-Ortega NH, Rojas-Gomez DM (2020) Adipogenesis regulation and endocrine disruptors: emerging insights in obesity. Biomed Res Int. https://doi.org/10.1155/2020/7453786
Kassotis CD, Nagel SC, Stapleton HM (2018) Unconventional oil and gas chemicals and wastewater-impacted water samples promote adipogenesis via PPARγ-dependent and independent mechanisms in 3T3-L1 cells. Sci Total Environ 640:1601–1610. https://doi.org/10.1016/j.scitotenv.2018.05.030
Doke M, Avecilla V, Felty Q (2018) Inhibitor of differentiation-3 and estrogenic endocrine disruptors: implications for susceptibility to obesity and metabolic disorders. BioMed Res Int. https://doi.org/10.1155/2018/6821601
Verbanck M, Canouil M, Leloire A, Dhennin V, Coumoul X, Yengo L, Froguel P, Poulain-Godefroy O (2017) Low-dose exposure to bisphenols A, F and S of human primary adipocyte impacts coding and non-coding RNA profiles. PLoS ONE 12(6):e0179583. https://doi.org/10.1371/journal.pone.0179583
Avecilla V, Doke M, Felty Q (2017) Contribution of inhibitor of dna binding/differentiation-3 and endocrine disrupting chemicals to pathophysiological aspects of chronic disease. Biomed Res Int. https://doi.org/10.1155/2017/6307109
Kim JT, Lee HK (2017) Childhood obesity and endocrine disrupting chemicals. Ann Pediatr Endocrinol Metab 22(4):219. https://doi.org/10.6065/apem.2017.22.4.219
Shoucri BM, Martinez ES, Abreo TJ, Hung VT, Moosova Z, Shioda T, Blumberg B (2017) Retinoid X receptor activation alters the chromatin landscape to commit mesenchymal stem cells to the adipose lineage. Endocrinology 158(10):3109–3125. https://doi.org/10.1210/en.2017-00348
Lejonklou MH, Dunder L, Bladin E, Pettersson V, Rönn M, Lind L, Waldén TB, Lind PM (2017) Effects of low-dose developmental bisphenol A exposure on metabolic parameters and gene expression in male and female Fischer 344 rat offspring. Environ Health Perspect 125(6):067018. https://doi.org/10.1289/EHP505
Green AJ, Hoyo C, Mattingly CJ, Luo Y, Tzeng J-Y, Murphy SK, Buchwalter DB, Planchart A (2018) Cadmium exposure increases the risk of juvenile obesity: a human and zebrafish comparative study. Int J Obes 42(7):1285–1295. https://doi.org/10.1038/s41366-018-0036-y
Haswell-Elkins M, Mcgrath V, Moore M, Satarug S, Walmby M, Ng J (2007) Exploring potential dietary contributions including traditional seafood and other determinants of urinary cadmium levels among indigenous women of a Torres Strait Island (Australia). J Eposure Sci Environ Epidemiol 17(3):298–306. https://doi.org/10.1038/sj.jes.7500547
Padilla MA, Elobeid M, Ruden DM, Allison DB (2010) An examination of the association of selected toxic metals with total and central obesity indices: NHANES 99–02. Int J Environ Res Public Health 7(9):3332–3347. https://doi.org/10.3390/ijerph7093332
Akinloye O, Ogunleye K, Oguntibeju OO (2010) Cadmium, lead, arsenic and selenium levels in patients with type 2 diabetes mellitus. Afr J Biotech 9(32):5189–5195
Nie X, Wang N, Chen Y, Chen C, Han B, Zhu C, Chen Y, Xia F, Cang Z, Lu M (2016) Blood cadmium in Chinese adults and its relationships with diabetes and obesity. Environ Sci Pollut Res 23(18):18714–18723. https://doi.org/10.1007/s11356-016-7078-2
Skalnaya MG, Tinkov AA, Demidov VA, Serebryansky EP, Nikonorov AA, Skalny AV (2014) Hair toxic element content in adult men and women in relation to body mass index. Biol Trace Elem Res 161(1):13–19. https://doi.org/10.1007/s12011-014-0082-9
El-Soud N, El-Laithy N, El-Saeed G, Wahby M, Khalil M, Morsy F, Shaffie N (2011) Antidiabetic activities of Foeniculum vulgare Mill. essential oil in streptozotocin-induced diabetic rats. Maced J Med Sci 4(2):139–146. https://doi.org/10.3889/MJMS.1857-5773.2011.0184
