
Abstract
Alzheimer’s disease (AD) is one of the most debilitating neurodegenerative diseases and is predicted to affect 1 in 85 people by 2050. Despite much effort to discover a therapeutic strategy to prevent progression or to cure AD, to date no effective disease-modifying agent is available that can prevent, halt, or reverse the cognitive and functional decline of patients with AD. Several underlying etiologies to this failure are proposed. First, accumulating evidence from past trials suggests a preventive as opposed to therapeutic paradigm, and the precise temporal and mechanistic relationship of β-amyloid (Aβ) and tau protein should be elucidated to confirm this hypothesis. Second, we are in urgent need of revised diagnostic criteria to support future trials. Third, various technical and methodological improvements are required, based on the lessons learned from previous failed trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
For some time, the β-amyloid (Aβ) theory of the pathogenesis of Alzheimer’s disease (AD) has led the development of therapeutic agents to combat the disease. However, more recently, tau protein has been recognized as a key component in AD pathogenesis. |
To ensure maximal protective or curative effects by therapeutic agents, it is critical to determine the precise temporal and mechanistic relationships between Aβ and tau. This will guide the choice of therapeutic agent (either anti-Aβ or anti-tau, alone or in combination) and the best time to initiate treatment. |
The AD diagnostic criteria currently used should be revised because they exclude prodromal or pre-symptomatic individuals at high risk of developing the disease, and do not include biomarkers, both of which fit better with the preventive paradigm. |
Errors in previous clinical trials of anti-Aβ therapies should be addressed and refined to guide future trials. |
1 β-Amyloid and Tau Pathologies: Gap and Interaction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the accumulation of misfolded proteins, both in synapses and in neurons, which ultimately result in synaptic loss, neuronal death, and cerebral atrophy, leading to impaired learning and memory with subsequent loss of higher cognitive functions. So far, the β-amyloid (Aβ) cascade has been the centerpiece of describing AD pathogenesis and has become the basis for the development of therapeutic agents to combat the disease. Indeed, plenty of evidence indicates that Aβ plaques and soluble Aβ oligomers directly confer toxicity to neurons and synapses, where the plaques act as a reservoir for the more toxic soluble Aβ oligomers [1–6]. However, efforts to develop therapeutic agents based on this premise, even when they effectively reduce Aβ load in the brain, have not demonstrated success in terms of clinical improvement.
Thus, attention has been focused towards the emerging role of tau protein as a consequence of downstream activation of Aβ cascade. Growing evidence indicates that tau protein, instead of Aβ, is the key event in AD pathogenesis. This observation is based on several factors. First, tau and the formation of its neurofibrillary tangles (NFTs), together with the number of synapses and neurons, have been shown to predict cognitive status both in patients with AD and in the murine model of the disease [7–11]. Second, tau hyperphosphorylation is associated with memory deficits in patients with AD, while Aβ deposition only correlates with functional network disruption as evaluated by magnetic resonance imaging (MRI) and electroencephalogram (EEG) [12]. Third, many clinical studies indicate that cognitive decline in previously healthy older individuals only occurs in the presence of phospho-tau and is not due to Aβ deposition, which is commonly found in the brains of these individuals [13]. In fact, the high Aβ load in older individuals does not always cause cognitive impairment [14]. In addition, the extent and perhaps the magnitude of Aβ deposition in patients with mild cognitive impairment (MCI) and AD are estimated to be similar, pointing to an alternative pathway of synapse loss and neuronal death [15]. Fourth, the time gap between the onset of detectable Aβ accumulation and the diagnosis of cognitive impairment and loss of hippocampal volume is relatively long [16], indicating that soluble Aβ oligomers may trigger tau hyperphosphorylation, with the latter being the ultimate key in initiating the devastating synapses and neuronal death in AD pathogenesis. Lastly, other neurodegenerative diseases (i.e., frontotemporal lobar degeneration with parkinsonism linked to chromosome 17 [FTLD17], Pick’s disease, and corticobasal degeneration) exert similar cognitive impairments in the absence of Aβ deposition but possess AD-like tau deposits in their brains due to tau mutation [17–20].
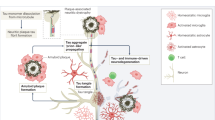
The precise mechanistic and temporal relationships between Aβ and tau protein have not been entirely elucidated. However, some clues exist. The mechanism of Aβ inducing tau hyperphosphorylation can be due to binding of Aβ molecules to various receptors, inducing inflammation. Various forms of Aβ oligomers when bound to several neuronal membrane receptors (α7nAChR, NMDAr, AMPAr, EphB2, insulin receptors, RAGE, PrR-r, LilrB2) can disturb intracellular signaling and upregulate GSK3β, which subsequently hyperphosphorylates tau protein, impairs synapse plasticity, induces neuronal excitation, and ultimately leads to cellular and synaptic death (Fig. 1). The second mechanism is through Aβ-induced inflammation as marked by astrocytosis and microgliosis, which accelerates tau pathology. In fact, chronic oxidative stress and astrocytic mediated-Aβ and caspase activation is known to independently cause tau hyperphosphorylation [21].

The critical and relevant pathogenesis of Alzheimer’s disease derived from the amyloid-beta cascade hypothesis. Soluble Aβ40 and Aβ42 monomers will coalesce to form the toxic amyloid-beta oligomers that are able to interact with metal ions to form insoluble plaques, which act as a ‘loose’ reservoir to constantly release amyloid-beta oligomers. The latter compound is proven to be toxic to the synapses by aberrantly increasing long-term potentiation and decreasing long-term depression simultaneously, thus causing memory impairment. Furthermore, to a certain extent, amyloid-beta oligomers can also activate tau hyperphosphorylation, which in turn causes tau to disintegrate from the microtubules and therefore impairs axonal transport and subsequent synaptic failures. Tau hyperphosphorylation, deposition, and accumulation in the neuron in the form of neurofibrillary tangles is the critical hallmark of clinical dementia in Alzheimer’s disease patients. Aβ amyloid-beta, AMPA-R α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, LTD long-term depression, LTP long-term potentiation, NFT neurofibrillary tangles, NMDA-R N-Methyl-d-aspartate receptor
It is important to understand this core issue, because future drug developments may target these missing points that have not been addressed previously. For instance, instead of finding a way to inhibit the production of soluble Aβ oligomers or aggressively clearing Aβ plaques, therapy could be directed to inhibiting these receptors. In fact, this approach has been applied and resulted in the discovery of the US FDA-approved AD drug, memantine (an NMDAr antagonist) [22]. In addition, a combined therapy that targets both Aβ and tau protein might be applied if we could localize the safest and most effective receptors in the interplay of these two proteins. Indeed, several compounds have been developed to inhibit GSK-3 activity, such as tideglusib and AZD1080. However, the former completed a phase II study without demonstrating significant clinical improvements in subjects with AD, whereas AZD1080 has completed a phase I trial with a confirmed target engagement and good safety and tolerability profiles [23].
