
Abstract
Histone deacetylases are key enzymes of epigenetic regulation and are therefore involved in crucial cellular events like transcription, differentiation, apoptosis and cell division. The deacetylation of many non-histone substrates contributes to these phenomena as well. Thus, these enzymes are an attractive target for anticancer drug development, and first inhibitors are already approved for treatment of cutaneous T-cell lymphoma. About 10–15 inhibitors are currently in clinical trials for a variety of diseases but as single-agent response rates have so far been limited, esp. in solid tumours. There are many open questions, such as the relevance of subtype selectivity in enzyme inhibition, the clinical relevance of non-histone substrates and proper combination regimens. Still, histone deacetylase inhibitors are a promising class of new anticancer agents, and here we review their anticancer properties with a focus on clinical trials from the last 3 years.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Reversible Histone Acetylation and Histone Deacetylases
One of the pivotal modifications associated with epigenetic gene regulation is the acetylation of histone proteins within chromatin. Two enzymes are responsible for the maintenance of the acetylation equilibrium. Histone acetyltransferases (HATs) mediate the transfer of an acetyl group to the ε-amino group of lysine residues in histones and other proteins by using the cofactor acetyl-CoA. Histone deacetylases (HDACs) catalyse the removal of the acetyl group. Upon acetylation, the positive charge of lysines is neutralised, and in the case of histones, the interaction with the negatively charged DNA backbone is diminished. This leads to a more open chromatin (euchromatin) that is available for binding of proteins like transcription factors. Deacetylation restores the positive charge, and the intensified interaction with the DNA backbone is leading to a more condensed form of the chromatin (heterochromatin). Generally, acetylation is associated with transcriptional activation, whereas deacetylation is associated with gene repression (Latham and Dent 2007). However, it was shown that HDACs are also located at active genes to reset the acetylation state in active genes and maintain an adequate level of histone acetylation (Wang et al. 2009).
Histone deacetylases can be divided into four classes based on phylogenetic comparison with yeast enzymes. The classes I, II and IV comprises zinc-dependent deacetylases (HDACs), whereas the class III enzymes have a NAD+-dependent mechanism and are generally referred to as sirtuins because of their homology to the yeast-silencing protein Sir2 (Sauve et al. 2006). Because of the different catalytic mechanism, clinically relevant inhibitors that are active on class I, II and IV HDACs do not target sirtuins and vice versa. Class I consists of HDAC 1, 2, 3 and 8 and is homologous to the yeast enzyme rpd3, and its members are predominantly located in the nucleus. Class II can be further subdivided into class IIa and class IIb. The isotypes 4, 5, 7 and 9 constitute the HDACs of class IIa, and the subtypes 6 and 10 belong to class IIb. HDAC 6 and 10 have two catalytic domains, and there is conflicting evidence on the relevance for in vivo enzyme activity. While some reports indicated that both are required for enzymatic activity (Verdin et al. 2003; Zhang et al. 2006), there is also evidence for the activity of only one site (Zou et al. 2006). The enzymes of class II are homologous to the yeast protein hda1 (Verdin et al. 2004) and shuttle between the cytoplasm and the nucleus (Yoo and Jones 2006). The only member of class IV is HDAC 11 which is located in the nucleus.
Besides histones many other proteins have been recognised as substrates for HDACs (Choudhary et al. 2009). Examples are transcription factors, hormone receptors, signal transducers, chaperones and proteins of the cytoskeleton. Protein acetylation and deacetylation influence a lot of processes besides transcription. An important example is protein stability via the non-histone substrates tubulin, which affects the aggresome pathway of protein degradation (Hideshima et al. 2005), and the chaperone hsp90 (Bali et al. 2005). Most of the non-histone proteins, e.g. the tumour suppressor p53 (Luo et al. 2000), are deacetylated by class I HDACs. α-Tubulin and hsp90 are targets of HDAC 6. Because of the many non-histone targets, HDACs are sometimes referred to as protein or lysine deacetylases (KDACs) rather than histone deacetylases. The application of subtype-specific HDACi might help to address the relevance of the different substrates in mechanistic studies, and the investigation of unselective inhibitors in comparison to selective inhibitors in the clinic will help to understand the underlying mechanisms (Glozak et al. 2005; Buchwald et al. 2009).
10.2 HDACs and Disease
In recent years HDACs have emerged as potential therapeutic targets because their inhibitors are able to reverse dysregulated epigenetic states associated with disease, esp. cancer. It could be shown that there is aberrant acetylation and altered expression of HDACs in cancer cells and tumour tissue (Bolden et al. 2006). Oncogenic fusion proteins, present in some forms of leukaemia, recruit HDAC-containing repressor complexes that constitutively repress expression of specific target genes. In acute promyelocytic leukaemia, fusion proteins of the retinoic acid receptor-α with other proteins that block transcription via recruitment of HDACs are responsible for pathogenesis of the disease on a molecular level (Lin et al. 2001; Pandolfi 2001). In diffuse large B-cell lymphomas, the transcription factor B-cell lymphoma 6 (BCL6) is highly overexpressed. BCL6 itself is hypoacetylated by HDACs and recruits HDAC 2 to repress growth-regulatory target genes. Treatment of this disease with HDACi results in hyperacetylation of BCL6, release of HDAC 2, reactivation of repressed target genes and tumour cell apoptosis (Pasqualucci et al. 2003). Overexpression of different HDACs has been reported in several cancers. In general, it was found that class I expression was high in advanced, strongly proliferating tumours and thus associated with negative prognosis in certain tumours. Class II HDACs were found to be downregulated in human tumours, and high expression predicted better outcome. These findings have to be further investigated because information about the acetylation status and HDAC expression in tumours compared to normal tissue might be an important marker of prognosis and response to treatment (Weichert 2009).
Several other diseases are associated with HDAC activity. HDACi might be able to overcome HIV latency by activation of HIV production from latently infected cells and thereby enhance elimination of these cells (Wightman et al. 2012). HDACs are also implicated to play a crucial role in cardiovascular diseases (Ohtani and Dimmeler 2011). HDAC inhibitors have been shown to be efficacious in preclinical models of heart failure (McKinsey 2012). Furthermore, altered histone acetylation is involved in neurodegeneration (Fischer et al. 2010). HDACi have shown neuroprotection in models for neurodegenerative diseases like Huntington’s disease or spinocerebellar ataxias (Gottesfeld and Pandolfo 2009).
Overall, HDACs play an important role in processes like apoptosis, differentiation and autophagy. Inhibitors targeting these enzymes are valuable to treat diseases that underlie a dysregulation in acetylation. Thus, a lot of effort has been put in the development of HDACi in the last few years. The preclinical pharmacology has been reviewed broadly, elsewhere (Khan and La Thangue 2012), and we will only highlight selected findings and focus mainly on the clinical studies.
10.3 HDAC Inhibitors (HDACi)
Originally, some HDACi have been discovered initially as inducers of cell differentiation (Leder et al. 1975; Riggs et al. 1977). They are able to cause cell cycle arrest in G1 and/or G2 phase, leading to inhibition of cell growth (Bolden et al. 2006). HDACi are also able to induce apoptosis by activating extrinsic (death receptor) and intrinsic (mitochondrial) pathways (Ma et al. 2009). The advantage of HDACi is that they show a high sensitivity towards transformed cells compared to normal cells (Parsons et al. 1997).
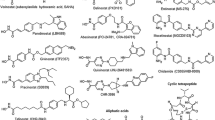
Because of these findings, the development of HDACi has become a major goal especially since clinical efficacy could be demonstrated for vorinostat and romidepsin (see below). So far inhibitors of four structurally different classes are in clinical development. All of them have a functional group that is responsible for chelating the zinc ion in the active centre and hence enzyme inactivation (see Fig. 10.1). Structural differences among the inhibitors lead to different HDAC subtype selectivity profiles. So far it is not clear whether pan-inhibitors or subtype-selective compounds are better for cancer treatment. This may vary with the disease indication. Additionally, it remains to be determined whether the chemical classification is associated with certain benefits or risks for one chemical group or the other.

