
Abstract
Purpose
Oraxol is an oral formulation of paclitaxel administered with a novel, minimally absorbed P-glycoprotein inhibitor encequidar (HM30181A). This phase Ib study was conducted to determine the maximum-tolerated dose (MTD) of Oraxol administered at a fixed dose for up to 5 consecutive days in patients with advanced malignancies.
Methods
Part 1 of this study utilized a 3 + 3 dose-escalation design to determine the MTD of oral paclitaxel 270 mg plus oral encequidar 15 mg administered daily. Dose escalation was achieved by increasing the number of consecutive dosing days per week (from 2 to 5 days per week). Dosing occurred for 3 consecutive weeks out of a 4-week cycle. Part 2 treated additional patients at the MTD to determine tolerability and recommended phase II dose (RP2D). Adverse events, tumor responses, and pharmacokinetic profiles were assessed.
Results
A total of 34 patients (n = 24 in Part 1, n = 10 in Part 2) received treatment. The MTD of Oraxol was determined to be 270 mg daily × 5 days per week per protocol definition and this was declared the RP2D. The most common treatment-related adverse events were fatigue, neutropenia, and nausea/vomiting. Hypersensitivity-type reactions were not observed. Of the 28 patients evaluable for response, 2 (7.1%) achieved partial response and 18 (64.3%) achieved stable disease. Pharmacokinetic analysis showed rapid absorption of paclitaxel when administered orally following encequidar. Paclitaxel daily exposure was comparable following 2–5 days dose levels.
Conclusion
The oral administration of encequidar with paclitaxel was safe, achieved clinically relevant paclitaxel levels, and showed evidence of anti-tumor activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Paclitaxel is a widely used anti-cancer agent and a cornerstone in the treatment for a variety of cancers, such as breast, ovarian, and lung. Currently, intravenous (IV) infusion is the only route of administration for paclitaxel in the clinic. The taxane has extremely low aqueous solubility and the initial formulation of paclitaxel contains the solvent Cremophor EL that is associated with hypersensitivity reactions and worsening neurotoxicity [1, 2]. In addition, IV administration of the Cremophor formulation of paclitaxel (Cre-paclitaxel) requires pre-medication and slower infusions, which can result in great inconvenience to the patients. Oral administration of paclitaxel is an attractive alternative delivery method that can overcome the difficulties with IV formulations. However, the previous efforts to develop oral formulations of paclitaxel have been limited due to its poor oral bioavailability. This is due to the activity of P-glycoproteins (P-gp) in the intestinal epithelial cells, which actively excrete and minimize the intestinal absorption of paclitaxel [3, 4]. Nonetheless, the development of oral paclitaxel remains an area of great interest and several strategies are under investigation to improve absorption of oral paclitaxel.
Oraxol (Athenex, USA) is an oral formulation of paclitaxel administered sequentially after encequidar (HM30181), a potent, selective, poorly absorbed P-gp inhibitor [5, 6]. Pre-dosing using the oral P-gp inhibitor enhances the gastrointestinal absorption of subsequently administered paclitaxel, thereby allowing the achievement of clinically relevant paclitaxel plasma levels. A preclinical study in rats showed that encequidar increased the oral bioavailability of paclitaxel from 3.4 to 41.3%, and the co-administration of oral paclitaxel and encequidar in a xenograft mouse model showed equivalent anti-tumor activity compared to IV paclitaxel [6]. HM-OXL-101 was a phase I trial that attempted to define the maximum-tolerated dose (MTD) for Oraxol in patients with advanced solid cancers [7]. Oral paclitaxel (liquid formulation) and encequidar were given on days 1, 8, and 15 of each 28-day cycle, with paclitaxel doses ranging from 60 to 420 mg/m2 and encequidar dosed at half of the paclitaxel doses (30–210 mg/m2). The MTD was not reached in this study and dose escalation was stopped because of non-linear pharmacokinetic (PK) at paclitaxel doses above 300 mg/m2. Another study, HM-OXL-201, assessed oral (hard capsule supplied as liquid-filled hard gelatin capsules containing 30 mg of paclitaxel active substance and the excipient polysorbate 80) paclitaxel doses of 90, 120, or 150 mg/m2 per day given on days 1, 2, 8, 9, 15, and 16 of each 28-day cycle in gastric cancer patients [8]. Encequidar was administered 15 mg on day 1. HM-OXL-201 determined oral paclitaxel 150 mg/m2 as the recommended dose and observed partial response in 9.3% of subjects.