Shao W, Liu Q, He X, Liu H, Gu A, Jiang Z (2017) Association between level of urinary trace heavy metals and obesity among children aged 6–19 years: NHANES 1999–2011. Environ Sci Pollut Res 24(12):11573–11581. https://doi.org/10.1007/s11356-017-8803-1
Jiang F, Zhi X, Xu M, Li B, Zhang Z (2018) Gender-specific differences of interaction between cadmium exposure and obesity on prediabetes in the NHANES 2007–2012 population. Endocrine 61(2):258–266. https://doi.org/10.1007/s12020-018-1623-3
Noor N, Zong G, Seely EW, Weisskopf M, James-Todd T (2018) Urinary cadmium concentrations and metabolic syndrome in US adults: The National Health and Nutrition Examination Survey 2001–2014. Environ Int 121:349–356. https://doi.org/10.1016/j.envint.2018.08.029
Huzior-Bałajewicz A, Pietrzyk J, Schlegel-Zawadzka M, Piatkowska E, Zachwieja Z (2001) The influence of lead and cadmium environmental pollution on anthropometric health factors in children. Przegl Lek 58(4):315
Qin YY, Leung CKM, Leung AOW, Wu SC, Zheng JS, Wong MH (2010) Persistent organic pollutants and heavy metals in adipose tissues of patients with uterine leiomyomas and the association of these pollutants with seafood diet, BMI, and age. Environ Sci Pollut Res 17(1):229–240
Adnan JA, Azhar SS, Hasni JM, Ahmad JS (2012) Urinary cadmium concentration and its risk factors among adults in Tanjung Karang, Selangor. Am-Eur J Toxicol Sci 4(2):80–88. https://doi.org/10.5829/idosi.aejts.2012.4.2.6331
Kelishadi R, Askarieh A, Motlagh ME, Tajadini M, Heshmat R, Ardalan G, Fallahi S, Poursafa P (2013) Association of blood cadmium level with cardiometabolic risk factors and liver enzymes in a nationally representative sample of adolescents: the CASPIAN-III study. J Environ Public Health. https://doi.org/10.1155/2013/142856
Park S, Lee B-K (2013) Body fat percentage and hemoglobin levels are related to blood lead, cadmium, and mercury concentrations in a Korean Adult Population (KNHANES 2008–2010). Biol Trace Elem Res 151(3):315–323. https://doi.org/10.1007/s12011-012-9566-7
Gonzalez-Reimers E, Martín-González C, Galindo-Martín L, Aleman-Valls M, Velasco-Vázquez J, Arnay-De-La-Rosa M, Pérez-Hernández O, Luis RH (2014) Lead, cadmium and zinc in hair samples: relationship with dietary habits and urban environment. Biol Trace Elem Res 157(3):205–210
Ahn B, Kim S-H, Park M-J (2017) Blood cadmium concentrations in Korean adolescents: from the Korea National Health and Nutrition Examination Survey 2010–2013. Int J Hyg Environ Health 220(1):37–42. https://doi.org/10.1016/j.ijheh.2016.10.003
Rotter I, Kosik-Bogacka D, Dołęgowska B, Safranow K, Lubkowska A, Laszczyńska M (2015) Relationship between the concentrations of heavy metals and bioelements in aging men with metabolic syndrome. Int J Environ Res Public Health 12(4):3944–3961. https://doi.org/10.3390/ijerph120403944
Ficková M, Eybl V, Kotyzová D, Mičková V, Möstbök S, Brtko J (2003) Long lasting cadmium intake is associated with reduction of insulin receptors in rat adipocytes. Biometals 16(4):561–566. https://doi.org/10.1023/A:1023485130767
Huang Y, Zhang W, Li H (2003) Effect of cadmium on body weight and organ coefficient of ovaries in female rats. Occup Health 19:7–9
Singh PK, Baxi D, Diwedi R, Ramachandran A (2012) Prior cadmium exposure improves glucoregulation in diabetic rats but exacerbates effects on metabolic dysregulation, oxidative stress, and hepatic and renal toxicity. Drug Chem Toxicol 35(2):167–177. https://doi.org/10.3109/01480545.2011.589450