HDAC inhibitors currently investigated in clinical trials
One group is the cyclic peptides like romidepsin (Istodax®). Romidepsin, a natural product isolated from Chromobacterium violaceum, is a prodrug that is activated in the cell by reduction of the disulfide and is a class I-selective inhibitor (Furumai et al. 2002). Romidepsin inhibits tumour growth in mouse models and humans (Ueda et al. 1994) and was approved by the FDA in 2009 for the treatment of refractory cutaneous T-cell lymphoma. Another important structural group is the hydroxamic acids. The first compound found to inhibit HDACs was the natural product trichostatin A (Yoshida et al. 1990). Further compounds like vorinostat, panobinostat, belinostat, givinostat and SB939 have shown great inhibitory activity and are currently under clinical investigation. Vorinostat (Zolinza®) was the first HDACi to be approved by the FDA for the treatment of cutaneous T-cell lymphoma in 2006. It has been shown that vorinostat is able to induce differentiation, cell growth arrest and apoptosis in numerous cancer cell lines at low micromolar concentrations and inhibits tumour growth with little toxicity in a wide range of animal models (Jones 2009). Panobinostat, an orally available HDACi, shows antiproliferative and cytotoxic activity in a variety of cancer cell lines and induces hyperacetylation of histone H3 and H4 (Atadja 2009). Belinostat is a potent (IC50 27 nM) HDACi that induces apoptosis in human tumour cell lines and xenografts (Plumb et al. 2003). Pracinostat (SB939) is a novel HDACi with improved pharmacokinetic properties. It shows a fourfold improved bioavailability and a threefold increased plasma half-life over vorinostat and accumulates in tumour tissue (Novotny-Diermayr et al. 2010). Furthermore quisinostat (JNJ-26481585), a hydroxamic acid-containing inhibitor with potent antitumoural activity and favourable pharmacodynamic properties was developed and is currently investigated in clinical studies (Arts et al. 2009). New pyrimidine hydroxamates with potent protein inhibition have been synthesised with CHR-3996 being the most promising one. CHR-3996 shows class I subtype selectivity, and good oral bioavailability as well as complete inhibition of growth in human tumour xenografts is described (Moffat et al. 2010). Another structurally different class of HDACi are the short-chain fatty acids like valproic acid and phenylbutyrate. These compounds show only low inhibitory (100–1000-fold weaker than romidepsin or vorinostat) effects but are applied already in the clinic for other indications. Valproic acid is already approved for the use as an antiepileptic drug and induces differentiation and hyperacetylation of histones (Göttlicher et al. 2001). Because of its well-characterised pharmacodynamic and pharmacokinetic profile and manageable side effects, it is investigated in different trials as antileukaemic agent, usually in combination with other drugs.
Benzamides like entinostat, mocetinostat and CS055 are another class of HDACi. Here the amino anilide group is responsible for enzyme inhibition and also confers a selectivity to class I HDACs (Bressi et al. 2010). Entinostat and mocetinostat are class I-selective inhibitors and cause cell cycle arrest and hyperacetylation of histone H4. Entinostat inhibits cell proliferation and growth in human tumour xenografts (Saito et al. 1999). Mocetinostat induces histone hyperacetylation and apoptosis and shows antiproliferative activities against several human cancer cell lines and xenografts (Fournel et al. 2008). CS055 has a similar chemical structure like entinostat. However, it displays lower toxicity, better tolerance and higher stability during administration to animals. It is a class I-selective inhibitor and induces growth arrest, apoptosis and differentiation of leukaemia cells (Gong et al. 2012).
HDACi are investigated in several clinical trials in haematological and solid malignancies as single agents, but in quite a number of cases, the efficacy was not satisfying. Therefore, in many studies HDACi are used in combination with other anticancer agents. The epigenetic combination HDACi and DNA-methyltransferase inhibitors like azacytidine in haematological malignancies is supported by the fact that in these malignancies, abnormal recruitment of HDACs to nuclear protein complexes takes place (Khan and La Thangue 2012). Furthermore, as outlined above there is a role of HDAC6 in protein degradation, and therefore, it was logic to combine HDACi with proteasome inhibitors that target the other major protein degradation pathway. This combination has shown efficacy, even in patients with relapsed/refractory disease who have previously received proteasome inhibitor treatment (Jagannath et al. 2010). In the light of the differentiation inducing abilities of HDACi, another obvious combination is one with other differentiating agents, such as retinoids. Additionally, many other mechanistically different agents are investigated. Also a combination with radiotherapy is based on preclinical evidence (Kim et al. 1999) and is applied clinically (see below).
Besides the question of mono- versus combination therapy, the question of class selectivity has to be addressed. It has to be proven whether class-specific HDACi will reveal greater clinical benefit or if pan-HDAC inhibition has greater efficacy. Due to the presence of HDACi in multi-protein complexes, there is also evidence for a complex rather than subtype selectivity of HDAC inhibitors (Bantscheff et al. 2011). A thorough overview of clinical trials of HDACi has been reported by us a few years ago (Wagner et al. 2010). Here we provide an update of the clinical trials reported between 2010 and the middle of 2012.
10.4 Clinical Trials of Vorinostat
Vorinostat was approved by the FDA for the treatment of refractory cutaneous T-cell lymphoma. It was the first HDACi to be approved and is currently under investigation in clinical trials as monotherapy as well as in combination with several other antitumour agents for various cancers (see Table 10.1).
Recently, five trials of vorinostat as single-agent therapy have been reported. In a phase I trial, the effect of vorinostat in patients suffering from advanced solid tumours and hepatic dysfunction was investigated. Fifty-seven patients were enrolled of which 42 had hepatic dysfunction. The recommended phase II dose for patients with mild, moderate and severe hepatic dysfunction was determined to be 300, 200 and 100 mg/day. Twelve patients experienced stabilisation of disease. Of five patients with adenoid cystic carcinoma, one patient had a partial response, and four patients had stable disease. As adenoid cystic carcinoma is usually refractory to chemotherapy, treatment with vorinostat is a concept to be further investigated in future trials (Ramalingam et al. 2010). In a phase I/II study, the impact of UDP-glucuronyltransferase 2B17 genotype on vorinostat metabolism and clinical outcome in Asian women with breast cancer was investigated. UGT2B17 is a key enzyme in the metabolism of vorinostat (Balliet et al. 2009). Vorinostat is glucuronidated by UGT2B17 and thereby inactivated. Patients received 400 mg/day vorinostat in a lead-in phase I followed by a phase II study. Patients were genotyped for UGT2B17 null genotype (UGT2B17*2), a deletion variant that reduced vorinostat glucuronidation. Wild-type homozygotes (UGT2B17*1/*1), heterozygotes (UGT2B17*1/*2) and homozygotes (UGT2B17*2/*2) for the deletion were compared. Patients who carried at least one copy of a functional UGT2B7 variant were expected to have clinically relevant enzymatic activity (UGT2B17*1/*1 or *1/*2), whereas patients who were homozygous for the null variant (UGT2B17*2/*2) were expected to possess minimal enzyme activity. UGT2B17*2 homozygotes were more likely to experience a serious adverse event, to derive clinical benefit and have longer progression-free survival compared to those who carried at least one copy of UGT2B17*1, although these differences were not statistically significant due to the small patient population. Twenty-six patients received treatment, one patient achieved a partial response and six patients had stable disease lasting for 12 weeks or more (Wong et al. 2011). Another phase I study of 16 patients with gastrointestinal cancer was reported. They were treated with vorinostat 300 mg bid for three consecutive days followed by four rest days per cycle or vorinostat 400 mg qd for 21 consecutive days per cycle. Five patients taking 300 mg bid and two patients taking 400 mg qd maintained stable disease for more than 8 weeks. The 300 mg bid dosing regimen was better tolerated in regard to hematologic toxicities. The most common drug-related adverse events were anorexia, nausea, fatigue and hyperglycaemia (Doi et al. 2013). In a phase II study, 35 patients with relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma were treated with vorinostat at a dose of 200 mg twice daily on days 1 through 14 of a 21-day cycle. Five patients had complete responses and five partial responses (Kirschbaum et al. 2011). In another phase II study, 25 patients with relapsed/refractory Hodgkin lymphoma received oral vorinostat, 200 mg twice daily, for 14 days on a 21-day cycle. The overall response rate was 4 %. One patient had a partial response and 12 had stable disease, seven of them remaining progression-free for more than a year (Kirschbaum et al. 2012). Taken together vorinostat as a single agent is well tolerated but shows in many cases only weak clinical activity, especially in solid tumours. Further studies of vorinostat in combination with other active agents will investigate the beneficial effect of a combination therapy. In the last few years, a lot of trials of vorinostat combination therapy for various cancers have been reported.