The second factor that matters is the temporal relationship between Aβ and tau proteins. It is believed that if we inhibit Aβ to a certain extent before it is sufficient enough to initiate tau hyperphosphorylation, then the subsequent irreversible damage can be prevented. Therefore, the timeframe of tau initiation by Aβ also plays an important part in deciding the best time to treat patients. It helps us in plotting the ‘golden period’ of treatment, where an initiation of therapeutic agents will confer maximal protective or even curative measures to the patient. A proposed refined Aβ cascade hypothesis by Karran et al. [24] can be considered excellent guidance in addressing this timeframe issue. In the corresponding hypothesis, the role of Aβ can be divided into three mutually exclusive conditions, i.e., Aβ as a trigger, threshold, or driver of the subsequent tau pathology (Fig. 2). The Aβ trigger scenario implies that once Aβ deposits reach a certain amount, a self-sustained, irreversible activation and acceleration of pathologic tau protein would occur. At this point, any efforts to reduce Aβ load would be of no value. The threshold scenario is different from the former in that tau activation is still reversible as long as the Aβ aggregate stress could be reduced to certain levels. Finally, the driver scenario presumes that Aβ deposition and tau activation is reversible regardless of its levels, and any therapeutic measure given at various timeframes would exert molecular and biochemical improvements in a dose-dependent manner.

Schematic diagram of a relationship between the extent of Aβ deposition and the progression of Alzheimer’s disease. Aβ deposits in the brain begin to accumulate as early as the prodromal or asymptomatic state where aggressive efforts to inhibit or clear Aβ may potentially prevent tau activation, thereby halting the disease before it becomes clinically manifested (Aβ trigger scenario). If no adequate therapeutic interference is given, there is still a critical period where an intervention to reduce Aβ might be of benefit to inhibit Aβ-dependent tau acceleration, despite that there may have been subtle cognitive decline as a consequence of neuronal and synaptic loss due to the role of toxic soluble Aβ oligomers (Aβ threshold scenario). Later, when the ‘golden period’ of administering an appropriate anti-Aβ agent has ended, tau phosphorylation and aggregation begin to accelerate significantly, becoming self-sustaining and independent of Aβ production and reduction rate or loads, which is simultaneously followed by marked cognitive and functional status decline. When the last event occurs, the natural history of the disease has reached the ‘point of no return’ where an administration of anti-Aβ agent could only, if anything, reduce clinical and biomarker parameters in a dose-dependent manner, without any chance to revert the disease severity (Aβ driver scenario). Aβ amyloid-beta, AD Alzheimer’s disease, MCI mild cognitive impairment
It is critical that the most likely Aβ–tau timeframe scenario be confirmed, since it will influence the selection criteria of patients with AD included in drug trials. One of the major problems behind the failure of most AD therapeutic trials is the selection of subjects who have already developed cognitive impairments (i.e. MCI, mild-to-moderate AD). This is considered too late, because once clinical symptoms are noticed, a linear degree of synapse and neuron destruction is already in place. In fact, the extent of Aβ plaques and NFT deposition correlates with the degree of cognitive impairment in a relatively dose- and anatomical-dependent manner [25]. When these plaques are reduced, no cognitive and functional improvements are seen, and the progression of the disease is ongoing. These findings apparently deny the Aβ driver scenario, thus emphasizing the importance of a preventive rather than a curative approach in managing AD.
Accumulating evidence thus favors the existence of either the Aβ trigger or threshold scenario. In fact, minute concentrations (sub-nanomolar range) of Aβ oligomers are proven to be able to induce neuritic cytoskeleton collapse [26], further supporting these scenarios. Interestingly, Aβ exerts its harmful effect via a tau-dependent mechanism. Furthermore, Aβ can induce tau hyperphosphorylation in the absence of plaques, and the administration of Aβ can reverse the cytoskeletal alteration and neuritic degeneration [26], suggesting that the trigger scenario might be true. However, with respect to the recent conducted research, no data yet exist that can confirm the independence and self-autonomy of tau hyperphosphorylation and propagation in the absence of Aβ once it is activated. One way to confirm this would be to conduct an animal study with a model that completely eliminates the Aβ (its levels and clearance activity can be measured with a metabolic labeling method [27]) once it activates tau protein, and see if the cytoarchitectural destruction continues.
Translating this into the design of future clinical trials, the most ideal candidates would be subjects who are at risk of suffering AD, particularly the known inherited autosomal-dominant amyloid precursor protein (APP), presenilin (PSEN)-1, and PSEN2 mutations but should have no trace of Aβ deposits when scanned using Pittsburgh compound B (PIB)-positron emission tomography (PET) imaging. Subsequently, these subjects should be monitored rigorously over a long-term cohort study in terms of clinical (cognitive and functional performances) and biological (plasma and cerebrospinal fluid [CSF] Aβ and tau levels and deposits, brain volume imaging) parameters.
2 Alzheimer’s Disease Diagnosis: A Proposal for Revision
The existing AD diagnostic criteria that have been used for more than 30 years emphasize the presence of cognitive impairments in order to make a probable diagnosis, and postmortem analysis is required for creating a definitive diagnosis. Given the evolution and current knowledge gained from AD pathogenesis and treatment, the criteria are considered somewhat irrelevant and might potentially hamper the progress of therapeutic research.
Several reasons exist for this issue. First, the current approach of targeting already symptomatic patients has failed to show cognitive improvements in many trials, indicating that once the disease manifests clinically, there may be no way to reverse it. Second, there is a long time gap between the onset of detectable Aβ accumulation and the diagnosis of cognitive impairment. Thus, therapy should be initiated early during this ‘latent’ pre-symptomatic period, instead of waiting until the clinical manifestation is apparent. Third, significant advancements have been made in the diagnostic field where Aβ deposition and tau hyperphosphorylation in the pre-symptomatic period can be detected using biofluid biomarkers and diagnostic modalities such as PET scan and functional MRI (fMRI) with high sensitivity and specificity. To maintain the scope of discussion, we only explore the third factor in this review.
Two main types of biomarkers are currently being developed in AD, i.e., fluid and imaging biomarkers. Fluid biomarkers are derived from the CSF by lumbar puncture, whereas imaging biomarkers involve the injection of a radioactive tracer and subsequent brain scan by PET, or the use of fMRI without radioactive tracer. Three proteins considered to be the ‘gold standard’ of CSF biomarkers are Aβ42, total tau (t-tau), and phosphorylated tau (p-tau). Generally, the presence of low Aβ42 levels and elevated t-tau and p-tau levels predict the progression of AD in a highly sensitive and specific fashion.
CSF Aβ42 is the core biomarker, where it usually decreases (by 50 %) in the presence of AD [28]. On the other hand, the levels of CSF t-tau and p-tau usually increase by up to 300 % in the same population. One of the potential explanations for the reduced levels of CSF Aβ42 is the incorporation of this soluble amyloid into plaques that tend to be ‘sticky’ in the brain [29]. Furthermore, these three biomarkers, when used in conjunction, can yield a higher diagnostic accuracy. One study found that the simultaneous, rather than individual, use of Aβ42 and t-tau can increase sensitivity to 86 % (vs. 78–84 % when used alone) and specificity to 97 % (vs. 84–90 % when used alone) [28]. Interestingly, p-tau is known to be more specific than Aβ42, because p-tau levels do not increase in brain inflammation other than AD (e.g. Creutzfeldt–Jakob disease, traumatic brain injury, or stroke) [28, 30]. Moreover, the p-tau elevations in pure tauopathy such as FTLD typically do not exceed those found in patients with AD, thus keeping this biomarker specific and reliable in predicting and distinguishing AD among other neurological diseases [31].