We conducted a population pharmacokinetic/pharmacodynamic (PK/PD) analysis to assess the relationship between paclitaxel exposure and absolute neutrophil count (ANC) nadir using data from the above two trials and published literature on IV paclitaxel. The results of the modeling showed that the percentage of patients who experienced grade 3 or 4 neutropenia is similar between Oraxol 150 mg/m2 given 5× per week and weekly IV paclitaxel 80 mg/m2, thus providing the rationale for the dose conversion of IV to oral dosing. Furthermore, the analysis did not identify significant patient factors affecting the interpatient variability in paclitaxel PK, suggesting that fixed-dosing should be comparable to BSA-based dosing (150 mg/m2 daily or 270 mg daily, using a BSA of 1.8 m2).
Based on these data, we designed and conducted a phase Ib dose-regimen finding study to determine the MTD, PK, and safety of a flat-dose approach to administering oral paclitaxel and encequidar in patients with advanced malignancies. Dose escalation was achieved by increasing the number of consecutive dosing days per week. For PK, even though all metabolites are non-active, their quantifications would help determine whether additional drug interaction studies with potential concomitant inhibitors/inducers, or organ-impairment studies, are justified.
Materials and methods
Study design
This was a multicenter, nonrandomized, open-label, single arm, phase Ib study of Oraxol in patients with advanced malignancies. The primary objective was to determine the MTD based on occurrence of dose-limiting toxicities (DLTs). Secondary objectives were to evaluate the recommended phase II dose (RP2D), safety, tolerability, pharmacokinetics, and activity of Oraxol. The study protocol was approved by the institutional review boards at each institution. Written informed consent was obtained from all patients (ClinicalTrials.gov identifier: NCT01967043).
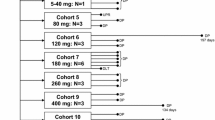
The study consisted 2 parts (Part 1 and 2). Part 1 was the dose-escalation phase to determine the MTD of Oraxol and consisted of 2 sequential arms: Arm 1 and 2 (Fig. 1). Dose escalation was achieved using the 3 + 3 dose-escalation design and the daily dosing of oral paclitaxel was fixed at 270 mg (approximately 150 mg/m2 assuming a body surface area of 1.8 m2). Dose escalation in successive cohorts was achieved by increasing the number of dosing days per week (2, 3, 4, and 5 days) for 3 consecutive weeks out of a 28-day cycle. Upon completion of Part 1, Part 2 proceeded to enroll additional ten patients to receive treatment at the MTD from Part 1 to determine the safety, tolerability, and RP2D.

Dose escalation plan in Part 1. PTX, paclitaxel
Arm 1 and 2 of Part 1 differed in the frequency at which the encequidar tablet was dosed. Arm 1 evaluated 15 mg encequidar methanesulfonate monohydrate (equivalent to 12.9 mg free base) given orally once per dosing week, whereas Arm 2 evaluated 15 mg encequidar given orally daily concurrently with paclitaxel under longer fasting conditions. After completion of the Arm 1 ‘3-Day’ cohort, Arm 1 was discontinued and Arm 2 was selected for patient enrollment. No DLTs were observed in Arm 1.
The MTD was defined as the highest total dose (most number of dosing days) for which no more than 1 of 6 treated patients experienced a DLT. If the MTD was not reached at the highest planned dose level (270 mg × 5 days per week), then that dose level was to be declared the MTD. Patients continued Oraxol treatment at their assigned dose for additional cycles until disease progression or discontinuation for other reasons.
Both encequidar and oral paclitaxel formulations were the same as those used in study HM-OXL-201 [8]. Patients were required to fast for at least 1 h before and 1 h after medication administration. Patients were able to receive anti-emetics if needed.