Treviño S, Waalkes MP, Hernández JAF, León-Chavez BA, Aguilar-Alonso P, Brambila E (2015) Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch Biochem Biophys 583:27–35. https://doi.org/10.1016/j.abb.2015.07.010
Kawakami T, Nishiyama K, Kadota Y, Sato M, Inoue M, Suzuki S (2013) Cadmium modulates adipocyte functions in metallothionein-null mice. Toxicol Appl Pharmacol 272(3):625–636. https://doi.org/10.1016/j.taap.2013.07.015
Zhang S, Jin Y, Zeng Z, Liu Z, Fu Z (2015) Subchronic exposure of mice to cadmium perturbs their hepatic energy metabolism and gut microbiome. Chem Res Toxicol 28(10):2000–2009. https://doi.org/10.1021/acs.chemrestox.5b00237
Yamamoto A, Wada O, Ono T, Ono H (1986) Cadmium stimulates glucose metabolism in rat adipocytes. J Inorg Biochem 27(3):221–226. https://doi.org/10.1016/0162-0134(86)80063-0
Kang D, Khil L-Y, Lee B-H, Moon C-K (2005) Effects of cadmium on glucose transport in 3T3-L1 adipocytes. Environ Health Toxicol 20(1):87–95
Harrison SA, Buxton JM, Clancy BM, Czech MP (1991) Evidence that erythroid-type glucose transporter intrinsic activity is modulated by cadmium treatment of mouse 3T3-L1 cells. J Biol Chem 266(29):19438–19449
Lachaal M, Liu H, Kim S-s, Jung CY (1996) Effects of cadmium on glucose transport in rat adipocytes and human erythrocytes: stimulation of GLUT1 catalytic activity. Exp Mol Med 28(1):33–40. https://doi.org/10.1038/emm.1996.6
Lachaal M, Liu H, Kim S-s, Spangler RA, Jung CY (1996) Cadmium increases GLUT1 substrate binding affinity in vitro while reducing its cytochalasin B binding affinity. Biochemistry 35(47):14958–14962. https://doi.org/10.1021/bi9617320
Han JC, Park SY, Hah BG, Choi GH, Kim YK, Kwon TH, Kim EK, Lachaal M, Jung CY, Lee W (2003) Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Arch Biochem Biophys 413(2):213–220
Moon J, Yoo B (2008) Protective effects of propolis on cadmium-induced cell death of 3T3-L1 adipocytes. Korean J Apic 23(4):289–294
Kawakami T, Sugimoto H, Furuichi R, Kadota Y, Inoue M, Setsu K, Suzuki S, Sato M (2010) Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology 267(1–3):20–26. https://doi.org/10.1016/j.tox.2009.07.022
Lee EJ, Moon JY, Yoo BS (2012) Cadmium inhibits the differentiation of 3T3-L1 preadipocyte through the C/EBPα and PPARγ pathways. Drug Chem Toxicol 35(2):225–231. https://doi.org/10.3109/01480545.2011.591401
Levy J, Gyarmati J, Lesko J, Adler R, Stevens W (2000) Dual regulation of leptin secretion: intracellular energy and calcium dependence of regulated pathway. Am J Physiol Endocrinol Metab 278:E892–E901. https://doi.org/10.1152/ajpendo.2000.278.5.E892
Planchart A, Green A, Hoyo C, Mattingly CJ (2018) Heavy metal exposure and metabolic syndrome: evidence from human and model system studies. Curr Environ Health Rep 5(1):110–124. https://doi.org/10.1007/s40572-018-0182-3
Cho N, Shaw J, Karuranga S, Huang Y, da Rocha FJ, Ohlrogge A, Malanda B (2018) IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138:271–281
Eze IC, Schaffner E, Foraster M, Imboden M, von Eckardstein A, Gerbase MW, Rothe T, Rochat T, Künzli N, Schindler C (2015) Long-term exposure to ambient air pollution and metabolic syndrome in adults. PLoS ONE 10(6):e0130337. https://doi.org/10.1371/journal.pone.0130337
Basner M, Riggs DW, Conklin DJ (2020) Environmental determinants of hypertension and diabetes mellitus: sounding off about the effects of noise. Am Heart Assoc. https://doi.org/10.1161/JAHA.120.016048