However, given the importance of disease prevention in asymptomatic individuals at high risk of developing AD, rather than simply establishing the diagnosis (which is considered too late for treatment), the use of biomarkers in the future will be of greatest value if they can detect the likelihood of someone getting dementia far before it happens, instead of confirming a diagnosis in the presence of overt clinical declines. The studies are ongoing, but several findings have confirmed this notion. For example, Aβ42 levels were used to predict future memory impairments in 57 healthy individuals 3 years before symptoms appeared [32]. Again, low levels of Aβ42 were associated with a greater likelihood of the disease. In this study, the combined use of Aβ42 and p-tau yielded a diagnostic sensitivity and specificity of 71.4 and 75.7 %, respectively. Similar findings are also found in other studies with longer durations of follow-up (8 years), indicating the consistency of the results [33, 34]. In addition, newer studies have used the tau/Aβ42 ratio in conjunction with Aβ42 and p-tau levels to increase the diagnostic yield, in which the higher the ratio, the greater the likelihood of someone experiencing cognitive decline [35, 36].
Besides fluid biomarkers, the development of imaging biomarkers for predicting AD is also promising. For instance, three amyloid tracers have currently been approved by the US FDA, i.e., florbetapir (Amyvid), flutemetamol (Vizamyl), and florbetaben (Neuraceq) [29]. PET scan with florbetapir, when compared with postmortem examinations, can yield a sensitivity and specificity of 96 and 100 %, respectively [37]. Interestingly, it also differentiates AD and FTLD with good accuracy, although to a lesser extent than in healthy subjects [38]. On the other hand, flutemetamol-based PET imaging is able to distinguish AD from older healthy controls with a sensitivity and specificity of 97.2 and 85.3 %, respectively [39].
PET scan is considered superior relative to fluid biomarkers because it can locate regional areas in the brain affected by the plaques. This is critical, since plaque formation follows a predictable pattern, thus different regions affected will exert different cognitive impairments [25]. Locating these affected regions may help specify the cognitive criteria used for detecting subtle cognitive changes, thus enhancing the performance of early detection.
The combination of fluid and imaging biomarkers can further strengthen the diagnostic performance over either method used alone. Several studies have confirmed their strong correlation [40, 41]. However, one head-to-head study has shown a better specificity of florbetapir-based PET scan compared with CSF Aβ42 [42].
In response to these diagnostic advances, two sets of novel AD diagnostic criteria have been proposed by the International Work Group (IWG) [43] and the National Institute on Aging and the Alzheimer’s Association (NIA-AA) working group [44]. Both have included the biomarker aids in the list, but only the criteria proposed by IWG require the presence of biomarker abnormalities to diagnose AD. In addition, despite both sets accepting MCI as part of the AD spectrum, only the NIA-AA criteria permit the diagnosis of AD in the absence of cognitive symptoms, and they also permit subjective impairment in memory and non-memory domains as part of the cognitive impairment in MCI individuals. Regardless of these differences, the addition of MCI, high-risk categories, and the use of biomarkers in the diagnostic sets will certainly increase the performance of early detection and prevention, and exert a positive impact on finding disease-modifying agents.
3 Critical Reviews on Current Drug Trials
To date, potential candidates for disease-modifying agents have been based on three key premises, i.e. inhibition of Aβ formation, prevention of Aβ aggregation, and increase in Aβ plaque clearance. To maintain the focus of this article, we limit the discussion to four types of therapeutic candidates, each with several prominent compounds described.
3.1 Beta-Secretase (BACE)-1 Inhibitors
The rationale for the development of beta-secretase (BACE)-1 inhibitors is valid and based on the amyloidocentric concept. However, most of these drugs have had disappointing results in clinical trials. Several factors should be taken into account. In terms of technical issues, the active site of the enzyme is large and complex, and various important physiological BACE-1 substrates exist (i.e. more than 100), which increases the likelihood of physiological disruption of other cellular machinery [45, 46]. Second, the agents were not used for long-term administration because of rapid metabolism and clearance in mice. This is true for LY2811376, the first oral non-peptidic BACE-1 inhibitor that entered human trials. Although the in vitro study demonstrated a tenfold binding selectivity of this agent toward BACE-1 over BACE-2, and over 50-fold specificity to bind BACE-1 over cathepsin D, pepsin, or renin [47, 48], testing of the drug was still aborted before phase II trials because of significant adverse effects in the form of enlargement of retinal epithelial cells and retinal photoreceptors and neuronal degeneration in LY2811376-treated rats [47]. The toxicity is thought to be due to the disruption of type 3 neuregulin-1, a known substrate that, besides APP, is responsible for the myelination process, as a result of a lack in or absence of BACE-1 [49, 50]. Similarly, a phase II trial of LY2886721 (NCT01561430) was suspended when four patients experienced abnormal liver function markers, suggesting a possible liver toxicity. The company suspected this adverse effect was off-target, unrelated to the BACE-1 inhibition. Nevertheless, another hypothesis proposed that the compound actually inhibited another BACE-1 substrate, β-galactoside α-2,6-sialyltransferase I (STGal6 I), thus contributing to the hepatotoxicity found in several subjects [51].
During pre-clinical studies, these drugs have a clear target engagement, high penetrance of the blood–brain barrier, and one (LY2811376) was shown to be very selective [47, 48]. However, the problem lies in organ toxicity resulting from inhibition of physiological processes. Moreover, administration in rats was not long-term due to its high metabolic clearance. This skipped step is important in differentiating whether the compound works more in a therapeutic or preventive manner, suggesting a gap in our understanding of its mechanism of action. Both of these issues should have been addressed before the compounds entered into human trials, since the failure might have been identified when these compounds were tested in higher complex mammals such as translational rhesus monkey models, which exhibit a relatively lower rate of clearance and drug metabolism than transgenic mice.
Second, from a diagnostic viewpoint, the biomarker used to evaluate disease progression is deemed insufficient. Although BACE-1 inhibitors work mainly to reduce the production of soluble Aβ (and thus decrease the production of CSFAβ42), it is possible that disease progression can be suppressed molecularly (i.e. reduced levels of synaptic and neuronal damage). Therefore, other biomarkers that better reflect the synaptic damage, such as t-tau and p-tau, should be measured at trial baseline and midpoint. These biomarkers, if included, are also beneficial in testing the Aβ hypothesis scenario of whether or not optimally inhibiting soluble Aβ can reverse disease progression (Aβ trigger, threshold, and driver), since BACE-1 activity was significantly reduced to 50–75 % by LY2886721 on day 14 [52], and the patients included in the trial were in the early stages of the disease spectrum (MCI and mild AD).
Third, from a philosophical viewpoint, the drug, despite its toxicity issues, might be potent if used early in the disease stage, i.e. when given to pre-symptomatic but high-risk individuals, and given in combination therapy with tau-targeted drugs. These notions, although unlikely for already discontinued drugs, may fuel the discovery of other compounds within the same class. Indeed, one promising BACE-1 inhibitor is currently undergoing phase III trials: MK-8931 (NCT01739348). MK-8931 has a good target engagement, clear pharmacokinetics and pharmacodynamics, and is currently undergoing two phase III trials (EPOCH and APECS trial) consisting of up to 1960 and 1500 patients, respectively [53, 54]. The APECS trial is considered to be more preventive because the patients recruited have mild MCI with positive Aβ deposits as confirmed by PET scan and CSF biomarkers (tau:Aβ42 ratio), as opposed to patients in the EPOCH trial, who have mild-to-moderate AD. However, neither trial should be anticipated with great optimism, since tau hyperphosphorylation might already have occurred, irreversibly damaging neurons and synapses, and thus, ensuring progression of the disease, assuming there is no point to the inhibition of Aβ once tau has been activated.
