
Abstract
The current clinical trial landscape targeting Alzheimer’s disease (AD) is reviewed in the context of studies completed from 2019 to 2021. This review focuses on available data for observational and phase II/III clinical trial results, which will have the most impact on the field. ClinicalTrials.gov, the United States (US) comprehensive federal registry, was queried to identify completed trials. There are currently 226 interventional clinical trials and 51 observational studies completed, suspended, terminated, or withdrawn within our selected time frame. This review reveals that the role of biomarkers is expanding and although many lessons have been learned, many challenges remain when targeting disease modification of AD through amyloid and tau. In addition, to halt or slow clinical progression of AD, new clinical and observational trials are focusing on prevention as well as the role of more diverse biological processes known to influence AD pathology.
Similar content being viewed by others
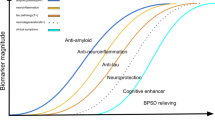
Avoid common mistakes on your manuscript.
Introduction
The goal of this paper is to review recent clinical trials, which were completed in the last 2 years (2019–2021). Special attention will be given to trials with published peer-reviewed results of phase II–III data. Select terminated trials, trials with unpublished results, and observational studies will be mentioned and classified according to intervention. We will also discuss ongoing studies with the potential to impact the field considerably.
There has been a rapid expansion of Alzheimer’s disease (AD) interventional therapeutic trials. Based on clinical trial activity as recorded in ClinicalTrials.gov, a comprehensive US government database, there were 226 interventional clinical trials completed, suspended, terminated, or withdrawn between 01/01/2019 and 05/01/2021 (Supplemental Table 1). Additionally, 51 observational studies were completed, suspended, terminated, or withdrawn within this same period (Supplemental Table 2). At the time of drafting this article, there were 783 active, recruiting, or enrolling studies related to AD.
The time course of the neuropathological changes (amyloid, tau, and neurodegeneration) in AD relative to their clinical outcome measures complicates clinical trials in disease-modifying therapies (DMTs). The build-up of amyloid typically occurs 5–20 years prior to the onset of symptoms and the tools used to measure clinical outcomes (cognitive testing) are not sensitive or specific enough to detect relevant early changes within the time frame of current clinical trials. In 2018, a biomarker-based biological definition of AD, the ATN framework [1], was introduced for research purposes to facilitate appropriate antemortem enrollment in AD clinical trials.
Defining AD based on the presence of amyloid and tau biomarkers is controversial but allows for intervention at preclinical stages of AD (i.e., before symptom onset) and in the earliest symptomatic stages. Prediction of symptom progression within clinical trials at these earlier stages remains an area of ongoing research [2, 3]. Given the controversy, the clinical diagnosis of AD is still governed by the criteria set by the National Institute on Aging in 2011 [4, 5]. In our view, the biomarker-based biological definition of AD is not at odds with the clinical–biological diagnosis of AD [3]. Ideally, biomarker classification will aid in our understanding of disease progression, creation of risk profiles for development of symptomatic AD, and proper clinical trial enrollment.
Alzheimer’s disease biomarkers
AD biomarkers have the potential to impact diagnosis, treatment [6], prognosis [7, 8], and clinical trial enrollment [3, 9]. In the clinical setting, academic medical centers are using structural MRIs, FDG-PET [10, 11], and cerebrospinal (CSF) biomarkers [12,13,14] for amyloid and tau with more frequency to improve diagnostic accuracy in atypical cases.
Amyloid and tau PET
Although three amyloid PET tracers [15, 16] and one tau PET ligand [17] have been granted clinical approval by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA), their use has mostly been limited to research studies at select academic centers. The limitation is secondary to cost and limited coverage by payers. The clinical role of neuroimaging biomarkers is likely to expand considerably as DMTs become a reality.
Cerebrospinal fluid based biomarkers
An important observational trial, the European Prevention of Alzheimer's Dementia (EPAD) showed that in a non-demented population, ATN profiles convey neuropsychological and structural information that may aid enrollment in secondary prevention trials. Data-driven models from EPAD confirm that the proposed CSF cut-off values for Aβ42 and p-tau181 are valid in a non-demented population [18]. In addition, CSF values for Aβ42/Aβ40 ratios predict amyloid PET positivity [19] and AD neuropathological diagnosis post-mortem [20, 21]. Despite their diagnostic benefit, testing is often expensive, perceived as invasive, and reserved for atypical cases. An accessible, less invasive, cost-effective method to facilitate treatment selection for DMTs and lower the cost of clinical trial screening is needed [22].
Plasma based biomarkers
Plasma biomarkers are one potential answer to these problems. Using samples from 6 different cohorts, a low plasma Aβ42/40 ratio was shown to significantly predict amyloid positivity on amyloid PET imaging or CSF testing, AUC-ROC (0.90; 95% CI = 0.87–0.93) [23]. The accuracy improved further when APOE ε4 copy number and age were included in the model [24, 25]. The currently enrolling prospective validation study (SEABIRD) will determine if these findings hold up in the general population. A serum-based p-tau181 biomarker is also being developed [26, 27] and plasma p-tau217 discriminated AD from other neurodegenerative diseases with significantly higher accuracy than other plasma- and MRI-based biomarkers [28]. This same group used the Swedish BioFINDER study and the Alzheimer’s Disease Neuroimaging Initiative (ADNI) to develop a 4-year prognostic model for conversion to AD utilizing combinations of these biomarkers and cognitive testing [29, 30]. A biomarker of neurodegeneration, plasma neurofilament light (NfL), has also shown promise in distinguishing psychiatric illness from neurodegenerative disease in two multicenter cohorts [31].
Amyloid β reduction strategies
Aβ is produced from the type-1 transmembrane glycoprotein, amyloid precursor protein (APP). APP undergoes cleavage by either α- or β-secretases. When cleaved by α-secretase the resultant fragments are an extracellular peptide and an intracellular peptide, which is further processed by γ-secretase catalytic subunits, presenilin proteins (PS1 and PS2). When cleaved by β-secretase the intracellular peptide is also processed by γ-secretase [32, 33]. The resulting peptide is typically a 40–42 amino acid (AA) in length [33], which is then released extracellularly. PS1/2 mutations lead to premature release of APP and can lead to longer, aggregation-prone Aβ peptides [4, 34]. While Aβ protofibrils and oligomers are known to be toxic [35, 36], it is now postulated that Aβ is also physiologically produced during neuronal activity [37, 38], augments synaptic plasticity [39] and functions in memory formation [38]. Amyloid plaques also increase with age even in cognitively unimpaired (CU) individuals and their pathogenic role is less certain [40,41,42]. One meta-analysis on the topic does put forward a rather convincing argument that the lack of efficacy of anti-amyloid therapies in general, may be a class effect, at least if administered during the early symptomatic phase of AD [43].
BACE (β-secretase) inhibitors
Several small molecules have been synthesized to inhibit the β-site APP cleaving enzyme-1 (BACE1) whose action represents the rate-limiting step in Aβ production. Throughout 2017–2019 multiple BACE inhibitor trials were terminated early and consistent with results from prior negative BACE inhibitor trials [44]. Atabecestat, verubecestat, umibecestat, lanabecestat, and elenbecestat were all discontinued due to cognitive worsening, reduced brain volumes, or side effects [45] (Table 1). Atabecestat was associated with dose-related cognitive worsening at 3 months and the presence of neuropsychiatric adverse events, although there was evidence of reversibility after 6 months off treatment [46]. These trial results argue that there is a considerable gap in our knowledge of the normal physiological function of APP, BACE, and Aβ.
Amyloid-β directed monoclonal antibodies (MAbs)
There have been 7 Aβ-directed MAbs with 14 phase II/III clinical trials completed or terminated early. Recent results from phase II/III clinical trials are summarized in Table 2. Biogen’s aducanumab recently received accelerated approval from the US FDA despite lackluster performance on clinical measures [47, 48]. Aducanumab had two simultaneous phase III trials (ENGAGE and EMERGE), which were both terminated early after futility analysis revealed that they were failing on their primary endpoint, the Clinical Dementia Rating–Sum of Boxes (CDR-SB). Aducanumab binds to amyloid plaques and oligomers and showed substantial dose- and treatment duration-related lowering of amyloid plaques [48,49,50]. Aducanumab must undergo further testing to show that the statistically significant benefit on clinical outcomes in one arm of a single clinical trial was not a type I error (false-positive).
Eisai’s BAN2401 (lecanemab) selectively binds to large, soluble Aβ protofibrils [51, 52]. Even though lecanemab did not meet its primary outcome in their phase IIb study [52], the MAb showed a reduction in amyloid and mild improvement in cognitive measures at 18 months [52]. Lecanemab’s phase III study, Clarity AD, is ongoing for early symptomatic AD. They are also testing it in pre-clinical stages of AD in their AHEAD 3–45 study, which is currently enrolling.
