
Abstract
Background and Objective
Henagliflozin, a selective inhibitor of the renal sodium glucose cotransporter-2, was developed for type 2 diabetes mellitus (T2DM). This study characterized single- and multiple-dose pharmacokinetics and pharmacodynamics of henagliflozin in Chinese patients with T2DM.
Methods
Thirty T2DM patients were randomized in a 4:1 ratio to orally receive either henagliflozin 5, 10, 20 mg/day or placebo for 10 days, except on day 2 and day 3. Pharmacokinetic and pharmacodynamic profiles were measured on day 1 and day 10.
Results
Henagliflozin exhibited dose-proportional plasma concentrations with a half-life ranging from 9.1 to 14 h. Steady-state plasma henagliflozin concentration was reached by day 7 in all active treatment groups. Henagliflozin decreased the 24-h mean plasma glucose by −0.3, −1.0 and −1.0 mmol/L with doses of 5, 10 and 20 mg on day 1, respectively. The corresponding values on day 10 were −0.8, −0.9 and −1.2 mmol/L. Twenty-four-hour urinary glucose excretion increased by 11, 65 and 82 times with doses of 5, 10 and 20 mg on day 1, respectively, with a similar trend on day 10. No treatment-related serious adverse events or discontinuations due to adverse events occurred.
Conclusions
The observed pharmacokinetic and pharmacodynamic profiles of henagliflozin support a once-daily dosing regimen in Chinese T2DM patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Steady-state plasma henagliflozin concentration was reached by day 7. Single- and multiple-dose administration resulted in dose-dependent decreases in mean plasma glucose. Taking the baseline values into account, 24-h urine glucose excretion increased in a dose proportional way. |
No treatment-related serious adverse events occurred. The observed pharmacokinetic and pharmacodynamic properties implied a once-daily dosing regimen in Chinese T2DM patients. |
1 Introduction
The increasing prevalence of type 2 diabetes mellitus (T2DM) represents one of the major health-economic challenges of the twenty-first century [1, 2]. The progressive nature of T2DM necessitates an intensification of treatment that may include increasing the dosage of oral antidiabetic agents and the introduction of additional medications in order to achieve treatment goals [3]. Novel antidiabetic agents, in particular those that work independently of insulin, are required.
The kidney contributes to maintaining normal blood glucose levels by reabsorbing about 180 g of glucose each day [4]. In the context of diabetes, blocking the reabsorption of glucose via the inhibition of sodium-glucose cotransporter-2 (SGLT2) has become an intriguing therapeutic strategy. Inhibition of SGLT2 is a rational insulin-independent therapeutic approach to lowering blood glucose through inhibition of renal glucose reabsorption [5, 6] resulting in a mandatory increase in urinary glucose and loss of calories.
Henagliflozin, an orally active inhibitor of SGLT2, is currently in development for the treatment of patients with T2DM. Henagliflozin is mainly metabolized by UDP-glucuronosyltrans-ferase in the liver with a plasma protein binding rate about 95 %. The main metabolites were three henagliflozin-glucuronide metabolites. The accumulative excretion rate of henagliflozin in urine is 3 %. In a previous pharmacokinetic study involving healthy Chinese subjects, henagliflozin was rapidly absorbed, with a T max (time at which the maximum concentration is observed) of 1.5–3.0 h and a half-life (T 1/2) of about 11.0–15.3 h. Doses of 20–50 mg provided near-maximal SGLT2 inhibition for 24 h, and systemic exposure increased dose proportionally. Henagliflozin did not reduce fasting plasma glucose in healthy people, and no serious adverse events (SAEs) were observed.
As indicated in the Permit from China Food and Drug Administration, the pharmacokinetic and pharmacodynamic properties of henagliflozin are required to be tested in T2DM patients. Therefore, the purpose of the current double-blind, randomized, placebo-controlled, single- and multiple-dose phase 1 study was to characterize the pharmacokinetics, pharmacodynamics, safety and tolerability of henagliflozin in Chinese T2DM patients (ClinicalTrials.gov: NCT02346175). The results will be applied to support the development of henagliflozin in China.
2 Methods
2.1 Study Population
The study was conducted in accordance with Good Clinical Practice as defined by the International Conference on Harmonisation, and Chinese State Food and Drug Administration, and Declaration of Helsinki. The study protocol and all amendments were reviewed and approved by the Clinical Research Ethics Committee of Chengdu Military General Hospital and Xijing Hospital. The investigators explained the nature and purpose of the study, participation/termination conditions, and the possible risks and benefits of treatment to the patients. All the patients provided informed consent.