Yang M, Cheng H, Shen C, Liu J, Zhang H, Cao J, Ding R (2020) Effects of long-term exposure to air pollution on the incidence of type 2 diabetes mellitus: a meta-analysis of cohort studies. Environ Sci Pollut Res 27(1):798–811. https://doi.org/10.1007/s11356-019-06824-1
Lei L-J, Chen L, Jin T-Y, Nordberg M, Chang X-L (2007) Estimation of benchmark dose for pancreatic damage in cadmium-exposed smelters. Toxicol Sci 97(1):189–195. https://doi.org/10.1093/toxsci/kfm016
Lei L, Guo J, Shi X, Kang H, Wang T, Zhang Z, Gao Y (2019) Relationship between urinary cadmium and type 2 diabetes mellitus in adults. Zhonghua Liu Xing Bing Xue Za Zhi 40(2):207–211
Guo F-F, Hu Z-Y, Li B-Y, Qin L-Q, Fu C, Yu H, Zhang Z-L (2019) Evaluation of the association between urinary cadmium levels below threshold limits and the risk of diabetes mellitus: a dose-response meta-analysis. Environ Sci Pollut Res 26(19):19272–19281
Trouiller-Gerfaux P, Podglajen E, Hulo S, Richeval C, Allorge D, Garat A, Matran R, Amouyel P, Meirhaeghe A, Dauchet L (2019) The association between blood cadmium and glycated haemoglobin among never-, former, and current smokers: a cross-sectional study in France. Environ Res 178:108673. https://doi.org/10.1016/j.envres.2019.108673
Xiao L, Zhou Y, Ma J, Cao L, Zhu C, Li W, Wang D, Fan L, Ye Z, Chen W (2019) Roles of C-reactive protein on the association between urinary cadmium and type 2 diabetes. Environ Pollut 255:113341. https://doi.org/10.1016/j.envpol.2019.113341
Bell R, Early J, Nonavinakere V, Mallory Z (1990) Effect of cadmium on blood glucose level in the rat. Toxicol Lett 54(2–3):199–205. https://doi.org/10.1016/0378-4274(90)90184-N
Merali Z, Singhal R (1980) Diabetogenic effects of chronic oral cadmium administration to neonatal rats. Br J Pharmacol 69(1):151–157. https://doi.org/10.1111/j.1476-5381.1980.tb10895.x
Chapatwala K, Rajanna E, Desaiah D (1980) Cadmium induced changes in gluconeogenic enzymes in rat kidney and liver. Drug Chem Toxicol 3(4):407–420. https://doi.org/10.3109/01480548009030129
Lei L-J, Jin T-Y, Zhou Y-F (2007) Insulin expression in rats exposed to cadmium. Biomed Environ Sci 20(4):295–301
Lei L-J, Jin T-Y, Zhou Y-F (2005) Effects of cadmium on levels of insulin in rats. Wei Sheng Yan Jiu 34(4):394–396
Lei L, Jin T, Zhou Y (2005) The toxic effects of cadmium on pancreas. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 23(1):45–49
Edwards JR, Prozialeck WC (2009) Cadmium, diabetes and chronic kidney disease. Toxicol Appl Pharmacol 238(3):289–293. https://doi.org/10.1016/j.taap.2009.03.007
Jacquet A, Arnaud J, Hininger-Favier I, Hazane-Puch F, Couturier K, Lénon M, Lamarche F, Ounnas F, Fontaine E, Moulis J-M (2018) Impact of chronic and low cadmium exposure of rats: sex specific disruption of glucose metabolism. Chemosphere 207:764–773. https://doi.org/10.1016/j.chemosphere.2018.05.099
Yau ET, Mennear JH (1977) Pancreatic metallothionein: protection against cadmium-induced inhibition of insulin secretory activity. Toxicol Appl Pharmacol 39(3):515–520
El Muayed M, Raja MR, Zhang X, MacRenaris KW, Bhatt S, Chen X, Urbanek M, O’Halloran TV, Lowe J, William L (2012) Accumulation of cadmium in insulin-producing β cells. Islets 4(6):405–416. https://doi.org/10.4161/isl.23101
Chang K-C, Hsu C-C, Liu S-H, Su C-C, Yen C-C, Lee M-J, Chen K-L, Ho T-J, Hung D-Z, Wu C-C (2013) Cadmium induces apoptosis in pancreatic β-cells through a mitochondria-dependent pathway: the role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS ONE 8(2):e54374. https://doi.org/10.1371/journal.pone.0054374