A phase I study of vorinostat combined with the proteasome inhibitor marizomib in patients with melanoma, pancreatic and lung cancer was conducted. Twenty-two patients received weekly marizomib in combination with 300 mg vorinostat daily for 16 days in 28-day cycles. No confirmed responses were reported, but of 18 evaluable patients, 11 had stable disease (61 %, all having melanoma). Combining marizomib with vorinostat in patients was seen to be feasible and tolerable (Millward et al. 2012). The MTD of vorinostat in combination with vinorelbine was investigated in a phase I study. Seven patients with advanced cancers were treated with a starting dose of 200 mg oral vorinostat once daily for 7 days every 21 days in combination with a weekly infusion of vinorelbine (25 mg/m2). This dosing was determined to be the MTD (Gandia et al. 2011). The safety, tolerability, pharmacokinetics and preliminary efficacy of vorinostat in combination with the DNA-methyltransferase inhibitor decitabine were also investigated in a phase I study in patients with advanced solid tumours and non-Hodgkin lymphomas. Forty-three patients were treated in a sequential or a concurrent dose schedule. Intravenous decitabine was administered on days 1–5 combined with oral vorinostat in a sequential (vorinostat starting on day six) or a concurrent schedule (vorinostat starting on day three), in 28-day cycle. The recommended phase II dose is decitabine 10 mg/m2/day on days 1–5 with 200 mg vorinostat twice daily on days 6–12 on a sequential schedule. The most frequent adverse events were neutropenia and thrombocytopenia. Of 38 patients evaluable for response, 11 had stable disease for 4 or more cycles of treatment (Stathis et al. 2011). The effect of vorinostat in combination with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer was studied in a phase II trial. Tamoxifen has antiproliferative effects on cancer cells. In combination with an HDACi, it could be shown that tamoxifen induces apoptosis rather than growth inhibition. Data from preclinical models suggest that the combination of tamoxifen with an HDACi resensitises hormone therapy-resistant breast cancer cells. Forty-three patients with ER-positive metastatic breast cancer progressing on endocrine therapy were enrolled and treated with 400 mg of vorinostat daily for 3 of 4 weeks and 20 mg tamoxifen daily, continuously. Eight patients had objective responses and nine patients had stable disease for more than 24 weeks. Histone H4 hyperacetylation in PBMCs was measured, and it was found that patients with a response or stable disease showed pronounced histone H4 hyperacetylation compared to nonresponders. This trial demonstrates that the combination of vorinostat and tamoxifen in patients with hormone receptor-positive breast cancer results in tumour regression or prolonged disease stabilisation in patients who had progressed on prior therapy and is worth to be further investigated (Munster et al. 2011). Two studies evaluated the effect of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumours. A phase I study determined the MTD of daily or twice daily vorinostat on 3 days when combined with fixed doses of 5-fluorouracil and leucovorin every 2 weeks. Forty-three patients were treated and the MTD of vorinostat in this combination was found to be 1,700 mg orally once daily on three consecutive days or 600 mg orally twice daily on three consecutive days every 2 weeks. Of 38 patients with 5-fluorouracil-refractory colorectal cancer, 21 had stable disease and one had a partial response (Fakih et al. 2010). In a phase II study, it was investigated whether the combination of relatively high doses of intermittent vorinostat overcomes resistance to 5-fluorouracil in refractory metastatic colorectal cancer. Fifteen patients received high-dose (1,400 mg) vorinostat, and 43 received low-dose (800 mg) vorinostat. The median progression-free survival and overall survival on the high-dose arm were 2.9 and 6.7 months, compared to 2.4 and 6.5 months on the low-dose arm, respectively. Accrual on the high-dose arm had to be halted because the number of patients without progression at 2 months did not reach the threshold. On the low-dose arm, one patient had a partial response and eight patients had stable disease. However, the progression-free survival rate did not reach the prespecified threshold of 27 out of 43 patients, and the combination was not deemed interesting enough for further evaluation (Fakih et al. 2012). Two trials have been reported using vorinostat in combination therapy for the treatment of recurrent glioblastoma. In a phase I trial, vorinostat was combined with bevacizumab and CPT-11. Nineteen patients were treated. The MTD of vorinostat was established at 400 mg on days 1–7 and 15–21 every 28 days when combined with bevacizumab and CPT-11. The median progression-free survival (PFS) among patients receiving higher-dose vorinostat had an improved response, although not statistically significant (Chinnaiyan et al. 2012). Furthermore, a phase II trial of vorinostat in combination with bortezomib was conducted. Thirty-seven patients were treated with vorinostat at a dose of 400 mg daily for 14 days of a 21-day cycle, and bortezomib was administered at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8 and 11 of the cycle. The trial failed to meet the interim analysis threshold for continuation. None of the 34 patients were progression-free at 6 months (Friday et al. 2012). Another phase I trial was reported to investigate the MTD of vorinostat in combination with bexarotene in patients with advanced (stage IB or higher) CTCL who were refractory to at least one prior systemic treatment and were suitable for bexarotene therapy. The study was divided into two parts. For patients enrolled in part I, up to three dose levels of vorinostat (200, 300 and 400 mg daily) and up to three dose levels of bexarotene (150, 225 and 300 mg/m2) were tested. For patients enrolled in part II, dosing began at dose level 6 with vorinostat at 400 mg once daily and bexarotene at 150 mg once daily. Four patients had partial responses and 15 patients had stable disease. The MTD of part I was established at vorinostat 200 mg/day plus bexarotene 300 mg/m2/day. The MTD for part II was not reached; a result of early study termination due to low enrolment. The efficacy of vorinostat and the retinoid bexarotene at an optimum dose and schedule could not be fully explored in patients with advanced CTCL because of the aggressive nature of the underlying disease. Furthermore, the number of patients treated in this study was small, and thus no general conclusions are possible regarding efficacy (Dummer et al. 2012). A phase II trial of vorinostat in combination with gemtuzumab ozogamicin (GO), as induction and post-remission therapy in older adults with previously untreated acute myeloid leukaemia, has been conducted. Patients received vorinostat 400 mg orally once daily on days 1–9 and GO 3 mg/m2 on day 8. Hydroxyurea was given to reduce the white blood cells to less than 10 × 109/L before treatment with vorinostat. Thirty-one patients have been enrolled of which six achieved complete remission and one achieved complete remission with incomplete platelet recovery. Four patients are in ongoing remission after 455, 496, 956 and 988 days (Walter et al. 2012). As already reviewed in (Wagner et al. 2010), vorinostat was also investigated in combination with radiotherapy (Ree et al. 2010).
10.5 Clinical Trials of Panobinostat
Panobinostat is a highly potent HDACi with antitumour activities at low nanomolar concentrations in several preclinical studies, and its clinical efficacy is currently under investigation in several clinical trials (see Table 10.2). There are several trials investigating the effect of panobinostat as single agent. In two phase I trials, the safety and tolerability of intravenous and oral panobinostat in solid tumours are studied. In a multicenter phase I dose-escalation study, 14 patients received intravenous panobinostat on days 1 and 8 of a 21-day cycle. Stable disease for more than 4 months was observed in six patients, and the MTD of 20 mg/m2 was thought to be safe and potentially effective in patients with advanced solid tumours (Morita et al. 2012). In another phase I, open-label, dose-escalation study, panobinostat was administered orally once daily on MWF weekly on a 28-day cycle to 13 patients with advanced solid tumours or CTCL. Seven patients had stable disease but the MTD was not reached (Fukutomi et al. 2012). The most frequently reported adverse event in both trials was thrombocytopenia, which could be observed for several other HDACi. Panobinostat was also administered as single agent in a phase II trial of patients with refractory metastatic renal cell carcinoma. Patients received 45 mg panobinostat twice a week. After 8 weeks of treatment, 12 patients had progressed disease, and because of the lack of efficacy, the trial was closed (Hainsworth et al. 2011). A phase II study of panobinostat in patients with low- or intermediate-risk myelodysplastic syndrome was conducted. Thirteen patients were treated to determine the clinical efficacy, safety and tolerability of oral panobinostat at a dose of 20 mg three times a week followed by 1 week of rest. One patient achieved a haematological improvement, and six patients had stable disease for a median duration of 6 months. Treatment was well tolerated but overall the study showed only limited clinical activity and was closed to further patient entry (Dimicoli et al. 2012). In a phase II study in patients with relapsed and/or refractory Hodgkin’s lymphoma after autologous stem-cell transplantation, the activity of panobinostat was examined. One hundred and twenty-nine patients were treated with 40 mg panobinostat orally three times per week in a 21-day cycle. Reductions in tumour size were observed in 74 % of the patients with 5 patients having complete response and 30 patients having a partial response lasting for 6.9 months in the median. Panobinostat monotherapy in patients who were heavily pretreated demonstrated antitumour activity, resulting in durable responses. This promising data suggest that further investigation especially in combination with other agents might be valuable to improve therapy (Younes et al. 2012). In a phase I study, treatment of patients with castration-resistant prostate cancer with panobinostat as single agent in comparison to panobinostat in combination with docetaxel was investigated. Eight patients received 20 mg panobinostat on 3 days a week for two consecutive weeks followed by a 1-week break. Eight patients received 15 mg panobinostat on the same schedule in combination with 75 mg/m2 docetaxel every 3 weeks and 5 mg prednisone orally twice a day. None of the patients in the panobinostat monotherapy arm responded. In the combination arm, 2 of 7 evaluable patients had a partial response and four patients had stable disease (Rathkopf et al. 2010). Thus, also panobinostat as a single agent shows limited clinical response so far. Therefore, panobinostat is also investigated in several trials in combination with various other antitumour agents.