3.2 γ-Secretase Inhibitors
To date, two prominent γ-secretase inhibitors (GSI) have been tested in human trials. Semagacestat (LY450139) is a potent GSI that acts as a noncompetitive inhibitor of the substrate. An in vitro study and two subsequent animal studies have confirmed the target engagement, mechanism of action, and pharmacokinetics of the agent [55–57]. However, based on preclinical data, the pharmacology of semagacestat is somewhat intricate, demonstrating initial lower plasma Aβ concentrations with high doses, but gradually increasing as the compound concentrations diminished (so-called plasma Aβ rebound phenomena) [58, 59]. This was thought to be due to a shift in the production of beta-C-terminal fragment (β-CTF), an intermediate byproduct of APP cleavage by BACE-1, as a consequence of γ-secretase inhibition [60]. It was further complicated by the fact that semagacestat also inhibits Notch signaling with a very thin margin as compared with γ-secretase inhibition, suggesting non-selective inhibition, despite its higher selectivity versus tarenflurbil [10, 61]. Notch signaling is essential in cell fate determination, and disruption of this process can cause tumor stem cell instability, acting as an oncogene, and promoting angiogenesis, and therefore may increase the risk of cancer [62–64]. These two problems should have been sufficient to suspend semagacestat prior to advancing into human trials. In reality, when continued until phase II trial, the drug’s predictable side effects related to Notch inhibition were observed (e.g. skin rashes, hair color change, gastrointestinal [GI] complaints). In fact, the phase III trial was terminated prematurely due to the significant adverse effects related to Notch inhibition.
Semagacestat development and testing was not thorough and systematic. In terms of pharmacokinetics, the plasma-rebound phenomena should have raised suspicion of a cleavage shift, and the byproduct effects on neurotoxicity should have been assessed carefully. In terms of its safety profile, semagacestat obviously inhibits Notch signaling and this has been shown in the phase II trial [65]. These adverse effects should have been addressed in a phase I study [65]. It seems that the phase I study lacked even the minimum criteria, wherein the compound was tested on healthy subjects (as opposed to patients with AD) on a single visit basis, involving observation for up to 12 h but no further follow-up. Therefore, target engagement, mechanism of action, pharmacokinetics, and safety profiles cannot be assessed. In addition, although an in vitro study raised concerns regarding Notch inhibition, this too was not addressed in the subsequent patient follow-up at phase I. Therefore, this phase I study seems to be a mere ‘formality’ rather than a prerequisite to gather qualified evidence.
The situation for avagacestat (BMS-708163) is similar. Despite claims that the drug is 193-fold more selective to APP than Notch [66], it also led to a higher discontinuation rate over placebo during the phase II trial, primarily due to skin and GI complaints. However, its development was more robust because its safety profile was tested in several preclinical studies and re-assessed more carefully in multiple phase I studies by administering multiple different doses to patients with AD, the right population with the right objectives [67–70]. After good therapeutic response with minimal adverse effects was observed, the drug was escalated to a phase II trial; again, with primary endpoints regarding the safety and tolerability of the compound [66]. Although the compound was eventually deemed to have failed, the phase II study is highly informative. For example, the drug only modestly reduced Aβ42 and elevated shorter forms of Aβ levels (Aβ14, Aβ15, Aβ16) due to a cleavage shift to C99 fragment by the α-secretase pathway [66, 71]. The associated cognitive decline among the subjects in the absence of brain atrophy (as measured by MRI), might be due to the accumulation of these shorter forms of Aβ, as they are also proven to be neurotoxic [72]. In addition, levels of t-tau and p-tau were reduced by 10 and 20 %, respectively. These reductions imply that avagacestat inhibition of Aβ can influence tau production as its downstream product, therefore confirming the Aβ–tau cascade hypothesis. If the results had been accompanied by cognitive improvement, we would have had robust evidence that pursuing amyloidocentric treatment is still worthwhile in attempts to modify the course of AD.
3.3 γ-Secretase Modulators
The concept of GSM was initially inferred when epidemiological studies indicated that the use of nonsteroidal anti-inflammatory drugs (NSAIDs) could become a protective factor for AD. Indeed, several NSAIDs, including sulindac, indomethacin, and ibuprofen, were proven to reduce Aβ42 production and shift toward shorter Aβ forms with a mechanism independent of cyclooxygenase (COX) inhibition [73–75]. This finding opened the door to a new therapeutic concept in which, instead of attempting to inhibit γ-secretase activity, the compound shifts the cleavage site of the corresponding enzyme N-terminally by one helical turn (3.6 amino acids) [76]. Thus, the compound does not interfere with other substrates like Notch and does not increase plasma Aβ40 concentrations. The first such compound ever tested in human trials was R-flurbiprofen (which was renamed ‘tarenflurbil’), a less potent COX inhibitor with the propensity to shift γ-secretase cleavage site, resulting in the production of shorter forms of Aβ. In vivo preclinical data demonstrated positive results toward reductions of brain Aβ40 with administration of 32 mg/kg/day for 9 days [77]. Similar findings were also seen with administration of 10 and 25 mg/kg/day; whereas one study did not show any reductions in brain Aβ40 and Aβ42 levels after flurbiprofen had been administered [78, 79].
Although less robust, these preclinical findings were translated into a phase I trial testing multiple dose regimens administered to 48 healthy elderly subjects for 21 days [80]. Although the drug was well tolerated, it did not reduce either plasma or CSF Aβ42 concentrations. A subsequent phase II study found a lower rate of decline in activities of daily living (ADL) in patients with mild AD when compared with placebo, whereas no significant effects were observed on the Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-cog) or the Clinical Dementia Rating scale Sum of Boxes (CDR-SB) in any group [81]. In addition, no information was provided regarding biomarker changes. Ultimately, an 18-month phase III trial was conducted, involving 1684 patients with mild AD receiving either tarenflurbil 800 mg twice a day or placebo [82]. No marked improvements were observed in cognitive or functional performance in the treatment group compared with placebo. The tarenflurbil group exhibited an increased incidence of dizziness, anemia, and infections.
In the case of tarenflurbil, the compound’s target engagement and mechanism of action were not clearly elucidated. The hypothesis was weakly supported and tended to overlap with the concept of anti-inflammation. The corresponding pharmacodynamic profile of the agent was not thoroughly assessed, and there were no indications that the drug would reduce brain Aβ at therapeutic dosages. Again, the phase II study was not equipped with biomarker information, which is essential because such a short trial will not affect cognitive change (if any) as rapidly as a change in biomarker levels. Moreover, the compound was not able to reach the CSF in an adequate concentration, i.e., theoretically sufficient to exert pharmacological effects. The drug should not have been escalated into clinical trials, whereby results were once again disappointing.
3.4 Immunotherapy
The concept of AD immunotherapy originated from the minimal Aβ plaque formation as well as minimal cyto- and synapse-architectural damage, observed after introducing a Aβ42 vaccine in APP transgenic mice, both before and after the onset of AD [83]. The immunization also induced better cognitive preservation and prevented learning and memory deficits when compared with older non-immunized transgenic mice [84]. Interestingly, cognitive impairment was reversed without apparent reduction of the total brain Aβ levels [85], suggesting that inhibition of Aβ in the presence of positive plaque is still effective in modifying the disease course, thereby supporting either Aβ threshold or driver scenarios.
