
Abstract
In addition to playing roles in the genesis and progression of cancer, mutant p53 also appears to play a significant role in the response to cancer therapy. In response to chemotherapy and radiation, two mainstays of cancer treatment, most cancer cells harboring p53 mutations show a reduced sensitivity compared to cells lacking p53 or those with wild type p53. However, there are also many instances where mutant p53 has shown no effect or enhances cellular sensitivity to chemotherapy and radiation. Similar to the in vitro cellular studies, the majority of clinical studies show a correlation between the presence of mutant p53 in patient tumors and adverse outcomes following treatment with chemotherapy agents or radiation in comparison to tumors with wild-type p53. However, it still remains unclear whether the presence of mutant p53 in tumors can serve as a reliable prognostic factor and aid in treatment planning. Thus, as genomic analysis of patient tumors becomes more cost effective, the role of mutant p53 in tumor responses from cancer therapy ultimately needs to be addressed. This chapter will discuss current mechanisms of how p53 mutations affect cellular responses to chemotherapy and radiation and discuss patient outcomes based on p53 status.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Part 1: The Role of p53 Status in Response to Chemotherapy and γ-Irradiation: In Vitro Studies and Mechanisms
Introduction
Some mutations in p53 are documented to impart “gain of function” properties to the cells that harbor them including enhanced oncogenesis, tumorigenesis, transformation, increased cell growth rates, and metastasis. Another gain of function property that has been well studied over the years is the response of cells to chemotherapy and radiotherapy. A large number of studies have examined the role of p53 mutants in the response to many commonly used anti-cancer drugs and to ionizing radiation in vitro. Using cell lines that express different forms of mutant p53, it has been possible to begin to characterize the role of mutant p53 in the response to chemotherapeutic agents or γ-irradiation. Moreover, these in vitro studies have also been extremely useful for identifying mechanisms of action of mutant p53 and have allowed for the identification of signaling pathways and genes that contribute directly or indirectly to the p53 gain of function phenotype.
In addition to the in vitro studies, a large number of clinical studies have been performed to determine if p53 status can be used to predict patient outcome from treatment with chemo or radiotherapy. For the patient that has developed cancer and for doctors treating these patients, this topic is of great importance as the presence of mutant p53 in tumors may influence outcome from cancer therapies. Therefore, this chapter is organized into two main parts. Part one focuses on the in vitro studies describing the role of mutant p53 in response to chemotherapeutic agents and γ-irradiation and underlining mechanisms that may contribute to the cellular responses. The second part of the chapter summarizes the role of p53 status in prediction of patient outcome following treatment with chemo or radiotherapy. These studies, organized by tumor site and treatment modality, aim to determine whether p53 status can be used as a prognostic factor in cancer treatment.
The Role of WT p53
Although this chapter does not focus on the role of WTp53 per se, its role obviously must be considered when discussing the effects of mutant p53. The WTp53 protein performs at least three major functions in response to a variety of forms of genotoxic stress: induction of apoptosis, cell-cycle arrest and growth control, and induction of DNA repair processes. Thus, lack of these cellular functions, through mutation of p53, will have profound affects on cellular responses to chemo and radiotherapy. As an example, it is well documented that treatment with chemotherapy agents or γ-irradiation results in the induction of p21Waf1, a mediator of the G1 cell cycle checkpoint in cells that contain WTp35 but not in cells with mutant p53 [1]. Thus, the absent functions of WTp53 when mutant p53 is present must be accounted for. Are the cellular effects observed in cells that contain mutant p53 due to the presence of mutant p53 or to the lack of WT p53? Indeed, many targets have been identified for WT p53 that are known to sensitize cells to killing by anticancer agents or radiation [2, 3]. Thus, these targets may not be activated if p53 is mutated. It is also possible that mutant p53 can heterodimerize with WTp53 proteins and act in a dominant negative fashion. So are the effects of mutant p53 due to the mutant proteins themselves or due to a dominant negative effect? These questions are not easily answered, especially in vivo, where it may not be possible to manipulate p53 genetics. The studies in vitro using isogenic p53 null cell lines have been more successful in this regard as mutant p53 can be directly compared to the absence of p53, or to WTp53.
The Nature of p53 Mutations
To understand how p53 mutations impact the response to chemo and radiotherapy, it is important to be aware of the different types of p53 mutations and the consequences of these mutations. Although a great many p53 mutations have been described, the majority of mutations in cancer are missense mutations located in the DNA-binding domain of p53. These mutations most often result in the production of full-length mutant protein that appears to exhibit effects over and above its loss of WT activity. Some of these effects include a dominant negative activity by interfering with WTp53, interference with the function of p63 and p73 [4, 5] and a ‘gain of function’ activity, in which p53 mutant proteins display oncogenic properties in their own right [6]. A large proportion of the missense mutations are associated with gain of function activities and arise in the ‘hotspot’ residues, of p53: R175, G245, R248, R273, R249 and R282. However, as might be expected, different p53 mutations do not have the same biological effects and it is likely due to the differential effects on protein function and conformation. For example, mutations at residues R248 and R273 interfere with DNA binding, whereas G245 and R249 mutations produce local distortion of the protein, and mutations at R175 and R282 produce global distortion of the protein structure [6]. Therefore, it is not surprising to see different results in the literature on the topic of the effects of mutant forms of p53. A comparison of the effects of two different p53 mutants may not be expected to yield similar results.
The Role of Mutant p53 in the Response to Chemotherapy
The p53 gene is one of the most frequently mutated genes in cancer. As such it is important to determine how it affects the response of tumors to chemotherapy. To this end, efforts have been directed to understand how p53 status impacts cell survival at the molecular level in response to a number of anticancer agents. It is important to note that the literature is not in agreement on the results of these studies and this perhaps is not surprising given all of the variables involved in these studies. One must consider cell type, chemotherapy agent and dose, p53 mutation, and genetic background of the cells in these experiments. It also important to note that p53 is only one gene and analysis of one gene may not be enough to predict cellular outcomes. The majority of reports in the literature show that mutant p53 confers decreased sensitivity to chemotherapy to the cells in which it is expressed [1, 7–15]. On the other hand, there are also reports that claim the opposite result; that is, that mutant p53 confers increased sensitivity to chemotherapy [16–18]. Still other reports suggest that WTp53 may confer increased resistance to chemotherapy agents [19]. A summary of the most significant reports is provided here.
Several studies provide evidence that cells that express mutant forms of p53 exhibit increased resistance (or decreased sensitivity) to chemotherapy agents. This phenomenon was described as early as 1997 by O’Connor and colleagues [1] and similar findings have been reported by several other research groups in different cell types [7, 9, 20–23]. O’Connor and colleagues conducted an extensive study of the role of p53 in a large spectrum of cancer cell lines against the growth inhibitory action of 123 anticancer agents [1]. A total of 39 p53 mutant and 18 wild type cell lines from a number of tissue types were examined. The overall conclusion was that cells with mutant p53 tended to exhibit less growth inhibition compared to cells with WTp53 when exposed to a number of clinically relevant anticancer agents including DNA cross-linking agents, antimetabolites, and topoisomerase inhibitors. Specifically, it was shown that mutant p53 conferred a median resistance to cisplatin, 5-FU, and bleomycin of 3–10 times to that of wild-type p53 cell lines [1]. Interestingly though, there was no difference observed between mutant p53 and WTp53 containing cells in response to treatment with the antimitotic drugs such as the Taxol family of agents [1].
Although a comparison of different cancer cell lines is a critically important piece of information as shown by the O’Connor study [1], differences in the genetic backgrounds of different cancer cell lines can obscure the overall role of p53. To address the question in isogenic cell lines, Blandino and colleagues introduced different p53 mutants into the p53-null H1299 lung adenocarcinoma cell line and measured their sensitivity to the chemotherapy agent, etoposide [9]. It was found that cells that expressed the R175H p53 mutant but not the R273H mutant exhibited a decreased sensitivity to etoposide compared to cells that expressed WTp53 or cells that expressed vector alone (null-p53). In contrast, both p53 mutants, R175H and R273H exerted similar effects with regard to cisplatin treatment, that is, both mutants conferred increased resistance to the drug [9]. Thus, not only are the effects of p53 mutants specific to the type of p53 mutant but also to the form of chemotherapeutic agent applied as well. Similar results were reported by Deb and colleagues who showed that introduction of p53 mutants into the H1299 cell line conferred decreased sensitivity to the chemotherapeutic agent etoposide [21]. However, in contrast to the study by Blandino [9], the R273H p53 mutant was also found to confer reduced sensitivity to etoposide treatment as measured by clonogenic assay [21].
Several additional studies have used different cell types and agents to address the question regarding the role of mutant p53 in the response to chemotherapy. Wong and colleagues found that expression of the p53 mutant, R273H, conferred doxorubicin resistance in an A431 human squamous carcinoma cell line [23]. It was also shown that expression of the R273H mutant into the p53-null human osteosarcoma cell line, Saos-2 reduced sensitivity to doxorubicin and methotrexate [23]. In another study it was shown that p53-null murine leukemic cells that expressed a temperature sensitive p53 mutant, V135A, exhibited decreased sensitivity to doxorubicin or cisplatin compared to cells expressing no p53 or WT p53 [7]. As p53 overexpression in cell lines could represent an artificial level of mutant p53 overexpression, the opposite technique, of reducing mutant p53 has also been explored. To validate this concept, the level of mutant p53 was reduced in human cancer cell lines by siRNA and it was found that depletion of mutant p53 reduced the resistance to anticancer drugs [24].