In a phase Ib study, the safety, tolerability and preliminary efficacy of panobinostat in combination with the immune modulator lenalidomide plus dexamethasone in patients with relapsed or refractory multiple myeloma were evaluated. Forty-six patients have been treated of which 30 were evaluable for response. One patient had a partial response, 7 had stable disease and 6 progressed on treatment. Some safety concerns were identified, but as preliminary efficacy was very encouraging, further studies with a lower dexamethasone dose and a noncontinuous panobinostat dosing schedule will be conducted (Mateos et al. 2010). A phase I study investigated the effect of panobinostat in combination with the nucleoside analogue gemcitabine in the treatment of solid tumours. Seventeen patients were enrolled, and after several dose de-escalations because of myelosuppression, the recommended doses for further studies were found to be intermittent oral panobinostat administered at a dose of 10 mg three times weekly for 2 weeks in combination with 800 mg/m2 gemcitabine administered intravenously on days 1 and 8 every 21 days. One patient with ovarian cancer had an unconfirmed partial response, and five patients had stable disease lasting more than 4 cycles (Jones et al. 2011). The combination of bevacizumab, everolimus and panobinostat was investigated in a phase I trial of 12 patients with advanced solid tumours. Patients received 10 mg of panobinostat three times weekly, 5 or 10 mg everolimus daily and bevacizumab at 10 mg/kg every 2 weeks. One patient with breast cancer had a partial response lasting for 2 months, and three patients with metastatic colorectal cancer had stable disease. HDAC activity of PBMCs was evaluated on day 1 and 15, but no significant difference was detected. These findings support 20 mg three times per week as the minimum dose level to ensure consistent HDAC inhibition. The addition of panobinostat to the combination bevacizumab/everolimus revealed added toxicity which compromised the tolerability of the full combination (Strickler et al. 2012). Panobinostat in combination with bevacizumab for the treatment of recurrent high-grade glioma was investigated in a phase I trial. Twelve patients received 10 mg/kg bevacizumab every 2 weeks and panobinostat at different dose levels. Three patients had partial response and 7 had stable disease. Although the MTD could not be found, a 30 mg dose in combination with bevacizumab was deemed to represent the maximum feasible dose (Drappatz et al. 2012). A phase II study to examine the efficacy and safety of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy was conducted. Patients received 1.3 mg/m2 bortezomib twice weekly and 20 mg panobinostat three times weekly during the first 2 weeks, followed by 9 days of rest. Seven patients were enrolled but the study had to be closed due to lack of treatment responses and early treatment-related toxicity (Wang et al. 2012). Panobinostat in combination with melphalan, thalidomide and prednisone was investigated in a phase II study in patients with relapsed and or refractory multiple myeloma. Thirty-one patients received a fixed dose of melphalan, thalidomide and prednisone with escalating doses of panobinostat three times weekly for 3 weeks, followed by a 9-day rest period. Two patients achieved a complete response, 12 patients achieved a partial response and 11 had stable disease, but 8 progressed on treatment. This study suggests that the combination of panobinostat plus melphalan, thalidomide and prednisone at this dose and schedule has no therapeutic benefit compared to the combination without HDACi but is more toxic. However, further studies with an improved dose schedule might improve efficacy (Offidani et al. 2012).
10.6 Clinical Trials of Romidepsin
Romidepsin was the second HDACi approved in 2009 by the FDA for the treatment of CTCL of patients who had received at least one systemic therapy. Besides this application, the effect of romidepsin is investigated in the last years in several trials against other haematologic cancers and solid tumours primarily as single agent (see Table 10.3).
A phase II study was conducted to confirm the efficacy of romidepsin in patients with treatment refractory cutaneous T-cell lymphoma. Ninety-six patients were treated with romidepsin as an intravenous infusion at a dose of 14 mg/m2 on days 1, 8 and 15 every 28 days. The overall response was 34 % with six patients having complete response. This study showed that romidepsin has significant activity as a single agent with durable responses in patients with refractory CTCL with an acceptable safety profile and is a valuable therapeutic agent for these patients (Whittaker et al. 2010). Two phase II studies investigated romidepsin in the treatment of patients with peripheral T-cell lymphoma. Piekarz et al. reported the treatment of 45 patients with 14 mg/m2 romidepsin on days 1, 8 and 15 of a 28-day cycle. Eight patients experienced complete responses and nine patients experienced partial responses (Piekarz et al. 2011). A further trial confirmed the efficacy of romidepsin in patients with relapsed or refractory peripheral T-cell lymphoma. One hundred and thirty patients received 14 mg/m2 romidepsin on days 1, 8 and 15 every 28 days. The overall response rate (ORR) was 25 % with 19 patients having complete response and 3 having stable disease (Coiffier et al. 2012). Furthermore, a phase II study was conducted investigating the efficacy of romidepsin in patients with multiple myeloma. Thirteen patients were treated with 13 mg/m2 romidepsin on days 1, 8 and 15 every 28 days. Five patients showed clinical benefit and two patients showed reduction of pain, but no patient achieved an objective response (Niesvizky et al. 2011). A phase II study investigated the effect of romidepsin in patients suffering from relapsed small-cell lung cancer. Sixteen patients were enrolled and received weekly infusions of romidepsin at 13 mg/m2 on days 1, 8 and 15 on a 4-week schedule. The best response seen was stable disease in three patients. The study was closed because it did not reach the target response rate (Otterson et al. 2010). Haigentz et al. reported a phase II trial of romidepsin in patients with recurrent/metastatic head and neck cancer. Fourteen patients were treated with 13 mg/m2 romidepsin on days 1, 8 and 15 of 28-day cycles. The best response seen was stable disease in two patients. Because the study lacked efficacy, it was terminated early (Haigentz et al. 2012). In a phase I/II trial, the effect of romidepsin in 50 patients with recurrent malignant glioma was studied. In phase I of the study, the maximum tolerated dose in patients receiving strong CYP3A4-inducing antiepileptic drugs (EIAEDs) was determined. Romidepsin is metabolised by cytochromes CYP3A4 and CYP3A514, and EIAEDs potently induce CYP3A4. It could be shown that EIAEDs did not affect romidepsin exposure. Because of the potential cardiotoxicity with doses of more than 17.7 mg/m2, the MTD for patients receiving EIAEDs was not defined. In phase II the PFS at 6 months was investigated. Phase II patients were treated with romidepsin at dosage of 13.3 mg/m2/day on days 1, 8 and 15 of each 28-day cycle. Among the 35 patients receiving romidepsin in phase II, one had progression-free survival for more than 6 months but developed tumour progression at 32 weeks. The trial showed that romidepsin had no significant clinical activity as a single agent in patients with recurrent glioma (Iwamoto et al. 2011).
A phase I trial of romidepsin in combination with gemcitabine in patients with advanced solid tumours was reported in 2012. Thirty-six patients with solid tumours were treated, and the MTD of 12 mg/m2 romidepsin and 800 mg/m2 gemcitabine was determined. Twenty-seven patients were evaluable of which 2 had a partial response and 14 had stable disease (Jones et al. 2012). The outcome of the described studies showed that the treatment with romidepsin is also effective in patients with peripheral T-cell lymphoma. However, in solid tumours a combination therapy with other agents might be valuable to improve efficacy.
10.7 Clinical Trials of Valproic Acid
Valproic acid is a well-tolerated drug already used in the treatment of epilepsy with a good characterised safety profile. Although it shows a 1,000-fold lesser HDAC activity compared to other HDACi like vorinostat, it is successfully applied in several trials against different cancers (see Table 10.4).
Preclinical data showed that Notch1 is a tumour suppressor in neuroendocrine tumours. Notch1 signalling is very minimal or nonexistent in neuroendocrine tumours, and the activation of Notch1 signalling leads to a decrease in tumour growth. Valproic acid has been shown to activate the Notch1 signalling pathway leading to a decrease in tumour markers. Because of these data, a phase I study to evaluate the effects of valproic acid on tumour marker production, tumour response, survival and Notch1 signalling has been conducted. Eight patients received a valproic acid dose of 500 mg orally, two times a day with a goal target serum level between 50 and 100 g/mL. Five patients had stable disease over the course of the treatment. It could be shown that the majority of patients experienced an improvement in their tumour markers and after treatment there was a tenfold induction of Notch1 mRNA compared to pretreatment levels (Mohammed et al. 2011).