Patients were eligible for inclusion in the studies if they met the following criteria: Chinese male and female subjects with a history of T2DM, aged 18–65 years with a body mass index (BMI) of 20–35 kg/m2. Eligible subjects were medically stable based on physical examination, medical history, laboratory results, vital sign measurements, and 12-lead electrocardiogram (ECG) at screening. In addition, eligible patients were on a stable regimen of antihyperglycemic therapy for at least 8 weeks prior to screening and had fasting plasma glucose (FPG) concentration <13.9 mmol/L at baseline. Stable antihyperglycemic regimens could include a single oral agent (e.g. metformin, a sulfonylurea, a thiazolidinediones ketone, a benzoic acid derivative, a dipeptidyl peptidase-4 inhibitor, or an α-glucosidase inhibitor) with glycated hemoglobin (HbA1c) between 7.0 and 10.0 %.
Major exclusion criteria included: pregnancy or breastfeeding; significant acute or chronic medical illness, including renal impairment (defined as serum creatinine >133 μmol/L for men and >126 μmol/L for women), or recent surgery; donation of blood or plasma within the 4 weeks prior to the start of the study; and/or prior exposure to any investigational drug or placebo within 4 weeks, or over-the-counter medications within 1 week before the start of the study; history of clinically significant diabetic complications, type 1 diabetes mellitus, or repeated severe hypoglycemic episodes.
2.2 Study Design
This was a two-center, double-blind, randomized, placebo-controlled, ascending-dose phase I study. Thirty patients were randomly assigned in a 4:1 ratio to receive either a multiple dose of 5, 10, or 20 mg henagliflozin (n = 8) or placebo (n = 2). Safety data were reviewed by the principal investigator and the sponsor. If a dose regimen (beginning with the lowest dose) was found to be safe and well tolerated, then the succeeding panel of eight subjects received the next higher dose of henagliflozin or placebo. Following a 3-week screening phase and a 3-week washout of antihyperglycemic agents for all subjects, eligible subjects entered the clinical research center on day −1 and underwent baseline safety and FPG and cumulative 24-h urine glucose excretion (UGE) assessments. Subjects were required to remain fasted for at least 10 h prior to administration of drugs and for 4 h after administration on the first day (day 1) and the last day (day 10). Intake of water was not allowed until 1 h after dosing. All subjects received standardized meals served at the study site at predetermined intervals.
Henagliflozin or the matching placebo were administered at the fasting state as oral tablets with 200 mL water on day 1 and day 4, then continued with the same daily dose for another 7 days (days 4–10). The time of dosing was recorded as 0 h. Pharmacokinetic profiles were determined in blood samples obtained immediately before 0 h, and at 0.25, 0.5, 1, 1.5, 2, 3, 6, 8, 10, 12, 24, 36, 48 and 72 h postdose commencing on day 1 (after a single dose) and commencing on day 10 (after multiple doses). At each time-point, 3 mL blood was drawn. Blood samples were also collected prior to dosing on days 7, 8 and 9 for the assessment of attainment of steady state on day 10. Blood was drawn from intravenous cannulae at 0 (immediately prior to dosing), 2, 4, 6, 8, 10, 12, 24 h postdose on day −1, day 1 and day 10 to measure plasma glucose. UGE was assessed from pooled 24-h urine collected at intervals ending at 4, 8, 12 and 24 h postdose on day −1, day 1 and day 10. Follow-up visits focusing on AE occurred 5–10 days after the last administration of study drug via phone call.
2.3 Bioanalysis
Analysis of plasma henagliflozin concentration was performed at Wuxi App Tec (Shanghai, China) using a validated LC/MS-MS [7] method and within the known period of drug stability in human plasma samples. All determinations of henagliflozin in plasma were generated in analytical runs using appropriate calibration curves and quality-control samples that met pre-established criteria, and were conducted in compliance with the applicable standard operating procedures in place at the time of analysis. The standard curve of henagliflozin in plasma ranged from 0.5 to 1000 ng/mL. The between-run variability and within-run variability for the analytical quality controls were less than 15 %. Glucose concentrations in plasma and urine were analyzed using the standardized hexokinase method at the study site. The estimated glomerular filtration rate (eGFR) was calculated using the following formula [8]:
2.4 Pharmacokinetic Measurements
Individual subject pharmacokinetic parameters including maximum observed plasma concentration (C max), time to maximum plasma concentration (T max), T 1/2, area under the plasma henagliflozin concentration–time curve from time zero to 24 h (AUC0–24) and to infinity (AUC∞) were derived by noncompartmental techniques (Phoenix WinNonlin 6.3; Pharsight Corporation). Accumulation ratio (AR) was defined as the ratio of AUC0–24 h at steady state (day 10) to AUC0–24 h after the first dose (day 1).