The Role of Mutant p53 and the Response to γ-Irradiation
Radiation is one of the main treatment modalities for cancer. However, a great number of tumor types exhibit resistance to radiation. As p53 is one of the most commonly mutated genes in cancer, it is important to determine the role of p53 status in radioresistance. The role of p53 GOF mutations and their response to radiation has been studied in a variety of cancer cell lines derived from different tissue types. A study of the role of p53 status and radiation in 60 different cancer cell lines was conducted by O’Connor and colleagues [1]. In this report it was shown that, perhaps as expected, the majority of cell lines expressing WTp53 showed a functional induction of mRNA for p21Waf1, GADD45 and Mdm2 in response to γ-irradiation whereas the majority of cell lines expressing mutant p53 did not [1]. This indicates that mutation of p53 abrogates, for the most part, the radiation induced G1 checkpoint in cell lines harboring mutant p53. But how does p53 status affect survival from radiation treatment? In this area, it is clear that the results of the studies are mixed. In some cells, evidence shows that the presence of mutant p53 reduces sensitivity to radiation. In other cells, no effect is reported or the presence of mutant p53 has been shown to increase radiosensitivity. There are number of factors that could contribute to the variability observed in these studies including difference in genetic background of the cell lines, the type of p53 mutation present, the cellular environment at the time of irradiation and the radiation dose. It is also clear that although p53 is involved in the cellular response to radiation, it is certainly not the only factor involved. These studies are summarized in several excellent reviews and the reader is directed to these for more information on radiation and p53 [25–27].
One of the earliest reports to examine the role of p53 mutants and radiosensitivity was by Lee and colleagues [28]. In this study, transgenic mice were generated that overexpressed the p53 mutants R193P or A135V. Hematopoietic cell lines derived from the transgenic mice were then compared to WT littermate cell lines for radiosensitivity. It was shown that overexpression of either R193P or A135V increased the radiation resistance of mouse hematopoietic cell lineages by 45–57 % [28]. Although the hematopoietic cells harboring the p53 mutants showed increased radioresistance, there was no difference compared to WT cells when treated with EMS, an alkylating agent [28]. In another early report, Li and colleagues utilized temperature-sensitive myeloid cell lines that allowed permissive expression of no p53, WT p53 or the p53 mutant, A135V [7]. It was shown that induction of WT p53 expression greatly enhanced γ-irradiation induced apoptosis relative to non-p53 producing cells. In contrast, induction of the p53 mutant A135V increased cell viability following irradiation 3-fold relative to non-p53 producing cells [7]. Interestingly, treatment with Actinomycin D, a potent inhibitor of transcription, abrogated the reduced apoptosis from γ-irradiation in the cells expressing mutant p53 [7]. This result was one of the first hints that mutant p53 may act at the transcriptional level to mediate its gain of function properties.
Bristow and colleagues employed rat embryo fibroblast (REF) clones to examine how mutant p53 contributes to the radiation response [29]. It was shown that the REF clones expressing p53 mutants (H273, N190, V135, P193, D236, A143) showed increased clonogenic resistance in response to γ-irradiation relative to the non-mutant p53 expression REF clones which expressed low levels of p53 [29]. In another report, Bristow and colleagues showed that cells that expressed the R193P p53 mutant were observed to have a significantly higher survival fraction after 2 gray (SF2) (0.86) than the parental p53-null cell line (0.65) [20]. However, with regard to mechanism, no differences were observed in apoptosis rates between the mutant p53 and control cell lines following doses of both 2 and 10 Gy [20]. Similarly, and unlike the report by Li and colleagues [7], the relative radio-resistance of the REF clones expressing mutant p53 compared to REF clones that did not express mutant p53 was not explained by decreased apoptosis based on a number of morphologic and biochemical end points [29]. The authors explain this finding by citing evidence, that in general, REF clones do not undergo apoptosis in response to treatment with γ-irradiation [29].
A few recent studies have addressed the role of mutant p53 and radiosensitivity on a much larger, more systematic scale. One of the first studies to compare the effect of different p53 GOF mutants on radiosensitivity was by Okaichi and colleagues. In this study, isogenic, stable cell lines were generated by transferring different p53 mutants into the Saos-2 cell line, an osteosarcoma, which is null for p53 [30]. A total of 16 different p53 mutants were analyzed (T123A, L130V, Q143A, V157F, H168R, R175H, I195T, C238Y, C242F, G244C, G245S, R273H, C277F, R280T, R282W, E286K) and compared to WTp53 and vector only (no p53). Cell lines were then treated with γ-irradiation and clonogenic assays performed to measure radiosensitivity. The parental Saos-2 cell line and vector only transformant (p53 null) were more radioresistant than Saos-2 transfected with WTp53. The p53 mutants exhibited a range of radiosensitivities [30]. The 175H, 244C, 245S, 273H and 282W transformants were similar in radiosensitivity to the parental and control vector transformants but much more radioresistant than WTp53 transformants. In contrast, the C242F transformants were similar to WTp53 in their radiosensitivity and the p53 mutants T123A, I195T and C238Y were actually more radiosensitive than WTp53. Thus, although most of the p53 mutants were more radioresistant than WTp53 (12/16), they were not more radioresistant than cells lacking p53 [30]. Therefore in the context of this particular cell line, the p53 mutants do not seem to exhibit a dramatic “gain of function” phenotype at least with regard to radioresistance. Although in this study only one cell type was examined, the use of isogenic cell lines was a major strength as analysis of cell lines with different genetic backgrounds can make identification of the role of mutant p53 difficult.
A comprehensive study examined the role of p53 in radiosensitivity using a wide range of tumor cell lines that varied in histological type [31]. In this study a total of 39 human tumor cell lines from 9 histological types were analyzed for p53 status, radiosensitivity by clonogenic assay, and level of p21 expression. On the basis of survival fraction after 2 GY (SF2), on average, cells that express WT53 exhibited more cell killing than cell lines that express mutant p53. However, when similar comparisons were made for cell killing at higher radiation doses, there was no significant difference between cells that expressed WTp53 versus mutant p53. When compared within each histological cell type, the cell lines expressing mutant p53 exhibited less cell killing (as measuring by SF2) than those cell lines that expressed WTp53 [31]. However, again when cell killing is measured at higher doses of irradiation, the differences between WT and mutant p53 expressing cell lines, even within the same histological type are not significant. In summary, the authors conclude that the cell lines fall into four radiosensitivity groups: very sensitive (VS), sensitive (S), resistant (R) and very resistant (VR). Using this classification, 16/17 cell lines with WTp53 were sensitive (S) and only 1/16 was very resistant (VR). In contrast, only 2/15 of the cell lines expressing mutant p53 were sensitive (S), while 13/17 were resistant (R) and 2/17 were very resistant (VR). The cell lines lacking p53 were also more radiosensitive than cell lines expressing mutant p53 suggesting some “gain of function” activity of mutant p53 in this setting [31].
In head and neck cancers, p53 is one of the most commonly altered genes. Thus, it has been examined with regard to p53 status. In a very recent report on head and neck cancer, Skinner and colleagues completed both a clinical study of patient outcome and p53 status as well as an in vitro study of HNSCC cell lines [32]. The clinical arm of this study found that patients with disruptive p53 mutations fared worse than those with WT p53 or non-disruptive p53 mutations with regard to local regional recurrence (LRR) and overall survival. The clinical aspects of this study are covered in more detail in part 2 of this chapter. In the second part of the study, a total of 38 HNSCC cell lines of known p53 status were analyzed by clonogenic assay for response to γ-irradiation. It was shown that cell lines harboring disruptive p53 mutations were more radioresistant than those with WTp53 or non-disruptive p53 mutations. Confirmation that the radioresistance was due to mutant p53 was confirmed by silencing mutant p53 in those cell lines expressing mutant p53 and showing that the radioresistance was reduced [32]. These data argue that a ‘gain of function’ activity for mutant p53, which has been observed for other cellular processes, also exists for the response to radiation. An interesting aspect to this study was that the in vitro radiosensitivity did not correlate with apoptosis or mitotic cell death but rather to radiation-induced senescence. Overall, the presence of disruptive p53 mutants strongly inhibited radiation induced senescence [32]. Thus, it may be important to analyze the correct response to radiation of a particular cell type when studying the role of p53 mutations.
