
Summary
Purpose This study investigated the maximum-tolerated dose (MTD), dose-limiting toxicity (DLT), and pharmacokinetic (PK) profiles of DHP107, a novel oral paclitaxel containing neither Cremophor EL nor P-glycoprotein (P-gp) inhibitor. Patients and methods Patients with advanced solid tumors refractory to all standard treatments were administered a single oral dose of DHP107 on a dose-escalating schedule (60–600 mg/m2) during the first chemotherapy cycle, and intravenous paclitaxel 175 mg/m2 during subsequent cycles. Cohorts of 3 patients were treated at each dose level provided no DLTs were observed. The pharmacokinetics of paclitaxel and its metabolites were investigated for oral DHP107 and intravenous paclitaxel. Results Thirty-four patients were enrolled. Dose-limiting toxicities were not observed, even at the highest dose level (600 mg/m2). Further dose escalation was not performed because pharmacokinetics did not increase proportionally at doses above 250 mg/m2. The coefficient of variance of AUClast DHP107 ranged from 11.8 % to 34.0 %, comparable to 24.4 % of intravenous paclitaxel 175 mg/m2. There were no grade 4 toxicities, whereas grade 3 toxicities included diarrhea (12.1 %), neutropenia (6.1 %) and fatigue (3.0 %). While no objective responses were observed, 11 patients (33.3 %) showed stable disease. Conclusions DHP107 was safe and feasible in patients with advanced malignancies. As exposure of paclitaxel plateau among patients receiving more than 250 mg/m2 of DHP107, the dose escalation of DHP107 may be limited to 250 mg/m2 in further clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Paclitaxel is an important agent used in the systemic treatment of various cancers, including breast, ovarian, gastric and non-small lung cancer [1–4]. As paclitaxel is poorly soluble in water, the marketed intravenous (IV) formulation contains a pharmaceutical solvent, Cremophor EL (BASF Corp, Ludwigshafen, Germany), which stabilizes emulsions of nonpolar paclitaxel in aqueous solvents. Followed by the need to treat with solvent-based paclitaxel, however, several problems including hypersensitivity and dosage limitations occur due to Cremophor EL [5, 6]. Another limit of IV paclitaxel is the inconvenience of continuous exposure despite its advantages. Continuous exposure to low concentrations of paclitaxel has been reported to have anti-tumor activity by inhibiting angiogenesis [7]. Using a mouse model of HT-29 human colon cancer, low dose metronomic paclitaxel has shown advantages [8]. Clinical trials of weekly paclitaxel resulted in better efficacy outcomes than the every 3-week administration in breast cancer patients [9].
Oral administration of paclitaxel has been an attractive candidate due to the issues associated with solvent-based IV paclitaxel. However, the development of oral paclitaxel has been limited by its poor bioavailability, which is related to the high affinity between paclitaxel and P-glycoprotein (P-gp), a multidrug transporter that limits the oral absorption and mediates the direct excretion of paclitaxel into the intestinal lumen. To increase systemic exposure to oral paclitaxel, this agent has been combined with a P-gp inhibitor, such as cyclosporin or ritonavir [10, 11]. These combinations, however, are associated with additional side effects, which may be due to drug-drug interactions and/or Cremophor EL-related issues.
DHP107 is a novel form of oral paclitaxel, containing mucoadhesive lipid free of Cremophor EL. Oral administration of DHP107 resulted in enhanced absorption and effective tissue distribution of paclitaxel without concomitant administration of P-gp inhibitors [12, 13]. The mucoadhesive lipid of DHP107 leads to enhanced drug delivery relevant to the physiologic processes of lipid digestion and absorption, which has been confirmed by the observation of accumulation of lipid in intestine and lipid droplets in intestinal epithelial cells and paclitaxel distribution after DHP107 administration in mouse models. A first in-human phase I and pharmacokinetic study of DHP107 for patients with advanced solid tumors was performed to explore the characteristics of DHP!07 in humans.
Patients and methods
Eligibiliy
Patients eligible for enrollment were ≥18 years of age; histologically confirmed unresectable or metastatic solid tumors with an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 2 or lower; failed all standard chemotherapeutic regimens; one or more measurable lesions as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0; and adequate hematologic, hepatic and renal functions.