Eligibility
The study enrolled patients who were age 18 years or older with histologically or cytologically confirmed solid tumor that was metastatic or unresectable and for which standard curative or palliative measures did not exist or were no longer effective. Other key eligibility requirements included: measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1; Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1; adequate bone marrow (absolute neutrophil count ≥ 1.5 × 109/L, platelet count ≥ 100 × 109/L, hemoglobin ≥ 9 g/L), liver (total bilirubin ≤ 1.5 mg/dL, alanine aminotransferase ≤ 3× upper limit of normal, alkaline phosphatase ≤ 3 × upper limit of normal), and renal function (serum creatinine ≤ 2 mg/dL); life expectancy ≥ 3 months; no concurrent use of medications known to be P-gp substrates, clinically significant inhibitors or inducers of P-gp or CYP2C8, or strong inhibitors or inducers of CYP3A4.
Key exclusion criteria included: uncontrolled intercurrent illness; significant cardiovascular disease or bleeding disorder; history of GI disease or major surgery to the upper GI tract that can impair oral drug absorption; pregnant or breast feeding; have not recovered to ≤ grade 1 toxicity from previous anti-cancer treatments; known history of allergy to paclitaxel. Of note, patients whose allergy was due to the IV solvent and not paclitaxel were eligible for this study. Patients who previously experienced severe toxicity to taxanes were eligible as long as they have recovered to ≤ grade 1 toxicity.
Safety and efficacy assessments
Safety was assessed throughout the study and up to 4 weeks after the last dose of study treatment. Adverse events (AEs) were graded per the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.03. Other safety assessments included regular monitoring of hematology, blood chemistry, and urinalysis; regular measurement of vital signs and electrocardiograms (ECGs); and the performance of physical examinations.
Tumor responses were assessed using imaging (computed tomography or magnetic resonance imaging) of the chest, abdomen, and pelvis based on RECIST v1.1 criteria [9]. Tumor assessment was conducted during the Screening Period (within 28 days of the first dose), Cycle 3 Day 1, and every 2 cycles thereafter.
Pharmacokinetic analyses
Blood samples to determine the plasma concentrations of paclitaxel and encequidar (as well as their metabolites) were collected predose and at 0.25, 0.5, 1, 2, 4, 6, and 8 h after the paclitaxel dose on days 1 and last day of dosing during Cycle 1 days 1–5. Samples were also collected predose and between 1 and 3 h post-dosing on Cycle 1 days 8 and 15. Blood samples were drawn into a collection tube with potassium (K2) EDTA and maintained on ice until centrifugation. Samples were centrifuged at ~ 2000 rpm at 4 °C for 10 min within 30 min of collection. Plasma was immediately harvested and stored at – 70 °C. Separate validated liquid chromatography–tandem mass spectroscopy assays were used to determine plasma concentrations of paclitaxel and its major metabolites 6α-hydroxy paclitaxel and 3’p-hydroxy paclitaxel, as well as HM30181 and its metabolite HM30181-M1 (Xenobiotic Laboratories, Plainsboro, NJ). The concentrations reported demonstrated acceptable performance during the assay. The lower limit of quantification (LLOQ) values are as follows: 1.0 ng/mL for paclitaxel, 0.25 ng/mL for 6α-hydroxy paclitaxel and 3’p-hydroxy paclitaxel, 0.5 ng/mL for HM30181, and 0.1 ng/mL for HM30181-M1. Plasma concentrations of both paclitaxel and encequidar PK parameters such as area under the plasma concentration–time curve (AUC), maximum (peak) plasma concentration (Cmax), and peak time (tmax) were estimated through non-compartmental analysis using Phoenix WinNonlin software (Pharsight, Princeton, NJ).