In another recent study of head and neck cancer, the role of mutant p53 was examined using an isogenic head and neck squamous carcinoma cell line pair generated to express WT p53 (HN30) or mutant p53 (HN31) respectively. It was shown that the HN31 cells demonstrated increased radioresistance compared with their wild type p53 (HN30) counterparts [33]. Interestingly, HNSCC cells expressing mutant p53 displayed decreased mitochondrial respiratory capacity and increased sensitivity to 2-DG inhibition of glycolysis [33]. This finding suggests that mutant p53 may impact mitochondrial function and that head and neck tumors expressing mutant p53 may be more susceptible to anti-metabolic strategies such as treatment with 2-DG. As there are no clinically available treatment strategies designed to specifically address mutant p53 containing head and neck tumors currently available, and the associated radioresistance, this finding may allow for the development of novel therapeutic approaches [33]. Although the above reports do support a role for mutant p53 in modulating the response to radiation, others do not [12, 17]. There is also some evidence that WTp53 can contribute to resistance from radiation [19].
Mechanisms of Mutant p53 in the Response to Chemotherapy or Radiation
An investigation into the mechanism of how mutant p53 affects cellular functions may allow for a better understanding of mutant p53 in cancer and promote the identification of new cancer targets. Thus, the underlying mechanism of mutant p53’s gain of function properties has been extensively investigated. However, in this section, we restrict the discussion of mutant p53’s mechanisms to those associated with alteration of the response to chemo or radiotherapy acknowledging that these mechanisms may not necessarily be dissociated from other gain of function properties of mutant p53.
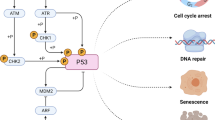
The mechanism of how the cellular response to chemo or radiotherapy is affected by p53 status has been widely investigated and hypothesized to be due to a number of factors. In theory, signaling pathways involved in mediating cell survival, growth, apoptosis, drug resistance or DNA repair could be involved. Indeed many of these pathways and the genes within these pathways have emerged as candidates for the action of mutant p53. For example, mutant p53 has been shown to be involved in regulating apoptosis [6, 7, 9, 23], genomic instability [34], DNA repair [11, 25, 29], senescence [32], autophagy [35], gene transcription [6, 21, 36–40], mitochondrial function [41], drug resistance [39, 42], protein kinase signaling [43, 44] and the microRNA pathway [45]. Some of the possible mechanisms for how mutant p53 could impart resistance to chemo or radiotherapy are summarized in Fig. 8.1.

A schematic of possible mechanisms of mutant p53 involved in the cellular response to chemotherapy agents or γ-irradiation is shown
As WTp53 is a critical regulator of apoptosis, perhaps it is not surprising that several reports have shown that expression of mutant p53 correlates with altered apoptotic pathways. As one example, it was shown that expression of the p53 mutant, R273H, in A431 cells correlated with doxorubicin resistance and lower rates of apoptosis [23]. Moreover, the drug resistance could be reduced by siRNA directed against p53, and this correlated with an increase in the expression of procaspase-3 and apoptosis [23]. Similar results were observed after introduction of p53-R273H into the p53-null human osteosarcoma cell line, Saos-2. Induction of expression of p53-273H in Saos-2 cells reduced sensitivity to doxorubicin and methotrexate, reduced procaspase-3 expression, and reduced DNA fragmentation, a marker of apoptosis [23]. Another link to apoptosis was established by Li and colleagues in myeloid cell lines [7]. It was shown that p53-null murine leukemic cells that expressed a temperature sensitive p53 mutant, V135A, exhibited decreased apoptosis rates in response to doxorubicin or cisplatin compared to cells expressing no p53 or WT p53 [7]. In a study by Blandino and colleagues, it was shown that expression of the p53 mutants 175H or 179H greatly reduced the rate of etoposide-induced apoptosis in H1299 cells compared to vector transfected controls [9]. Other p53 mutants, such as 273H and 248W had a much milder protective effect. Considering the evidence for dysregulation of apoptosis in mutant p53 expressing cells, it is not surprising that several genes related to apoptosis have also been found to be altered in their expression levels in these cells. Notable examples include upregulation of BAG-1 [46] and NF-κB2 [21] and downregulation of FAS [47] and MST-1 [48]. For a more comprehensive review of genes dysregulated by mutant p53, the reader is directed to the review by Brosh and Rotter [6].
The biological assay used to measure p53 function is critically important in determining if a ‘gain of function’ effect of mutant p53 is present. For example, Skinner and colleagues found that two modes of cell death commonly associated with irradiation, apoptosis and mitotic cell death, were unaffected by p53 status [32]. However, if radiation-induced senescence was assayed, then p53 status played a significant role [32]. It was found that the presence of p53 mutations correlating with decreased radiation-induced senescence, p21 expression, and release of ROS [32]. In addition to apoptosis, a role for mutant p53 in modulating autophagy was identified as well [35]. In this study, it was shown that although γ-irradiation increased the level of autophagy in the p53-null lung cell line, H1299, expression of the R175H p53 mutant in H1299 cells greatly attenuated the level of γ-irradiation induced autophagy. Consistent with this result, the expression of Beclin-1, a marker of autophagy, also increased in H1299 cells in response to γ-irradiation but not in H1299 p53-R175H expressing cells [35]. These results suggest a gain of function role for p53 mutants through inhibition of autophagy in response to γ-irradiation [35].
Another possibility that has been explored is that mutant p53 alters gene expression of a wide array of genes involved in cancer pathways. Early evidence that transcription dysregulation may be one mechanism of mutant p53 included the finding that Actinomycin D, a potent inhibitor of transcription, blocked the reduction in apoptosis rate mediated by mutant p53 in response to treatment with doxorubicin or cisplatin [7]. One of the first studies to address the role of gene transcription in the mechanism of action of p53 GOF mutants was reported by Deb and colleagues in 2005 [21]. To avoid difficulties inherent in comparing different cancer cell lines, Deb and colleagues generated isogenic stable cell lines of a non-small cell cancer cell line, H1299, which is devoid of p53, that expressed either vector alone, or the GOF p53 mutants R175H, R273H or D281G. Relative to vector-transfected cells, H1299 cells expressing mutant forms of p53 showed a survival advantage when treated with etoposide as measured by clonogenic assay [21]. Interestingly, however, cells expressing the transactivation-deficient triple mutant p53-D281G (L22Q/W23S) had significantly lower resistance to etoposide. As the L22Q/W23S mutant is shown to be deficient in transactivation, this result suggested that the p53 mutants were acting at the level of gene transcription. To explore this further, RNA was extracted from H1299 or 21PT stable cell lines that expressed the p53 mutants R175H, R273H, D281G, or vector alone and analyzed for gene expression using an Affymetrix gene array chip. Analysis of the gene array data indicated that all three p53 mutants upregulated a common set of genes involved in a diverse array of processes including in cell cycle control, oncogenesis, invasion, metastasis, DNA replication, cell survival, and transcription. One of the genes found to be upregulated by p53 GOF proteins was NF-κB2 (p100/p52), a member of a family of sequence specific DNA binding transcription factors. This result raised the possibility that transcription factors themselves may be altered by mutant p53 leading to a secondary level of gene induction or repression. To explore the role of NF-κB2 in chemosensitivity, it was overexpressed in H1299 cells. It was also shown that H1299 cells overexpressing NF-κB2 were less sensitive to etoposide and siRNAs directed against NF-κB2 increased etoposide sensitivity [21]. As all three p53 mutants activated the NF-κB2 pathway, one possible pathway through which p53 mutants induce loss of drug sensitivity is via upregulation of the NF-κB2 pathway. Although evidence suggests that mutant p53 may act to induce the expression of genes apart from WTp53, the mechanism of transactivation by mutant p53 is not yet clear. One hypothesis is that mutant p53 may interact with other transcription factors and activate different promoters than when WTp53 is present. Evidence in support of this theory was shown by using ChIP assays [38]. In this report, the NF-κB2 promoter showed increased interaction with CBP and STAT2 in the presence of mutant p53 [38]. Thus, in H1299 cells, mutant p53 may induce gain of function activities by enhancing recruitment of CBP and STAT2 on the promoters of target genes [38].
In another approach to uncover mechanisms underlying p53 GOF mutant activities, a proteomic analysis was used to identify mutant p53 interacting proteins [41, 49]. From this analysis, MCM7, a protein involved in DNA replication, was shown to specifically interact with mutant p53 but not WTp53 [49]. Another protein, Tim50, which forms part of the mitochondrial protein import machinery, although not shown to physically interact with mutant p53, was found to be highly overexpressed in cancer cell lines that also express p53 mutants. Analysis of the Tim50 promoter revealed that mutant p53, but not WTp53 was able to upregulate Tim50 transcription [41]. Interestingly, reduction of Tim50 expression by siRNA reduced the resistance of cells harboring the p53 mutant, R175H, to paclitaxel but had no effect upon cells lacking p53. These findings identify the Tim50 gene as a transcriptional target of mutant p53 and suggest a novel mechanism by which p53 mutants enhance chemoresistance [41].