In several trials valproic acid is applied in combination with other therapeutic agents. Two studies in combination with cytarabine in acute myeloid leukaemia were conducted. A phase II study investigated the effect of valproate in acute myeloid leukaemia (AML) and refractory anaemia with excess of blasts (RAEB). Thirty-one patients were treated with subsequent courses of low-dose cytarabine (20 mg) twice daily for 8 days and valproic acid with a starting dose of 5 mg/kg. Dose escalation of valproic acid was performed according to patient tolerance until the therapeutic range of 50–100 μg/mL was reached. Eight patients had complete response, with nearly complete clearing of marrow blasts and normalisation of blood counts at a median of 5 months. Three patients showed haematologic improvement. It could be shown that low-dose cytarabine in combination with valproic acid is well tolerated and shows good therapeutic activity in elderly patients with AML/RAEB (Corsetti et al. 2011). In another trial 15 patients were treated with a starting dose of 200 mg valproic acid three times a day. The dose was increased according to patient tolerance to achieve serum valproate levels of 50–100 μg/mL. Additionally, patients received 10 mg/m2 cytarabine daily for the first 14 days of therapy. In contrast to the study mentioned before, no responses were observed (Lane et al. 2012). A possible explanation might be that the dose of valproic acid could not be escalated due to poor tolerance of this patient population. In the trial reported by Corsetti et al., the patients received a higher dose of cytarabine with a longer duration, and the patients had a better prognosis at the time of inclusion of the study. In a phase II study, valproate in combination with the DNA-methyltransferase inhibitor hydralazine was investigated in 12 patients with myelodysplastic syndrome. Patients received 83/182 mg hydralazine and 30 mg/kg valproic acid on a daily schedule. Overall response was seen in six patients, including 1 complete response and 1 partial response and 4 haematological improvements. A follow-up after about 14 months showed that only two patients progressed to AML. Overall this study shows that the combination of valproate and hydralazine may be an effective and safe combination in the treatment of MDS; however, so far only a small number of patients were treated (Candelaria et al. 2011). In another trial valproate in combination with hydralazine was administered to eight patients with chronic myeloid leukaemia who were refractory to imatinib. Patients received 83/182 mg hydralazine and 30 mg/kg valproic acid on a daily schedule and continued receiving imatinib at the same dose they were receiving at the time of progression. Two patients had a complete response and 3 had stable disease and only 1 did not respond. This trial gives evidence that therapy using an epigenetic agent can overcome imatinib resistance. However, the number of patients in this study is too small to make a clear statement (Cervera et al. 2012).
Besides haematological malignancies, the efficacy of valproic acid was studied in two trials in solid tumours. In a phase I study, valproic acid in combination with ATRA-IV is investigated in solid tumours refractory to prior therapy. One patient had stable disease lasting for 16 weeks. The MTD of both drugs in combination could not be established due to early closure of the trial (David et al. 2010). In another trial the combination of valproic acid with doxorubicin in patients with mesothelioma was examined. In the phase II trial, 45 patients were treated with oral valproic acid until a serum valproate level of 50–100 μg/mL was reached. Then patients were treated additionally with doxorubicin at 60 mg/m2 every 3 weeks. Valproic acid administration continued during the whole treatment. Seven patients had a partial response. In recurrent mesothelioma after first-line cisplatin-based chemotherapy, this treatment regimen seems to be effective and warrants further trials (Scherpereel et al. 2011).
10.8 Clinical Trials of Belinostat
Belinostat is a hydroxamic acid-based HDACi that is currently investigated in clinical trials (see Table 10.5).
Two trials investigated the efficacy of belinostat as single agent. Cashen et al. reported the application of belinostat in the treatment of myelodysplastic syndrome. Twenty-one patients were enrolled and treated with 1,000 mg/m2 belinostat on days 1–5 of a 21-day cycle. One patient received a partial response lasting for 2.1 months. The study was closed after the first stage of enrolment (Cashen et al. 2012).
Another trial using belinostat as single agent was reported in 2011 by Giaccone et al. In this phase II study, patients with recurrent or refractory advanced thymic epithelial tumours were treated with 1 g/m2 on days 1–5 of a 21-day cycle. Forty-one patients were enrolled of which 25 had thymoma and 16 had thymic carcinoma. Two patients achieved a partial response and 25 had stabilisation of disease lasting for 5.8 months (median), and treatment was well tolerated. In general, patients with thymic carcinoma had significantly shorter survival than those with thymoma. Protein hyperacetylation was analysed, but no correlation between hyperacetylation and response could be found (Giaccone et al. 2011). Several studies describe the combination of belinostat with other agents in solid tumours. In a phase I study, the combination of belinostat with carboplatin and/or paclitaxel was investigated in patients having solid tumours. Belinostat was administered in escalating doses of 600, 800 and 1,000 mg/m2/day on days 1–5 of a 21-day cycle. Carboplatin and paclitaxel were administered on day 3. Treatment was well tolerated and the recommended dose of belinostat was 1,000 mg/m2/day. The pharmacokinetics of belinostat, paclitaxel and carboplatin were unaltered by the concurrent administration. There was one complete CA-125 response, two patients had a partial response and six patients had stable disease for more than 6 months (Lassen et al. 2010). Another phase II study investigated the effect of the combination belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube or primary peritoneal carcinoma. Twenty-seven patients received belinostat in a dose of 1,000 mg/m2 for 5 days every 3 weeks and carboplatin on day 3. There was one complete response, one partial response and 12 patients had stable disease. Because the overall response rate did not meet the criteria for further development, the study was closed at the first stage (Dizon et al. 2012a).
The activity of belinostat, carboplatin, and paclitaxel in women with previously treated ovarian cancer was also investigated by Dizon et al. Thirty-five patients were treated with belinostat (1,000 mg/m2) daily for 5 days; carboplatin and paclitaxel were given on day 3 of a 21-day cycle. Three patients had a complete response, and 12 had a partial response. The overall response rate among resistant patients was 44 and 63 % among sensitive patients. This study could show that the addition of belinostat to standard therapy could enhance the outcome and is worth to be further investigated (Dizon et al. 2012b).
10.9 Clinical Trials of Mocetinostat
Preclinical data have shown that mocetinostat, an oral class I-selective HDAC inhibitor, has potent antiproliferative activity against a wide range of cancers (Fournel et al. 2008). To investigate its clinical efficacy, several clinical trials for haematologic malignancies and solid tumours were conducted (Wagner et al. 2010). In the last year a phase II trial of mocetinostat in patients with relapsed Hodgkin’s lymphoma was reported (see Table 10.6). Fifty-one patients were treated with Mocetinostat three times a week and a dose of 85 mg was found to be the best tolerated. Two patients had a complete response, 12 had a partial response and one had stable disease. These results show that mocetinostat has significant clinical efficacy and its use either as single agent or in combination should be further investigated (Younes et al. 2011).
10.10 Clinical Trials of Entinostat
The ability of HDACi to resensitise tumour cells to retinoids was investigated in a phase I trial of entinostat in combination with 13-cis retinoic acid in patients with solid tumours (see Table 10.6). Nineteen patients were treated with entinostat orally once a week and 13-cis retinoic acid orally twice a day (1 mg/kg) for 21 days every 4 weeks. The MTD was determined at 4 mg/m2 entinostat. There were no complete or partial responses. However, seven patients had stable disease (Pili et al. 2012).
10.11 Clinical Trials of Givinostat
The safety and efficacy of givinostat in the treatment of JAK2V617F-positive chronic myeloproliferative neoplasms, including polycythaemia vera, essential thrombocythaemia and myelofibrosis, was studied in a phase II trial (see Table 10.6). Twenty-nine patients received oral givinostat for 24 weeks at a dose of 50 mg twice daily. Among the patients having polycythaemia vera or essential thrombocythaemia, one patient had a complete haematological response, and six patients had a partial response. Furthermore, complete control of pruritus was observed in all but one patient. Among the patients having myelofibrosis, three patients had achieved a complete response. JAK2V617F tumour allele burden was monitored during treatment and a progressive decrease of the JAK2V617F mutated allele during treatment could be found. The treatment was well tolerated with the most common side effects being mild gastrointestinal disorders like diarrhoea, nausea and gastric or abdominal pain. This study confirmed that givinostat shows clinical activity. In vitro experiments implied that the efficacy of givinostat could be improved by combining it with hydroxycarbamide or other JAK2V617F inhibitors (Rambaldi et al. 2010).
10.12 Clinical Trials of Chidamide
Chidamide is a new orally available benzamide-containing HDACi. Preclinical data have shown that chidamide has in vitro and in vivo antitumour activity against several cancer cell lines. In a phase I trial, tolerability and dose-limiting toxicities of chidamide in patients with advanced solid tumours or lymphomas were determined (see Table 10.6). Thirty-one patients received oral doses of 5, 10, 17.5, 25, 32.5 or 50 mg chidamide either twice (BIW) or three times (TIW) per week for four consecutive weeks every 6 weeks. Five patients had a partial response and 11 patients had stable disease. Chidamide is well tolerated, exhibited a relatively long half-life of 17–18 h and showed a long-lasting histone H3 acetylation (Dong et al. 2012). This data suggest that chidamide is a promising new HDACi and its clinical efficacy should be further evaluated.