Eli Lilly’s donanemab is directed at an N-terminal pyroglutamate Aβ epitope in established amyloid plaques. In its phase II trial, TRAILBLAZER-ALZ, donanemab showed a reduction in amyloid plaques, tau neurofibrillary tangles (NFTs), and met its primary clinical endpoint, the Integrated Alzheimer’s Disease Rating Scale (iADRS). Enthusiasm was tempered by the lack of statistical significance on multiple standard measures of cognitive decline including the CDR-SB, although all the secondary measures appeared to be trending towards a positive effect [53]. Based on the accelerated approval of aducanumab and their phase II results, both donanemab and lecanemab have obtained the FDA's breakthrough therapy designation. They are poised for FDA accelerated approval if the precedent set by aducanumab is carried forward.
Roche’s crenezumab binds Aβ monomers and oligomers. It was designed to minimize amyloid-related imaging abnormalities (ARIA) [54]. Both phase III trials of crenezumab (CREAD 1 and 2) were discontinued after futility analysis but are currently still in a prevention trial through the Dominantly Inherited Alzheimer's Network Trials Unit (DIAN-TU) for preclinical dominantly-inherited AD.
Roche’s gantenerumab and Eli Lilly’s solanezumab were also a part of DIAN-TU. Neither gantenerumab nor the solanezumab arm reached its primary clinical outcome. Development of solanezumab has been halted in symptomatic patients due to its failure to reduce decline in cognition or function in 3 phase III trials (EXPEDITION 1–3) [55, 56]. The Anti-Amyloid Treatment in Asymptomatic Alzheimer's (A4) study of solanezumab is still ongoing in amyloid positive CU individuals.
Roche’s prodromal AD study of gantenerumab, SCarlet RoAD, initially failed futility analysis, and they converted both gantenerumab phase III trials (SCarlet RoAD and Marguerite RoAD) [57] into a combined SCarlet/Marguerite RoAD open-label extension (OLE) cohort to learn more about the therapy’s response. Post-hoc analyses suggested there had been a dose-dependent slowing of cognitive decline and that higher doses may be needed [58]. This was used to improve the design of the ongoing, phase III trials (GRADUATE 1 and 2).
Amyloid-β vaccines
Most MAb therapies activate the immune system to remove specific Aβ fragments, so a vaccine targeted to these fragments is a logical target for disease modification. Unfortunately, six percent of vaccinated patients developed aseptic meningoencephalitis with the first-generation vaccine and the risk was deemed too high to continue development [59]. Second-generation vaccines seem to have a better risk profile. Grifols’ ABvac40 vaccine targets the C terminus of Aβ40 [60]. An ongoing phase II study in patients with early AD is due for completion in 2022. United Neuroscience’s UB-311 is a synthetic peptide vaccine against the Aβ1–14 sequence [61]. Preliminary results reported that patients with mild AD declined less than controls on CDR-SB and phase III development is underway.
AC Immune’s ACI-24 is a liposomal vaccine, which elicits antibody responses to the truncated Aβ1-15 sequence closer to the C-terminus, thereby avoiding pro-inflammatory T-cell activation [62]. It showed promise in a mouse model of Down syndrome (DS) [63]. Immunogenicity and safety in the DS population were reported as positive but not yet available for review. ACI-24 is also currently being tested in a phase 2 clinical trial in patients with mild AD.
Amyloid-β aggregation inhibitors
Aβ aggregation inhibitors have not seen success in prior phase II/III trials. A phase II study of GV-971 (sodium oligomannate) showed a positive trend in the primary outcome, Alzheimer’s disease assessment scale—Cognitive Subscale 12 (ADAS-cog12) but it did not reach statistical significance [64]. It received conditional marketing approval in China to improve cognitive function in mild to moderate AD. A phase III trial in China showed a positive outcome on ADAS-cog12, but possible bias and confounding issues have been raised. An international phase III trial called Green Memory just started enrolling in 2021.
Alzheon’s ALZ-801 is a prodrug of tramiprosate, a central GABA partial receptor agonist that helps stabilize Aβ42 monomers, reducing oligomeric and fibrillar amyloid aggregation [65]. ALZ-801 is thought to increase the amount of tramiprosate that reaches the brain. Although tramiprosate failed its phase II and III primary endpoints, a later subgroup analysis reported slowing of cognitive decline in APOE ε4 homozygotes [66, 67]. The US FDA granted ALZ-801 fast track designation for a phase III trial (APOLLOE4) in homozygous APOE ε4 individuals with early AD based on the prior tramiprosate trial data [68,69,70].
Tau reduction strategies
The tau protein is an integral component of neurons, providing microtubule stability and transport of key proteins across varying axon lengths. The disruption of this cytoskeleton and the failure of key protein transport leads to impaired neuroplasticity, cellular dysfunction, and cell death. Pathological tau can be identified decades before the onset of clinical symptoms, in the locus coeruleus [71] and the entorhinal cortex [72]. In the amnestic version of AD, pathological aggregates of tau follow a stereotypical pattern of deposition, termed Braak staging [73]. There is accumulating evidence that a cascading network failure [74], a prion-like spread [75], or a combination of both through synaptic uptake in highly connected brain networks [76] is responsible for clinical progression. The location of tau pathology correlates with symptoms and disease severity [77], even across distinct phenotypical variants of AD [78, 79]. This makes tau a principal target for DMTs.
Post-translational modifications
Post-translational modifications are key to tau protein structure and function. Over 100 sites for modifications have been identified and the complexity of these events is beyond the scope of this paper. Small molecules that inhibit kinase activities responsible for abnormal tau phosphorylation are suggested therapeutic targets.
Lithium, a GSK-3β kinase inhibitor, showed promise in animal models but has had little success in human trials [80]. However, results of a clinical trial in 2019 by Forlenza et al. suggested that amnestic mild cognitive impairment (MCI) participants progressed at a slower rate over four years and showed positive effects on CSF Aβ levels [81]. A current phase IV study, LATTICE is actively recruiting.
Saracatinib and nilotinib, small molecule inhibitors of Fyn and Abl, respectively, are repurposed tyrosine kinase inhibitors approved in cancer treatment. Saracatinib’s phase II trial in participants with mild AD did not meet its primary or secondary outcomes [82]. Nilotinib’s phase II trial suggested better tolerability with positive findings against relevant AD biomarkers [83]. These potentially positive results warrant further investigation by a multicenter study aiming for a larger enrollment and longer duration.
Acetylation of tau residues can prevent intracellular clearance of abnormal tau by ubiquitin and other mechanisms [84]. One specific lysine residue, K174, is integral for tau homeostasis and is blocked by a non-steroidal anti-inflammatory drug, salsalate [85]. The first of two planned phase I/II trials testing salsalate in mild to moderate AD (SAL-AD) are expected to result in 2022.
Stabilization of microtubules
When tau is abnormally hyperphosphorylated, the binding of microtubules is impaired, resulting in defective cellular trafficking and poor cytoskeleton structure. To rescue this loss of function, microtubule-stabilizing agents have been proposed as therapeutic targets. Previous trials have been discontinued due to tolerability and lack of efficacy. The most recent failed trial was published in 2020 where TPI-287, a taxane derivative, did not reach adequate levels in the CSF and likely did not reach the target. Additionally, participants with AD had more severe side effects and worsening of cognition across the tauopathies with escalating doses [86]. The idea of tau stabilization therapies has been questioned by Qiang et al. in 2018, who suggested tau stabilizers may be harmful. Tau does not simply stabilize microtubules but helps axonal microtubules remain labile and dynamic, adding and subtracting length as cellular demands fluctuate [87].
Tau aggregation prevention
In addition to the suggested loss of function, increasing levels of abnormal tau in the cytosol, leaves critical microtubule-binding regions that are prone to aggregation exposed. This may lead to a gain of function toxicity to the cell as NFTs mature in a templating manner. Blocking these critical binding regions is a proposed target. Methylene Blue is a phenothiazine that can disrupt aggregation of tau-tau bonds to prevent aggregation. It was modified and rebranded by TauRx Therapeutics as LMTM (including LMT-X and Trx0237) and multiple phase II and phase III studies have provided mostly negative results with various hypotheses for the cause [88]. These hypotheses are being further tested with a low-dose LMTM monotherapy phase III clinical trial set to result in 2023.
Immunologic clearance
As discussed above, tau phosphorylation sites are plentiful including N-terminal, C-terminal, and inner microtube-binding regions. The proper site for immunologic targeting is crucial but is an ongoing area of research. Intracellular targeting of tau remains difficult. However, an extracellular tau species termed eTau, often found as a truncated form, is considered pathologic and is an easier target. The theory of prion-like spreading in communicating neurons and across connected neuronal networks has increasing evidence, and the eTau form is thought to play a significant pathologic role in this process [89].