Statistical analysis
Statistical analyses for this phase Ib study were descriptive and exploratory. Baseline characteristics, PK, and safety analysis included all patients who received ≥ 1 dose of Oraxol, and efficacy analysis included patients who completed baseline and at least one post-baseline tumor assessment. Best overall response was defined as the biggest tumor size reduction or the smallest tumor size increase (if tumor size reduction did not happen) based on the sum of baseline target lesion diameters from the start of treatment until disease progression or withdrawal from the study for any other reason. Descriptive statistics were used to summarize patient characteristics, safety, and tumor response. Continuous variables were summarized using the mean, standard deviation (SD), median, minimum, and maximum. Categorical variables were summarized by counts and by percentage of patients in corresponding categories. Statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics and disposition
The study was conducted between 2013 and 2016 at three study sites in the US. A total of 34 patients were treated in the study; 24 patients in Part 1 and 10 patients in Part 2. Baseline patient characteristics are summarized in Table 1. Median age was 63.5 years (range: 32–79 years) and the majority of the patients were male (58.8%) and White (88.2%). More than half of the patients had received > 2 lines of prior chemotherapy (58.8%), including prior exposure to taxanes (52.9%). A majority of the patients completed Cycle 1 (n = 26; 76%). The most common reason for discontinuation from the study was disease progression (n = 26; 76%).
Safety and tolerability
Oraxol had an acceptable safety profile at doses up to encequidar 15 mg daily + paclitaxel 270 mg daily for 5 days, which was the highest weekly dose tested in this study. At this dose level, only one out of six patients experienced a DLT. Therefore, per protocol definition, the highest dose tested was determined to be the MTD. The single DLT (febrile neutropenia) occurred in the Part 1 Arm 2 (5 days) group during Cycle 1.
In Part 1 Arm 1, safety was evaluated in six patients who received encequidar 15 mg weekly + paclitaxel 270 mg daily for 2 or 3 consecutive days. No grade ≥ 3 treatment-related adverse events (TRAEs) were observed in this cohort of patients. Grade 2 TRAEs included anorexia, nausea/vomiting, fatigue, dysgeusia, and hyperbilirubinemia.
In Part 1 Arm 2, safety was evaluated in 18 patients who received encequidar 15 mg daily + paclitaxel 270 mg daily for 2, 3, 4, or 5 consecutive days. In Part 2, safety was evaluated in additional ten patients who received the 5-day dosing. Most of the TRAEs were grade 2 or 3 in severity and most were observed with the 5-day dosing. At the highest dosing level, the most frequently reported TRAEs were neutropenia (41.2%), nausea (41.2%), fatigue (35.3%), and anorexia (29.4%). Nine patients (32.1%) experienced grade ≥ 3 TRAEs, 1 in the 4-day group (3.5%) and 8 in the 5-day group (28.6%). Grade ≥ 3 TRAEs included neutropenia, anemia, febrile neutropenia, dehydration, hypophosphatemia, nausea/vomiting, and pneumonia (Table 2). The study observed no grade 5 TRAEs, no episodes of treatment-related hypersensitivity-type reactions, and no treatment-related neuropathy.
Pharmacokinetics
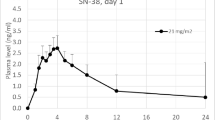
The PK analysis included all of the 24 patients from Part 1 and 10 patients from Part 2. Paclitaxel PK parameters at all assessment days are summarized in Table 3. Following Oraxol administration, paclitaxel showed rapid absorption, followed by a biexponential decay in plasma concentration (Fig. 2a). Across all dosing regimens, median time of maximum plasma concentration (Tmax) ranged from 1 to 2 h and mean Cmax on day 1 ranged from 147 to 246 ng/mL. After 5 consecutive days of dosing, mean Cmax was approximately 140 ng/mL and ranged from 25 to 263 ng/mL, suggesting minimal paclitaxel accumulation. Across all cohorts, mean (± SD) paclitaxel plasma exposures (AUC0–8 h) on day 1 ranged from 367 (± 293) to 792 (± 654) ng*hr/mL. Minimal changes in paclitaxel exposure were observed in patients when the frequency of encequidar dosing was altered and prandial status was controlled, suggesting no additional increase in plasma exposure and no accumulation following 2–5 consecutive days of treatment.