10.13 Clinical Trials of Pracinostat (SB939)
Because of the favourable preclinical pharmacological properties of pracinostat, the maximum tolerated dose, the pharmacokinetics, the pharmacodynamics and preliminary efficacy of pracinostat in patients with advanced solid malignancies were investigated in a phase I trial (see Table 10.6). Thirty patients received oral pracinostat (10–80 mg/day) three times a week for 3 weeks in a 4-week cycle. Five patients had stable disease. The maximum tolerated dose using this regimen was determined at 80 mg/day, and the recommended dose for phase II studies is 60 mg/day. Although no partial responses were seen, prolonged nonprogression of breast cancer, follicular thyroid carcinoma, neuroblastoma and hepatocellular carcinoma were promising observations and should be further evaluated (Yong et al. 2011).
Another phase I trial was reported by Razak et al. 38 patients received oral pracinostat. The maximal administered dose was 90 mg, and the recommended phase II dose was 60 mg given five consecutive days every 2 weeks. No objective tumour responses were observed, but ten patients showed stable disease for 5.7 months (median) (Razak et al. 2011).
The most common adverse events were toxicities that included fatigue, nausea, vomiting, anorexia and diarrhoea and are common among HDACi application.
10.14 Clinical Trials of CHR-3996
CHR-3996 is a new, orally available, class I-selective HDACi that has shown promising activity against a wide range of cancer cell lines. In a phase I study, the pharmacokinetics and pharmacodynamics of CHR-3996 were investigated in patients with refractory solid tumours (see Table 10.6). Thirty-eight patients received CHR-3996 (5–160 mg) once a day. One patient had a partial response and nine patients had stable disease. The recommended phase II dose was 40 mg/day. The most common seen DLTs were thrombocytopenia, fatigue and elevated plasma creatinine. Because of the manageable toxicity profile and favourable pharmacokinetic and pharmacodynamic properties, CHR-3996 should be investigated in further studies (Banerji et al. 2012).
10.15 Conclusions
HDACi are antiproliferative agents with manageable side effects, successfully applied in the clinic for the treatment of mainly haematological diseases. Besides vorinostat and romidepsin which are already approved by the FDA, several other potent HDACi are currently investigated for their clinical efficacy in the treatment of cancer.
Even though some promising results could be observed, especially in combination with other anticancer agents, the treatment of solid malignancies with HDACi remains to be unsatisfactory and has to be further investigated in clinical trials. Several trials could demonstrate that HDACi can be administered in combination with standard therapy without decrease of the doses and without additional side effects. Improvement of the dosing and the schedule as well as the optimisation of combination therapies might help to achieve an increased response especially in solid tumours.
Especially interesting in that regard is the question of surrogate biomarkers for HDAC inhibitor dosing. Diagnostic as well as prognostic biomarkers are valuable tools to optimise therapy and improve safety. Although biomarkers like the acetylation of tumour tissue and PBMCs to correlate treatment and response are used in many trials, a clear correlation could not be verified yet. Standard procedures are the analysis of histone hyperacetylation by western blotting, ELISA or mass spectrometry (Chung et al. 2006) Alternatively, total cellular histone deacetylase activity measured with a cell-permeable small molecule substrate has been suggested (Hoffmann et al. 1999; Bonfils et al. 2008).
Another question that has to be addressed is if class-selective inhibitors can increase efficacy or reduce side effects. To answer that, more information about the role of specific isoforms and their function in cancer is needed. Some experiments using knockout mice have been done to elucidate this (Haberland et al. 2009), but a lot still needs to be learned to fully understand the distinct roles of the HDAC subtypes. In this context the role of protein versus histone acetylation and the clinical application has to be investigated. This will be especially true for the planned clinical development of HDAC6-selective inhibitors.
Altogether, despite many years of HDAC research, there are still many fundamental issues that we have to understand better. Still, HDAC inhibitors are already used in the clinic with some success, and we expect to exploit their potential even better in the upcoming years.
Abbreviations
- bid:
-
Two times daily
- CPT-11:
-
Irinotecan
- CR:
-
Complete response
- CTCL:
-
Cutaneous T-cell lymphoma
- DLT:
-
Dose-limiting toxicity
- EIAEDs:
-
Enzyme-inducing antiepileptic drugs
- FDA:
-
Food and Drug Administration
- HDAC:
-
Histone deacetylase
- HDACi:
-
Histone deacetylase inhibitor
- HI:
-
Haematological improvement
- MTD:
-
Maximum tolerated dose
- MWF:
-
Monday, Wednesday, Friday
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PBMC:
-
Peripheral blood mononuclear cell
- PFS:
-
Progression free survival
- PR:
-
Partial response
- qd:
-
Once daily
- RP2D:
-
Recommended phase II dose
- SD:
-
Stable disease
References
Arts J, King P, Marien A, Floren W, Belien A, Janssen L, Pilatte I, Roux B, Decrane L, Gilissen R, Hickson I, Vreys V, Cox E, Bol K, Talloen W, Goris I, Andries L, Du Jardin M, Janicot M, Page M, van Emelen K, Angibaud P (2009) JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res 15:6841–6851
Atadja P (2009) Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 280:233–241
Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O et al (2009) Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res 15:5250–5257
Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K (2005) Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280:26729–26734
Balliet RM, Chen G, Gallagher CJ, Dellinger RW, Sun D, Lazarus P (2009) Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT phenotype. Cancer Res 69:2981–2989
Banerji U, van Doorn L, Papadatos-Pastos D, Kristeleit R, Debnam P, Tall M, Stewart A, Raynaud F, Garrett MD, Toal M, Hooftman L, De Bono JS, Verweij J, Eskens FA (2012) A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin Cancer Res 18:2687–2694
Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol 29:255–265
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–784
Bonfils C, Kalita A, Dubay M, Siu LL, Carducci MA, Reid G, Martell RE, Besterman JM, Li Z (2008) Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin Cancer Res 14:3441–3449
Bressi JC, Jennings AJ, Skene R, Wu Y, Melkus R, De Jong R, O’Connell S, Grimshaw CE, Navre M, Gangloff AR (2010) Exploration of the HDAC2 foot pocket: synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett 20:3142–3145
Buchwald M, Kramer OH, Heinzel T (2009) HDACi–targets beyond chromatin. Cancer Lett 280:160–167
Candelaria M, Herrera A, Labardini J, Gonzalez-Fierro A, Trejo-Becerril C, Taja-Chayeb L, Perez-Cardenas E, de la Cruz-Hernandez E, Arias-Bofill D, Vidal S, Cervera E, Duenas-Gonzalez A (2011) Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann Hematol 90:379–387
Cashen A, Juckett M, Jumonville A, Litzow M, Flynn PJ, Eckardt J, LaPlant B, Laumann K, Erlichman C, DiPersio J (2012) Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann Hematol 91:33–38
Cervera E, Candelaria M, Lopez-Navarro O, Labardini J, Gonzalez-Fierro A, Taja-Chayeb L, Cortes J, Gordillo-Bastidas D, Duenas-Gonzalez A (2012) Epigenetic therapy with hydralazine and magnesium valproate reverses imatinib resistance in patients with chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk 12:207–212
Chinnaiyan P, Chowdhary S, Potthast L, Prabhu A, Tsai YY, Sarcar B, Kahali S, Brem S, Yu HM, Rojiani A, Murtagh R, Pan E (2012) Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro Oncol 14:93–100
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840
Chung EJ, Lee MJ, Lee S, Trepel JB (2006) Assays for pharmacodynamic analysis of histone deacetylase inhibitors. Expert Opin Drug Metab Toxicol 2:213–230
Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, Pinter-Brown L, Padmanabhan S, Shustov A, Nichols J, Carroll S, Balser J, Balser B, Horwitz S (2012) Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30:631–636
Corsetti MT, Salvi F, Perticone S, Baraldi A, De Paoli L, Gatto S, Pietrasanta D, Pini M, Primon V, Zallio F, Tonso A, Alvaro MG, Ciravegna G, Levis A (2011) Hematologic improvement and response in elderly AML/RAEB patients treated with valproic acid and low-dose Ara-C. Leuk Res 35:991–997
David KA, Mongan NP, Smith C, Gudas LJ, Nanus DM (2010) Phase I trial of ATRA-IV and Depakote in patients with advanced solid tumor malignancies. Cancer Biol Ther 9:678–684