To date, several compounds have successfully entered clinical trials. The first active second-generation immunotherapy is AN1792, which consists of Aβ42 peptide as the immunogen, along with QS21, a potent adjuvant that induced strong T-helper (Th)-1 responses. This was administered to patients with mild to moderate AD in a phase I trial [86, 87]. After being deemed safe and tolerable in phase I, it was tested in a phase II trial to determine the safety, tolerability, and pilot efficacy of the compound; however, development was halted due to a significantly higher incidence of meningoencephalitis in the treatment group [88]. The addition of polysorbate 80 during the final period of the phase I trial was thought to be the underlying trigger for the inflammatory process, since the agent caused a shift from Th-2 to the pro-inflammatory Th-1 activation [89].
Subjects who were vaccinated with AN1792, including those who were later diagnosed with Lewy body dementia, demonstrated decreased, or even a lack of, parenchymal Aβ plaque mainly in the cerebral cortex, both frontal and temporal [90–93]. Mechanistically, Aβ clearance was predominantly due to phagocytosis by microglia and macrophages, suggesting cellular, rather than humoral immune responses [90–92]. This was not the anticipated result, because the objective of the vaccine was supposed to be the activation of the opposite mechanism, thus generating a specific and long-term clearance and neuroprotection. If humoral response is prioritized, then attempts to minimize Th-1 activation should be sought. This can be done, for example, by replacing Aβ42 self-antigen, an antigen that is used in AN1792 development, with a foreign synthetic epitope such as pan human leukocyte antigen DR-binding peptide (PADRE), while keeping Aβ42-derived B epitope intact [94, 95]. In addition, total soluble Aβ was increased in gray and white matter [96], suggesting that the toxic Aβ oligomers were trapped in the cortex. This implies that the vaccination approach is not suitable for treating late AD cases wherein Aβ plaque already exists in abundance [96]. Alternatively, future vaccines must also assist in clearance and this should be confirmed in terms of target engagement and a proof-of-concept mechanism of action before it progresses to human trials. Another issue is the unaltered densities of NFTs, neuropil threads, and cerebral amyloid angiopathy in the cortical areas [90–92, 97], implying that the vaccine did not promote specific clearance of plaques other than Aβ. However, t-tau and p-tau levels should be measured if we are to address the direct impact of Aβ inhibition on the new onset of tau hyperphosphorylation.
Overall, clinically, the treatment group demonstrated small but significant improvements in cognitive decline and a relatively slower rate of disease progression [86, 98]. Interestingly, almost 5 years after the vaccination period, responders still had low but detectable AN1792 antibodies and were proven to have significantly reduced functional decline when compared with placebo, along with a similar rate of brain loss within the two groups [99]. AN1792, along with the concept of active vaccination, is actually an excellent therapeutic candidate when compared with the previous drug approaches discussed, whereby it is the only agent to date that has markedly reduced Aβ plaques. However, the main problem is the strong induction of cellular-mediated immunity and that the vaccine was administered in the ‘late’ AD cases.
With regards to minimizing side effects, efforts to boost the immune system and plaque clearance should be better assayed pre-clinically. Any compound that is intended to boost the immune response should be rationally designed, have clear target engagement, and be properly screened, thus decreasing the possibility of excessive trigger of the immune system. Furthermore, the combined vaccine should also be first tested in canine models to better represent the human APP sequence and cleavage processing [100, 101]. Furthermore, the vaccine should be appropriately given to the right population, i.e. pre-symptomatic people at high risk of developing AD in the future. In that way, it acts in a preventive pre-exposure prophylaxis paradigm, which is the essence of an active vaccination strategy.
In response to AN1792 studies, three active immunizations that aim to elicit strong humoral immune responses while avoiding excessive Th activation have been developed. CAD106, a combination of Aβ1-6 derived from N-terminal and Qβ virus-like particles have passed a phase IIa trial with sustained antibody titers and good safety profiles, although paradoxically it showed increased total plasma Aβ concentration with no difference in t-tau and p-tau from baseline levels [102]. The other agent, ACC-001 (vanutide cridificar) also has passed phase I study with good antibody response and safety profiles, without any meaningful serious adverse effects due to Th1 activation, despite using QS21 [103]. The agent is being further evaluated in a phase II study (ACCTION), incorporating PET scan, brain volumetric MRI, and CSF biomarkers as diagnostic and monitoring adjuvants. In addition, affitope (AD-02) has also progressed to phase II trial after the previous phase I study confirmed its safety profile. However, given that all these trials involve patients with mild AD (instead of the pre-symptomatic high-risk AD), promising results, other than more lessons to learn, might not be anticipated.
Passive immunotherapy was developed as an alternative approach because of the strong immune response elicited by active immunization, and the difficulty of finding the most appropriate Aβ epitope to target with maximum clearance. Bapineuzumab is a humanized 3D6, a highly specific mouse monoclonal antibody (mAb) (immunoglobulin [Ig]-G2b) targeting Aβ amino acid residues 1–5 [104]. A preclinical study demonstrated that the administration of bapineuzumab into APP transgenic mice for 6 months could reduce Aβ plaques by 86 % [105]. The proposed mechanism of action is likely due to Fc-mediated microglial phagocytosis [104], although it was later suggested that bapineuzumab tends to prevent the aggregation, and thus deposition of, Aβ, rather than modifying the existing plaques [106]. Furthermore, although a murine study revealed that 3D6 induced overt microhemorrhage, the compound was escalated to phase I trial, in which three subjects who had been given the highest dose of the compound (5 mg/kg) developed vasogenic edema (ARIA-E), and one of them suffered from microhemorrhage (ARIA-M) [107]. In this phase I study, brain Aβ levels as represented in the CSF were not measured, instead plasma Aβ levels were used as the parameter of increased clearance. Plasma Aβ can also be produced by other organs, which may lead to an erroneous interpretation.
In conclusion, the side effects seen in an animal study, which may occur in a dose-dependent manner, should have been investigated seriously. However, bapineuzumab was advanced into human trials where there was also no proof-of-concept regarding mechanism of action of whether it could clear existing Aβ plaques. Given the less equipped information gathered from the phase I study, it was no surprise when the phase II and III trials failed to demonstrate any cognitive improvements in the treatment group, although the levels of p-tau were found to decrease [108, 109]. The decrease in p-tau suggested that by targeting the new Aβ formation or the less likely enhancement of its clearance, one can expect to see the decrease of p-tau as the downstream cascade in the amyloidocentric hypothesis, even in the presence of overt cognitive impairments. However, the decrease in p-tau does not mean that cognitive functions are also improved. In other words, even when we are able to reduce the levels and activities of hyperphosphorylated tau as the prime culprit of cognitive decline, a certain threshold of Aβ deposition exists from which, once reached, there is no return.
Solanezumab, another passive immunotherapy, is a humanized IgG1 mAb derived from m266. It is recognized for its ability to bind soluble Aβ but not the already established plaques [110–112]. Solanezumab demonstrated a clear target engagement and mechanism of action pre-clinically by binding to Aβ 16–24 epitope and capturing peripheral Aβ. It was also deemed safe in the animal study with no apparent induction of microhemorrhage. Indeed, a phase I study confirmed its safety profile with no evidence of microhemorrhage and its associated vasogenic edema. The phase II trial also demonstrated similar results, confirming peripheral efflux of Aβ from the CSF and no evidence of adverse effects, yet no improvements were seen in terms of cognitive and functional performance [104, 113]. In this case, a clear target engagement and mechanism of action, plus good biomarker changes were not translated into clinical improvements. The problem may not lie with the vaccine, but rather that the subjects involved had mild-to-moderate AD. Given the compound’s main action is to clear the newly formed Aβ but not the existing plaques, the underlying pathological process and subsequent effects on cognition remains unaltered. Predictably, two phase III trials (Expedition 1 and Expedition 2) failed to show any cognitive and functional improvements among treatment groups when compared with placebo, along with unaltered Aβ and tau levels [114].