Mohammadi P, Rahimifard M, Baeeri M, Abdollahi M, Mostafalou S (2019) Mechanistic assessment of cadmium toxicity in association with the functions of estrogen receptors in the Langerhans islets. Iranian J Basic Med Sci 22(4):445. https://doi.org/10.22038/ijbms.2019.33939.8076
Kanis JA, Melton LJ III, Christiansen C, Johnston CC, Khaltaev N (1994) The diagnosis of osteoporosis. J Bone Miner Res 9(8):1137–1141
Wallin M, Barregard L, Sallsten G, Lundh T, Karlsson MK, Lorentzon M, Ohlsson C, Mellström D (2016) Low-level cadmium exposure is associated with decreased bone mineral density and increased risk of incident fractures in elderly men: the MrOS Sweden Study. J Bone Miner Res 31(4):732–741. https://doi.org/10.1002/jbmr.2743
Scimeca M, Feola M, Romano L, Rao C, Gasbarra E, Bonanno E, Brandi ML, Tarantino U (2017) Heavy metals accumulation affects bone microarchitecture in osteoporotic patients. Environ Toxicol 32(4):1333–1342. https://doi.org/10.1002/tox.22327
Johri N, Jacquillet G, Unwin R (2010) Heavy metal poisoning: the effects of cadmium on the kidney. Biometals 23(5):783–792
Chen X, Zhu G, Jin T, Lei L, Liang Y (2011) Bone mineral density is related with previous renal dysfunction caused by cadmium exposure. Environ Toxicol Pharmacol 32(1):46–53. https://doi.org/10.1016/j.etap.2011.03.007
Rodríguez J, Mandalunis PM (2018) A review of metal exposure and its effects on bone health. J Toxicol. https://doi.org/10.1155/2018/4854152
Ibrahim KS, Beshir S, Shahy EM, Shaheen W (2016) Effect of occupational cadmium exposure on parathyroid gland. Open Access Maced J Med Sci 4(2):302. https://doi.org/10.3889/oamjms.2016.042
Nishijo M, Nambunmee K, Suvagandha D, Swaddiwudhipong W, Ruangyuttikarn W, Nishino Y (2017) Gender-specific impact of cadmium exposure on bone metabolism in older people living in a cadmium-polluted area in Thailand. Int J Environ Res Public Health 14(4):401. https://doi.org/10.3390/ijerph14040401
Wallin M, Sallsten G, Fabricius-Lagging E, Öhrn C, Lundh T, Barregard L (2013) Kidney cadmium levels and associations with urinary calcium and bone mineral density: a cross-sectional study in Sweden. Environ Health 12(1):22. https://doi.org/10.1186/1476-069X-12-22
Kazantzis G (2004) Cadmium, osteoporosis and calcium metabolism. Biometals 17(5):493–498. https://doi.org/10.1023/b:biom.0000045727.76054.f3
Åkesson A, Bjellerup P, Lundh T, Lidfeldt J, Nerbrand C, Samsioe G, Skerfving S, Vahter M (2006) Cadmium-induced effects on bone in a population-based study of women. Environ Health Perspect 114(6):830–834. https://doi.org/10.1289/ehp.8763
Chen X, Wang Z, Zhu G, Nordberg GF, Jin T, Ding X (2019) The association between cumulative cadmium intake and osteoporosis and risk of fracture in a Chinese population. J Eposure Sci Environ Epidemiol 29(3):435–443. https://doi.org/10.1038/s41370-018-0057-6
Malin Igra A, Vahter M, Raqib R, Kippler M (2019) Early-life cadmium exposure and bone-related biomarkers: a longitudinal study in children. Environ Health Perspect 127(3):037003. https://doi.org/10.1289/EHP3655
Li X, Li R, Yan J, Song Y, Huo J, Lan Z, Chen J, Zhang L (2020) Co-exposure of cadmium and lead on bone health in a southwestern Chinese population aged 40–75 years. J Appl Toxicol 40(3):352–362. https://doi.org/10.1002/jat.3908
Brzóska MM, Moniuszko-Jakoniuk J (2004) Low-level exposure to cadmium during the lifetime increases the risk of osteoporosis and fractures of the lumbar spine in the elderly: studies on a rat model of human environmental exposure. Toxicol Sci 82(2):468–477
Brzóska MM, Moniuszko-Jakoniuk J (2005) Disorders in bone metabolism of female rats chronically exposed to cadmium. Toxicol Appl Pharmacol 202(1):68–83