Anti-tau antibodies targeting C-terminal regions have fewer studies and current studies are in phase I at the time of this article. Gosuranemab (BIIB092), tilavonemab (ABBV-8E12), and semorinemab (R07105705) are IgG4 monoclonal anti-tau antibodies that target N-terminal tau sites (Table 3). In the last 2 years, Biogen and AbbVie posted negative results of primary clinical and secondary biomarker endpoints in phase II trials for progressive supranuclear palsy (PSP) and AD, despite good target engagement. Most recently, semorinemab also missed primary clinical endpoints and did not show a reduction in secondary tau-PET biomarker endpoints. Roche has another phase II trial of R07105705 set to post results by the end of 2021.
Several mid-region anti-tau antibodies are in the pipeline in phase I or phase II trials. Lilly’s compound, zagotenemab (LY3303560), is a hybrid that binds to mid- and N-terminus regions. It should conclude its phase II trial before the end of 2021. Janssen’s JNJ-63733657 mid-region antibody reported positive preliminary phase I data showing dose-dependent reductions in pTau CSF measures with a phase II trial starting in 2021. Four additional mid-region anti-tau compounds are undergoing phase I trials with the hope that the field is zeroing in on the most important region for this key AD protein. Additionally, three anti-tau vaccines are set to release data from phase II trials later this year (Table 4).
Other potential disease-modifying pathways
Other potentially modifiable processes, which may contribute to AD's neuropathology include inflammation [90], oxidative stress [91], metabolism, and excitotoxicity [92]. They have also been hampered by negative results. Despite this, researchers remain resolute, and the lessons learned are being utilized to refine trials and identify the correct target populations.
Inflammation reduction
Inflammation is recognized as playing multiple important roles in neurodegeneration. Individuals with AD pathology and high levels of TREM2’s (triggering receptor expressed on myeloid cells 2) soluble portion in CSF progress more slowly [93]. TREM2 stimulation is thought to reduce microglial activity and inflammatory response to amyloid plaques [94]. AL002, Alector and AbbVie’s MAb to microglial lipid receptor TREM2, met its phase I (INVOKE) endpoint and is recruiting for phase II (INVOKE-2). Alector and AbbVie’s other collaboration, the anti-CD33 antibody, AL003 has entered phase I [95].
ALZT-OP1 [96], a combination of cromolyn, a mast-cell stabilizer that suppresses cytokine release, and ibuprofen (NSAID) confirmed safety in a phase I study [97]. We are now awaiting the results of a phase III clinical trial (COGNITE), completed in 2020.
Cassava Sciences’ sumifilam (PTI-125) reduces inflammation and tau phosphorylation in animal models through binding filamin, thereby preventing the binding of Aβ42 to α7 nicotinic acetylcholine receptor (α7nAChR) [98]. The phase II study did not meet its primary endpoint but had a positive effect on biomarkers. The company plans to start two phase III trials later this year.
In transgenic mice, granulocyte–macrophage colony-stimulating factor (GM-CSF) is associated with microglial activation and reduction in amyloid [99]. A phase II trial in mild AD provided evidence of safety and cognitive benefit on Mini-Mental State Exam (MMSE) [100]. In the phase II MADE trial minocycline failed to modify cognition in early AD [101].
Lenalidomide is a chemotherapy agent for multiple hematological cancers and has known anti-inflammatory immune responses in animal models [102, 103]. A phase II study is currently underway in patients with amnestic MCI secondary to AD (MCLENA-1) [104].
Masitinib is a selective tyrosine kinase inhibitor that inhibits mast-cells. The company claims the phase IIb/III study met its primary endpoint (ADAS-Cog), but results have yet to be published and the phase III study is ongoing.
A phase IIb/III study of plasma exchange with albumin replacement showed that patients with moderate AD had less decline than controls but there was no statistically significant difference in mild AD [105]. Plasma exchange purportedly functions through amyloid reduction and the anti-inflammatory effect of albumin, but it is hampered by its invasive nature and cost. Other anti-inflammatory medications with pleiotropic disease-modifying effects target the “brain-gut” axis, such as rifaximin [106], or modifying interactions with infectious resident organisms as with atuzaginstat, an irreversible inhibitor of gingipain [107, 108] or valacyclovir in HSV 1/2 [109].
Metabolism and bioenergetics
Brain metabolism, or bioenergetics, is largely mediated through mitochondria. Metabolism declines with aging and is accelerated in AD [110,111,112,113,114]. These changes are often demonstrated clinically on FDG-PET before neurodegeneration is seen on structural MRI. Declining brain bioenergetics likely contributes to disease-specific neuropathology and represents a potential therapeutic target.
Multiple agents attempting to positively impact brain bioenergetics are currently under investigation [115]. Semaglutide, a glucagon-like peptide 1 (GLP-1) agonist, is enrolling two large phase III trials. Liraglutide, another GLP-1 agonist has an ongoing phase II trial (ELAD) [116]. The Metformin in Alzheimer’s Dementia Prevention (MAP) study is looking into the effects of metformin plus exercise as well as diet [117] and the DAPA trial of a sodium-glucose co-transporter-2 (SGLT-2) inhibitor in non-diabetic patients is currently recruiting.
Rasagiline is a monoamine oxidase inhibitor (MAO-I) that improves mitochondrial function and reduces amyloid accumulation, tau hyperphosphorylation, NFT formation, and neuron loss [118,119,120]. A recent phase II trial showed less decline on FDG-PET in participants receiving rasagiline but no difference in clinical outcome measures [121]. In a phase II trial, riluzole was shown to reduce the decline of regional cerebral glucose metabolism in AD as measured with FDG-PET [122].
Symptom reducing trials
Mild behavioral impairment is increasingly recognized as a change in AD that may precede cognitive symptoms by several years [123] and correlates with tau-PET measures [124]. Escitalopram is a selective serotonin reuptake inhibitor (SSRI) with limited off-target binding and is well tolerated in older adults. Escitalopram improved CSF Aβ42 levels in CU individuals relative to placebo [125] although the clinical significance of this is unclear. A phase I trial with escitalopram was terminated in 2019 due to insufficient enrollment. Additionally, it is being tested in a phase III trial for behavioral symptoms of dementia.
Acadia’s pimavanserin (Nuplazid) met its primary outcome in a phase II trial of AD with psychosis [126]. The follow-up HARMONY trial, which included multiple dementia subtypes, also met its primary outcome, and pimavanserin was submitted to the FDA for approval as a treatment for dementia-related psychosis [127]. The US FDA rejected the initial application, and they are in further discussions with Acadia.
Primary prevention and lifestyle modifications
The 2020 Lancet Commission report added three new risk factors for dementia including air pollution, traumatic brain injury, and excess alcohol consumption. The report suggests 40% of dementia may involve 12 modifiable risk factors starting with increased education in early life, reducing vascular risk factors in midlife, and treating depression and social isolation in later life [128]. Social isolation, compared to high engagement in social activities, in later life is associated with cognitive trajectory in a recent meta-analysis review [129]. A randomized controlled trial on blood pressure modification, SPRINT-MIND, had an overall reduced occurrence of MCI [130]. The higher intensity target (systolic < 120 mmHg) had a lower incidence compared to the systolic < 140 mmHg group, but this did not reach statistical significance due to low power with limited incident MCI cases [130].
In 2021 the Alzheimer’s Prevention through Exercise (APEX) trial published their aerobic exercise intervention in CU individuals with elevated amyloid [131]. There was no appreciable difference in amyloid load after one year, but there was improved hippocampal blood flow in the APOE ε4 individuals [132]. This finding parallels a recent pathology paper indicating that higher levels of physical activity correlated with better cognition, not because of reduced AD pathology [133] but due to preserved white matter integrity and brain tissue microstructure in the hippocampus [134].
Secondary analysis data from the Age-Well trial were published in 2020 lending further evidence to untreated sleep apnea’s association with higher brain amyloid burden and as a risk factor for AD [135]. Similarly, a retrospective study looking at Medicare data suggested positive airway pressure (PAP) adherence was associated with lower odds of incident AD diagnosis (OR 0.65) [136].
DNA/RNA-based primary prevention
APOE ε4 is the most common genetic risk factor in AD and is present in up to 65% of patients with late-onset AD. A 5000-person neuropathological study has confirmed that there is a low likelihood of Alzheimer's dementia in APOE ε2 homozygotes and being an APOE ε2 carrier has a protective effect [137]. A gene therapy approach to AD may also be on the horizon for those who are APOE ε4 carriers. A phase I trial delivering an adeno-associated virus carrying the gene for APOE ε2 (AAVrh.10hAPOE2) directly into the subarachnoid cisternae of 15 patients with early to late-stage AD, who inherited two copies of APOE ε4, is ongoing. A phase II trial of a tau anti-sense oligonucleotide (BIIB080) is also set to complete in 2022 (Table 4).