Mean (± SD) plasma concentration–time profiles of paclitaxel (a) and encequidar (b) following Oraxol administrations, stratified by cohort and day. a1 Paclitaxel (day-8 sample is the last timepoint of days 2–5 last-day dosing). a2 X-axis truncated at 10 h for visualization of absorption profiles, including tmax. C1-A1 Part 1 Arm 1 × 2 days; C1-A2 Part 1 Arm 2 × 2 days, C2-A1 Part 1 Arm 1 × 3 days, C2-A2 Part 1 Arm 2 × 3 days, C3-A2 Part 1 Arm 2 × 4 days, C4-A2 Part 1 Arm 2 × 5 days
Plasma exposures of paclitaxel metabolites, 3’p-Asdhydroxy paclitaxel and 6α-hydroxy paclitaxel, were relatively low following Oraxol administration. The mean (± SD) metabolic ratios of 3’p-hydroxy paclitaxel and 6α-hydroxy paclitaxel across all cohorts and days ranged from 0.140 (± 0.064) to 0.471 (± 0.250) and 0.0382 (± 0.0060) to 0.209 (± 0.364), respectively.
The systemic exposure of encequidar was low with the 15-mg oral dose (Fig. 2b). Mean (± SD) Cmax of metabolites HM30181 and HM30181-M1 ranged from 0.738 (± 0.248) to 2.08 (± 0.660) ng/mL and 0.130 (± 0.0276) to 0.480 (± 0.00) ng/mL, respectively, across all cohorts and days of Oraxol dosing. A few plasma concentrations were measurable, and thus, no other PK parameters could be estimated based on PK rules.
Anti-tumor activity
Of the 28 patients evaluable for efficacy, 2 patients (7.1%) achieved a partial response and 18 patients (64.3%) achieved stable disease as their best response per RECIST v1.1. A summary of the responses is shown in Fig. 3. Of the 20 patients who achieved a partial response or stable disease, 11 patients (55%), including one of the patients who achieved a partial response, had prior exposures to taxanes. Median duration of response for patients with stable disease was 3.02 months. Both of the patients with partial response were in 5 day dose cohorts. One of the patients had salivary gland carcinoma and had received no prior systemic therapies. The duration of response for this patient was 6.1 months. The other patient who achieved partial response had ovarian cancer and was heavily pretreated with > 5 lines of anti-neoplastic agents, including IV paclitaxel. The duration of response for this patient was 5.58 months.

Waterfall plot of best percentage change from baseline in tumor size by dose level. aPatient with partial response, ovarian cancer. bPatient with partial response, salivary gland carcinoma
Fourteen patients with prior exposure to taxanes were evaluable for efficacy. Of these, ten patients (71.4%) achieved stable disease, three patients (21.4%) had progressive disease, and one (7.2%) achieved partial response, as described above.
Discussion
Oral administration of paclitaxel is an attractive therapeutic option for patients given the convenience and potentials for a more favorable toxicity profile compared to the IV formulations. A few approaches have been tested clinically, including inhibiting P-gp efflux pump using cyclosporin (CsA) [10,11,12,13,14,15] and using lipid-based formulation (e.g., DHP107) [16,17,18,19]. CsA is a calcineurin inhibitor used as an immunosuppressant in solid organ transplant and had been used at pharmacologically active doses to enhance the oral bioavailability of paclitaxel. However, CsA is also a strong inhibitor of CYP3A, BCRP, MRP2, and OATP enzymes that may lead to potentially problematic drug-drug interactions with concomitant medications [20]. The systemic absorption of CsA may also lead to serious toxicities, e.g., nephro- and neuro-toxicities, when used over multiple cycles [21]. In contrast, encequidar is a highly selective and potent P-gp inhibitor with poor oral bioavailability that, when used over multiple cycles, avoids the potential toxicities and drug–drug interactions associated with systemic absorption; as such, the sequential administration with paclitaxel (Oraxol) provides an attractive approach to clinical development.
Here, we successfully evaluated the approach of inhibiting the P-gp efflux pump using oral encequidar to enhance the gastrointestinal absorption of oral paclitaxel. Paclitaxel dose escalation was achieved by increasing the number of consecutive dosing days per dosing week. The daily dosing of encequidar in tandem with paclitaxel dosing was used for the majority of the study.
Oraxol demonstrated an acceptable safety profile up to the highest planned dose, which was encequidar 15 mg daily + paclitaxel 270 mg daily for 5 days per week for 3 out of 4 weeks. Based on the protocol definition, the highest tested dose was declared to be the MTD. No treatment-related hypersensitivity-type reactions were observed. Interestingly, treatment-related or worsening of pre-existing peripheral neuropathy was not observed in enrolled patients. GI toxicities such as nausea/vomiting were commonly observed in our study. While the majority of observed GI toxicities were grade 2, they may have significant impact on patients’ quality of life. The use of pre-medication with anti-emetics and as-needed anti-diarrheals are recommended.