Major exclusion criteria were prior exposure to taxanes; concurrent use of P-glycoprotein inhibitors; chronic use of proton pump inhibitors or H2-receptor antagonists; and prior cholecystectomy or total gastrectomy which could cause abnormal bile acid secretion. Additional exclusion criteria were any unresolved toxicity from previous treatment, including peripheral neuropathy higher than grade 2.
Study design
This single-center, open-label, dose-escalation phase 1 trial, conducted at Asan Medical Center, Seoul, Korea was performed to investigate the maximum-tolerated dose (MTD), dose-limiting toxicity (DLT), of oral DHP107 and compare the safety and PK of paclitaxel in different formulations, oral DHP107 and intravenous paclitaxel. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and all patients provided informed consent. The protocol was approved by the Korea Food and Drug Administration and the Institutional Review Board of Asan Medical Center, Korea.
The first cycle consisted of single escalating doses of oral DHP107 on day 1 and the second and subsequent cycles, 3 weeks later, consisted of IV paclitaxel, 175 mg/m2 over 3 h, until progression or intolerable toxicity. DHP107 was stored under refrigerated conditions and thawed at 40–60 °C just prior to administration. It was administered with a half glass of water (100 ml) 2 h after a light breakfast and followed by fasting for 2 h. DHP107 was provided by DAE HWA Pharmaceutical Co., Ltd.
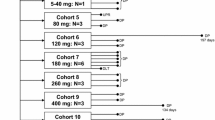
The initial dose of DHP107 was 60 mg/m2 (level 1), with doses escalated to 120 mg/m2 (level 2), 200 mg/m2 (level 3), and subsequently increased by 50 mg/m2 from level 4 (250 mg/m2) to the level at which the dose-limiting toxicity (DLT) occurred. The maximal tolerated dose (MTD) was defined as the dose of DHP107 that produced DLT in two or more of six patients. Three patients were entered into each dose level; if DLT occurred in one or two of the first three patients, three additional patients were treated with the same dose of DHP107. If no DLT was observed in the initial three patients or in only one of six patients, the dose was increased to the next level. Intra-patient dose escalation was not allowed.
Safety assessments
Determination of safety and toxicity of DHP107 was based on the first cycle of treatment and compared with second cycle of IV paclitaxel. Intrapatient dose escalation was not allowed. Treatment-related adverse events were evaluated according to the Common Terminology Criteria for Adverse Events (CTCAE) of the National Cancer Institute, version 3.0. Dose-limiting toxicity (DLT) was defined as: grade 4 neutropenia lasting more than 7 days; grade 3 or higher febrile neutropenia or thrombocytopenia; any other grade 3 or higher non-hematologic toxicity, including nausea, vomiting, and diarrhea, that did not improve within 2 days after the start of appropriate management; or a treatment interruption lasting more than 2 weeks.
Pharmacokinetic sampling and analysis
Serial blood samples were collected during the first cycle (before administration of DHP107, and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 32, and 48 h after administration of DHP107) and the second cycle (before administration of paclitaxel, 1.5 h after starting administration, and 0.5, 1, 2, 4, 6, 8, 12, 24, 32, and 48 h after finishing administration of paclitaxel). Serial urine samples were collected before and, at 24 and 48 h after administration of DHP107 or paclitaxel.
The pharmacokinetics of paclitaxel and it metabolites were analyzed using non-compartmental methods in WinNonlin® 5.2 (Pharsight Corporation, Mountain View, CA). The area under the concentration time curve (AUC) was calculated by the linear trapezoidal method. The peak plasma concentration (Cmax) was obtained directly from the concentration versus time data. The terminal elimination rate constant (λz) was estimated by least squares regression analysis of the terminal phase of the log-linear plot of concentration versus time, and individual terminal half-life (t1/2) values were calculated as 0.693/λz.