Dimicoli S, Jabbour E, Borthakur G, Kadia T, Estrov Z, Yang H, Kelly M, Pierce S, Kantarjian H, Garcia-Manero G (2012) Phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in patients with low or intermediate-1 risk myelodysplastic syndrome. Am J Hematol 87:127–129
Dizon DS, Blessing JA, Penson RT, Drake RD, Walker JL, Johnston CM, Disilvestro PA, Fader AN (2012a) A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 125:367–371
Dizon DS, Damstrup L, Finkler NJ, Lassen U, Celano P, Glasspool R, Crowley E, Lichenstein HS, Knoblach P, Penson RT (2012b) Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int J Gynecol Cancer 22:979–986
Doi T, Hamaguchi T, Shirao K, Chin K, Hatake K, Noguchi K, Otsuki T, Mehta A, Ohtsu A (2013) Evaluation of safety, pharmacokinetics, and efficacy of vorinostat, a histone deacetylase inhibitor, in the treatment of gastrointestinal (GI) cancer in a phase I clinical trial. Int J Clin Oncol 18(1):87–95
Dong M, Ning ZQ, Xing PY, Xu JL, Cao HX, Dou GF, Meng ZY, Shi YK, Lu XP, Feng FY (2012) Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother Pharmacol 69:1413–1422
Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, Gerard M, Schiff D, Chi AS, Batchelor TT, Doherty LM, Ciampa AS, Lafrankie DC, Ruland S, Snodgrass SM, Raizer JJ, Wen PY (2012) Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol 107:133–138
Dummer R, Beyer M, Hymes K, Epping MT, Bernards R, Steinhoff M, Sterry W, Kerl H, Heath K, Ahern JD, Hardwick JS, Garcia-Vargas J, Baumann K, Rizvi S, Frankel SR, Whittaker SJ, Assaf C (2012) Vorinostat combined with bexarotene for treatment of cutaneous T-cell lymphoma: in vitro and phase I clinical evidence supporting augmentation of retinoic acid receptor/retinoid X receptor activation by histone deacetylase inhibition. Leuk Lymphoma 53(8):1501–1508
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109:31–39
Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M (2013) Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur J Cancer 49:386–394
Fakih MG, Fetterly G, Egorin MJ, Muindi JR, Espinoza-Delgado I, Zwiebel JA, Litwin A, Holleran JL, Wang K, Diasio RB (2010) A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin Cancer Res 16:3786–3794
Fakih MG, Groman A, McMahon J, Wilding G, Muindi JR (2012) A randomized phase II study of two doses of vorinostat in combination with 5-FU/LV in patients with refractory colorectal cancer. Cancer Chemother Pharmacol 69:743–751
Fischer A, Sananbenesi F, Mungenast A, Tsai LH (2010) Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci 31:605–617
Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, Gillespie J, Wang JJ, Ste-Croix H, Rahil J, Lefebvre S, Moradei O, Delorme D, Macleod AR, Besterman JM, Li Z (2008) MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther 7:759–768
Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E, Jaeckle K, Galanis E (2012) Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol 14:215–221
Fukutomi A, Hatake K, Matsui K, Sakajiri S, Hirashima T, Tanii H, Kobayashi K, Yamamoto N (2012) A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest New Drugs 30:1096–1106
Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S (2002) FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res 62:4916–4921
Gandia P, Arellano C, Chalret du Rieu Q, Lochon I, Campone M, Pierga JY, Poublanc M, Hennebelle I, Filleron T, Chatelut E, Delord JP (2011) Unexpected high levels of vorinostat when combined with vinorelbine in patients with advanced cancer. Curr Clin Pharmacol 6:274–279
Giaccone G, Rajan A, Berman A, Kelly RJ, Szabo E, Lopez-Chavez A, Trepel J, Lee MJ, Cao L, Espinoza-Delgado I, Spittler J, Loehrer PJ Sr (2011) Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J Clin Oncol 29:2052–2059
Glozak MA, Sengupta N, Zhang X, Seto E (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23
Gong K, Xie J, Yi H, Li W (2012) CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J 443:735–746
Gottesfeld JM, Pandolfo M (2009) Development of histone deacetylase inhibitors as therapeutics for neurological disease. Future Neurol 4:775–784
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20:6969–6978
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42
Haigentz M Jr, Kim M, Sarta C, Lin J, Keresztes RS, Culliney B, Gaba AG, Smith RV, Shapiro GI, Chirieac LR, Mariadason JM, Belbin TJ, Greally JM, Wright JJ, Haddad RI (2012) Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol 48(12):1281–1288
Hainsworth JD, Infante JR, Spigel DR, Arrowsmith ER, Boccia RV, Burris HA (2011) A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Invest 29:451–455
Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC (2005) Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA 102:8567–8572
Hoffmann K, Brosch G, Loidl P, Jung M (1999) A non-isotopic assay for histone deacetylase activity. Nucleic Acids Res 27:2057–2058
Iwamoto FM, Lamborn KR, Kuhn JG, Wen PY, Yung WK, Gilbert MR, Chang SM, Lieberman FS, Prados MD, Fine HA (2011) A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro Oncol 13:509–516
Jagannath S, Dimopoulos MA, Lonial S (2010) Combined proteasome and histone deacetylase inhibition: a promising synergy for patients with relapsed/refractory multiple myeloma. Leuk Res 34:1111–1118
Jones P (2009) Histone deacetylase inhibitors. In: Sippl W, Jung M (eds) Epigenetic targets in drug discovery. Wiley VCH, New York, pp 185–223
Jones SF, Bendell JC, Infante JR, Spigel DR, Thompson DS, Yardley DA, Greco FA, Murphy PB, Burris HA 3rd (2011) A phase I study of panobinostat in combination with gemcitabine in the treatment of solid tumors. Clin Adv Hematol Oncol 9:225–230
Jones SF, Infante JR, Spigel DR, Peacock NW, Thompson DS, Greco FA, McCulloch W, Burris Iii HA (2012) Phase 1 results from a study of romidepsin in combination with gemcitabine in patients with advanced solid tumors. Cancer Invest 30:481–486
Khan O, La Thangue NB (2012) HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 90:85–94
Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M (1999) Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem 274:31127–31130
Kirschbaum M, Frankel P, Popplewell L, Zain J, Delioukina M, Pullarkat V, Matsuoka D, Pulone B, Rotter AJ, Espinoza-Delgado I, Nademanee A, Forman SJ, Gandara D, Newman E (2011) Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol 29:1198–1203
Kirschbaum MH, Goldman BH, Zain JM, Cook JR, Rimsza LM, Forman SJ, Fisher RI (2012) A phase 2 study of vorinostat for treatment of relapsed or refractory Hodgkin lymphoma: Southwest Oncology Group Study S0517. Leuk Lymphoma 53:259–262
Lane S, Gill D, McMillan NA, Saunders N, Murphy R, Spurr T, Keane C, Fan HM, Mollee P (2012) Valproic acid combined with cytosine arabinoside in elderly patients with acute myeloid leukemia has in vitro but limited clinical activity. Leuk Lymphoma 53:1077–1083
Lassen U, Molife LR, Sorensen M, Engelholm SA, Vidal L, Sinha R, Penson RT, Buhl-Jensen P, Crowley E, Tjornelund J, Knoblauch P, de Bono JS (2010) A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer 103:12–17
Latham JA, Dent SY (2007) Cross-regulation of histone modifications. Nat Struct Mol Biol 14:1017–1024
Leder A, Orkin S, Leder P (1975) Differentiation of erythroleukemic cells in the presence of inhibitors of DNA synthesis. Science 190:893–894
Lin RJ, Sternsdorf T, Tini M, Evans RM (2001) Transcriptional regulation in acute promyelocytic leukemia. Oncogene 20:7204–7215
Luo J, Su F, Chen D, Shiloh A, Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408:377–381
Ma X, Ezzeldin HH, Diasio RB (2009) Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs 69:1911–1934
Mateos M, Spencer A, Taylor K, Lonial S, De La Rubia J, Facon T, Bengoudifa B, Hazell K, Bourquelot PM, San-Miguel JF (2010) Phase Ib study of oral panobinostat (LBH589) plus lenalidomide (LEN) plus dexamethasone (DEX) in patients (Pts) with relapsed (Rel) or Rel and refractory (Ref) multiple myeloma (MM). J Clin Oncol (Meeting Abstracts) 28(Suppl):8030
McKinsey TA (2012) Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol 52:303–319
Millward M, Price T, Townsend A, Sweeney C, Spencer A, Sukumaran S, Longenecker A, Lee L, Lay A, Sharma G, Gemmill RM, Drabkin HA, Lloyd GK, Neuteboom ST, McConkey DJ, Palladino MA, Spear MA (2012) Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest New Drugs 30(6):2303–2317
Moffat D, Patel S, Day F, Belfield A, Donald A, Rowlands M, Wibawa J, Brotherton D, Stimson L, Clark V, Owen J, Bawden L, Box G, Bone E, Mortenson P, Hardcastle A, van Meurs S, Eccles S, Raynaud F, Aherne W (2010) Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo[3.1.0]hex-3-yl)-N-hydroxypyrim idine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J Med Chem 53:8663–8678