The incidence of severe toxicities compares favorably to Cre-paclitaxel, which caused ≥ grade 3 neutropenia in at least 52% of patients, anemia in 16% of patients, hypersensitivity-type reaction in 2% of patients, and grade ≥ grade 3 peripheral neuropathy in 3–18% of patients [22]. Of note, in patients with metastatic disease, peripheral neuropathy is often the dose and duration limiting toxicity. The lack of peripheral neuropathy in our study is exciting and consistent with the existing data that neuropathy is uncommon with Oraxol [8, 23]. The incidence of neutropenia across all dose levels is also comparable to albumin-bound paclitaxel [24].
The PK aspect was the secondary objective in the current study, as the full characterization of oral paclitaxel (with encequidar) PK in comparison with IV paclitaxel was performed in a separate study [25]. Hence, to minimize patient visits, the PK profile was limited to 8 h post-dose (the 8 h AUC is 75% of 24 h AUC) [25]. The PK analyses revealed that paclitaxel was rapidly absorbed following Oraxol administration. Oraxol formulation achieved mean Cmax of 147–246 ng/mL and mean AUC0-8 h of 367–792 ng*hr/mL on day 1 of oral paclitaxel dosing at 270 mg. In this study, Oraxol delivered lower paclitaxel exposure than published oral CsA/paclitaxel PK studies [26,27,28]. For example, Britten et al. reported mean paclitaxel AUC 3238 ng*hr/mL following a single dose of 180 mg paclitaxel orally [26]. The higher paclitaxel exposure using CsA is likely secondary to effects on other metabolic enzymes besides P-gp such as CYP3A4.
Paclitaxel exposure was comparable across consecutive days of dosing, suggesting no accumulation following 2–5 days of administration. For the P-gp inhibitor, the plasma concentrations of HM30181 and HM30181-M1 were minimal when encequidar was administered orally at 15 mg daily, suggesting poor gastrointestinal absorption. For PK metabolites, their ratios to parent paclitaxel appeared to be significant to suggest that additional drug interaction studies with potential concomitant inhibitors/inducers, or organ-impairment studies, are justified.
Anti-tumor efficacy was observed with Oraxol particularly at higher dose levels. Partial response and stable disease were achieved as the best overall response per RECIST v1.1 criteria in 2 (7.1%) and 18 (64.3%) patients, respectively. Both partial responses were observed at the 5-day dose, as were 9 of the 18 patients who had stable disease. Clinical activity was seen in multiple tumor types including gynecologic cancers (ovarian cancer, mixed muellerian tumor of uterus, and peritoneal high-grade papillary serous carcinoma), prostate cancer, head and neck cancers (adenoid cystic carcinoma, salivary gland carcinoma, and esthesioneuroblastoma), and sarcoma. Of note, the majority who previously received taxanes achieved stable disease (71.4%) and partial response was observed in one patient. This suggests that Oraxol may have clinical activity in patients who were previously treated using taxanes.
Overall, this study demonstrates that fixed-dosing administration of Oraxol is feasible, safe, and tolerable, and the observed efficacy supports further development. Additionally, Oraxol has the potential to be an additional treatment option for patients who were previously intolerant of or progressed on taxanes. Moving forward, our next step will be to study this novel oral formulation of paclitaxel in tumor types that are known to be responsive to IV paclitaxel. Oraxol is already being evaluated in metastatic breast cancer and angiosarcoma (NCT02594371, NCT03544567), as well as in combination regimens with angiogenic and immune checkpoint inhibitors (NCT02970539, NCT03588039).
Data availability
The data that support the findings of this study are available on request from the corresponding author upon reasonable request.