Chemoresistance may also be attributed, at least in part, by the action of mutant p53 in the dysregulation of the microRNA pathway. It was shown that expression of the p53 mutant, R275H, in the p53-null lung cell line H1299, resulted in the down regulation of the expression of miR-223. Moreover, in a colon and breast cancer cell line that expressed the p53 mutant, R273H, down-regulation of mutant p53 by shRNAi increased miR-223 expression [45]. Chromatin immunoprecipitation (ChIP) assays showed that mutant p53 was capable of binding to the miR-223 promoter [45]. Consistent with these results, overexpression of miR-223 sensitized cells to cisplatin or 5-fluorouracil. Moreover, down-regulation of mutant p53 also upregulated the levels of the Stathmin protein, a known target of miR-223. Thus from this data a model was proposed whereby mutant p53, through the down-regulation of miR-223, upregulated the Stathmin protein which contributed to chemoresistance [45].
Mutant p53 may also act to alter protein phosphorylation signaling pathways. One of the genes found to be upregulated in H1299 cells following expression of the p53 mutants R175H, R273H, and D281G was the protein tyrosine kinase Axl [44]. Consistent with this result, knockdown of endogenous mutant p53 in two different human lung cancer cell lines, H1048 (p53-R273C) and H1437 (p53-R267P) reduced Axl expression [44]. It was suggested that mutant p53 may act directly at the Axl promoter as ChIP assays demonstrated the presence of mutant p53 at the Axl promoter [44]. As it is known that Axl expression affects some of the same pathways as mutant p53 such as apoptosis, cell adhesion, and motility, the role of mutant p53 was investigated in these processes. Interestingly, knockdown of Axl by RNAi resulted in a reduction of mutant p53 gain of function activities in lung cancer cells expressing endogenous mutant p53, including growth rate and cellular motility. Taken together, these results suggest that mutant p53 may act to upregulate the Axl protein tyrosine kinase which then executes, at least in part, some of the p53 gain of function activities.
Conclusions and Possible Reasons for Discrepancies Among the Studies
The results of the studies described here, which notably represents only a small fraction of the total studies in the literature on mutant p53, gives a somewhat mixed verdict with regard to how mutant p53 affects the cellular response to chemotherapeutic agents or γ-irradiation. Although the majority of mutant p53 studies suggest that the presence of mutant p53 reduces sensitivity to chemotherapy or radiation, there are many studies that show no effect or show the opposite effect; that is, that mutant p53 enhances sensitivity to these agents. Thus, it is important to examine some of the possible reasons for these discrepancies. First, it is important to note that unless isogenic cancer cell lines containing mutant p53 are compared to each other, the same result after treatment with chemo or radiotherapy should not be expected. Cancer cell lines that are derived from different tissues are known to respond differently to different chemotherapeutic agents regardless of p53 status. This is because the cancer cell lines, much like different patient tumors, show a great amount of genetic variability. Another important point, as mentioned earlier, is that p53 gain of function mutants act differently depending upon the location and type of mutation in p53. It is well documented that the degree of chemoresistance depends critically on the type of mutation present in p53 [1]. Another important variable to consider is the type of biological assay performed to measure “gain of function” of p53 in response to treatment with chemo or radiotherapy. For some p53 mutants and cell types, a clonogenic assay may be appropriate. Chemo or radiotherapy responses are typically measured using this assay which measures the reproductive integrity of cells regardless of the specific mode of cell death involved [50]. For this reason, the clonogenic assay has long been the ‘gold standard’ for measuring responses to chemo or radiotherapy. Unfortunately, not all cell lines form colonies in vitro. For example, some HNSCC cell lines do not form colonies and therefore other viability assays, such as the MTT growth assay, are employed. Although most of the studies discussed here employ the clonogenic assay, in the cases where it is not used, it is important to recognize that this could be one source of variability in the results observed. Finally, there may also simply be variability due to how different investigators conduct their experiments. The larger question that remains is what is the significance of these in vitro studies and the mechanisms uncovered for the action of mutant p53 in a clinical setting? Inevitably, as these new targets are discovered, it will become important to evaluate the significance of these findings in animal models or in clinical settings.
Part 2: The Role of p53 Status in Response to Chemotherapy and γ-Irradiation: Clinical Studies and Patient Outcome
Introduction
The p53 gene and its protein product have been extensively studied in vitro and in animal models. But how can what is known about p53 at a molecular level translate to a clinical setting? Numerous studies have been conducted to analyze the relevance of a tumor’s p53 status in regards to patient outcome. Special attention has been, and continues to be, paid towards understanding the implications of having WT versus mutant p53 in response to cancer therapy. Ultimately, if we are able to establish a relationship between p53 mutational status and response to therapy, we will be able to sequence each patient’s tumor and ascertain which patients will respond to which therapies. In so doing, the morbidity associated with ineffective treatment modalities, and the delay in achieving response with effective ones, can be avoided.
This chapter aims to review what has been shown with respect to p53 mutation status and the response to treatment of various tumor types. The review is limited specifically to p53 mutation, not p53 over-expression, deletion, or loss of function. It is also limited to chemotherapy and radiation therapy as the treatment modalities studied. Unlike surgery, which acts to physically remove cancer cells, chemo and radiation rely on intrinsic and extrinsic cellular pathways, involving genes such as p53, to cause cancer cell death.
With very few exceptions, this chapter is concerned exclusively with studies that analyze p53 status via gene sequencing methods, as opposed to immunohistochemical staining (IHC). Studies involving IHC to assess p53 status were avoided because IHC lacks standardization and is subject to a number of biases including observer bias and variation in scoring methods among institutions. IHC is also more likely to result in false positive or false negative results and is therefore not as sensitive for detecting p53 mutations as is direct gene sequencing [51]. Another limitation of IHC is that it relies on a small number of pre-defined protein markers per tissue section to discover the presence or absence of the mutated protein. In order to develop a clinically relevant molecular indicator of response to therapy, a standardized, unbiased, and sensitive mechanism for detecting the status of the molecular marker, in the present case p53, is needed.
Gynecological Malignancies
Changes in p53 are the most frequent genetic event described in advanced ovarian cancer [52]. Understanding the role of mutant p53 in response to therapy, therefore, may be of particular importance in the treatment of this cancer. In advanced disease, where chemotherapy is the mainstay of treatment, knowledge of the relationship between p53 status and response to therapy could help to determine which chemotherapeutic agent would be most effective and which agents should be avoided in a particular patient.
Taxane- and platinum-based chemotherapeutic agents are routinely used in the treatment of ovarian cancer. There is evidence, however, that the presence of mutant p53 within tumor cells can impact a patient’s response to these drugs. Existing data seems to show, however, that the effect of p53 mutational status on response to each of these therapies is not concordant. With respect to platinum-based chemotherapy, several studies have shown that tumors harboring a p53 mutation are more likely to be resistant to treatment [53–57]. Indeed, not only the presence of mutant p53, but also the specific type of mutation, has been shown to play a role in response to platinum-based chemotherapy [53]. However, despite the large number of studies that have found a correlation between p53 status and response to platinum-based chemotherapy, there is some data suggesting that no such association exists [58]. In the case of taxol-based chemotherapy, on the other hand, it has been shown that harboring a p53 mutation in tumor cells predicts a favorable response to treatment [59]. These results imply that certain patients, namely those without a mutant p53, will respond better to platinum-based chemotherapy, while those who harbor a mutation are more likely to respond to taxol-based agents. More research in this area will enable clinicians to tailor each patient’s therapy in order to avoid subjecting cancer patients to taxing chemotherapeutic regimens which confer no clinical benefit.
The role of p53 status in other gynecological malignancies has also been studied, however, results are less conclusive than in the case of ovarian cancer. In cervical cancer, for instance, there remains a controversial correlation between p53 status and response to therapy [60]. Moreover, the data that is available assesses p53 status via IHC and gene expression as opposed to gene sequencing [60] which makes it difficult to draw meaningful conclusions given operator bias and lack of standardization, as previously discussed. There is some evidence, however, that cervical cancers with mutant p53 detected by gene sequencing are more likely to be radio-resistant [61]. Radiation therapy may, therefore, not be the appropriate modality of treatment for patients with these tumors. However, further investigation is needed before meaningful conclusions can be drawn.
Breast Cancer
Breast cancer is the most common non-cutaneous cancer among women and the second most common cause of cancer death among women. As in the case of many other malignancies, p53 is the most commonly mutated gene in breast cancer [62]. There are a number of treatment approaches that can be used in the management of breast cancer, some of which are interchangeable. For instance, lumpectomy with radiation is equivalent to mastectomy in many cases of early stage cancer. In the case of larger tumors, neoadjuvant chemotherapy can be used to decrease tumor volume in an attempt to avoid what may otherwise be a disfiguring surgery. Given this arsenal of treatment approaches and the morbidity associated with each one – including toxic radiation and chemotherapy – the ability to predict response to therapy would be invaluable in the management of patients with breast cancer. Determining the relationship between p53 mutational status and response to these various therapies could save many patients from undergoing unsuccessful treatments, and their associated complications, from the outset.
Neo-adjuvant chemotherapy is often employed in the management of locally-advanced invasive breast cancer; however, response to chemo varies and is often unpredictable [63]. Furthermore, there exist a number of agents to choose from, some of which may be more successful than others. Patients who fail to respond to a particular neo-adjuvant regimen are not only subjected to the toxic effects of an ineffective therapy, but, their tumors are potentially given time to grow and spread until an effective regimen is initiated. Predicting a response to chemotherapy in the neoadjuvant setting via assessment of tumor markers, such as p53, would be beneficial to a great number of patients. The studies published to date which utilized gene sequencing to assess p53 status and response to chemotherapy in the neoadjuvant setting, however, have unfortunately shown conflicting results [64–66].