Mohammed TA, Holen KD, Jaskula-Sztul R, Mulkerin D, Lubner SJ, Schelman WR, Eickhoff J, Chen H, Loconte NK (2011) A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist 16:835–843
Morita S, Oizumi S, Minami H, Kitagawa K, Komatsu Y, Fujiwara Y, Inada M, Yuki S, Kiyota N, Mitsuma A, Sawaki M, Tanii H, Kimura J, Ando Y (2012) Phase I dose-escalating study of panobinostat (LBH589) Administered intravenously to Japanese patients with advanced solid tumors. Invest New Drugs 30(5):1950–1957
Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE (2011) A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 104:1828–1835
Niesvizky R, Ely S, Mark T, Aggarwal S, Gabrilove JL, Wright JJ, Chen-Kiang S, Sparano JA (2011) Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 117:336–342
Novotny-Diermayr V, Sangthongpitag K, Hu CY, Wu X, Sausgruber N, Yeo P, Greicius G, Pettersson S, Liang AL, Loh YK, Bonday Z, Goh KC, Hentze H, Hart S, Wang H, Ethirajulu K, Wood JM (2010) SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther 9:642–652
Offidani M, Polloni C, Cavallo F, Marina Liberati A, Ballanti S, Pulini S, Catarini M, Alesiani F, Corvatta L, Gentili S, Caraffa P, Boccadoro M, Leoni P, Palumbo A (2012) Phase II study of melphalan, thalidomide and prednisone combined with oral panobinostat in patients with relapsed/refractory multiple myeloma. Leuk Lymphoma 53(9):1722–1727
Ohtani K, Dimmeler S (2011) Epigenetic regulation of cardiovascular differentiation. Cardiovasc Res 90:404–412
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S (2007) Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25:3109–3115
Otterson GA, Hodgson L, Pang H, Vokes EE (2010) Phase II study of the histone deacetylase inhibitor Romidepsin in relapsed small cell lung cancer (Cancer and Leukemia Group B 30304). J Thorac Oncol 5:1644–1648
Pandolfi PP (2001) Histone deacetylases and transcriptional therapy with their inhibitors. Cancer Chemother Pharmacol 48(Suppl 1):S17–S19
Parsons PG, Hansen C, Fairlie DP, West ML, Danoy PA, Sturm RA, Dunn IS, Pedley J, Ablett EM (1997) Tumor selectivity and transcriptional activation by azelaic bishydroxamic acid in human melanocytic cells. Biochem Pharmacol 53:1719–1724
Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R (2003) Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood 101:2914–2923
Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH (2009) Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 27:5410–5417
Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, Jaffe ES, Ling A, Turner M, Peer CJ, Figg WD, Steinberg SM, Smith S, Joske D, Lewis I, Hutchins L, Craig M, Fojo AT, Wright JJ, Bates SE (2011) Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 117:5827–5834
Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, Carducci MA, Rudek MA (2012) Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer 106:77–84
Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R (2003) Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther 2:721–728
Ramalingam SS, Kummar S, Sarantopoulos J, Shibata S, LoRusso P, Yerk M, Holleran J, Lin Y, Beumer JH, Harvey RD, Ivy SP, Belani CP, Egorin MJ (2010) Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol 28:4507–4512
Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, Gattoni E, Salmoiraghi S, Finazzi MC, Di Tollo S, D’Urzo C, Vannucchi AM, Barosi G, Barbui T (2010) A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol 150:446–455
Rathkopf D, Wong BY, Ross RW, Anand A, Tanaka E, Woo MM, Hu J, Dzik-Jurasz A, Yang W, Scher HI (2010) A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol 66:181–189
Razak AR, Hotte SJ, Siu LL, Chen EX, Hirte HW, Powers J, Walsh W, Stayner LA, Laughlin A, Novotny-Diermayr V, Zhu J, Eisenhauer EA (2011) Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br J Cancer 104:756–762
Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, Abrahamsen TW, Flatmark K (2010) Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol 11:459–464
Riggs MG, Whittaker RG, Neumann JR, Ingram VM (1977) n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 268:462–464
Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O (1999) A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 96:4592–4597
Sauve AA, Wolberger C, Schramm VL, Boeke JD (2006) The biochemistry of sirtuins. Annu Rev Biochem 75:435–465
Scherpereel A, Berghmans T, Lafitte JJ, Colinet B, Richez M, Bonduelle Y, Meert AP, Dhalluin X, Leclercq N, Paesmans M, Willems L, Sculier JP (2011) Valproate-doxorubicin: promising therapy for progressing mesothelioma. A phase II study. Eur Respir J 37:129–135
Stathis A, Hotte SJ, Chen EX, Hirte HW, Oza AM, Moretto P, Webster S, Laughlin A, Stayner LA, McGill S, Wang L, Zhang WJ, Espinoza-Delgado I, Holleran JL, Egorin MJ, Siu LL (2011) Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin Cancer Res 17:1582–1590
Strickler JH, Starodub AN, Jia J, Meadows KL, Nixon AB, Dellinger A, Morse MA, Uronis HE, Marcom PK, Zafar SY, Haley ST, Hurwitz HI (2012) Phase I study of bevacizumab, everolimus, and panobinostat (LBH-589) in advanced solid tumors. Cancer Chemother Pharmacol 70(2):251–258
Ueda H, Manda T, Matsumoto S, Mukumoto S, Nishigaki F, Kawamura I, Shimomura K (1994) FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. J Antibiot (Tokyo) 47:315–323
Verdin E, Dequiedt F, Kasler HG (2003) Class II histone deacetylases: versatile regulators. Trends Genet 19:286–293
Verdin E, Dequiedt F, Fischle W, Frye R, Marshall B, North B (2004) Measurement of mammalian histone deacetylase activity. Methods Enzymol 377:180–196
Wagner JM, Hackanson B, Lubbert M, Jung M (2010) Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics 1:117–136
Walter RB, Medeiros BC, Powell BL, Schiffer CA, Appelbaum FR, Estey EH (2012) Phase II trial of vorinostat and gemtuzumab ozogamicin as induction and post-remission therapy in older adults with previously untreated acute myeloid leukemia. Haematologica 97:739–742
Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138:1019–1031
Wang H, Cao Q, Dudek AZ (2012) Phase II study of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy. Anticancer Res 32:1027–1031
Weichert W (2009) HDAC expression and clinical prognosis in human malignancies. Cancer Lett 280:168–176
Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH (2010) Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol 28:4485–4491
Wightman F, Ellenberg P, Churchill M, Lewin SR (2012) HDAC inhibitors in HIV. Immunol Cell Biol 90:47–54
Wong NS, Seah E, Wang LZ, Yeo WL, Yap HL, Chuah B, Lim YW, Ang PC, Tai BC, Lim R, Goh BC, Lee SC (2011) Impact of UDP-gluconoryltransferase 2B17 genotype on vorinostat metabolism and clinical outcomes in Asian women with breast cancer. Pharmacogenet Genomics 21:760–768
Yong WP, Goh BC, Soo RA, Toh HC, Ethirajulu K, Wood J, Novotny-Diermayr V, Lee SC, Yeo WL, Chan D, Lim D, Seah E, Lim R, Zhu J (2011) Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, SB939, in patients with refractory solid malignancies. Ann Oncol 22:2516–2522
Yoo CB, Jones PA (2006) Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 5:37–50
Yoshida M, Kijima M, Akita M, Beppu T (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265:17174–17179
Younes A, Oki Y, Bociek RG, Kuruvilla J, Fanale M, Neelapu S, Copeland A, Buglio D, Galal A, Besterman J, Li Z, Drouin M, Patterson T, Ward MR, Paulus JK, Ji Y, Medeiros LJ, Martell RE (2011) Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol 12:1222–1228
Younes A, Sureda A, Ben-Yehuda D, Zinzani PL, Ong TC, Prince HM, Harrison SJ, Kirschbaum M, Johnston P, Gallagher J, Le Corre C, Shen A, Engert A (2012) Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J Clin Oncol 30:2197–2203
Zhang Y, Gilquin B, Khochbin S, Matthias P (2006) Two catalytic domains are required for protein deacetylation. J Biol Chem 281:2401–2404
Zou H, Wu Y, Navre M, Sang BC (2006) Characterization of the two catalytic domains in histone deacetylase 6. Biochem Biophys Res Commun 341:45–50
Acknowledgments
The authors thank the Deutsche Forschungsgemeinschaft for funding (Ju 295/9-1 within the priority programme SPP1463 Epigenetic regulation of normal hematopoiesis and its dysregulation in myeloid neoplasia; coordinators M. Lübbert, Freiburg, C. Plass, Heidelberg).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Keller, K., Jung, M. (2014). Histone Deacetylase (HDAC) Inhibitors in Recent Clinical Trials for Cancer Therapy. In: Lübbert, M., Jones, P. (eds) Epigenetic Therapy of Cancer. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-38404-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-642-38404-2_10
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-38403-5
Online ISBN: 978-3-642-38404-2
eBook Packages: MedicineMedicine (R0)