Despite disappointing results from the previous studies, several mAbs have been developed and progressed into clinical trials. Gantenerumab, a humanized mAb targeting the N-terminal and central amino acids of Aβ is being tested in 770 patients with prodromal AD during the ongoing phase II/III trial. Although pre-clinically the compound does not alter plasma Aβ, it preferentially binds to cerebral Aβ and reduces plaques by cell-mediated immune response [115]. However, the potential side effects of ARIA among the treatment groups are still of concern [116]. Another potential compound being developed is BAN2401, a humanized version of mAb158 which is 1000-fold more selective in binding Aβ protofibrils than monomers, thus ensuring that the agent works mainly by inducing a humoral immune response [117]. Indeed, it has passed the safety evaluations in phase I and IIa trials, and now is under investigation in a phase IIb trial.
Lastly, crenezumab (MABT5102 or RG7412) is a humanized mAb IgG4 targeting Aβ1–15 [118]. The compound was claimed to have immunogenicity against a wide range of multiple Aβ forms, including protofibrils and oligomers, exerting its effect through inhibiting aggregation and promoting plaque disaggregation. Of note, IgG4 backbone was chosen to minimize the over-activation of Fc-gamma receptor that would recruit the microglia and potentially result in harmful side effects [119]. Although the target engagement is unclear, the candidate has passed the safety evaluation of a phase I trial with no evidence of inducing ARIA-E or ARIA-M and has been tested in two phase II trials (ABBY and BLAZE trials). The ABBY trial involved 431 patients with mild-to-moderate AD given 15 mg/kg/month of the compound, while the BLAZE trial was a second smaller phase II study that involved 91 patients with mild-to-moderate AD [120]. The compound is also being tested in pre-symptomatic carriers of autosomal dominant pre-senilin mutations in comparison with placebo for a study duration of 5 years. The subjects do not exhibit any signs of MCI at the time of enrollment, while its primary outcome is to evaluate cognitive decline in both groups, including essential secondary outcomes, like time to progression to MCI, brain Aβ load, and imaging biomarkers.
The results were disappointing as the ABBY trial failed to meet its primary endpoints (improvement in ADAS-Cog12 and CDR-SB score) [121]. However, analyses of individuals with milder symptoms demonstrated a statistically significant 35.4 % reduction of cognitive decline in the treatment group when compared with placebo. This finding, again, stresses the importance of commencing the therapeutic effort as early as possible. Meanwhile, the BLAZE trial did not meet any of the study outcomes.
4 Conclusions and Further Directions
Efforts to find an effective disease-modifying therapy for AD are still ongoing but, to date, no single agent has been found to work. Several factors are proposed to play a role in this failure, including lack of understanding of several points in the Aβ cascade, currently ‘outdated’ diagnostic criteria, and various technical and methodological issues underlying therapeutic agent development and testing. Our understanding of several points in AD pathogenesis is still limited, mainly the precise temporal and mechanistic relationship between Aβ and tau protein. Getting relevant and adequate information about this is critical to the success of developing future therapeutic candidates.
It is important to note that despite aberrant tau phosphorylation playing a key role in cognitive decline, AD is a totally different disease from pure tauopathy, wherein the former is initiated by toxic Aβ formation and deposition with different pathophysiological processes. P-tau levels, for example, are markedly increased in AD when compared with FTLD. Moreover, p-tau levels that can be substantially reduced by a certain anti-Aβ (for example, bapineuzumab) do not correlate with cognitive and functional improvements in subjects with AD. Therefore, while it is acceptable to pursue tau-related drug development, we should not totally abandon amyloidocentric-based efforts. Instead, gaps that existed in past drug development should be learned from.
In addition, AD is a disease with multiple risk factors, and ‘peripheral’ pathologies in the Aβ cascade also contribute to the disease. Among these, cardiovascular risk factors are thought to play roles. Indeed, accumulating evidence suggests that hypertension and dyslipidemia are risk factors for developing AD in the future [122–124]. Therefore, aggressive approaches to treat the underlying cardiovascular risk factors should be thoroughly investigated to determine whether or not they also prevent the development of AD. In fact, several trials have investigated this (PreDIVA and FINGER trials), and one has found a large proportion of elderly have one or more cardiovascular risk factors and thus become the potential target to be controlled for also lowering the risk of dementia [125, 126]. Modifications of these risk factors with medications and lifestyle changes can readily be combined and vigorously emphasized in AD prevention, along with other potential anti-Aβ compounds being investigated.
With regards to technical issues, any therapeutic candidate should ideally undergo a standardized and thorough investigation in a systematic style (Fig. 3). In order to gain meaningful outcomes, the drug should ideally be developed in a stepwise fashion, from in vitro testing, cell-based assay and in vivo testing, to subsequent phase I–III trials. Each step has its own parameters, i.e., in vitro testing evaluates target engagement and potency, and cell-based assays review the mechanism of action, confirm target engagement, record the penetrance of drugs into the blood–brain barrier, and identify selectivity. In animal studies, all of these parameters should be reconfirmed if possible, plus additional but critical evaluations performed such as assessment of safety profiles, dose conversion from animals to humans, and preliminary efficacy studies [104]. Later, in a phase I trial, the pharmacokinetics, pharmacodynamics, safety profiles, and secondary targets such as efficacy should be rigorously checked and conclusions from this trial should be carefully extrapolated and reconfirmed in the phase II trial, wherein safety and efficacy become the main concerns. Lastly, a phase III trial is supposed to confirm promising findings from the previous trials regarding efficacy and safety profiles with the addition of tolerability factors if the drug does not seriously affect the health, plus development of a drug regimen that would be useful in the clinical setting.

Several critical technical and methodological aspects that need to be addressed when conducting a search and test of a novel Alzheimer’s disease therapeutic candidate. Aβ amyloid-beta, AD Alzheimer’s disease, CSF cerebrospinal fluid, MRI magnetic resonance imaging, PiB-PET Pittsburgh compound B-positron emission tomography
With regards to the revision of diagnostic criteria, in the personal view of the authors, the NIA-AA criteria provide a better diagnostic timeframe and sensitivity than those of the IWG. They increase awareness that individuals with pathologic abnormalities in the absence of cognitive deterioration have already suffered from AD. Furthermore, it will accommodate future drug trials with more confidence when including and treating pre-symptomatic individuals in the absence of cognitive impairments, further emphasizing the importance of the preventive paradigm rather than waiting for the disease to become manifest. Of course, such a policy will raise ethical and legal dilemmas on the basis of treating individuals without overt clinical symptoms, not to mention if any serious adverse effects occur with long-term use. An aggressive and early treatment policy will also increase a nation’s financial expenditure, especially in countries with high numbers of aging people.
It is therefore very important to continuously refine our understanding of the pathogenesis of AD, in line with pharmacological developments in this area. For example, the dynamics of late-onset AD pathogenesis should be clearly elucidated and readily predicted with high accuracy using various tools, so that individuals at high risk can be treated. Such a concept is not new, and various diseases such as dyslipidemia and diabetes mellitus have been aggressively treated with anti-cholesterol and glucose-lowering agents before the occurrence of fatal complications like myocardial infarction, stroke, and renal failure. In terms of the cost, it is prudent to assume that preventing a disease in high-risk individuals is much more efficient than letting someone get AD, which is known to be expensive, both financially and in terms of its impact on quality of life.