Hormonal therapy is another treatment modality often used in breast cancer therapy. Tamoxifen, which is commonly used, works by binding to estrogen receptors, decreasing DNA synthesis, and inhibiting estrogen effects. Because it is involved in regulating the cell cycle, it is highly plausible that a mutation in p53 may play a role in response to the drug. The current consensus in the literature seems to be that mutated p53 causes resistance to tamoxifen [62, 67, 68]. Currently, whether or not tamoxifen is prescribed to patients is largely predicated on a tumor’s hormone receptor status. However, even in patients whose tumors are highly estrogen or progesterone receptor positive, tamoxifen therapy has been known to fail, thus exposing women to a drug that can cause osteoporosis and increase the risk of uterine cancer, while conferring no benefit in the management of their disease. It would, therefore, be valuable to find an additional means of predicting response to this drug. One of these may be via assessment of p53 status.
There is limited data in the literature that studies the role of p53 mutation in response to radiation therapy in breast cancer, another mainstay in the treatment of this disease. Moreover, the available studies seem to have conflicting results. In the case of combined treatment, for instance, involving neoadjuvant chemo, surgery, and radiation therapy, it was found that a mutation in p53 predicted a poor response to therapy [69], while in the case of radiation therapy alone, a mutant p53 was shown to sensitize tumors to therapy [70]. More studies are needed before conclusions linking p53 mutation to treatment response can be drawn.
Head and Neck Cancer
Genetic changes in p53 have been reported to occur in approximately 45 % of head and neck cancers [71, 72]. Treatment for head and neck cancer is selected on the basis of site and stage of disease. Patients’ response to therapy can vary widely for any given site at any given stage, however. Due to the prevalence of p53 mutations in head and neck cancers and the heterogeneity in treatment response, there has been significant interest in attempting to find molecular markers that can predict response to therapy. In the case of neoadjuvant chemotherapy and the response of patients with various stages of head and neck cancer, it has been found that p53 mutations predict failure to respond to treatment [14]. In patients treated with radiation therapy, either in the adjuvant setting or as primary therapy, it has also been found on numerous occasions that a mutation in p53 makes tumor cells less likely to respond to treatment [15, 32, 73, 74]. Finally, in one study in which both treatment modalities were assessed, p53 mutational status again predicted an unfavorable response to therapy [75].
While, in head and neck cancers overall, p53 mutation appears to portend poor response to therapy, some studies focusing on specific sites have found that no correlation exists between treatment and p53 status. For instance, in the case of nasopharyngeal cancers, one study found that p53 mutations were infrequent and were not associated with failure of radiation therapy [76]. Another study that focused only on laryngeal cancers found that, while p53 mutation was a common occurrence in these cancers, the presence of a mutation had no bearing on response to radiation [77]. In the case of oropharyngeal cancers, on the other hand, p53 mutation was associated with radiation resistance [78]. These findings may imply that the effect of p53 mutation on response to various therapies varies by site. Further investigation is needed to determine for which tumor sites the knowledge of p53 mutational status would be applicable. More evidence is also needed to assess whether mutation in p53 predicts a favorable or unfavorable response to therapy in those sites where it plays a role.
Prostate Cancer
Like breast cancer in women, prostate cancer is the most common non-cutaneous cancer in men and the second most common cause of cancer-related death among men. P53 mutations, however, have only been reported in approximately twenty percent of prostate cancers [79]. This may, at least in part, explain why there is so little data regarding the role of p53 mutations in response to therapy for this disease. With respect to prostate cancer therapy, radiation or surgery are the first-line therapies used to treat the disease. However, there is a subset of patients who fail radiation therapy and subsequently need to undergo salvage prostatectomy. Conversely, patients who fail surgery undergo salvage radiation which can result in good tumor control. It would, therefore, be beneficial to determine whether or not a particular patient’s tumor is radiosensitive prior to initiating therapy.
There is little available data regarding prostate cancer, p53 status by sequencing techniques, and treatment outcomes. Most of the available studies have analyzed p53 status by IHC, to look for the presence of mutated p53 [79, 80]. One study that used both genetic sequencing and IHC to determine p53 status found that p53 immunoreactivity by IHC predicted failure to respond to radiation therapy, while the presence of a true gene mutation had no significant association with response to treatment [81]. This result has been replicated, seeming to indicate that in the case of prostate cancer, p53 mutational status does not predict response to radiation therapy [82]. More research is needed, however, before it can be concluded that no association between p53 and response to therapy exists.
Hematologic Malignancies
Hematologic malignancies encompass those tumors that affect the blood, bone marrow, and the lymph nodes. They affect both pediatric and adult populations. Chemotherapy is the mainstay of treatment for these malignancies but response to therapy is unpredictable. While some patients achieve disease-free survival using certain chemotherapeutic regimens, other patients with the same disease fail to adequately respond to the same treatment regimen.
The frequency of p53 mutations in hematologic malignancies has been reported as anywhere from 5 to 50 % [83]. The disparity in frequency of mutations among researchers might explain why results regarding the effect of p53 mutation status on response to treatment are also varied. In one study that looked at patients with acute myelogenous leukemia (AML), myelodysplastic syndrome (MDS), and chronic lymphoblastic leukemia (CLL), p53 mutation predicted a favorable response to chemotherapy in all three diseases [84]. Another study involving exclusively patients with CLL found that p53 mutations were significantly correlated with poor response to treatment [85]. In the case of lymphomas, studies have failed to show a significant relationship between p53 status and response to treatment [86, 87]. There are a number of explanations as to why these results are so varied. One explanation is that p53 status and response to therapy in hematologic malignancies is dependent upon the specific disease entity - ALL versus CLL versus CML, and so on. Another explanation may be that it depends on the specific chemotherapeutic agents used. Ultimately, the answer is most likely to be a combination of both.
Pulmonary Malignancies
Non-small cell lung cancer is the most common non-cutaneous cancer world-wide and it is the number one cause of cancer death worldwide. In certain cases, the response to therapy can be as low as fifty percent. This is especially true in advanced disease, which portends the worst prognosis. Not only are responses to therapy among patients varied, but there are a number of different chemotherapeutic regimens that can be employed in treating the disease. Therefore, identifying tumor markers that predict response to therapy in the case of pulmonary malignancies, such as non-small cell lung cancer, would be of great benefit to patients and clinicians, alike.
In locally advanced disease, chemotherapy with or without the addition of radiation is increasingly being used in the neo-adjuvant setting but response to treatment is unpredictable at the present time. Furthermore, there are different options that can be used for induction chemotherapy, among them are cisplatinum and paclitaxel. Results in the literature have shown that while cisplatinum-based induction chemotherapy either alone [88] or in combination with radiation therapy [89] is ineffective in the case of p53-mutated cancers, response to paclitaxel either alone [90] or in combination with radiation therapy [91] was unrelated to p53 status. These results indicate that, for patients whose tumors harbor a p53 mutation, cisplatinum-based regimens should be avoided. Meanwhile, the fact that p53 status is unrelated to response to paclitaxel suggests that the drug can be used regardless of whether or not a mutation is present. It also signals a need to identify more tumor markers that can help us guide treatment.
Tumors of the Central Nervous System
In the case of central nervous system (CNS) tumors, which encompasses a number of different tumor histologies, data analyzing p53 mutational status and response to therapy are limited. This may be explained by the fact that p53 mutation is a highly infrequent event in certain CNS tumors [92, 93]. Another explanation may be that that mutation status is difficult to assess. CNS tumors are frequently unresected, as in the case of primary lymphoma, or are unresectable, as in the case of brainstem gliomas, so there is limited tissue available for study.
It is important to realize that p53 may not play a role in the tumorigenesis of every tumor type. Therefore, determining the mutational status of these tumors would not be beneficial in predicting response to therapy. For example, Yeung et al. attempt to summarize what is known about the mechanism of radioresistance in vestibular Schwannomas (VS) because radiation therapy has emerged as an alternative treatment modality to surgery for these tumors [94]. The authors hoped that in determining the mechanisms of radioresistance in VS, tumor markers could eventually be identified to guide treatment planning. In identifying markers that predict tumor response to treatment, patients with radioresistant tumors could eventually be identified prior to initiating radiation therapy where treatment failure is likely to ensue. These patients could be selected for microsurgical resection instead. However, the authors found that in these tumors, p53 mutations do not contribute to tumor pathogenesis and, therefore, mutational status is unlikely to be a helpful marker in predicting treatment response.
In other tumor types, p53 mutation may be so infrequent that knowing a tumor’s p53 status may be of low yield in terms of predicting response to therapy. In a study looking at glioblastomas and response to either radiation therapy or temazolomide, p53 mutations were only observed in 15 % of tumors out of a total of 301 tumors analyzed [95]. The authors found a trend towards increased response to temazolomide in the presence of p53 mutations, but their finding was not statistically significant, likely because of the small number of p53 mutated tumors.