The plasma concentrations of paclitaxel and its metabolites were determined using high-performance liquid chromatography (Agilent 1100 series; Agilent Technologies, Santa Clara, CA) with tandem mass spectrometry (API 3000; ABSciex, Foster City, CA) after sample preparation by liquid–liquid extraction. In brief, samples were removed from the freezer and allowed to thaw at room temperature (25 °C). A total of 1 ml of the thawed plasma sample was mixed with 1 ml sodium hydrogen phosphate solution (0.1 mol/l, pH 7.48), and 30 μl internal standard (paclitaxel) for 1 min, followed by the addition of 5 ml of 1-chlorobutane, mixing for 5 min and centrifugation at 1,238 g for 10 min. The upper layer was transferred to a culture tube and evaporated to dryness under a speed vacuum, at 45 °C for 2 h. The dry residue was reconstituted in 100 μl methanol and transferred to a vial for analysis by liquid chromatography–tandem mass spectrometry.23 The lower limit of quantification was 0.03 ng/ml. The calibration curve was linear over the range of 0.03–40.0 ng/ml, with the coefficients of determination (R2) greater than 0.997 for all patients. The intra- and interassay precision levels, determined for 0.05, 0.5, 1.0, and 1.5 ng/ml fentanyl, were 3.72–5.73 % and 2.53–4.10 %, respectively, and their accuracies were 85.24–96.22 % and 96.11–102.5 %, respectively.
Results
Patients and treatment
Between February 2008 and September 2009, 34 patients were enrolled; their clinical characteristics are shown in Table 1. Median patient age was 54 years (range, 20–68 years); 24 patients (70.6 %) were male and 10 (29.4 %) female; and 29 (85.3 %) had good performance status (PS) of 0 or 1 at study entry. Of these 34 patients, 25 (73.5 %) had colorectal cancers, 2 (5.9 %) had stomach cancer, 1 each (2.9 %) had pancreatic, neuroendocrine and urinary bladder cancer, and 4 (11.8 %) had unidentified primary sites. The most common site of metastasis was the liver, observed in 22 patients (64.7 %), followed by the lung in 11 (32.4 %) and lymph nodes in 8 (23.5 %).
Thirty-three evaluable patients were entered into the 11 dose levels of DPH107; 4 patients were enrolled at level 9 because one was not evaluable for toxicity due to rapid clinical deterioration prior to administration of DHP107. DLT did not occur, even at the highest DHP107 dosage (level 11, 600 mg/m2/day). However, the dose of DHP107 was not further increased because pharmacokinetic analysis showed that the plasma concentration of DHP107 plateau among patients receiving more than 250 mg/m2 and the increasing interindividual variability in pharmacokinetics.
Toxicity
Dose-limiting toxicity
Thirty-three patients were evaluable for toxicity after the first cycle of DHP107; their toxicities are listed in Table 2. No NCI-CTCAE grade 4 toxicity was observed. Grade 3 toxicities included diarrhea in 4 patients (12.1 %), neutropenia in 2 (6.1 %), and fatigue in 1 (3.0 %). Frequently observed all grade toxicities included diarrhea in 18 patients (54.5 %) and abdominal pain in 12 (36.4 %). Thirty-one patients were evaluable for toxicity after the second cycle of IV paclitaxel. There was no grade 3 toxicity, but grade 4 neutropenia was observed in one patient.
Comparison of toxicity profiles between DHP107 and paclitaxel
Toxicities of all grades observed more frequently after DHP107 included diarrhea (18/33, 54.5 % vs 2/31, 6.5 %) and abdominal pain (12/33, 36.4 % vs 8/31, 25.8 %), whereas toxicities observed more frequently after paclitaxel included myalgia (22/31, 71.0 % vs 5/33, 15.2 %) and alopecia (19/31, 61.3 % vs 3/33, 9.1 %).