References
Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24(4):219–24.
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155(3):853–62.
Dahlgren KN, Manelli AM, Stine WB Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277(35):32046–53.
Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, et al. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci. 2003;100(11):6370–5.
Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, et al. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. 2000;275(26):19439–42.
Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–57.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80.
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–9.
Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–500.
Giacobini E, Gold G. Alzheimer disease therapy–moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9(12):677–86.
Stancu IC, Ris L, Vasconcelos B, Marinangeli C, Goeminne L, Laporte V, et al. Tauopathy contributes to synaptic and cognitive deficits in a murine model for Alzheimer’s disease. FASEB J. 2014;28(6):2620–31.
Pievani M, de Haan W, Wu T, Seeley WW, Frisoni GB. Functional network disruption in the degenerative dementias. Lancet Neurol. 2011;10(9):829–43.
Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, et al. Amyloid-beta–associated clinical decline occurs only in the presence of elevated P-tau. Arch Neurol. 2012;69(6):709–13.
Corrada MM, Berlau DJ, Kawas CH. A population-based clinicopathological study in the oldest-old: the 90+ study. Curr Alzheimer Res. 2012;9(6):709–17.
Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–28.
Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5.
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59.
Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–72.
Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov. 2009;8(10):783–93.
Reddy PH. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011;1415:136–48.
Cosman KM, Boyle LL, Porsteinsson AP. Memantine in the treatment of mild-to-moderate Alzheimer’s disease. Expert Opin Pharmacother. 2007;8(2):203–14.
Georgievska B, Sandin J, Doherty J, Mortberg A, Neelissen J, Andersson A, et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem. 2013;125(3):446–56.
Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712.
Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68(1):1–14.
Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci. 2011;108(14):5819–24.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774.
Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–44.
Fagan AM. CSF biomarkers of Alzheimer’s disease: impact on disease concept, diagnosis, and clinical trial design. Adv Geriatr. 2014;2014:14.
Hampel H, Blennow K, Shaw LM, Hoessler YC, Zetterberg H, Trojanowski JQ. Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp Gerontol. 2010;45(1):30–40.
Irwin DJ, Trojanowski JQ, Grossman M. Cerebrospinal fluid biomarkers for differentiation of frontotemporal lobar degeneration from Alzheimer’s disease. Front Aging Neurosci. 2013;5:6.
Stomrud E, Hansson O, Blennow K, Minthon L, Londos E. Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord. 2007;24(2):118–24.
Gustafson DR, Skoog I, Rosengren L, Zetterberg H, Blennow K. Cerebrospinal fluid beta-amyloid 1-42 concentration may predict cognitive decline in older women. J Neurol Neurosurg Psychiatry. 2007;78(5):461–4.
Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K. Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dement Geriatr Cogn Disord. 2003;15(3):169–76.
Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64(3):343–9.
Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69(7):631–9.
Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol. 2012;11(8):669–78.
Kobylecki C, Langheinrich T, Hinz R, Vardy ER, Brown G, Martino ME, et al. 18F-florbetapir PET in patients with frontotemporal dementia and Alzheimer disease. J Nucl Med. 2015;56(3):386–91.
Hatashita S, Yamasaki H, Suzuki Y, Tanaka K, Wakebe D, Hayakawa H. [18F]Flutemetamol amyloid-beta PET imaging compared with [11C]PIB across the spectrum of Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2014;41(2):290–300.
Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1(8–9):371–80.
Landau SM, Lu M, Joshi AD, Pontecorvo M, Mintun MA, Trojanowski JQ, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol. 2013;74(6):826–36.
Mattsson N, Insel PS, Landau S, Jagust W, Donohue M, Shaw LM, et al. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer’s disease. Ann Clin Transl Neurol. 2014;1(8):534–43.
Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger-Gateau P, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9(11):1118–27.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement J Alzheimer’s Assoc. 2011;7(3):263–9.
Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS One. 2009;4(12):e8477.
Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31(14):3157–68.
May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, et al. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci. 2011;31(46):16507–16.
Vassar R, Kuhn PH, Haass C, Kennedy ME, Rajendran L, Wong PC, et al. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem. 2014;130(1):4–28.
Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9(12):1520–5.
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, et al. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314(5799):664–6.
Lahiri DK, Maloney B, Long JM, Greig NH. Lessons from a BACE1 inhibitor trial: off-site but not off base. Alzheimer’s Dement. 2014;10:S411–9.
Alzforum. LY2886721. 2013. Available from: http://www.alzforum.org/therapeutics/ly2886721. Accessed 19 June 2015.
An Efficacy and Safety Trial of MK-8931 in Mild to Moderate Alzheimer’s Disease (P07738) (EPOCH) [database on the Internet]. Clinicaltrials.gov. 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT01739348. Accessed 19 June 2015.
Efficacy and Safety Trial of MK-8931 in Participants With Prodromal Alzheimer’s Disease (MK-8931-019) (APECS) [database on the Internet]. Clinicaltrials.gov. 2015. Available from: https://clinicaltrials.gov/ct2/show/NCT01953601. Accessed 19 June 2015.
Morgan D, Gitter BD. Evidence supporting a role for anti-Abeta antibodies in the treatment of Alzheimer’s disease. Neurobiol Aging. 2004;25(5):605–8.
May PC, Yang Z, Li W-Y, Hyslop PA, Siemers E, Boggs LN. O3-06-07 Multi-compartmental pharmacodynamic assessment of the functional gamma-secretase inhibitor LY450139 in PDAPP transgenic mice and non-transgenic mice. Neurobiol Aging. 2004;25:S65.
Hyslop PA, May PC, Audia JE, Calligaro DO, McMillian CL, Garner CO, et al. P1-180 Reduction in a-beta(1-40) and A-beta(1-42) in CSF and plasma in the beagle dog following acute oral dosing of the gamma secretase inhibitor, LY450139. Neurobiol Aging. 2004;25:S147.
Henley DB, May PC, Dean RA, Siemers ER. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin Pharmacother. 2009;10(10):1657–64.
Lanz TA, Karmilowicz MJ, Wood KM, Pozdnyakov N, Du P, Piotrowski MA, et al. Concentration-dependent modulation of amyloid-beta in vivo and in vitro using the gamma-secretase inhibitor, LY-450139. J Pharmacol Exp Ther. 2006;319(2):924–33.
Li T, Huang Y, Jin S, Ye L, Rong N, Yang X, et al. Gamma-secretase modulators do not induce Abeta-rebound and accumulation of beta-C-terminal fragment. J Neurochem. 2012;121(2):277–86.
Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31(10):2261–74.
Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012;30(19):2307–13.
Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–71.
Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16(2):196–208.
Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, et al. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin Neuropharmacol. 2007;30(6):317–25.
Coric V, van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012;69(11):1430–40.
Dockens R, Wang JS, Castaneda L, Sverdlov O, Huang SP, Slemmon R, et al. A placebo-controlled, multiple ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of avagacestat (BMS-708163) in healthy young and elderly subjects. Clin Pharmacokinet. 2012;51(10):681–93.
Tong G, Wang JS, Sverdlov O, Huang SP, Slemmon R, Croop R, et al. A contrast in safety, pharmacokinetics and pharmacodynamics across age groups after a single 50 mg oral dose of the gamma-secretase inhibitor avagacestat. Br J Clin Pharmacol. 2013;75(1):136–45.