Knowing a tumor’s p53 status may be helpful, not only in predicting who will respond to which therapies, but also in identifying those patients who no longer require additional therapy. Choroid plexus tumors are pediatric tumors with poor survival rates which can be treated with chemotherapy, radiation therapy, or both. In a study by Tabori et al. [96], the authors found that patients without p53 mutations who were treated only with chemotherapy had excellent survival rates. The authors concluded that adding radiation therapy to the treatment regimen was unnecessary. If other studies were conducted also supporting this finding, that tumors with mutant p53 have an excellent response to chemotherapy, children diagnosed with choroid plexus tumors could be spared an additional, unnecessary treatment modality that increases morbidity with no survival benefit.
Tumors of the Gastrointestinal Tract
P53 status has been widely studied in tumors of the gastrointestinal tract. These focus primarily on colon cancer, which is the third most frequently diagnosed cancer in the United States in both men and women. Unfortunately, however, a great number of these studies analyze p53 status by IHC rather than by gene sequencing, as with many of the other tumors sites we have seen. Again, because IHC is a less sensitive means of detecting p53 mutation it is not very helpful in determining a clinically useful means of assessing the relationship between p53 and response to therapy. Thus, these individual studies will not be discussed here.
A number of review articles and meta-analyses have been published attempting to uncover the potential role of p53 status as predictive of GI tumors’ response to therapy. These review articles highlight the fact that studies using gene sequencing to determine p53 status are few and far between. In their review of the available literature which assesses predictors of histological response to neo-adjuvant radiation and chemo-radiation, for instance, Smith et al. note that there are three times as many studies using IHC to detect p53 mutational status as there are studies using either gene sequencing or single strand conformational polymorphism analysis. Another review article by Peterson et al. [97] similarly found that, when either chemotherapy or radiation was the treatment modality used, analysis by IHC was much more frequently employed than gene sequencing. Not only is detection by IHC less reliable, but the use of this method in place of gene sequencing can result in different, even conflicting, results. In a review by Munro et al. [51], for instance, the authors found that when studies used IHC to detect p53 abnormalities, they found no relationship between p53 and treatment response to chemo, while they found that p53 mutation was associated with a poor response to radiation therapy. The same review article found that when studies used gene sequencing to detect alterations in the gene, p53 mutation was associated with an unfavorable response to both chemotherapy and to radiation.
While many of these review articles focus on the relationship between p53 mutation and treatment outcome, another approach is to draw conclusions regarding response to therapy in wild-type tumors. In a review of the available literature studying p53 status as a predictive biomarker in response to neo-adjuvant chemo-radiation, Chen et al. [98] found that wild-type tumors were associated with good response to neo-adjuvant treatment. Unfortunately, it is not possible to extrapolate the data and conclude that having a mutated p53 would therefore predict a poor response to therapy. This is not possible because in certain cases, as we have seen with tumors in other organ systems, there may be no predictable relationship between p53 mutation and treatment response and other tumor markers may be needed.
More research is undoubtedly needed before any definitive conclusions can be drawn. However, in looking at the actual studies included in the reviews and the meta-analyses mentioned above using gene sequencing, the current evidence shows that p53 mutation is associated with poor response to therapy in the case of gastrointestinal tumors. In the case of neo-adjuvant radiation, the presence of a p53 mutation detected by gene sequencing has been found to predict a poor response to treatment [99, 100]. Adjuvant chemotherapy, too, in the presence of a p53 mutation has been shown to be less effective [101]. Finally, in the case of chemotherapy given for advanced, Stage IV colorectal cancer, studies have shown a poor response to treatment when p53 is mutated in tumor cells [102, 103].
P53 and response to treatment has also been studied in other gastrointestinal tumors, such as esophageal carcinoma. Like with many other cancers, the treatment of esophageal carcinoma employs a number of treatment modalities, none of which have been overwhelmingly effective in controlling this disease. There is, therefore, an interest in trying to identify tumor characteristics which can predict treatment response in order to avoid unnecessary, costly, and taxing therapies. In one study looking at esophageal carcinoma alone, p53 mutation was associated with poor response to chemo-radiation [104]. More studies are needed to support this finding.
Conclusions
Tumor markers are increasingly being studied as a means to determine tumor response to therapy. If tumor markers could be correctly identified which predict therapeutic response, unnecessary treatments, and the cost and morbidity associated with them, could be avoided. Effective therapies could also be identified early on in the management of aggressive tumors, allowing for more effective tumor control. Knowledge of tumor markers and response to therapy can, therefore, be used to guide treatment selection, resulting in better patient outcomes. In the case of p53, this review has shown that in the majority of tumor sites including ovarian, breast, head and neck, lung and gastrointestinal tumors, research has shown that p53 mutation predicts an unfavorable response to both chemotherapy and radiation (Please refer to Table 8.1 for a summary of the data.). Despite all that has been done thus far, much more research is needed before p53 mutational status can be adapted to the clinical setting. However, with further research, sequencing of the p53 gene may allow us to identify those patients harboring p53 mutations and spare them from unnecessary, costly, taxing, and ultimately ineffective treatments.
References
O’Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, Scudiero DA, Monks A, Sausville EA et al (1997) Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res 57:4285–4300
MacLachlan TK, El-Deiry WS (2002) Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci U S A 99:9492–9497
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS (2002) BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 4:842–849
Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A et al (2002) Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem 277:18817–18826
Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L, Levine AJ, Sacchi A, Cesareni G et al (2000) Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem 275:29503–29512
Brosh R, Rotter V (2009) When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 9:701–713
Li R, Sutphin PD, Schwartz D, Matas D, Almog N, Wolkowicz R, Goldfinger N, Pei H, Prokocimer M, Rotter V (1998) Mutant p53 protein expression interferes with p53-independent apoptotic pathways. Oncogene 16:3269–3277
Fan S, el-Deiry WS, Bae I, Freeman J, Jondle D, Bhatia K, Fornace AJ Jr, Magrath I, Kohn KW, O’Connor PM (1994) p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res 54:5824–5830
Blandino G, Levine AJ, Oren M (1999) Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 18:477–485
Asada N, Tsuchiya H, Tomita K (1999) De novo deletions of p53 gene and wild-type p53 correlate with acquired cisplatin-resistance in human osteosarcoma OST cell line. Anticancer Res 19:5131–5137
Fan J, Bertino JR (1999) Modulation of cisplatinum cytotoxicity by p53: effect of p53-mediated apoptosis and DNA repair. Mol Pharmacol 56:966–972
Bartussek C, Naumann U, Weller M (1999) Accumulation of mutant p53(V143A) modulates the growth, clonogenicity, and radiochemosensitivity of malignant glioma cells independent of endogenous p53 status. Exp Cell Res 253:432–439
Chang FL, Lai MD (2001) Various forms of mutant p53 confer sensitivity to cisplatin and doxorubicin in bladder cancer cells. J Urol 166:304–310
Cabelguenne A, Blons H, de Waziers I, Carnot F, Houllier AM, Soussi T, Brasnu D, Beaune P, Laccourreye O, Laurent-Puig P (2000) p53 alterations predict tumor response to neoadjuvant chemotherapy in head and neck squamous cell carcinoma: a prospective series. J Clin Oncol 18:1465–1473
Koch WM, Brennan JA, Zahurak M, Goodman SN, Westra WH, Schwab D, Yoo GH, Lee DJ, Forastiere AA, Sidransky D (1996) p53 mutation and locoregional treatment failure in head and neck squamous cell carcinoma. J Natl Cancer Inst 88:1580–1586