Youness ER, Mohammed NA, Morsy FA (2012) Cadmium impact and osteoporosis: mechanism of action. Toxicol Mech Methods 22(7):560–567. https://doi.org/10.3109/15376516.2012.702796
Rodríguez J, Mandalunis PM (2016) Effect of cadmium on bone tissue in growing animals. Exp Toxicol Pathol 68(7):391–397. https://doi.org/10.1016/j.etp.2016.06.001
Lv Y-J, Wei Q-Z, Zhang Y-C, Huang R, Li B-S, Tan J-B, Wang J, Ling H-T, Wu S-X, Yang X-F (2019) Low-dose cadmium exposure acts on rat mesenchymal stem cells via RANKL/OPG and downregulate osteogenic differentiation genes. Environ Pollut 249:620–628. https://doi.org/10.1016/j.envpol.2019.03.027
He S, Zhuo L, Cao Y, Liu G, Zhao H, Song R, Liu Z (2020) Effect of cadmium on osteoclast differentiation during bone injury in female mice. Environ Toxicol 35(4):487–494. https://doi.org/10.1002/tox.22884
Chen X, Zhu G, Gu S, Jin T, Shao C (2009) Effects of cadmium on osteoblasts and osteoclasts in vitro. Environ Toxicol Pharmacol 28(2):232–236. https://doi.org/10.1016/j.etap.2009.04.010
Chen X, Zhu G, Jin T, Zhou Z, Gu S, Qiu J, Xiao H (2012) Cadmium stimulates the osteoclastic differentiation of RAW264. 7 cells in presence of osteoblasts. Biol Trace Elem Res 146(3):349–353. https://doi.org/10.1007/s12011-011-9256-x
Papa V, Bimonte V, Wannenes F, D’Abusco A, Fittipaldi S, Scandurra R, Politi L, Crescioli C, Lenzi A, Di Luigi L (2015) The endocrine disruptor cadmium alters human osteoblast-like Saos-2 cells homeostasis in vitro by alteration of Wnt/β-catenin pathway and activation of caspases. J Endocrinol Invest 38(12):1345–1356. https://doi.org/10.1007/s40618-015-0380-x
Hu K-H, Li W-X, Sun M-Y, Zhang S-B, Fan C-X, Wu Q, Zhu W, Xu X (2015) Cadmium induced apoptosis in MG63 cells by increasing ROS, activation of p38 MAPK and inhibition of ERK 1/2 pathways. Cell Physiol Biochem 36(2):642–654
Al-Ghafari A, Elmorsy E, Fikry E, Alrowaili M, Carter WG (2019) The heavy metals lead and cadmium are cytotoxic to human bone osteoblasts via induction of redox stress. PLoS ONE 14(11):e0225341. https://doi.org/10.1371/journal.pone.0225341
Wu L, Wei Q, Lv Y, Xue J, Zhang B, Sun Q, Xiao T, Huang R, Wang P, Dai X (2019) Wnt/β-Catenin pathway is involved in cadmium-induced inhibition of osteoblast differentiation of bone marrow mesenchymal stem cells. Int J Mol Sci 20(6):1519. https://doi.org/10.3390/ijms20061519
WHO (2017). https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
Pope CA III, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ (2004) Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation 109(1):71–77. https://doi.org/10.1161/01.CIR.0000108927.80044.7F
Tellez-Plaza M, Jones MR, Dominguez-Lucas A, Guallar E, Navas-Acien A (2013) Cadmium exposure and clinical cardiovascular disease: a systematic review. Curr Atheroscler Rep 15(10):356. https://doi.org/10.1007/s11883-013-0356-2
Kukongviriyapan U, Apaijit K, Kukongviriyapan V (2016) Oxidative stress and cardiovascular dysfunction associated with cadmium exposure: beneficial effects of curcumin and tetrahydrocurcumin. Tohoku J Exp Med 239(1):25–38. https://doi.org/10.1620/tjem.239.25
Bagchi D, Vuchetich P, Bagchi M, Hassoun E, Tran M, Tang L, Stohs S (1997) Induction of oxidative stress by chronic administration of sodium dichromate [chromium VI] and cadmium chloride [cadmium II] to rats. Free Radical Biol Med 22(3):471–478. https://doi.org/10.1016/s0891-5849(96)00352-8
Liu F, Jan K-Y (2000) DNA damage in arsenite-and cadmium-treated bovine aortic endothelial cells. Free Radical Biol Med 28(1):55–63. https://doi.org/10.1016/s0891-5849(99)00196-3