Albright CF, Dockens RC, Meredith JE Jr, Olson RE, Slemmon R, Lentz KA, et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J Pharmacol Exp Ther. 2013;344(3):686–95.
Tong G, Castaneda L, Wang JS, Sverdlov O, Huang SP, Slemmon R, et al. Effects of single doses of avagacestat (BMS-708163) on cerebrospinal fluid Abeta levels in healthy young men. Clin Drug Investig. 2012;32(11):761–9.
Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, et al. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2011;32(6):1090–8.
Broersen K, Rousseau F, Schymkowitz J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: oligomer size or conformation? Alzheimers Res Ther. 2010;2(4):12.
Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, et al. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003;278(34):31831–7.
Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, et al. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Investig. 2003;112(3):440–9.
Townsend KP, Pratico D. Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19(12):1592–601.
Selkoe DJ. The therapeutics of Alzheimer’s disease: where we stand and where we are heading. Ann Neurol. 2013;74(3):328–36.
Imbimbo BP, Del Giudice E, Cenacchi V, Volta R, Villetti G, Facchinetti F, et al. In vitro and in vivo profiling of CHF5022 and CHF5074 Two beta-amyloid1-42 lowering agents. Pharmacol Res. 2007;55(4):318–28.
Peretto I, Radaelli S, Parini C, Zandi M, Raveglia LF, Dondio G, et al. Synthesis and biological activity of flurbiprofen analogues as selective inhibitors of beta-amyloid(1)(-)(42) secretion. J Med Chem. 2005;48(18):5705–20.
Lanz TA, Fici GJ, Merchant KM. Lack of specific amyloid-beta(1-42) suppression by nonsteroidal anti-inflammatory drugs in young, plaque-free Tg2576 mice and in guinea pig neuronal cultures. J Pharmacol Exp Ther. 2005;312(1):399–406.
Galasko DR, Graff-Radford N, May S, Hendrix S, Cottrell BA, Sagi SA, et al. Safety, tolerability, pharmacokinetics, and Abeta levels after short-term administration of R-flurbiprofen in healthy elderly individuals. Alzheimer Dis Assoc Disord. 2007;21(4):292–9.
Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: a randomised phase II trial. Lancet Neurol. 2008;7(6):483–93.
Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557–64.
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–7.
Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982–5.
Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. A[beta] peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408(6815):979–82.
Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology. 2005;64(1):94–101.
Wisniewski T, Frangione B. Immunological and anti-chaperone therapeutic approaches for Alzheimer disease. Brain Pathol. 2005;15(1):72–7.
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54.
Pride M, Seubert P, Grundman M, Hagen M, Eldridge J, Black RS. Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis. 2008;5(3–4):194–6.
Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9(4):448–52.
Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65(11):1040–8.
Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, et al. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64(1):129–31.
Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, et al. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. 2007;64(4):583–7. doi:10.1001/archneur.64.4.583.
Davtyan H, Petrushina I, Ghochikyan A. Immunotherapy for Alzheimer’s disease: DNA- and protein-based epitope vaccines. Methods Mol Biol. 2014;1143:259–81.
Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H, et al. Alzheimer’s disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Abeta species in amyloid precursor protein transgenic mice. J Neurosci. 2007;27(46):12721–31.
Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, et al. Amyloid-β Peptide Remnants in AN-1792-Immunized Alzheimer’s Disease Patients. Am J Pathol. 2006;169(3):1048–63.
Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14(1):11–20.
Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38(4):547–54.
Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009;6(2):144–51.
Robinson SR, Bishop GM, Lee HG, Munch G. Lessons from the AN 1792 Alzheimer vaccine: lest we forget. Neurobiol Aging. 2004;25(5):609–15.
Wisniewski T, Konietzko U. Amyloid-beta immunisation for Alzheimer’s disease. Lancet Neurol. 2008;7(9):805–11.
Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM. Active immunotherapy options for Alzheimer’s disease. Alzheimers Res Ther. 2014;6(1):7.
Arai H, Suzuki H, Yoshiyama T, Lobello K, Peng Y, Liu E, et al. Safety, tolerability and immunogenicity of an immunotherapeutic vaccine (vanutide cridificar [ACC-001]) and the QS-21 adjuvant in Japanese individuals with mild-to-moderate Alzheimer’s disease: a phase IIa, multicenter, randomized, adjuvant and placebo clinical trial. Alzheimer’s Dement. 9(4):P282.
Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2013;76(2):185–205.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–9.
Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, et al. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron. 2012;76(5):908–20.
Black RS, Sperling RA, Safirstein B, Motter RN, Pallay A, Nichols A, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24(2):198–203.
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24):2061–70.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–33.
Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25(3):629–36.
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 2001;98(15):8850–5.
Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, et al. Antibody capture of soluble Abeta does not reduce cortical Abeta amyloidosis in the PDAPP mouse. Neurodegener Dis. 2008;5(2):65–71.
Imbimbo BP, Ottonello S, Frisardi V, Solfrizzi V, Greco A, Seripa D, et al. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev Clin Immunol. 2012;8(2):135–49.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–21.
Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, et al. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimers Dis. 2012;28(1):49–69.
Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207.
Lannfelt L, Moller C, Basun H, Osswald G, Sehlin D, Satlin A, et al. Perspectives on future Alzheimer therapies: amyloid-beta protofibrils—a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther. 2014;6(2):16.
Moreth J, Mavoungou C, Schindowski K. Passive anti-amyloid immunotherapy in Alzheimer’s disease: what are the most promising targets? Immun Ageing. 2013;10(1):18.
Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;32(28):9677–89.
Watt AD, Crespi GA, Down RA, Ascher DB, Gunn A, Perez KA, et al. Do current therapeutic anti-Abeta antibodies for Alzheimer’s disease engage the target? Acta Neuropathol. 2014;127(6):803–10.
Melville NA. Crenezumab fails in Alzheimer’s but is there a silver lining? WebMD. 2014. http://www.medscape.com/viewarticle/828883. Accessed 26 Aug 2014.
Skoog I, Nilsson L, Persson G, Lernfelt B, Landahl S, Palmertz B, et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 347(9009):1141–5.
Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ. 2001;322(7300):1447–51.
Breteler MMB. Vascular risk factors for Alzheimer’s disease. Neurobiol Aging. 1996;21(2):153–60.
Richard E, Van den Heuvel E, Moll van Charante EP, Achthoven L, Vermeulen M, Bindels PJ, et al. Prevention of dementia by intensive vascular care (PreDIVA): a cluster-randomized trial in progress. Alzheimer Dis Assoc Disord. 2009;23(3):198–204.
Kivipelto M, Solomon A, Ahtiluoto S, Ngandu T, Lehtisalo J, Antikainen R, et al. The Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER): study design and progress. Alzheimer’s Dement J Alzheimer’s Assoc. 2000;9(6):657–65.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to assist in the preparation of this review.
Conflict of interest
Andreas Soejitno, Anastasia Tjan, and Thomas E. Purwata declare that they have no conflicts of interest that are relevant to the contents of this review.
Rights and permissions
About this article

Cite this article
Soejitno, A., Tjan, A. & Purwata, T.E. Alzheimer’s Disease: Lessons Learned from Amyloidocentric Clinical Trials. CNS Drugs 29, 487–502 (2015). https://doi.org/10.1007/s40263-015-0257-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-015-0257-8