2.5 Pharmacodynamic Measurements
Pharmacodynamic measurements included FPG, 24-h mean plasma glucose (MPG), and UGE.
2.6 Safety Evaluation
The determination of AEs was based on the medical review of AE reports and the results of vital sign measurements, 12-lead ECGs, physical examinations, and clinical laboratory tests. Subjects were closely monitored for AEs throughout the study. Treatment-emergent adverse events (TEAEs) were defined as AEs that were new in onset or increased in severity or frequency following administration of study drug. Serious adverse events (SAEs) were defined as those that resulted in death; required inpatient hospitalization or prolongation of existing hospitalization; resulted in persistent or significant disability, congenital anomaly, or birth defect; or that were life threatening.
2.7 Statistical Methods
All subjects who received henagliflozin or placebo were included in the safety and pharmacodynamic populations. All subjects who received henagliflozin were included in the pharmacokinetic population. The pharmacokinetic and pharmacodynamic estimates were summarized by henagliflozin doses and study days. Analysis of variance (ANOVA) was used to investigate pharmacodynamic effects of henagliflozin. Paired t test was applied for the within-subject differences before and after treatment. Statistical significance was achieved if P value was <0.05.
3 Results
3.1 Demographics
Of the 30 patients, 24 were evenly randomized to receive a single oral dose of henagliflozin 5, 10 and 20 mg. Another six patients were assigned to receive placebo. Mean age was 50.8 years, and seven (23.3 %) were women (Table 1). Laboratory measurements, including plasma glucose concentration, HbA1c, and liver and kidney function at screening, were listed in Table 1.
3.2 Previous Antihyperglycemic Agents
As shown in Table 1, before screening, the numbers of patients who took metformin were 1 in the placebo group, and 3 in the 5 and 10 mg dose groups. In the placebo group, two patients used sulfonylurea or α-glucosidase inhibitor. One patient in the 10 mg dose group took sulfonylurea. The corresponding number was 2 in the 20 mg dose group.
3.3 Pharmacokinetic Properties
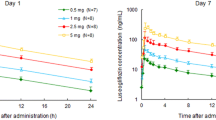
Single- and multiple-dose pharmacokinetic results for henagliflozin are summarized in Table 2. Median T max values of henagliflozin range from 1.00 to 2.00 h across the dose range. T 1/2 of henagliflozin were 9.1–14.0 and 10.3–13.8 h on day 1 and day 10, respectively. In all active treatment groups, steady-state plasma concentrations of henagliflozin were reached by day 7. At steady state, accumulative ratios of henagliflozin range from 1.16 to 1.24. C max, AUC0–24 and AUC∞ on both day 1 and day 10 increased dose proportionally.
Figure 1 shows the mean plasma henagliflozin concentration-time profiles for the 5, 10, and 20 mg dose groups on day 1 and day 10. Mean plasma henagliflozin concentrations increased in a dose-dependent manner. The concentrations of henagliflozin after administration of each dose on day 1 were similar to that after administration of the same dose daily for 6 days (days 4–10).

Plasma henagliflozin concentration-time profiles on day 1 and day 10
3.4 Pharmacodynamic Properties
FPG, MPG, and UGE measured at baseline, on day 1 and day 10, are summarized in Table 3. FPG decreased in the 5 mg group (−1.5 mmol/L, p < 0.001 on day 1; −1.5 mmol/L, p < 0.05 on day 10), 10 mg group (−0.5 mmol/L, p > 0.05 on day 1; −0.7, p > 0.05 on day 10) and in the 20 mg group (−2.7 mmol/L, p < 0.001 on day 1; −2.5 mmol/L, p > 0.05 on day 10). Twenty-four-hour MPG decreased by −0.3 mmol/L in the 5 mg group (p > 0.05), −1.0 mmol/L in the 10 mg (p > 0.05) and 20 mg groups (p < 0.05) on day 1. The corresponding values were −0.8, −0.9 and −1.2 mmol/L on day 10 (p > 0.05 for all). There were significant increases (p ≤ 0.05 for all) in UGE for all dose groups of henagliflozin on day 1 and day 10. The UGE increased by 54.2 and 56.8 g with 5 mg, 90.0 and 85.0 g with 10 mg, and 49.7 and 50.4 g with 20 mg on day 1 and day 10, respectively. UGE also increased in placebo; however, significance was only observed on day 10 (p = 0.01).