Rani A, Kumar A, Lal A, Pant M (2014) Cellular mechanisms of cadmium-induced toxicity: a review. Int J Environ Health Res 24(4):378–399
Messner B, Knoflach M, Seubert A, Ritsch A, Pfaller K, Henderson B, Shen YH, Zeller I, Willeit J, Gn L (2009) Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler Thromb Vasc Biol 29(9):1392–1398. https://doi.org/10.1161/ATVBAHA.109.190082
Knoflach M, Messner B, Shen YH, Frotschnig S, Liu G, Pfaller K, Wang X, Matosevic B, Willeit J, Kiechl S (2011) Non-toxic cadmium concentrations induce vascular inflammation and promote atherosclerosis. Circ J 75(10):2491–2495. https://doi.org/10.1253/circj.cj-11-0196
Myong J-P, Kim H-R, Jang T-W, Lee HE, Koo J-W (2014) Association between blood cadmium levels and 10-year coronary heart disease risk in the general Korean population: the Korean National Health and Nutrition Examination Survey 2008–2010. PLoS ONE 9(11):e111909
Barregard L, Sallsten G, Fagerberg B, Borné Y, Persson M, Hedblad B, Engström G (2016) Blood cadmium levels and incident cardiovascular events during follow-up in a population-based cohort of Swedish adults: the Malmö Diet and Cancer Study. Environ Health Perspect 124(5):594–600. https://doi.org/10.1289/ehp.1509735
Fagerberg B, Bergström G, Borén J, Barregard L (2012) Cadmium exposure is accompanied by increased prevalence and future growth of atherosclerotic plaques in 64-year-old women. J Intern Med 272(6):601–610. https://doi.org/10.1111/j.1365-2796.2012.02578.x
Fagerberg B, Kjelldahl J, Sallsten G, Barregard L, Forsgard N, Österberg K, Hultén LM, Bergström G (2016) Cadmium exposure as measured in blood in relation to macrophage density in symptomatic atherosclerotic plaques from human carotid artery. Atherosclerosis 249:209–214. https://doi.org/10.1016/j.atherosclerosis.2016.01.011
Howard DP, Van Lammeren GW, Rothwell PM, Redgrave JN, Moll FL, de Vries J-PP, De Kleijn DP, Den Ruijter HM, De Borst GJ, Pasterkamp G (2015) Symptomatic carotid atherosclerotic disease: correlations between plaque composition and ipsilateral stroke risk. Stroke 46(1):182–189
Liberda EN, Zuk AM, Tsuji LJ (2019) Complex contaminant mixtures and their associations with intima-media thickness. BMC Cardiovasc Disord 19(1):289. https://doi.org/10.1186/s12872-019-1246-5
Ilmiawati C, Reza M, Yanni M, Rusjdi DA (2020) Blood Cd levels and carotid intima-media thickness in young adults living in Padang, Indonesia. BMC Res Notes 13:1–7. https://doi.org/10.1186/s13104-020-05042-0
Hecht EM, Landy DC, Ahn S, Hlaing WM, Hennekens CH (2013) Hypothesis: cadmium explains, in part, why smoking increases the risk of cardiovascular disease. J Cardiovasc Pharmacol Ther 18(6):550–554. https://doi.org/10.1177/1074248413494815
Solenkova NV, Newman JD, Berger JS, Thurston G, Hochman JS, Lamas GA (2014) Metal pollutants and cardiovascular disease: mechanisms and consequences of exposure. Am Heart J 168(6):812–822. https://doi.org/10.1016/j.ahj.2014.07.007
Choi S, Kwon J, Kwon P, Lee C, Jang S-I (2020) Association between blood heavy metal levels and predicted 10-year risk for a first atherosclerosis cardiovascular disease in the general Korean population. Int J Environ Res Public Health 17(6):2134. https://doi.org/10.3390/ijerph17062134
Kolluru GK, Tamilarasan K, Priya SG, Durgha N, Chatterjee S (2006) Cadmium induced endothelial dysfunction: consequence of defective migratory pattern of endothelial cells in association with poor nitric oxide availability under cadmium challenge. Cell Biol Int 30(5):427–438