References
Singla AK, Garg A, Aggarwal D (2002) Paclitaxel and its formulations. Int J Pharm 235:179–192. https://doi.org/10.1016/S0378-5173(01)00986-3
Gelderblom H, Verweij J, Nooter K, Sparreboom A (2001) Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer 37:1590–1598. https://doi.org/10.1016/S0959-8049(01)00171-X
Sparreboom A, van Asperen J, Mayer U et al (1997) Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A 94:2031–2035. https://doi.org/10.1073/pnas.94.5.2031
Bardelmeijer HA, Beijnen JH, Brouwer KR et al (2000) Increased oral bioavailability of paclitaxel by GF120918 in mice through selective modulation of P-glycoprotein. Clin Cancer Res 6:4416LP-4421LP
Paek IB, Kim SY, Kim MS et al (2007) Characterization of human liver cytochrome P-450 enzymes Involved in the O-demethylation of a new P-glycoprotein Inhibitor HM-30181. J Toxicol Environ Heal Part A 70:1356–1364. https://doi.org/10.1080/15287390701434307
Kwak J-O, Lee SH, Lee GS et al (2010) Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel. Eur J Pharmacol 627:92–98. https://doi.org/10.1016/j.ejphar.2009.11.008
Lee HJ, Heo D-S, Cho J-Y et al (2014) A phase I study of oral paclitaxel with a novel P-Glycoprotein inhibitor, HM30181A, in patients with advanced solid cancer. Cancer Res Treat 46:234–242. https://doi.org/10.4143/crt.2014.46.3.234
Lee K-W, Lee KH, Zang DY et al (2015) Phase I/II study of weekly oraxol for the second-line treatment of patients with metastatic or recurrent gastric cancer. Oncologist 20:896–897. https://doi.org/10.1634/theoncologist.2015-0202
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumors: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Malingré MM, Meerum Terwogt JM, Beijnen JH et al (2000) Phase I and pharmacokinetic study of oral paclitaxel. J Clin Oncol 18:2468–2475. https://doi.org/10.1200/JCO.2000.18.12.2468
Malingré MM, Beijnen JH, Rosing H et al (2001) A phase I and pharmacokinetic study of bi-daily dosing of oral paclitaxel in combination with cyclosporin A. Cancer Chemother Pharmacol 47:347–354. https://doi.org/10.1007/s002800000226
Kruijtzer CMF, Schellens JHM, Mezger J et al (2002) Phase II and pharmacologic study of weekly oral paclitaxel plus cyclosporine in patients with advanced non-small-cell lung cancer. J Clin Oncol 20:4508–4516. https://doi.org/10.1200/JCO.2002.04.058
Kruijtzer CMF, Boot H, Beijnen JH et al (2003) Weekly oral paclitaxel as first-line treatment in patients with advanced gastric cancer. Ann Oncol 14:197–204. https://doi.org/10.1093/ANNONC/MDG078
Veltkamp SA, Thijssen B, Garrigue JS et al (2006) (2006) A novel self-microemulsifying formulation of paclitaxel for oral administration to patients with advanced cancer. Br J Cancer 956(95):729–734. https://doi.org/10.1038/sj.bjc.6603312
Veltkamp SA, Alderden-Los C, Sharma A et al (2007) A pharmacokinetic and safety study of a novel polymeric paclitaxel formulation for oral application. Cancer Chemother Pharmacol 59:43–50. https://doi.org/10.1007/S00280-006-0245-2/TABLES/4
Hong JW, Lee I-H, Kwak YH et al (2007) Efficacy and tissue distribution of DHP107, an oral paclitaxel formulation. Mol Cancer Ther 6:3239–3247. https://doi.org/10.1158/1535-7163.MCT-07-0261
Hong YS, Kim KP, Lim HS et al (2012) (2012) A phase I study of DHP107, a mucoadhesive lipid form of oral paclitaxel, in patients with advanced solid tumors: crossover comparisons with intravenous paclitaxel. Investig New Drugs 313(31):616–622. https://doi.org/10.1007/S10637-012-9841-7
Kang YK, Ryu MH, Park SH et al (2018) Efficacy and safety findings from DREAM: a phase III study of DHP107 (oral paclitaxel) versus i.v. paclitaxel in patients with advanced gastric cancer after failure of first-line chemotherapy. Ann Oncol 29:1220–1226. https://doi.org/10.1093/ANNONC/MDY055
Ryu M, Ryoo B, Kim TW et al (2017) A phase I/IIa study of DHP107, a novel oral paclitaxel formulation, in patients with advanced solid tumors or gastric cancer. Oncologist 22:129-e8. https://doi.org/10.1634/theoncologist.2016-0273
Yang Y, Li P, Zhang Z et al (2020) Prediction of cyclosporin-mediated drug interaction using physiologically based pharmacokinetic model characterizing interplay of drug transporters and enzymes. Int J Mol Sci 21:7023. https://doi.org/10.3390/ijms21197023
Graham RM (1994) Cyclosporine: mechanisms of action and toxicity. Cleve Clin J Med 61:308LP-313LP
Pfizer. Paclitaxel [Package insert]. US food and drug administration website. www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf.