Figure 2 shows the changes in FPG, MPG and UGE from baseline. MPG decreased dose dependently, while FPG did not. Compared with baseline, UGE increased by 11, 65, and 82 times with doses of 5, 10, and 20 mg on day 1, respectively. Corresponding values on day 10 were 21, 82, and 110 times. Therefore, 24-h UGE increased with henagliflozin doses.

Changes in pharmacodynamic profiles of henagliflozin from baseline to day 1 and day 10
3.5 Safety and Tolerability
There were no treatment-related SAEs. No subjects discontinued the study (5–20 mg henagliflozin) due to AEs. TEAEs were reported by 16.7 % (1/6) of subjects in the placebo group and by 16.7 % (4/24) of subjects treated with henagliflozin (all doses), without apparent dose relationship. All TEAEs were considered to be mild in intensity. No episodes of hypoglycemia were reported. None of the TEAEs reported during the study were considered by the investigator to be very likely or probably related to the study drug. Two reports of increased urine β2 microglobulin in the henagliflozin 5 and 10 mg groups and one report of pruritus in the henagliflozin 20 mg group were considered to be possibly related to the study drug. A mild increase in urine red blood cells was observed in one patient receiving placebo, which was not considered to be as the result of the drug administration. Treatment with henagliflozin had no clinically meaningful effects on laboratory test results, vital signs, ECG parameters, or physical examination measurements.
4 Discussion
This is the first study that has assessed the safety, tolerability, and the pharmacokinetic and pharmacodynamic properties of henagliflozin in Chinese T2DM patients. Henagliflozin was well tolerated in Chinese T2DM patients within the dose range from 5 to 20 mg. No SAEs or discontinuations as a result of AEs were reported at any dose in this phase I study. These findings are consistent with that reported in the previous single- and multiple-dose study of Chinese nondiabetic subjects (data not published). All doses of henagliflozin induced improvements in glycemic parameters in Chinese T2DM patients. The pharmacokinetic and pharmacodynamic results support a once-daily dosing regimen.
Rapid oral absorption of henagliflozin was observed in this study with median T max values ranging from 1.00 to 2.00 h. The C max, AUC0–24 and AUC∞ values for henagliflozin increased in proportion with the increase in dose over the 5–20 mg range. The pharmacokinetic parameters of henagliflozin on day 1 were similar to those on day 10, indicating a time-independent pattern. The mean T 1/2 of henagliflozin was 9.1–14.0 and 10.3–13.8 h on day 1 and day 10, respectively. The accumulative ratio ranged from 1.16 to 1.24 upon repeated daily dose of henagliflozin on day 10 within each dose panel.
In the current study, all doses of henagliflozin induced improvements in glycemic parameters in Chinese T2DM patients. Single dose decreases the 24-h MPG by −0.3 mmol/L in the 5 mg group, −1.0 mmol/L in 10 mg (p > 0.05) and 20 mg groups on day 1. The corresponding values were −0.8, −0.9 and −1.2 mmol/L on day 10. The FPG changes were not dose proportional, although larger changes in FPG were observed in all three dosage groups compared to placebo group. This may be due to the inter-individual and inter-group variability. In our previous study in healthy Chinese subjects, henagliflozin did not reduce FPG level (data not published). This observation reflects the mechanism of action of henagliflozin, which is independent of insulin action but dependent on the filtered glucose load, and suggests a low propensity to cause hypoglycemia.
In all active treated groups, UGE increased significantly from baseline. The highest absolute change in UGE was observed in the 10 mg group with a value of 90.0 and 85.0 g on day 1 and day 10, respectively. Taking the baseline value into account, UGE increased most significantly in the 20 mg group (by 82–110 times), followed by the 10 mg group (65–82 times) and by the 5 mg group (11–21 times). Therefore, 24-h UGE increased in a dose proportional way. The pharmacodynamic effect of henagliflozin on UGE was comparable with those of dapagliflozin and canagliflozin. The mean values of 24-h UGE were 113 and 111 g following 300 mg of canagliflozin [7] on day 1 and day 7, and 68.4 and 71.41 g following 10 mg of dapagliflozin [9].