Future outlook
One thing is clear from recent clinical trials, AD pathology is complex and many fundamental questions remain unanswered. Judging by the lack of success in clinical trial outcomes, a greater understanding of the underlying biology contributing to normal aging and neurodegeneration is sorely needed. In addition, the field needs to address the heterogeneity of AD, both in clinical presentation and disease progression. Models and biomarkers that acknowledge the heterogeneity of disease progression are vital for interpreting complex clinical trial results. Despite the abundance of failed trials, Aβ and tau continue to be the dominant target in AD therapeutics research. However, the field must consider sporadic AD as a multifactorial pathology that requires a multi-faceted approach to therapy. Increased attention to the genetic and environmental risk factors for AD will improve our primary and secondary prevention strategies. With our aging population, a focus on prevention will be essential in the path forward while we wait for a much-needed pharmacological breakthrough.
References
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu EC, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Elliott C, Masliah E, Ryan L, Silverberg N (2018) NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14:535–562. https://doi.org/10.1016/j.jalz.2018.02.018
ten Kate M, Ingala S, Schwarz AJ, Fox NC, Chételat G, van Berckel BNM, Ewers M, Foley C, Gispert JD, Hill D, Irizarry MC, Lammertsma AA, Molinuevo JL, Ritchie C, Scheltens P, Schmidt ME, Visser PJ, Waldman A, Wardlaw J, Haller S, Barkhof F (2018) Secondary prevention of Alzheimer’s dementia: neuroimaging contributions. Alzheimer’s Res Ther 10:112. https://doi.org/10.1186/s13195-018-0438-z
Dubois B, Villain N, Frisoni GB, Rabinovici GD, Sabbagh M, Cappa S, Bejanin A, Bombois S, Epelbaum S, Teichmann M, Habert M-O, Nordberg A, Blennow K, Galasko D, Stern Y, Rowe CC, Salloway S, Schneider LS, Cummings JL, Feldman HH (2021) Clinical diagnosis of Alzheimer’s disease: recommendations of the International Working Group. Lancet Neurol 20:484–496. https://doi.org/10.1016/S1474-4422(21)00066-1
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, van der Flier WM (2021) Alzheimer’s disease. Lancet 397:1577–1590. https://doi.org/10.1016/s0140-6736(20)32205-4
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:263–269
Rabinovici GD, Gatsonis C, Apgar C, Chaudhary K, Gareen I, Hanna L, Hendrix J, Hillner BE, Olson C, Lesman-Segev OH (2019) Association of amyloid positron emission tomography with subsequent change in clinical management among Medicare beneficiaries with mild cognitive impairment or dementia. JAMA 321:1286–1294
Hanseeuw BJ, Betensky RA, Jacobs HI, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC (2019) Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol 76:915–924
Guo T, Korman D, Baker SL, Landau SM, Jagust WJ, AsDN I (2021) Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer’s Disease pathophysiology. Biol Psychiat 89:786–794
Spallazzi M, Barocco F, Michelini G, Immovilli P, Taga A, Morelli N, Ruffini L, Caffarra P (2019) CSF biomarkers and amyloid PET: concordance and diagnostic accuracy in a MCI cohort. Acta Neurol Belg 119:445–452. https://doi.org/10.1007/s13760-019-01112-8
Levin F, Ferreira D, Lange C, Dyrba M, Westman E, Buchert R, Teipel SJ, Grothe MJ, For the Alzheimer’s Disease Neuroimaging I (2021) Data-driven FDG-PET subtypes of Alzheimer’s disease-related neurodegeneration. Alzheimer Res Ther 13:49. https://doi.org/10.1186/s13195-021-00785-9
Chételat G, Arbizu J, Barthel H, Garibotto V, Law I, Morbelli S, van de Giessen E, Agosta F, Barkhof F, Brooks DJ, Carrillo MC, Dubois B, Fjell AM, Frisoni GB, Hansson O, Herholz K, Hutton BF, Jack CR, Lammertsma AA, Landau SM, Minoshima S, Nobili F, Nordberg A, Ossenkoppele R, Oyen WJG, Perani D, Rabinovici GD, Scheltens P, Villemagne VL, Zetterberg H, Drzezga A (2020) Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol 19:951–962. https://doi.org/10.1016/S1474-4422(20)30314-8
Zetterberg H, Bendlin BB (2021) Biomarkers for Alzheimer’s disease-preparing for a new era of disease-modifying therapies. Mol Psychiatry 26:296–308. https://doi.org/10.1038/s41380-020-0721-9
Zetterberg H, Blennow K (2021) Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol Neurodegener 16:10. https://doi.org/10.1186/s13024-021-00430-x
Budelier MM, Bateman RJ (2019) Biomarkers of Alzheimer disease. J Appl Lab Med 5:194–208. https://doi.org/10.1373/jalm.2019.030080
Villemagne VL, Doré V, Bourgeat P, Burnham SC, Laws S, Salvado O, Masters CL, Rowe CC (2017) Aβ-amyloid and Tau Imaging in Dementia. Semin Nucl Med 47:75–88. https://doi.org/10.1053/j.semnuclmed.2016.09.006
van Waarde A, Marcolini S, de Deyn PP, Dierckx R (2021) PET agents in dementia: an overview. Semin Nucl Med 51:196–229. https://doi.org/10.1053/j.semnuclmed.2020.12.008
Jie CV, Treyer V, Schibli R, Mu L (2021) TauvidTM: the First FDA-Approved PET Tracer for Imaging Tau Pathology in Alzheimer’s disease. Pharmaceuticals 14:110
Ingala S, De Boer C, Masselink LA, Vergari I, Lorenzini L, Blennow K, Chételat G, Di Perri C, Ewers M, van der Flier WM, Fox NC, Gispert JD, Haller S, Molinuevo JL, Muniz-Terrera G, Mutsaerts HJ, Ritchie CW, Ritchie K, Schmidt M, Schwarz AJ, Vermunt L, Waldman AD, Wardlaw J, Wink AM, Wolz R, Wottschel V, Scheltens P, Visser PJ, Barkhof F (2021) Application of the ATN classification scheme in a population without dementia: findings from the EPAD cohort. Alzheimers Dement 17:1189–1204. https://doi.org/10.1002/alz.12292
Schindler SE, Gray JD, Gordon BA, Xiong C, Batrla-Utermann R, Quan M, Wahl S, Benzinger TLS, Holtzman DM, Morris JC, Fagan AM (2018) Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement 14:1460–1469. https://doi.org/10.1016/j.jalz.2018.01.013
Therriault J, Benedet AL, Pascoal TA, Savard M, Ashton NJ, Chamoun M, Tissot C, Lussier F, Kang MS, Bezgin G (2021) Determining Amyloid-β positivity using 18F-AZD4694 PET imaging. J Nucl Med 62:247–252
Baiardi S, Abu-Rumeileh S, Rossi M, Zenesini C, Bartoletti-Stella A, Polischi B, Capellari S, Parchi P (2019) Antemortem CSF Aβ42/Aβ40 ratio predicts Alzheimer’s disease pathology better than Aβ42 in rapidly progressive dementias. Ann Clin Transl Neurol 6:263–273. https://doi.org/10.1002/acn3.697
Mattke S, Cho SK, Bittner T, Hlávka J, Hanson M (2020) Blood-based biomarkers for Alzheimer’s pathology and the diagnostic process for a disease-modifying treatment: projecting the impact on the cost and wait times. Alzheimers Dement 12:e12081. https://doi.org/10.1002/dad2.12081
West T, Kirmess KM, Meyer MR, Holubasch MS, Knapik SS, Hu Y, Contois JH, Jackson EN, Harpstrite SE, Bateman RJ, Holtzman DM, Verghese PB, Fogelman I, Braunstein JB, Yarasheski KE (2021) A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegener 16:30. https://doi.org/10.1186/s13024-021-00451-6
West T, Kirmess KM, Meyer MR, Holubasch MS, Knapik SS, Hu Y, Contois JH, Jackson EN, Harpstrite SE, Bateman RJ (2021) A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol Neurodegener 16:1–12
Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, Holtzman DM, Morris JC, Benzinger TL, Xiong C (2019) High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 93:e1647–e1659
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, Chai X, Proctor NK, Eichenlaub U, Zetterberg H, Blennow K, Reiman EM, Stomrud E, Dage JL, Hansson O (2020) Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 26:379–386. https://doi.org/10.1038/s41591-020-0755-1
Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, Chamoun M, Savard M, Kang MS, Therriault J, Schöll M, Massarweh G, Soucy JP, Höglund K, Brinkmalm G, Mattsson N, Palmqvist S, Gauthier S, Stomrud E, Zetterberg H, Hansson O, Rosa-Neto P, Blennow K (2020) Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 19:422–433. https://doi.org/10.1016/s1474-4422(20)30071-5
Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, Su Y, Chen Y, Serrano GE, Leuzy A, Mattsson-Carlgren N, Strandberg O, Smith R, Villegas A, Sepulveda-Falla D, Chai X, Proctor NK, Beach TG, Blennow K, Dage JL, Reiman EM, Hansson O (2020) Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 324:772–781. https://doi.org/10.1001/jama.2020.12134
Palmqvist S, Tideman P, Cullen N, Zetterberg H, Blennow K, Dage JL, Stomrud E, Janelidze S, Mattsson-Carlgren N, Hansson O (2021) Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med 27:1034–1042
Cullen NC, Leuzy A, Janelidze S, Palmqvist S, Svenningsson AL, Stomrud E, Dage JL, Mattsson-Carlgren N, Hansson O (2021) Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun 12:3555. https://doi.org/10.1038/s41467-021-23746-0
Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, Benedet AL, Pascoal TA, Lleó A, Parnetti L, Galimberti D, Bonanni L, Pilotto A, Padovani A, Lycke J, Novakova L, Axelsson M, Velayudhan L, Rabinovici GD, Miller B, Pariante C, Nikkheslat N, Resnick SM, Thambisetty M, Schöll M, Fernández-Eulate G, Gil-Bea FJ, López de Munain A, Al-Chalabi A, Rosa-Neto P, Strydom A, Svenningsson P, Stomrud E, Santillo A, Aarsland D, van Swieten JC, Palmqvist S, Zetterberg H, Blennow K, Hye A, Hansson O (2021) A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun 12:3400. https://doi.org/10.1038/s41467-021-23620-z
Dinkel F, Trujillo-Rodriguez D, Villegas A, Streffer J, Mercken M, Lopera F, Glatzel M, Sepulveda-Falla D (2020) Decreased deposition of beta-amyloid 1–38 and increased deposition of beta-amyloid 1–42 in brain tissue of presenilin-1 E280A familial Alzheimer’s disease patients. Front Aging Neurosci 12:220. https://doi.org/10.3389/fnagi.2020.00220
De Strooper B, Annaert W (2010) Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu Rev Cell Dev Biol 26:235–260. https://doi.org/10.1146/annurev-cellbio-100109-104117
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN (2017) Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170:443–456.e414
Yakupova EI, Bobyleva LG, Shumeyko SA, Vikhlyantsev IM, Bobylev AG (2021) Amyloids: the history of toxicity and functionality. Biology. https://doi.org/10.3390/biology10050394
Johannesson M, Sahlin C, Söderberg L, Basun H, Fälting J, Möller C, Zachrisson O, Sunnemark D, Svensson A, Odergren T, Lannfelt L (2021) Elevated soluble amyloid beta protofibrils in Down syndrome and Alzheimer’s disease. Mol Cell Neurosci 114:103641. https://doi.org/10.1016/j.mcn.2021.103641
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB (1992) Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359:322–325
Kent SA, Spires-Jones TL, Durrant CS (2020) The physiological roles of tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol 140:417–447. https://doi.org/10.1007/s00401-020-02196-w
Finnie PS, Nader K (2020) Amyloid beta secreted during consolidation prevents memory malleability. Current Biology 30:1934–1940.e1934
Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Aalten P, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BN, Bibeau K, Blennow K, Brooks DJ, van Buchem MA, Camus V, Cavedo E, Chen K, Chetelat G, Cohen AD, Drzezga A, Engelborghs S, Fagan AM, Fladby T, Fleisher AS, van der Flier WM, Ford L, Förster S, Fortea J, Foskett N, Frederiksen KS, Freund-Levi Y, Frisoni GB, Froelich L, Gabryelewicz T, Gill KD, Gkatzima O, Gómez-Tortosa E, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Hildebrandt H, Ishihara L, Ivanoiu A, Jagust WJ, Johannsen P, Kandimalla R, Kapaki E, Klimkowicz-Mrowiec A, Klunk WE, Köhler S, Koglin N, Kornhuber J, Kramberger MG, Van Laere K, Landau SM, Lee DY, de Leon M, Lisetti V, Lleó A, Madsen K, Maier W, Marcusson J, Mattsson N, de Mendonça A, Meulenbroek O, Meyer PT, Mintun MA, Mok V, Molinuevo JL, Møllergård HM, Morris JC, Mroczko B, Van der Mussele S, Na DL, Newberg A, Nordberg A, Nordlund A, Novak GP, Paraskevas GP, Parnetti L, Perera G, Peters O, Popp J, Prabhakar S, Rabinovici GD, Ramakers IH, Rami L, Resende de Oliveira C, Rinne JO, Rodrigue KM, Rodríguez-Rodríguez E, Roe CM, Rot U, Rowe CC, Rüther E, Sabri O, Sanchez-Juan P, Santana I, Sarazin M, Schröder J, Schütte C, Seo SW, Soetewey F, Soininen H, Spiru L, Struyfs H, Teunissen CE, Tsolaki M, Vandenberghe R, Verbeek MM, Villemagne VL, Vos SJ, van Waalwijk van Doorn LJ, Waldemar G, Wallin A, Wallin Å K, Wiltfang J, Wolk DA, Zboch M, Zetterberg H, (2015) Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313:1924–1938. https://doi.org/10.1001/jama.2015.4668
Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BN, Scheltens P, Visser PJ, Verfaillie SC, Zwan MD, Adriaanse SM, Lammertsma AA, Barkhof F, Jagust WJ, Miller BL, Rosen HJ, Landau SM, Villemagne VL, Rowe CC, Lee DY, Na DL, Seo SW, Sarazin M, Roe CM, Sabri O, Barthel H, Koglin N, Hodges J, Leyton CE, Vandenberghe R, van Laere K, Drzezga A, Forster S, Grimmer T, Sánchez-Juan P, Carril JM, Mok V, Camus V, Klunk WE, Cohen AD, Meyer PT, Hellwig S, Newberg A, Frederiksen KS, Fleisher AS, Mintun MA, Wolk DA, Nordberg A, Rinne JO, Chételat G, Lleo A, Blesa R, Fortea J, Madsen K, Rodrigue KM, Brooks DJ (2015) Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 313:1939–1949. https://doi.org/10.1001/jama.2015.4669
DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14:32. https://doi.org/10.1186/s13024-019-0333-5
Ackley SF, Zimmerman SC, Brenowitz WD, Tchetgen Tchetgen EJ, Gold AL, Manly JJ, Mayeda ER, Filshtein TJ, Power MC, Elahi FM, Brickman AM, Glymour MM (2021) Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ 372:n156. https://doi.org/10.1136/bmj.n156
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, Mohs R (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369:341–350. https://doi.org/10.1056/NEJMoa1210951
Miranda A, Montiel E, Ulrich H, Paz C (2021) Selective secretase targeting for Alzheimer’s disease therapy. J Alzheimers Dis 81:1–17. https://doi.org/10.3233/jad-201027
Sperling R, Henley D, Aisen PS, Raman R, Donohue MC, Ernstrom K, Rafii MS, Streffer J, Shi Y, Karcher K, Raghavan N, Tymofyeyev Y, Bogert J, Brashear HR, Novak G, Thipphawong J, Saad ZS, Kolb H, Rofael H, Sanga P, Romano G (2021) Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: a truncated randomized phase 2b/3 clinical trial. JAMA Neurol 78:293–301. https://doi.org/10.1001/jamaneurol.2020.4857
Knopman DS, Jones DT, Greicius MD (2021) Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement 17:696–701. https://doi.org/10.1002/alz.12213
Dunn B, Stein P, Cavazzoni P (2021) Approval of aducanumab for Alzheimer disease—the FDA’s perspective. JAMA Intern Med. https://doi.org/10.1001/jamainternmed.2021.4607
Salloway S, Cummings J (2021) Aducanumab, amyloid lowering, and slowing of Alzheimer disease. Neurology. https://doi.org/10.1212/wnl.0000000000012451
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537:50–56. https://doi.org/10.1038/nature19323
Tucker S, Möller C, Tegerstedt K, Lord A, Laudon H, Sjödahl J, Söderberg L, Spens E, Sahlin C, Waara ER, Satlin A, Gellerfors P, Osswald G, Lannfelt L (2015) The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis 43:575–588. https://doi.org/10.3233/jad-140741
Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, Reyderman L, Berry DA, Berry S, Gordon R, Kramer LD, Cummings JL (2021) A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 13:80. https://doi.org/10.1186/s13195-021-00813-8
Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M (2021) Donanemab in early Alzheimer’s disease. N Engl J Med 384:1691–1704
Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, Ward M, Friesenhahn M, Rabe C, Brunstein F, Quartino A, Honigberg LA, Fuji RN, Clayton D, Mortensen D, Ho C, Paul R (2018) ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90:e1889–e1897. https://doi.org/10.1212/wnl.0000000000005550
Schwarz AJ, Sundell KL, Charil A, Case MG, Jaeger RK, Scott D, Bracoud L, Oh J, Suhy J, Pontecorvo MJ (2019) Magnetic resonance imaging measures of brain atrophy from the EXPEDITION3 trial in mild Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv 5:328–337
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378:321–330
Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, Klein G, Andjelkovic M, Dubois B, Boada M, Blennow K, Santarelli L, Fontoura P (2017) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9:95. https://doi.org/10.1186/s13195-017-0318-y
Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, Andjelkovic M, Ristic S, Wang G, Bateman R, Kerchner GA, Baudler M, Fontoura P, Doody R (2019) Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther 11:101. https://doi.org/10.1186/s13195-019-0559-z
Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M (2009) Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res 6:144–151
Lacosta AM, Pascual-Lucas M, Pesini P, Casabona D, Pérez-Grijalba V, Marcos-Campos I, Sarasa L, Canudas J, Badi H, Monleón I, San-José I, Munuera J, Rodríguez-Gómez O, Abdelnour C, Lafuente A, Buendía M, Boada M, Tárraga L, Ruiz A, Sarasa M (2018) Safety, tolerability and immunogenicity of an active anti-Aβ(40) vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res Ther 10:12. https://doi.org/10.1186/s13195-018-0340-8
Wang CY, Wang PN, Chiu MJ, Finstad CL, Lin F, Lynn S, Tai YH, De Fang X, Zhao K, Hung CH, Tseng Y, Peng WJ, Wang J, Yu CC, Kuo BS, Frohna PA (2017) UB-311, a novel UBITh(®) amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement 3:262–272. https://doi.org/10.1016/j.trci.2017.03.005
Zampar S, Wirths O (2020) Immunotherapy targeting Amyloid-β peptides in Alzheimer’s disease. In: Huang X (ed) Alzheimer’s disease: drug discovery. Exon Publications Brisbane, Australia, pp 23–42
Belichenko PV, Madani R, Rey-Bellet L, Pihlgren M, Becker A, Plassard A, Vuillermot S, Giriens V, Nosheny RL, Kleschevnikov AM (2016) An anti-β-amyloid vaccine for treating cognitive deficits in a mouse model of down syndrome. PLoS ONE 11:e0152471
Wang T, Kuang W, Chen W, Xu W, Zhang L, Li Y, Li H, Peng Y, Chen Y, Wang B, Xiao J, Li H, Yan C, Du Y, Tang M, He Z, Chen H, Li W, Lin H, Shi S, Bi J, Zhou H, Cheng Y, Gao X, Guan Y, Huang Q, Chen K, Xin X, Ding J, Geng M, Xiao S (2020) A phase II randomized trial of sodium oligomannate in Alzheimer’s dementia. Alzheimers Res Ther 12:110. https://doi.org/10.1186/s13195-020-00678-3
Kocis P, Tolar M, Yu J, Sinko W, Ray S, Blennow K, Fillit H, Hey JA (2017) Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: integrating molecular analytical methods, pharmacokinetic and clinical data. CNS Drugs 31:495–509. https://doi.org/10.1007/s40263-017-0434-z
Abushakra S, Porsteinsson A, Vellas B, Cummings J, Gauthier S, Hey J, Power A, Hendrix S, Wang P, Shen L (2016) Clinical benefits of tramiprosate in Alzheimer’s disease are associated with higher number of APOE4 alleles: the “APOE4 gene-dose effect.” J Prev Alz Dis 3:219–228
Abushakra S, Porsteinsson A, Scheltens P, Sadowsky C, Vellas B, Cummings J, Gauthier S, Hey JA, Power A, Wang P, Shen L, Tolar M (2017) Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer's disease suggest disease modification potential. J Prev Alzheimers Dis 4:149–156. https://doi.org/10.14283/jpad.2017.26
Tolar M, Abushakra S, Sabbagh M (2020) The path forward in Alzheimer’s disease therapeutics: reevaluating the amyloid cascade hypothesis. Alzheimers Dement 16:1553–1560. https://doi.org/10.1016/j.jalz.2019.09.075
Sabbagh MN (2017) Clinical effects of oral tramiprosate in APOE4/4 homozygous patients with mild Alzheimer's disease suggest disease modification. J Prev Alzheimers Dis 4:136–137. https://doi.org/10.14283/jpad.2017.24
Manzano S, Agüera L, Aguilar M, Olazarán J (2020) A Review on tramiprosate (homotaurine) in Alzheimer’s disease and other neurocognitive disorders. Front Neurol 11:614. https://doi.org/10.3389/fneur.2020.00614
Mather M, Harley CW (2016) The Locus Coeruleus: essential for maintaining cognitive function and the aging brain. Trends Cognit Sci 20:214–226. https://doi.org/10.1016/j.tics.2016.01.001
Van Hoesen GW, Hyman BT, Damasio AR (1991) Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1:1–8. https://doi.org/10.1002/hipo.450010102
Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16:271–278. https://doi.org/10.1016/0197-4580(95)00021-6 (Discussion 278–284)
Jones DT, Graff-Radford J, Lowe VJ, Wiste HJ, Gunter JL, Senjem ML, Botha H, Kantarci K, Boeve BF, Knopman DS, Petersen RC, Jack CR Jr (2017) Tau, amyloid, and cascading network failure across the Alzheimer’s disease spectrum. Cortex 97:143–159. https://doi.org/10.1016/j.cortex.2017.09.018
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. https://doi.org/10.1016/j.neuron.2014.04.047
Raj A, LoCastro E, Kuceyeski A, Tosun D, Relkin N, Weiner M (2015) Network diffusion model of progression predicts longitudinal patterns of atrophy and metabolism in Alzheimer’s disease. Cell Rep 10:359–369. https://doi.org/10.1016/j.celrep.2014.12.034
Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, Rawal B, Parisi JE, Petersen RC, Kantarci K, Ross OA, Duara R, Knopman DS, Jack CR Jr, Dickson DW (2015) Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 138:1370–1381. https://doi.org/10.1093/brain/awv050
Hoenig MC, Bischof GN, Seemiller J, Hammes J, Kukolja J, Onur ÖA, Jessen F, Fliessbach K, Neumaier B, Fink GR, van Eimeren T, Drzezga A (2018) Networks of tau distribution in Alzheimer’s disease. Brain 141:568–581. https://doi.org/10.1093/brain/awx353
Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, La Joie R, Aksman LM, Grothe MJ, Iturria-Medina Y, Pontecorvo MJ, Devous MD, Rabinovici GD, Alexander DC, Lyoo CH, Evans AC, Hansson O (2021) Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat Med 27:871–881. https://doi.org/10.1038/s41591-021-01309-6
Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A, Frölich L, Schröder J, Schönknecht P, Riepe MW, Kraft I, Gasser T, Leyhe T, Möller HJ, Kurz A, Basun H (2009) Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 70:922–931
Forlenza OV, Radanovic M, Talib LL, Gattaz WF (2019) Clinical and biological effects of long-term lithium treatment in older adults with amnestic mild cognitive impairment: randomised clinical trial. Br J Psychiatry 215:668–674. https://doi.org/10.1192/bjp.2019.76
van Dyck CH, Nygaard HB, Chen K, Donohue MC, Raman R, Rissman RA, Brewer JB, Koeppe RA, Chow TW, Rafii MS, Gessert D, Choi J, Turner RS, Kaye JA, Gale SA, Reiman EM, Aisen PS, Strittmatter SM (2019) Effect of AZD0530 on cerebral metabolic decline in Alzheimer disease: a randomized clinical trial. JAMA Neurol 76:1219–1229. https://doi.org/10.1001/jamaneurol.2019.2050
Turner RS, Hebron ML, Lawler A, Mundel EE, Yusuf N, Starr JN, Anjum M, Pagan F, Torres-Yaghi Y, Shi W, Mulki S, Ferrante D, Matar S, Liu X, Esposito G, Berkowitz F, Jiang X, Ahn J, Moussa C (2020) Nilotinib effects on safety, tolerability, and biomarkers in Alzheimer’s disease. Ann Neurol 88:183–194. https://doi.org/10.1002/ana.25775
Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:5–21. https://doi.org/10.1038/nrn.2015.1
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21:1154–1162. https://doi.org/10.1038/nm.3951