Fan S, Smith ML, Rivet DJ 2nd, Duba D, Zhan Q, Kohn KW, Fornace AJ Jr, O’Connor PM (1995) Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res 55:1649–1654
Bradford CR, Zhu S, Ogawa H, Ogawa T, Ubell M, Narayan A, Johnson G, Wolf GT, Fisher SG, Carey TE (2003) P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck 25:654–661
Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T et al (2013) p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21:793–806
Martinez-Rivera M, Siddik ZH (2012) Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem Pharmacol 83:1049–1062
Bristow RG, Peacock J, Jang A, Kim J, Hill RP, Benchimol S (2003) Resistance to DNA-damaging agents is discordant from experimental metastatic capacity in MEF ras-transformants-expressing gain of function MTp53. Oncogene 22:2960–2966
Scian MJ, Stagliano KE, Anderson MA, Hassan S, Bowman M, Miles MF, Deb SP, Deb S (2005) Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol Cell Biol 25:10097–10110
Vikhanskaya F, Lee MK, Mazzoletti M, Broggini M, Sabapathy K (2007) Cancer-derived p53 mutants suppress p53-target gene expression – potential mechanism for gain of function of mutant p53. Nucleic Acids Res 35:2093–2104
Wong RP, Tsang WP, Chau PY, Co NN, Tsang TY, Kwok TT (2007) p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol Cancer Ther 6:1054–1061
Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A (2006) Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 25:304–309
Bristow RG, Benchimol S, Hill RP (1996) The p53 gene as a modifier of intrinsic radiosensitivity: implications for radiotherapy. Radiother Oncol 40:197–223
El-Deiry WS (2003) The role of p53 in chemosensitivity and radiosensitivity. Oncogene 22:7486–7495
Dahm-Daphi J (2000) p53: biology and role for cellular radiosensitivity. Strahlenther Onkol 176:278–285
Lee JM, Bernstein A (1993) p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A 90:5742–5746
Bristow RG, Hu Q, Jang A, Chung S, Peacock J, Benchimol S, Hill R (1998) Radioresistant MTp53-expressing rat embryo cell transformants exhibit increased DNA-dsb rejoining during exposure to ionizing radiation. Oncogene 16:1789–1802
Okaichi K, Ide-Kanematsu M, Izumi N, Morita N, Okumura Y, Ihara M (2008) Variations in sensitivity to ionizing radiation in relation to p53 mutation point. Anticancer Res 28:2687–2690
Williams JR, Zhang Y, Zhou H, Gridley DS, Koch CJ, Russell J, Slater JS, Little JB (2008) A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int J Radiat Biol 84:253–264
Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK, Myers JN (2012) TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res 18:290–300
Sandulache VC, Skinner HD, Ow TJ, Zhang A, Xia X, Luchak JM, Wong LJ, Pickering CR, Zhou G, Myers JN (2012) Individualizing antimetabolic treatment strategies for head and neck squamous cell carcinoma based on TP53 mutational status. Cancer 118:711–721
Xu Y (2008) Induction of genetic instability by gain-of-function p53 cancer mutants. Oncogene 27:3501–3507
Cheng G, Kong D, Hou X, Liang B, He M, Liang N, Ma S, Liu X (2013) The tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother Radiopharm 28:153–159
Kim E, Deppert W (2004) Transcriptional activities of mutant p53: when mutations are more than a loss. J Cell Biochem 93:878–886
Scian MJ, Stagliano KE, Ellis MA, Hassan S, Bowman M, Miles MF, Deb SP, Deb S (2004) Modulation of gene expression by tumor-derived p53 mutants. Cancer Res 64:7447–7454
Vaughan CA, Singh S, Windle B, Sankala HM, Graves PR, Andrew Yeudall W, Deb SP, Deb S (2012) p53 mutants induce transcription of NF-kappaB2 in H1299 cells through CBP and STAT binding on the NF-kappaB2 promoter and gain of function activity. Arch Biochem Biophys 518:79–88
Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD (2001) Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 276:39359–39367
Strano S, Dell’Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blandino G (2007) Mutant p53: an oncogenic transcription factor. Oncogene 26:2212–2219
Sankala H, Vaughan C, Wang J, Deb S, Graves PR (2011) Upregulation of the mitochondrial transport protein, Tim50, by mutant p53 contributes to cell growth and chemoresistance. Arch Biochem Biophys 512:52–60
Strauss BE, Shivakumar C, Deb SP, Deb S, Haas M (1995) The MDR1 downstream promoter contains sequence-specific binding sites for wild-type p53. Biochem Biophys Res Commun 217:825–831
Valenti F, Fausti F, Biagioni F, Shay T, Fontemaggi G, Domany E, Yaffe MB, Strano S, Blandino G, Di Agostino S (2011) Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle 10:4330–4340
Vaughan CA, Singh S, Windle B, Yeudall WA, Frum R, Grossman SR, Deb SP, Deb S (2012) Gain-of-function activity of mutant p53 in lung cancer through up-regulation of receptor protein tyrosine kinase Axl. Genes Cancer 3:491–502
Masciarelli S, Fontemaggi G, Di Agostino S, Donzelli S, Carcarino E, Strano S, Blandino G (2013) Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene 32:1–8
Yang X, Pater A, Tang SC (1999) Cloning and characterization of the human BAG-1 gene promoter: upregulation by tumor-derived p53 mutants. Oncogene 18:4546–4553
Zalcenstein A, Stambolsky P, Weisz L, Muller M, Wallach D, Goncharov TM, Krammer PH, Rotter V, Oren M (2003) Mutant p53 gain of function: repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants. Oncogene 22:5667–5676
Zalcenstein A, Weisz L, Stambolsky P, Bar J, Rotter V, Oren M (2006) Repression of the MSP/MST-1 gene contributes to the antiapoptotic gain of function of mutant p53. Oncogene 25:359–369
Deb S, Graves PR (2013) Identification of novel mutant p53 interacting proteins by proteomic analysis. Methods Mol Biol 962:85–94
Ramamoorthy M, Vaughan C, Deb S, Deb SP (2012) Measurement of chemosensitivity and growth rate in p53 expressing cells. Methods Mol Biol 962:127–133
Munro AJ, Lain S, Lane DP (2005) P53 abnormalities and outcomes in colorectal cancer: a systematic review. Br J Cancer 92:434–444
Havrilesky L, Darcy kM, Hamdan H, Priore RL, Leon J, Bell J, Berchuck A (2003) Prognostic significance of p53 mutation and p53 overexpression in advanced epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol 21:3814–3825
Righetti SC, Della Torre G, Pilotti S, Menard S, Ottone F, Colnaghi MI, Pierotti MA, Lavarino C, Cornarotti M et al (1996) A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res 56:689–693
Buttitta F, Marchetti A, Gadducci A, Pellegrini S, Morganti M, Carnicelli V, Cosio S, Gagetti O, Genazzani AR, Bevilacqua G (1997) p53 alterations are predictive of chemoresistance and aggressiveness in ovarian carcinomas: a molecular and immunohistochemical study. Br J Cancer 75:230–235
Reles A, Wen WH, Schmider A, Gee C, Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C et al (2001) Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res 7:2984–2997
Calvert AH, Ghokul S, Al-Azraqi A, Wright J, Lind M, Bailey N, Highley M, Siddiqui N, Lunec J et al (1999) Carboplatin and paclitaxel, alone and in combination: dose escalation, measurement of renal function, and role of the p53 tumor suppressor gene. Semin Oncol 26:90–94
Kigawa J, Sato S, Shimada M, Takahashi M, Itamochi H, Kanamori Y, Terakawa N (2001) p53 gene status and chemosensitivity in ovarian cancer. Hum Cell 14:165–171
Bauerschlag DO, Schem C, Weigel MT, Von Kaisenberg C, Strauss A, Bauknecht T, Maass N, Meinhold-Heerlein I (2010) The role of p53 as a surrogate marker for chemotherapeutical responsiveness in ovarian cancer. J Cancer Res Clin Oncol 136:79–88
Lavarino C, Pilotti S, Oggionni M, Gatti L, Perego P, Bresciani G, Pierotti MA, Scambia G, Ferrandina G et al (2000) p53 gene status and response to platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma. J Clin Oncol 18:3936–3945
Petera J, Sirak I, Beranek M, Vosmik M, Drastikova M, Paulikova S, Soumarova R (2012) Molecular predictive factors of outcome of radiotherapy in cervical cancer. Neoplasma 58:469–475
Ishikawa H, Mitsuhashi N, Sakurai H, Maebayashi K, Niibe H (2001) The effects of p53 status and human papillomavirus infection on the clinical outcome of patients with stage IIIB cervical carcinoma treated with radiation therapy alone. Cancer 91:80–89
Berns EM, Foekens JA, Vossen R, Look MP, Devilee P, Henzen-Logmans SC, van Staveren IL, van Putten WL, Inganas M et al (2000) Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res 60:2155–2162
Chen MB, Zhu YQ, Xu JY, Wang LQ, Liu CY, Ji ZY, Lu PH (2012) Value of TP53 status for predicting response to neoadjuvant chemotherapy in breast cancer: a meta-analysis. PLoS One 7:e39655
Kandioler-Eckersberger D, Ludwig C, Rudas M, Kappel S, Janschek E, Wenzel C, Schlagbauer-Wadl H, Mittlbock M, Gnant M et al (2000) TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin Cancer Res 6:50–56