There were no treatment-related SAEs. No subjects withdrew from the study due to AEs. Two subjects reported increased urine β2 microglobulin in the 5 and 10 mg dose groups, and one reported pruritus in the 20 mg dose group. These were considered to be possibly related to the study drug. As reported by Kasichayanula et al. [9], there were eight adverse events that were considered to be related to the administration of dapagliflozin in the multiple ascending dose study. These were ‘protein urine present’, observed in two patients each receiving 2.5 and 20 mg; ‘thirst’ and ‘pollakiuria’ in one patient receiving 2.5 mg; ‘nocturia’ and ‘blood alkaline phosphatase increased’ in one patient receiving 10 mg. There were no adverse events leading to discontinuation. In the single and multiple ascending dose study of canagliflozin in T2DM patients [7], no treatment-related adverse events were reported.
The main limitation of the current study is that the renal function parameters were not evaluated using eGFR, and the urinary glucose excretion was not corrected by urinary creatinine. As the efficacy of SGLT2 inhibitors is dependent on renal function, the pharmacodynamic profiles of henagliflozin might be influenced. This might explain the non-dose-proportional relation of pharmacodynamic measurements such as FPG. In addition, we did not assess the pharmacokinetic and pharmacodynamic properties of henagliflozin in other ethnic cohorts. Previous studies showed that the pharmacokinetic profiles of dapagliflozin in Japanese healthy people and T2DM patients were similar to those in US cohorts [10–12]. Further study might be required before administration of henagliflozin to other ethnic groups.
5 Conclusion
In conclusion, henagliflozin was generally well tolerated in Chinese T2DM patients. All doses of henagliflozin induced improvements in glycemic parameters in Chinese T2DM patients. The observed pharmacokinetic and pharmacodynamic profiles of henagliflozin support a once-daily dosing regimen.
References
Freeman H, Cox RD. Type-2 diabetes: a cocktail of genetic discovery. Hum Mol Genet. 2006;15(2):R202–9.
Naser KA, Gruber A, Thomson GA. The emerging pandemic of obesity and diabetes: are we doing enough to prevent a disaster? Int J Clin Pract. 2006;60(9):1093–7.
Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the study of diabetes. Diabetes Care. 2009;32(5):193–203.
Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med. 2007;261(1):32–43.
Katsuno K, Fujimori Y, Takemura Y, Hiratochi M, Itoh F, Komatsu Y, et al. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther. 2007;320(1):323–30.
Oku A, Ueta K, Arakawa K, Kano-Ishihara T, Matsumoto M, Adachi T, et al. Antihyperglycemic effect of T-1095 via inhibition of renal Na+-glucose cotransporters in streptozotocin-induced diabetic rats. Biol Pharm Bull. 2000;23(12):1434–7.
Devineni D, Curtin CR, Polidori D, Gutierrez MJ, Murphy J, Rusch S, et al. Pharmacokinetics and pharmacodynamics of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in subjects with type 2 diabetes mellitus. J Clin Pharmacol. 2013;53(6):601–10.
US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for industry. Pharmacokinetics in patients with impaired renal function-study design, data analysis, and impact on dosing and labeling. 2010; Clincial Pharmacology; Revision 1.
Kasichayanula S, Chang M, Hasegawa M, Liu X, Yamahira N, LaCreta FP, et al. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetes Obes Metab. 2011;13(4):357–65.
Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85(5):513–9.
Komoroski B, Vachharajani N, Boulton D, Kornhauser D, Geraldes M, Li L, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Ther. 2009;85(5):520–6.
Kasichayanula S, Liu X, LaCreta F, Griffen SC, Boulton DW. Clincial pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transport type 2. Clin Pharmacokinet. 2014;53(1):17–27.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Jiangsu Hengrui Medicine Co., Ltd.
Conflict of interest
Haiyan Liu, Yan-Ping Liu, Ying Chen, Minquan Wang, Jia Li and Huaqiong Shen were full-time employees of Jiangsu Hengrui Medicine Co., Ltd., when the study was conducted. All other authors have no conflicts of interest.
Ethical approval
The study was conducted in accordance with Good Clinical Practice as defined by the International Conference on Harmonisation, and Chinese State Food and Drug Administration, and Declaration of Helsinki. The study protocol and all amendments were reviewed and approved by the Clinical Research Ethics Committee of Chengdu Military General Hospital and Xijing Hospital.
Informed consent
All subjects were informed of the nature and purpose of the study, and gave written consent to participate before any screening procedures.
Rights and permissions
About this article

Cite this article
Yong, X., Wen, A., Liu, X. et al. Pharmacokinetics and Pharmacodynamics of Henagliflozin, a Sodium Glucose Co-Transporter 2 Inhibitor, in Chinese Patients with Type 2 Diabetes Mellitus. Clin Drug Investig 36, 195–202 (2016). https://doi.org/10.1007/s40261-015-0366-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-015-0366-7