Tsai RM, Miller Z, Koestler M, Rojas JC, Ljubenkov PA, Rosen HJ, Rabinovici GD, Fagan AM, Cobigo Y, Brown JA, Jung JI, Hare E, Geldmacher DS, Natelson-Love M, McKinley EC, Luong PN, Chuu EL, Powers R, Mumford P, Wolf A, Wang P, Shamloo M, Miller BL, Roberson ED, Boxer AL (2020) Reactions to multiple ascending doses of the microtubule sabilizer TPI-287 in patients with Alzheimer disease, progressive supranuclear palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol 77:215–224. https://doi.org/10.1001/jamaneurol.2019.3812
Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, Baas PW (2018) Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol 28:2181-2189.e2184. https://doi.org/10.1016/j.cub.2018.05.045
Wilcock GK, Gauthier S, Frisoni GB, Jia J, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Schelter BO, Wischik DJ, Davis CS, Staff RT, Vuksanovic V, Ahearn T, Bracoud L, Shamsi K, Marek K, Seibyl J, Riedel G, Storey JMD, Harrington CR, Wischik CM (2018) Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: cohort analysis as modified primary outcome in a phase III clinical trial. J Alzheimers Dis 61:435–457. https://doi.org/10.3233/jad-170560
Gibbons GS, Lee VMY, Trojanowski JQ (2019) Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol 76:101–108. https://doi.org/10.1001/jamaneurol.2018.2505
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement 12:719–732. https://doi.org/10.1016/j.jalz.2016.02.010
Butterfield DA (2002) Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic Res 36:1307–1313. https://doi.org/10.1080/1071576021000049890
Wang R, Reddy PH (2017) Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis 57:1041–1048. https://doi.org/10.3233/jad-160763
Edwin TH, Henjum K, Nilsson LNG, Watne LO, Persson K, Eldholm RS, Saltvedt I, Halaas NB, Selbæk G, Engedal K, Strand BH, Knapskog AB (2020) A high cerebrospinal fluid soluble TREM2 level is associated with slow clinical progression of Alzheimer’s disease. Alzheimers Dement 12:e12128. https://doi.org/10.1002/dad2.12128
Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S, Ibrahim A, Rhinn H, Tassi I, Rosenthal A, Schwabe T, Colonna M (2020) Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. https://doi.org/10.1084/jem.20200785
Estus S, Shaw BC, Devanney N, Katsumata Y, Press EE, Fardo DW (2019) Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol 138:187–199. https://doi.org/10.1007/s00401-019-02000-4
Hori Y, Takeda S, Cho H, Wegmann S, Shoup TM, Takahashi K, Irimia D, Elmaleh DR, Hyman BT, Hudry E (2015) A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J Biol Chem 290:1966–1978. https://doi.org/10.1074/jbc.M114.586602
Brazier D, Perry R, Keane J, Barrett K, Elmaleh DR (2017) Pharmacokinetics of cromolyn and ibuprofen in healthy elderly volunteers. Clin Drug Investig 37:1025–1034. https://doi.org/10.1007/s40261-017-0549-5
Burns L, Wang HY (2017) Altered filamin A enables amyloid beta-induced tau hyperphosphorylation and neuroinflammation in Alzheimer’s disease. NN 4:263
Sanchez-Ramos J, Song S, Sava V, Catlow B, Lin X, Mori T, Cao C, Arendash GW (2009) Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer’s mice. Neuroscience 163:55–72. https://doi.org/10.1016/j.neuroscience.2009.05.071
Potter H, Woodcock JH, Boyd TD, Coughlan CM, O’Shaughnessy JR, Borges MT, Thaker AA, Raj BA, Adamszuk K, Scott D, Adame V, Anton P, Chial HJ, Gray H, Daniels J, Stocker ME, Sillau SH (2021) Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer’s disease. Alzheimers Dement 7:e12158. https://doi.org/10.1002/trc2.12158
Howard R, Zubko O, Bradley R, Harper E, Pank L, O’Brien J, Fox C, Tabet N, Livingston G, Bentham P, McShane R, Burns A, Ritchie C, Reeves S, Lovestone S, Ballard C, Noble W, Nilforooshan R, Wilcock G, Gray R (2020) Minocycline at 2 different dosages vs placebo for patients with mild Alzheimer disease: a randomized clinical trial. JAMA Neurol 77:164–174. https://doi.org/10.1001/jamaneurol.2019.3762
Decourt B, Drumm-Gurnee D, Wilson J, Jacobson S, Belden C, Sirrel S, Ahmadi M, Shill H, Powell J, Walker A, Gonzales A, Macias M, Sabbagh MN (2017) Poor safety and tolerability hamper reaching a potentially therapeutic dose in the use of thalidomide for Alzheimer’s disease: results from a double-blind, placebo-controlled trial. Curr Alzheimer Res 14:403–411. https://doi.org/10.2174/1567205014666170117141330
Decourt B, Lahiri DK, Sabbagh MN (2017) Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr Alzheimer Res 14:412–425. https://doi.org/10.2174/1567205013666160930110551
Decourt B, Wilson J, Ritter A, Dardis C, DiFilippo FP, Zhuang X, Cordes D, Lee G, Fulkerson ND, St Rose T, Hartley K, Sabbagh MN (2020) MCLENA-1: a phase II clinical trial for the assessment of safety, tolerability, and efficacy of lenalidomide in patients with mild cognitive impairment due to Alzheimer’s disease. Open Access J Clin Trials 12:1–13. https://doi.org/10.2147/oajct.s221914
Boada M, López OL, Olazarán J, Núñez L, Pfeffer M, Paricio M, Lorites J, Piñol-Ripoll G, Gámez JE, Anaya F, Kiprov D, Lima J, Grifols C, Torres M, Costa M, Bozzo J, Szczepiorkowski ZM, Hendrix S, Páez A (2020) A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: primary results of the AMBAR Study. Alzheimers Dement 16:1412–1425. https://doi.org/10.1002/alz.12137
Cerovic M, Forloni G, Balducci C (2019) Neuroinflammation and the gut microbiota: possible alternative therapeutic targets to counteract Alzheimer’s disease? Front Aging Neurosci 11:284. https://doi.org/10.3389/fnagi.2019.00284
Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J (2019) Porphyromonas gingivalis in Alzheimer's disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5:eaau3333. https://doi.org/10.1126/sciadv.aau3333
Costa MJF, de Araújo IDT, da Rocha AL, da Silva RL, Dos Santos CP, Borges BCD, de Aquino Martins ARL, de Vasconcelos Gurgel BC, Lins R (2021) Relationship of porphyromonas gingivalis and Alzheimer’s disease: a systematic review of pre-clinical studies. Clin Oral Investig 25:797–806. https://doi.org/10.1007/s00784-020-03764-w
Devanand DP, Andrews H, Kreisl WC, Razlighi Q, Gershon A, Stern Y, Mintz A, Wisniewski T, Acosta E, Pollina J, Katsikoumbas M, Bell KL, Pelton GH, Deliyannides D, Prasad KM, Huey ED (2020) Antiviral therapy: Valacyclovir Treatment of Alzheimer’s Disease (VALAD) Trial: protocol for a randomised, double-blind, placebo-controlled, treatment trial. BMJ Open 10:e032112. https://doi.org/10.1136/bmjopen-2019-032112
Swerdlow RH (2020) The mitochondrial hypothesis: dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int Rev Neurobiol 154:207–233. https://doi.org/10.1016/bs.irn.2020.01.008
Swerdlow RH (2018) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 62:1403–1416. https://doi.org/10.3233/jad-170585
Swerdlow RH, Koppel S, Weidling I, Hayley C, Ji Y, Wilkins HM (2017) Mitochondria, cybrids, aging, and Alzheimer’s disease. Prog Mol Biol Transl Sci 146:259–302. https://doi.org/10.1016/bs.pmbts.2016.12.017
Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH (2018) Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement 4:28–36. https://doi.org/10.1016/j.trci.2017.11.002
Koppel SJ, Swerdlow RH (2018) Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem Int 117:114–125. https://doi.org/10.1016/j.neuint.2017.05.019
Ballard C, Nørgaard CH, Friedrich S, Mørch LS, Gerds T, Møller DV, Knudsen LB, Kvist K, Zinman B, Holm E, Torp-Pedersen C, Hansen CT (2020) Liraglutide and semaglutide: Pooled post hoc analysis to evaluate risk of dementia in patients with type 2 diabetes. Alzheimers Dement 16:e042909. https://doi.org/10.1002/alz.042909