Pharmacokinetics
Serial plasma concentration data for non-compartmental pharmacokinetic analysis were obtained from 33 patients for DHP107 and from 30 for IV paclitaxel. Following oral administration of DHP107, paclitaxel was rapidly absorbed, with a median Tmax 3.7 h (1.5–6.1), and a bi-exponential decay in plasma concentration (Fig. 1a and b). Plasma paclitaxel AUClast and Cmax increased proportionally to DHP107 dose ranging from 60 to 250 mg/m2. However, at doses ≥300 mg/m2, AUClast and Cmax increased less than proportionally, with inter-individual variability larger than at doses of 60–250 mg/m2 (Table 3). The coefficient of variance (CV) of AUClast in groups treated with 60–250 mg/m2 DHP107 ranged from 11.8 % to 34.0 %, comparable to the 24.4 % observed for 175 mg/m2 IV paclitaxel (Table 3). The mean terminal elimination half-lives of DHP107 were 12.2–24.2 h and showed no trend by dose groups (Table 3).

Mean plasma paclitaxel and metabolites (p’-3-hydroxypaclitaxel, 6α-hydroxypaclitaxel) concentrations over time (metabolites were measured only in 600 mg/m2 group)
Free paclitaxel concentration in plasma was measured with the same batch of ultrafiltration kit in 10 patients (3 on DHP107 and 3 on IV paclitaxel). The unbound fraction of paclitaxel (%) was calculated from free and total paclitaxel concentration. Overall, the unbound fraction was 1.7-fold higher for DHP107 than for IV paclitaxel.
The two main metabolites of paclitaxel, p’-3-hydroxypaclitaxel and 6α-hydroxypaclitaxel, were measured in 3 patients after 600 mg/m2 DHP107 and again after 175 mg/m2 IV paclitaxel. The concentrations of both metabolites were lower than the concentrations of paclitaxel in both regimens. While p’-3-hydroxypaclitaxel concentration was higher than that of 6α-hydroxypaclitaxel after DHP107, the reverse was true after IV paclitaxel (Fig. 1c and d).
Urine for PK analysis was obtained after administration of 200–600 mg/m2 DHP107 (n = 26) and IV paclitaxel (n = 25). There was no definite linear relationship between doses based on body surface area and the amount of paclitaxel excreted as unchanged into urine until 48 h (Ae0-48h) and the fraction excreted into urine unchanged (Fe) tended to decrease with dose increment based on body surface area. Ae0-48h (2.15 mg vs 4.37–9.62 mg) and Fe (1.2 vs 1.2–7.3) were lower after IV paclitaxel 175 mg/m2 than after 200–600 mg/m2 DHP107.
Discussion
DHP107 is an efficient lipid-based oral paclitaxel that is systemically absorbed in the absence of P-gp inhibitors or Cremophor EL. In this first-in-human phase I study, DHP107 showed tolerable clinical features and pharmacokinetic profiles, which were dose linear in the range of 60–250 mg/m2 with inter-individual variations comparable to those of IV paclitaxel.
Despite the widespread use of paclitaxel, the present IV formulation, which includes Cremophor EL, is often problematic, due to hypersensitivity to Cremophor EL and the wide inter-individual variability of paclitaxel. In addition, more frequent dosing, including weekly or metronomic administration, has been reported advantageous compared with once every 3 weeks. These features have led to efforts to develop an oral form of paclitaxel.
Early animal studies showed that the poor bioavailability of paclitaxel was related to P-gp and CYP3A4. Strategies to inhibit P-gp and CYP3A4 and increase the systemic absorption of oral paclitaxel include the addition of cyclosporine, ritonavir or GF120918 to paclitaxel [10, 11, 14–16]. Although use of these agents resulted in an 8- to 10-fold increase in systemic exposure to paclitaxel, issues related to immunosuppression and drug interaction arose [11, 15–17]. DHP 107 may therefore have advantages over other formulations of oral paclitaxel that require P-gp and/or CYP3A4 inhibitors.
The absence of Cremophor EL is another advantage of DHP107. Although Cremophor EL is absorbed from the gastrointestinal tract, it limits paclitaxel absorption from the intestine by trapping it in micelles [18]. This has often resulted in non-linear pharmacokinetics of paclitaxel. We found that, at doses of 60–250 mg/m2, DHP107 showed linear pharmacokinetic properties. At higher DHP107 doses, however, AUC and Cmax did not show dose linearity and their inter-individual variations became larger. This was likely due to limited absorption of DHP107 at high dose levels rather than to its increased clearance by dose. Despite these escalating doses of DHP107, the clearance and terminal elimination half-lives of paclitaxel were similar without definite trends among dose levels. The tendency of Fe to decrease as DHP107 dose increased was also likely due to limited absorption, rather than to decreased renal excretion of paclitaxel at higher doses. These results suggest that DHP107 exposure should be increased by interval changes rather than dose escalation in doses exceeding 250 mg/m2, which is similar to the case of other oral anti-cancer agents [19].