Everett CJ, Frithsen IL (2008) Association of urinary cadmium and myocardial infarction. Environ Res 106(2):284–286
Tellez-Plaza M, Navas-Acien A, Crainiceanu CM, Guallar E (2008) Cadmium exposure and hypertension in the 1999–2004 National Health and Nutrition Examination Survey (NHANES). Environ Health Perspect 116(1):51–56. https://doi.org/10.1289/ehp.10764
Tellez-Plaza M, Navas-Acien A, Crainiceanu CM, Sharrett AR, Guallar E (2010) Cadmium and peripheral arterial disease: gender differences in the 1999–2004 US National Health and Nutrition Examination Survey. Am J Epidemiol 172(6):671–681. https://doi.org/10.1093/aje/kwq172
Peters JL, Perlstein TS, Perry MJ, McNeely E, Weuve J (2010) Cadmium exposure in association with history of stroke and heart failure. Environ Res 110(2):199–206. https://doi.org/10.1016/j.envres.2009.12.004
Li H, Fagerberg B, Sallsten G, Borné Y, Hedblad B, Engström G, Barregard L, Andersson EM (2019) Smoking-induced risk of future cardiovascular disease is partly mediated by cadmium in tobacco: Malmö Diet and Cancer Cohort Study. Environ Health 18(1):56. https://doi.org/10.1186/s12940-019-0495-1
Santos-Gallego CG, Jialal I (2016) Cadmium and atherosclerosis: Heavy metal or singing the blues? Atherosclerosis 249:230–232. https://doi.org/10.1016/j.atherosclerosis.2016.01.041
Satarug S, Garrett SH, Sens MA, Sens DA (2010) Cadmium, environmental exposure, and health outcomes. Environ Health Perspect 118(2):182–190. https://doi.org/10.1289/ehp.0901234
Yuan Z, Luo T, Liu X, Hua H, Zhuang Y, Zhang X, Zhang L, Zhang Y, Xu W, Ren J (2019) Tracing anthropogenic cadmium emissions: from sources to pollution. Sci Total Environ 676:87–96. https://doi.org/10.1016/j.scitotenv.2019.04.250
Agency EM (2013) EMA/491185/2013 Committee for Medicinal Products for Human Use (CHMP) Giotrif—CHMP assessment report International non-proprietary name: afatinib Procedure No. EMEA/H/C/002280. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002280/WC500152394.pdf
Schaefer HR, Dennis S, Fitzpatrick S (2020) Cadmium: Mitigation strategies to reduce dietary exposure. J Food Sci 85(2):260–267. https://doi.org/10.1111/1750-3841.14997
Kusznierewicz B, Bączek-Kwinta R, Bartoszek A, Piekarska A, Huk A, Manikowska A, Antonkiewicz J, Namieśnik J, Konieczka P (2012) The dose-dependent influence of zinc and cadmium contamination of soil on their uptake and glucosinolate content in white cabbage (Brassica oleracea var. capitata f. alba). Environ Toxicol Chem 31(11):2482–2489. https://doi.org/10.1002/etc.1977
Mostofa MG, Rahman M, Ansary M, Uddin M, Fujita M, Tran L-SP (2019) Interactive effects of salicylic acid and nitric oxide in enhancing rice tolerance to cadmium stress. Int J Mol Sci 20(22):5798. https://doi.org/10.3390/ijms20225798
Habib R, Wahdan SA, Gad AM, Azab SS (2019) Infliximab abrogates cadmium-induced testicular damage and spermiotoxicity via enhancement of steroidogenesis and suppression of inflammation and apoptosis mediators. Ecotoxicol Environ Saf 182:109398. https://doi.org/10.1016/j.ecoenv.2019.109398
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflicts of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants and/or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bimonte, V.M., Besharat, Z.M., Antonioni, A. et al. The endocrine disruptor cadmium: a new player in the pathophysiology of metabolic diseases. J Endocrinol Invest 44, 1363–1377 (2021). https://doi.org/10.1007/s40618-021-01502-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-021-01502-x