Femminella GD, Frangou E, Love SB, Busza G, Holmes C, Ritchie C, Lawrence R, McFarlane B, Tadros G, Ridha BH, Bannister C, Walker Z, Archer H, Coulthard E, Underwood BR, Prasanna A, Koranteng P, Karim S, Junaid K, McGuinness B, Nilforooshan R, Macharouthu A, Donaldson A, Thacker S, Russell G, Malik N, Mate V, Knight L, Kshemendran S, Harrison J, Hölscher C, Brooks DJ, Passmore AP, Ballard C, Edison P (2019) Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials 20:191. https://doi.org/10.1186/s13063-019-3259-x
Campbell JM, Stephenson MD, de Courten B, Chapman I, Bellman SM, Aromataris E (2018) Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J Alzheimers Dis 65:1225–1236. https://doi.org/10.3233/jad-180263
Bar-Am O, Amit T, Weinreb O, Youdim MB, Mandel S (2010) Propargylamine containing compounds as modulators of proteolytic cleavage of amyloid-beta protein precursor: involvement of MAPK and PKC activation. J Alzheimers Dis 21:361–371. https://doi.org/10.3233/jad-2010-100150
Jenner P, Langston JW (2011) Explaining ADAGIO: a critical review of the biological basis for the clinical effects of rasagiline. Mov Disord 26:2316–2323. https://doi.org/10.1002/mds.23926
Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K (2020) Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement 6:e12050–e12050. https://doi.org/10.1002/trc2.12050
Matthews DC, Ritter A, Thomas RG, Andrews RD, Lukic AS, Revta C, Kinney JW, Tousi B, Leverenz JB, Fillit H, Zhong K, Feldman HH, Cummings J (2021) Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer’s dementia. Alzheimers Dement 7:e12106. https://doi.org/10.1002/trc2.12106
Matthews DC, Mao X, Dowd K, Tsakanikas D, Jiang CS, Meuser C, Andrews RD, Lukic AS, Lee J, Hampilos N, Shafiian N, Sano M, David Mozley P, Fillit H, McEwen BS, Shungu DC, Pereira AC (2021) Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain. https://doi.org/10.1093/brain/awab222
Mallo SC, Ismail Z, Pereiro AX, Facal D, Lojo-Seoane C, Campos-Magdaleno M, Juncos-Rabadán O (2019) Assessing mild behavioral impairment with the mild behavioral impairment checklist in people with subjective cognitive decline. Int Psychogeriatr 31:231–239. https://doi.org/10.1017/s1041610218000698
Johansson M, Stomrud E, Insel PS, Leuzy A, Johansson PM, Smith R, Ismail Z, Janelidze S, Palmqvist S, van Westen D, Mattsson-Carlgren N, Hansson O (2021) Mild behavioral impairment and its relation to tau pathology in preclinical Alzheimer’s disease. Transl Psychiatry 11:76. https://doi.org/10.1038/s41398-021-01206-z
Sheline YI, Snider BJ, Beer JC, Seok D, Fagan AM, Suckow RF, Lee JM, Waligorska T, Korecka M, Aselcioglu I, Morris JC, Shaw LM, Cirrito JR (2020) Effect of escitalopram dose and treatment duration on CSF Aβ levels in healthy older adults: a controlled clinical trial. Neurology 95:e2658–e2665. https://doi.org/10.1212/wnl.0000000000010725
Ballard C, Banister C, Khan Z, Cummings J, Demos G, Coate B, Youakim JM, Owen R, Stankovic S (2018) Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol 17:213–222. https://doi.org/10.1016/s1474-4422(18)30039-5
Cummings J (2021) New approaches to symptomatic treatments for Alzheimer’s disease. Mol Neurodegener 16:2. https://doi.org/10.1186/s13024-021-00424-9
Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, Brayne C, Burns A, Cohen-Mansfield J, Cooper C, Costafreda SG, Dias A, Fox N, Gitlin LN, Howard R, Kales HC, Kivimäki M, Larson EB, Ogunniyi A, Orgeta V, Ritchie K, Rockwood K, Sampson EL, Samus Q, Schneider LS, Selbæk G, Teri L, Mukadam N (2020) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396:413–446. https://doi.org/10.1016/S0140-6736(20)30367-6
Evans IEM, Martyr A, Collins R, Brayne C, Clare L (2019) Social isolation and cognitive function in later life: a systematic review and meta-analysis. J Alzheimers Dis 70:S119-s144. https://doi.org/10.3233/jad-180501
Williamson JD, Pajewski NM, Auchus AP, Bryan RN, Chelune G, Cheung AK, Cleveland ML, Coker LH, Crowe MG, Cushman WC, Cutler JA, Davatzikos C, Desiderio L, Erus G, Fine LJ, Gaussoin SA, Harris D, Hsieh MK, Johnson KC, Kimmel PL, Tamura MK, Launer LJ, Lerner AJ, Lewis CE, Martindale-Adams J, Moy CS, Nasrallah IM, Nichols LO, Oparil S, Ogrocki PK, Rahman M, Rapp SR, Reboussin DM, Rocco MV, Sachs BC, Sink KM, Still CH, Supiano MA, Snyder JK, Wadley VG, Walker J, Weiner DE, Whelton PK, Wilson VM, Woolard N, Wright JT Jr, Wright CB (2019) Effect of intensive vs standard blood pressure control on probable Dementia: a randomized clinical trial. JAMA 321:553–561. https://doi.org/10.1001/jama.2018.21442
Vidoni ED, Morris JK, Watts A, Perry M, Clutton J, Van Sciver A, Kamat AS, Mahnken J, Hunt SL, Townley R, Honea R, Shaw AR, Johnson DK, Vacek J, Burns JM (2021) Effect of aerobic exercise on amyloid accumulation in preclinical Alzheimer’s: a 1-year randomized controlled trial. PLoS ONE 16:e0244893–e0244893. https://doi.org/10.1371/journal.pone.0244893
Kaufman CS, Honea RA, Pleen J, Lepping RJ, Watts A, Morris JK, Billinger SA, Burns JM, Vidoni ED (2021) Aerobic exercise improves hippocampal blood flow for hypertensive Apolipoprotein E4 carriers. J Cereb Blood Flow Metab: 271678x21990342 https://doi.org/10.1177/0271678x21990342
Buchman AS, Yu L, Wilson RS, Lim A, Dawe RJ, Gaiteri C, Leurgans SE, Schneider JA, Bennett DA (2019) Physical activity, common brain pathologies, and cognition in community-dwelling older adults. Neurology 92:e811–e822. https://doi.org/10.1212/wnl.0000000000006954
Dawe RJ, Yu L, Leurgans SE, James BD, Poole VN, Arfanakis K, Schneider JA, Bennett DA, Buchman AS (2021) Physical activity, brain tissue microstructure, and cognition in older adults. PLoS ONE 16:e0253484. https://doi.org/10.1371/journal.pone.0253484
André C, Rehel S, Kuhn E, Landeau B, Moulinet I, Touron E, Ourry V, Le Du G, Mézenge F, Tomadesso C, de Flores R, Bejanin A, Sherif S, Delcroix N, Manrique A, Abbas A, Marchant NL, Lutz A, Klimecki OM, Collette F, Arenaza-Urquijo EM, Poisnel G, Vivien D, Bertran F, de la Sayette V, Chételat G, Rauchs G (2020) Association of sleep-disordered breathing with Alzheimer disease biomarkers in community-dwelling older adults: a secondary analysis of a randomized clinical trial. JAMA Neurol 77:716–724. https://doi.org/10.1001/jamaneurol.2020.0311
Dunietz GL, Chervin RD, Burke JF, Conceicao AS, Braley TJ (2021) Obstructive sleep apnea treatment and dementia risk in older adults. Sleep. https://doi.org/10.1093/sleep/zsab076
Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, Chen Y, Su Y, Myers AJ, Hardy J, Paul Vonsattel J, Younkin SG, Bennett DA, De Jager PL, Larson EB, Crane PK, Keene CD, Kamboh MI, Kofler JK, Duque L, Gilbert JR, Gwirtsman HE, Buxbaum JD, Dickson DW, Frosch MP, Ghetti BF, Lunetta KL, Wang LS, Hyman BT, Kukull WA, Foroud T, Haines JL, Mayeux RP, Pericak-Vance MA, Schneider JA, Trojanowski JQ, Farrer LA, Schellenberg GD, Beecham GW, Montine TJ, Jun GR (2020) Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun 11:667. https://doi.org/10.1038/s41467-019-14279-8
Author information
Authors and Affiliations
Contributions
This was an invited review. Dr. JP performed the literature search and data analysis. Dr. JP and Dr. RT, drafted and critically revised the work.
Corresponding author
Ethics declarations
Conflicts of interest
Dr. Townley is a site principal investigator for the following related studies: AHEAD 3-45, TRC-PAD, and Vaccinex SIGNAL-AD. Dr. Pleen has nothing to disclose and no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pleen, J., Townley, R. Alzheimer’s disease clinical trial update 2019–2021. J Neurol 269, 1038–1051 (2022). https://doi.org/10.1007/s00415-021-10790-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10790-5