We found that the concentration of p’-3-hydroxypaclitaxel was higher than that of 6α-hydroxypaclitaxel after DHP107 administration, whereas the opposite was observed after IV paclitaxel. This may be related to the fact that expression of CYP3A4 is higher than that of CYP2C8 in the gastrointestinal tract. However, since these two metabolites are inactive, this difference in metabolite profile was not considered clinically significant [20, 21].
Our study is characterized by its design, which performed intra-subject comparison of pharmacokinetic parameters of oral and IV formulations. The CV of AUClast in patients administered 60–250 mg/m2 DHP107 ranged from 11.8 % to 34.0 %, which is comparable to the 24.4 % of 175 mg/m2 IV paclitaxel. The variability of oral exposure may be smaller than previous oral formulations of paclitaxel [22]. The limited variability of DHP107 may be related to it being a lipid-based, semisolid formulation of oral paclitaxel without P-gp or CYP3A4 inhibitors and without Cremophor EL or ethanol [12]. Compared to previous oral paclitaxel formulations, DHP107 seemed to reveal less interindividual pharmacokinetic variability. The reason for the increased variability at higher dose level is not clear, but we assume that the mucoadhesive lipid contribute to most of the lower dose absorption while higher doses may involve other pathways including P-gp. In order to clarify the role of P-gp in transport of paclitaxel from DHP107, clinical studies of drug-drug interaction of DHP107 and P-gp inhibitors such as ketoconazole or cyclosporine will be required to clarify the potential of drug-drug interaction.
DHP107 was safe and well-tolerated, with no incidence of grade 4 toxicity. Significant grade 3 toxicities included diarrhea in 4 patients (12.1 %), neutropenia in 2 (6.1 %), and fatigue in 1 (3.0 %). When compared with IV paclitaxel, DHP107 showed less frequent alopecia and myalgia. Diarrhea of all grades was the most frequently observed toxicity of DHP107 (18/33, 54.5 %). This high incidence of diarrhea may be due to the specific formulation of DHP107 or single high-dose administration; the frequency and intensity of diarrhea and abdominal pain seemed to increase at higher doses (≥ 400 mg/m2). Characteristically, diarrhea and abdominal pain developed 2–4 h after administration of DHP107 and subsided approximately one day later.
Despite the low exposure of paclitaxel with the DHP107 formulation, various schedules including weekly or low-dose metronomic administration may be further investigated. At present, another phase I and pharmacokinetic study of weekly DHP107 is ongoing.
Our study had several limitations. The contributions of P-gp and CYP3A4 to DHP107 absorption were not investigated. Studies of drug-drug interactions are required to fully characterize the mechanism of absorption and the effects of various concomitant medications. Although this study was designed in a crossover fashion, which is optimal for comparison of oral and IV pharmacokinetic parameters, the IV dose could not parallel oral dose because of issues related to benefits for patients. However, as more parenteral chemotherapeutic agents become developed into oral formulations, we believe that the approach described here may be utilized for their evaluation.
In conclusion, DHP107, a novel oral paclitaxel either without Cremophor EL or concomitant P-gp inhibitor, was safe and feasible for patients with advanced malignancies in this study. Favorable pharmacokinetic profiles including tolerability and limited variability warrants further investigation of DHP107 in larger populations.