Oshima K, Naoi Y, Kishi K, Nakamura Y, Iwamoto T, Shimazu K, Nakayama T, Kim SJ, Baba Y et al (2011) Gene expression signature of TP53 but not its mutation status predicts response to sequential paclitaxel and 5-FU/epirubicin/cyclophosphamide in human breast cancer. Cancer Lett 307:149–157
Geisler S, Lonning PE, Aas T, Johnsen H, Fluge O, Haugen DF, Lillehaug JR, Akslen LA, Borresen-Dale AL (2001) Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res 61:2505–2512
Bergh J, Norberg T, Sjogren S, Lindgren A, Holmberg L (1995) Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat Med 1:1029–1034
Aas T, Borresen AL, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug JE, Akslen LA, Lonning PE (1996) Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med 2:811–814
Faille A, De Cremoux P, Extra JM, Linares G, Espie M, Bourstyn E, De Rocquancourt A, Giacchetti S, Marty M, Calvo F (1994) p53 mutations and overexpression in locally advanced breast cancers. Br J Cancer 69:1145–1150
Jansson T, Inganas M, Sjogren S, Norberg T, Lindgren A, Holmberg L, Bergh J (1995) p53 status predicts survival in breast cancer patients treated with or without postoperative radiotherapy: a novel hypothesis based on clinical findings. J Clin Oncol 13:2745–2751
Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa RA, Correo R, Eby YJ, Ruppert JM, Sidransky D (1993) The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res 53:4477–4480
Caamano J, Zhang SY, Rosvold EA, Bauer B, Klein-Szanto AJ (1993) p53 alterations in human squamous cell carcinomas and carcinoma cell lines. Am J Pathol 142:1131–1139
Alsner J, Sorensen SB, Overgaard J (2001) TP53 mutation is related to poor prognosis after radiotherapy, but not surgery, in squamous cell carcinoma of the head and neck. Radiother Oncol 59:179–185
Gallo O, Chiarelli I, Boddi V, Bocciolini C, Bruschini L, Porfirio B (1999) Cumulative prognostic value of p53 mutations and bcl-2 protein expression in head-and-neck cancer treated by radiotherapy. Int J Cancer 84:573–579
Hegde PU, Brenski AC, Caldarelli DD, Hutchinson J, Panje WR, Wood NB, Leurgans S, Preisler HD, Taylor SG 4th et al (1998) Tumor angiogenesis and p53 mutations: prognosis in head and neck cancer. Arch Otolaryngol Head Neck Surg 124:80–85
Chang KP, Hao SP, Lin SY, Tsao KC, Kuo TT, Tsai MH, Tseng CK, Tsang NM (2002) A lack of association between p53 mutations and recurrent nasopharyngeal carcinomas refractory to radiotherapy. Laryngoscope 112:2015–2019
Saunders ME, MacKenzie R, Shipman R, Fransen E, Gilbert R, Jordan RC (1999) Patterns of p53 gene mutations in head and neck cancer: full-length gene sequencing and results of primary radiotherapy. Clin Cancer Res 5:2455–2463
Obata A, Eura M, Sasaki J, Saya H, Chikamatsu K, Tada M, Iggo RD, Yumoto E (2000) Clinical significance of p53 functional loss in squamous cell carcinoma of the oropharynx. Int J Cancer 89:187–193
Grignon DJ, Caplan R, Sarkar FH, Lawton CA, Hammond EH, Pilepich MV, Forman JD, Mesic J, Fu KK et al (1997) p53 status and prognosis of locally advanced prostatic adenocarcinoma: a study based on RTOG 8610. J Natl Cancer Inst 89:158–165
Prendergast NJ, Atkins MR, Schatte EC, Paulson DF, Walther PJ (1996) p53 immunohistochemical and genetic alterations are associated at high incidence with post-irradiated locally persistent prostate carcinoma. J Urol 155:1685–1692
Rakozy C, Grignon DJ, Li Y, Gheiler E, Gururajanna B, Pontes JE, Sakr W, Wood DP Jr, Sarkar FH (1999) p53 gene alterations in prostate cancer after radiation failure and their association with clinical outcome: a molecular and immunohistochemical analysis. Pathol Res Pract 195:129–135
Incognito LS, Cazares LH, Schellhammer PF, Kuban DA, Van Dyk EO, Moriarty RP, Wright GL Jr, Somers KD (2000) Overexpression of p53 in prostate carcinoma is associated with improved overall survival but not predictive of response to radiotherapy. Int J Oncol 17:761–769
Imamura J, Miyoshi I, Koeffler HP (1994) p53 in hematologic malignancies. Blood 84:2412–2421
Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P (1994) p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 84:3148–3157
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, Dearden CE, Richards SM, Catovsky D, Morgan GJ (2012) Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol 29:2223–2229
Preudhomme C, Dervite I, Wattel E, Vanrumbeke M, Flactif M, Lai JL, Hecquet B, Coppin MC, Nelken B et al (1995) Clinical significance of p53 mutations in newly diagnosed Burkitt’s lymphoma and acute lymphoblastic leukemia: a report of 48 cases. J Clin Oncol 13:812–820
Klumb CE, Furtado DR, de Resende LM, Carrico MK, Coelho AM, de Meis E, Maia RC, Rumjanek FD (2003) DNA sequence profile of TP53 gene mutations in childhood B-cell non-Hodgkin’s lymphomas: prognostic implications. Eur J Haematol 71:81–90
Kandioler-Eckersberger D, Kappel S, Mittlbock M, Dekan G, Ludwig C, Janschek E, Pirker R, Wolner E, Eckersberger F (1999) The TP53 genotype but not immunohistochemical result is predictive of response to cisplatin-based neoadjuvant therapy in stage III non-small cell lung cancer. J Thorac Cardiovasc Surg 117:744–750
Kandioler D, Stamatis G, Eberhardt W, Kappel S, Zochbauer-Muller S, Kuhrer I, Mittlbock M, Zwrtek R, Aigner C et al (2008) Growing clinical evidence for the interaction of the p53 genotype and response to induction chemotherapy in advanced non-small cell lung cancer. J Thorac Cardiovasc Surg 135:1036–1041
King TC, Akerley W, Fan AC, Moore T, Mangray S, Hsiu Chen M, Safran H (2000) p53 mutations do not predict response to paclitaxel in metastatic nonsmall cell lung carcinoma. Cancer 89:769–773
Safran H, King T, Choy H, Gollerkeri A, Kwakwa H, Lopez F, Cole B, Myers J, Tarpey J, Rosmarin A (1996) p53 mutations do not predict response to paclitaxel/radiation for nonsmall cell lung carcinoma. Cancer 78:1203–1210
Irving RM, Moffat DA, Hardy DG, Barton DE, Xuereb JH, Maher ER (1993) Molecular genetic analysis of the mechanism of tumorigenesis in acoustic neuroma. Arch Otolaryngol Head Neck Surg 119:1222–1228
Monoh K, Ishikawa K, Yasui N, Mineura K, Andoh H, Togawa K (1998) p53 tumor suppressor gene in acoustic neuromas. Acta Otolaryngol Suppl 537:11–15
Yeung AH, Sughrue ME, Kane AJ, Tihan T, Cheung SW, Parsa AT (2009) Radiobiology of vestibular schwannomas: mechanisms of radioresistance and potential targets for therapeutic sensitization. Neurosurg Focus 27:E2
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M, Schackert G, Simon M et al (2009) Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27:5743–5750
Tabori U, Shlien A, Baskin B, Levitt S, Ray P, Alon N, Hawkins C, Bouffet E, Pienkowska M et al (2010) TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol 28:1995–2001
Petersen S, Thames HD, Nieder C, Petersen C, Baumann M (2001) The results of colorectal cancer treatment by p53 status: treatment-specific overview. Dis Colon Rectum 44:322–333. discussion 333–334
Chen MB, Wu XY, Yu R, Li C, Wang LQ, Shen W, Lu PH (2012) P53 status as a predictive biomarker for patients receiving neoadjuvant radiation-based treatment: a meta-analysis in rectal cancer. PLoS One 7:e45388
Kandioler D, Zwrtek R, Ludwig C, Janschek E, Ploner M, Hofbauer F, Kuhrer I, Kappel S, Wrba F et al (2002) TP53 genotype but not p53 immunohistochemical result predicts response to preoperative short-term radiotherapy in rectal cancer. Ann Surg 235:493–498
Rebischung C, Gerard JP, Gayet J, Thomas G, Hamelin R, Laurent-Puig P (2002) Prognostic value of P53 mutations in rectal carcinoma. Int J Cancer 100:131–135
Westra JL, Schaapveld M, Hollema H, de Boer JP, Kraak MM, de Jong D, ter Elst A, Mulder NH, Buys CH et al (2005) Determination of TP53 mutation is more relevant than microsatellite instability status for the prediction of disease-free survival in adjuvant-treated stage III colon cancer patients. J Clin Oncol 23:5635–5643
Benhattar J, Cerottini JP, Saraga E, Metthez G, Givel JC (1996) p53 mutations as a possible predictor of response to chemotherapy in metastatic colorectal carcinomas. Int J Cancer 69:190–192
Lenz HJ, Hayashi K, Salonga D, Danenberg KD, Danenberg PV, Metzger R, Banerjee D, Bertino JR, Groshen S et al (1998) p53 point mutations and thymidylate synthase messenger RNA levels in disseminated colorectal cancer: an analysis of response and survival. Clin Cancer Res 4:1243–1250
Ribeiro U Jr, Finkelstein SD, Safatle-Ribeiro AV, Landreneau RJ, Clarke MR, Bakker A, Swalsky PA, Gooding WE, Posner MC (1998) p53 sequence analysis predicts treatment response and outcome of patients with esophageal carcinoma. Cancer 83:7–18
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Tchelebi, L., Ashamalla, H., Graves, P.R. (2014). Mutant p53 and the Response to Chemotherapy and Radiation. In: Deb, S., Deb, S. (eds) Mutant p53 and MDM2 in Cancer. Subcellular Biochemistry, vol 85. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9211-0_8
Download citation
DOI: https://doi.org/10.1007/978-94-017-9211-0_8
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9210-3
Online ISBN: 978-94-017-9211-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)