Ravi V, Wagner M, Chen TW-W et al (2020) A phase II study of oraxol in the treatment of unresectable cutaneous angiosarcoma. J Clin Oncol 38:11517. https://doi.org/10.1200/JCO.2020.38.15_suppl.11517
Celgene Corporation. Abraxane [Package insert]. US food and drug administration website. www.accessdata.fda.gov/drugsatfda_docs/label/2020/021660s047lbl.pdf
Jackson CGCA, Hung T, Segelov E et al (2021) Oral paclitaxel with encequidar compared to intravenous paclitaxel in patients with advanced cancer: a randomised crossover pharmacokinetic study. Br J Clin Pharmacol 87:4670–4680. https://doi.org/10.1111/bcp.14886
Britten CD, Baker SD, Denis LJ et al (2000) Oral paclitaxel and concurrent cyclosporin A: targeting clinically relevant systemic exposure to paclitaxel1. Clin Cancer Res 6:3459–3468
Helgason HH, Kruijtzer CMF, Huitema ADR et al (2006) Phase II and pharmacological study of oral paclitaxel (Paxoral) plus ciclosporin in anthracycline-pretreated metastatic breast cancer. Br J Cancer 95:794–800. https://doi.org/10.1038/sj.bjc.6603332
Chu Z, Chen J-S, Liau C-T et al (2008) Oral bioavailability of a novel paclitaxel formulation (Genetaxyl) administered with cyclosporin A in cancer patients. Anticancer Drugs 19:275–281. https://doi.org/10.1097/cad.0b013e3282f3fd2e
Acknowledgements
We would like to thank the patients, their families, and all the investigators who participated in this clinical trial.
Funding
Athenex, Inc provided the funding and drug for the clinical trial.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
NSA is a paid consultant for Mirati and QED; she receives institutional funding from Agios, Inc., Array, Atlas, Bayer HealthCare, BMS, Celgene, Debio, Eli Lilly and Company, EMD Serono, Incyte Corporation, Intensity, Merck & Co., Inc. and Taiho Pharmaceuticals Co., Ltd.; she participates on advisory boards for Incyte, QED, and Glaxo Smith Kline. JRD receives institutional research funding from Adlai Norte, Takeda, Gilead, Merck, AstraZeneca, Astellas, Abbvie, BMS, OnKure, Deciphera, Bayer, Hutchison, and Genentech; she is a consultant for Gilead and OnKure; she owns stock options in OnKure Therapeutics. MO receives research funding from Eli Lilly and Pfizer; he is a consultant for AstraZeneca and Novartis. AJ receives institutional research funding from Pfizer, Merck, SQZ Biotech, Moderna, Iovance, Khar, DebioPharm, Cantargia, and Sanofi; he owns stock options in Champions Oncology and Suvica. JZ, DK, WKC, DC, and RW are employees of Athenex, Inc. DC owns stock in Athenex, Inc. and Merck & Co., Inc. No conflicts of interests were disclosed by the other authors.
Ethical approval
This study was approved by the institutional review boards at each institution and was performed in accordance with the principles of the Declaration of Helsinki.
Consent to participate
Written informed consent to participate in the study was obtained from all participants included in the study.
Consent to publish
Written informed consent for publication of clinical data was obtained from all participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, W.W., Li, J.J., Azad, N.S. et al. A phase Ib study of Oraxol (oral paclitaxel and encequidar) in patients with advanced malignancies. Cancer Chemother Pharmacol 90, 7–17 (2022). https://doi.org/10.1007/s00280-022-04443-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-022-04443-1