References
Vergote I, Trope CG, Amant F et al (2010) Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med 363:943–53
Kang HJ, Chang HM, Kim TW et al (2008) A phase II study of paclitaxel and capecitabine as a first-line combination chemotherapy for advanced gastric cancer. Br J Cancer 98:316–22
Di Leo A, Gomez HL, Aziz Z et al (2008) Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol 26:5544–52
Sandler A, Gray R, Perry MC et al (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355:2542–50
Weiss RB, Donehower RC, Wiernik PH et al (1990) Hypersensitivity reactions from taxol. J Clin Oncol 8:1263–8
Liebmann J, Cook JA, Mitchell JB (1993) Cremophor EL, solvent for paclitaxel, and toxicity. Lancet 342:1428
Kerbel RS, Kamen BA (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4:423–36
Zhang M, Tao W, Pan S et al (2009) Low-dose metronomic chemotherapy of paclitaxel synergizes with cetuximab to suppress human colon cancer xenografts. Anticancer Drugs 20:355–63
Seidman AD, Berry D, Cirrincione C et al (2008) Randomized phase III trial of weekly compared with every-3-weeks paclitaxel for metastatic breast cancer, with trastuzumab for all HER-2 overexpressors and random assignment to trastuzumab or not in HER-2 nonoverexpressors: final results of Cancer and Leukemia Group B protocol 9840. J Clin Oncol 26:1642–9
Malingre MM, Beijnen JH, Rosing H et al (2001) Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients. Br J Cancer 84:42–7
Malingre MM, Beijnen JH, Rosing H et al (2001) A phase I and pharmacokinetic study of bi-daily dosing of oral paclitaxel in combination with cyclosporin A. Canc Chemother Pharmacol 47:347–54
Hong JW, Lee IH, Kwak YH et al (2007) Efficacy and tissue distribution of DHP107, an oral paclitaxel formulation. Mol Cancer Ther 6:3239–47
Shin BS, Kim HJ, Hong SH et al (2009) Enhanced absorption and tissue distribution of paclitaxel following oral administration of DHP 107, a novel mucoadhesive lipid dosage form. Canc Chemother Pharmacol 64:87–94
Britten CD, Baker SD, Denis LJ et al (2000) Oral paclitaxel and concurrent cyclosporin A: targeting clinically relevant systemic exposure to paclitaxel. Clin Cancer Res 6:3459–68
Veltkamp SA, Alderden-Los C, Sharma A et al (2007) A pharmacokinetic and safety study of a novel polymeric paclitaxel formulation for oral application. Canc Chemother Pharmacol 59:43–50
Veltkamp SA, Rosing H, Huitema AD et al (2007) Novel paclitaxel formulations for oral application: a phase I pharmacokinetic study in patients with solid tumours. Cancer Chemother Pharmacol 60:635–42
Malingre MM, Terwogt JM, Beijnen JH et al (2000) Phase I and pharmacokinetic study of oral paclitaxel. J Clin Oncol 18:2468–75
Bardelmeijer HA, Ouwehand M, Malingre MM et al (2002) Entrapment by Cremophor EL decreases the absorption of paclitaxel from the gut. Canc Chemother Pharmacol 49:119–25
Kantarjian H, Giles F, Wunderle L et al (2006) Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 354:2542–51
Rahman A, Korzekwa KR, Grogan J et al (1994) Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2 C8. Cancer Res 54:5543–6
Sparreboom A, Huizing MT, Boesen JJ et al (1995) Isolation, purification, and biological activity of mono- and dihydroxylated paclitaxel metabolites from human feces. Canc Chemother Pharmacol 36:299–304
Chu Z, Chen JS, Liau CT et al (2008) Oral bioavailability of a novel paclitaxel formulation (Genetaxyl) administered with cyclosporin A in cancer patients. Anticancer Drugs 19:275–81
Acknowledgment
This study was supported by a grant from DAE HWA Pharmaceutical Co. and Gangwon Leading Industry Office by Korean government
Conflict of Interest
Hyeyoun Kim works for DAE HWA Pharmaceutical Co., Ltd. as a commissioner, and has a leadership position to disclose.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yong Sang Hong and Kyu-pyo Kim are equally contributed to this work.
Rights and permissions
About this article
Cite this article
Hong, Y.S., Kim, Kp., Lim, HS. et al. A phase I study of DHP107, a mucoadhesive lipid form of oral paclitaxel, in patients with advanced solid tumors: Crossover comparisons with intravenous paclitaxel. Invest New Drugs 31, 616–622 (2013). https://doi.org/10.1007/s10637-012-9841-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-012-9841-7