
Abstract
Bevacizumab is frequently used to treat patients with recurrent high-grade glioma (HGG), but responses are generally not durable. Panobinostat is a histone deacetylase inhibitor with anti-neoplastic and anti-angiogenic effects and may work synergistically with VEGF inhibitors. We performed a phase I study to evaluate the safety and tolerability of the combination of orally administered panobinostat with bevacizumab in patients with recurrent HGG. Patients with recurrent HGG were treated on a 3 + 3 trial design. Patients received bevacizumab 10 mg/kg every other week in combination with oral panobinostat. The starting dose of panobinostat was 20 mg three times per week, weekly (cohort 1). Due to concerns for thrombocytopenia with the weekly dosing regimen, the protocol was amended to examine an every other week regimen. Cohort 2 received panobinostat 20 mg three times per week, every other week, and cohort 3 received 30 mg three times per week, every other week. Dose-limiting toxicity during the first 30 days was used to determine the maximum-tolerated dose. Twelve patients (median age 50, median KPS 90) with recurrent HGG were enrolled. One dose-limiting toxicity (DLT) (Grade 3 thrombocytopenia) was observed in cohort 1. No DLTs were observed in cohorts 2 and 3. The following grade 3 toxicities were seen in one patient each: thrombocytopenia, hypophosphatemia, esophageal hemorrhage, and deep venous thrombosis. There were no grade 4 or 5 toxicities. There were three patients with partial responses and seven with stable disease. The recommended doses for further study are oral panobinostat 30 mg three times per week, every other week, in combination with bevacizumab 10 mg/kg every other week. A phase II clinical trial in recurrent HGG is underway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High-grade gliomas (HGG) are the most common malignant primary brain tumors [1] and are associated with a poor prognosis [2]. Bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor (VEGF), is frequently used to treat HGG patients at recurrence. In phase II clinical trials of bevacizumab monotherapy for recurrent GBM, response rates were 20–26%, six-month progression free survivals (PFS6) were 29–42.6% and median overall survivals (OS) were 7.1–9.2 months [3, 4]. Based on these results, bevacizumab received accelerated approval by the United States Food and Drug Administration (FDA) as single agent use in recurrent GBM. Phase II studies in recurrent anaplastic gliomas similarly suggest that bevacizumab in combination with irinotecan also has good activity in this patient population with PFS6 of 56–61% and median OS of 65 weeks [5, 6].
Even though the results of these trials are encouraging, patients eventually progress despite bevacizumab. HGGs can develop resistance to treatment within months of starting therapy and some patients may not even respond to bevacizumab treatment [7]. Once patients progress on one bevacizumab-containing regimen, median progression-free survival on a second bevacizumab-containing regimen is only 30–38 days [4, 8]. This suggests that targeting VEGF alone may not be sufficient for durable responses.
Panobinostat (LBH589) has potent histone deacetylase (HDAC) inhibitory activity at low nanomolar concentrations against Class I, II, and IV purified recombinant HDAC enzymes. Biodistribution studies suggest some brain penetration. Panobinostat is at least a 10-fold more potent pan-HDAC inhibitor than vorinostat [9]. There is strong preclinical evidence to suggest that HDAC inhibitors (HDACi) have anti-neoplastic and anti-angiogenic effects in HGG. HDAC inhibition promotes growth arrest and apoptotic cells death in glioma models [10–17]. Vorinostat, an HDACi, demonstrated modest single-agent activity in a phase II clinical trial for recurrent GBM, with a subpopulation achieving prolonged disease stability [18]. In addition, HDACi, including LBH589, reduce VEGF secretion and modulate the expression of the other VEGF family members via inhibition of HIF-1α, and therefore may inhibit angiogenesis in glioblastoma [19–22]. Although the predominant mechanisms of resistance to anti-angiogenic therapies are still being elucidated, signaling by the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1α (SDF-1α) may be an important mediator of vasculogenesis and tumor cell invasion in GBM. In a phase II clinical trial of cediranib, an oral pan-VEGF receptor (VEGFR) small molecule inhibitor, for recurrent GBM, plasma obtained at the time of tumor recurrence showed statistically significant increases in plasma levels of 1α (SDF-1α) [23]. Panobinostat depletes CXCR4 levels and signaling [24]. Furthermore, preclinical studies have further established that inhibition of CXCR4 results in diminished growth of orthotopic GBM xenografts [25, 26]. The addition of panobinostat to bevacizumab may help abrogate this putative resistance mechanism to anti-VEGF therapies.
Based on this preclinical rationale, a Phase I trial was performed to study the safety and tolerability of combining panobinostat with bevacizumab in patients with recurrent high-grade glioma.
Patients and methods
The study was approved by the institutional review board at Dana-Farber/Harvard Cancer Center (Dana Farber Cancer Institute Protocol 08-342). All patients gave signed informed consent as per institutional guidelines. The study was designed to determine the maximum-tolerated dose (MTD) and dose-limiting toxicities (DLT) of oral LBH589 in combination with intravenous bevacizumab 10 mg/kg every two weeks.
Patient eligibility
Eligibility criteria included histologically confirmed GBM, gliosarcoma, anaplastic astrocytomas (AA), anaplastic oligodendrogliomas AO or anaplastic oligoastrocytomas AOA with documented radiographic progression with any number of prior relapses, age ≥ 18 years old, Karnofsky performance status (KPS) ≥ 60, and adequate bone marrow reserve and organ function. When the protocol was amended to examine an every other week regimen (as described below), the protocol was also amended to exclude patients with a history of grade 2 thrombocytopenia or grade 3 neutropenia on any prior regimen. Prior treatments with VEGF targeted therapies and/or HDACi were not permitted. Patients on enzyme inducing anticonvulsants, valproic acid, warfarin, or QT-prolonging medications were excluded, as were patients with clinically significant cardiovascular events, cardiac arrhythmias, QT-prolonging conditions, or significant intratumoral or peritumoral hemorrhage.
Treatment and study design
Panobinostat was supplied by Novartis Pharmaceuticals Corporation (East Hanover, NJ). The starting dose was 20 mg orally three times per week, every week (Days 1, 3, 5, 8, 10, 12, 15, 17, 19, 22, 24, 26 of every 28-day cycle). Due to the concerns for thrombocytopenia with the weekly dosing regimen, the protocol was amended to examine an every other week regimen. This every other week regimen had been previously evaluated in adult patients with advanced solid tumor or hematologic malignancies and appeared to result in less thrombocytopenia and dose reductions and consequently higher exposures [27]. Subsequent cohorts were treated at 20 mg three times per week, every other week (Days 1, 3, 5, 15, 17, 19, of every 28-day cycle) or 30 mg three times per week, every other week (Days 1, 3, 5, 15, 17, 19, of every 28-day cycle). Based on pooled safety data from a total of 559 patients with advanced hematologic and non-hematologic malignancies enrolled across 3 Phase I and 6 Phase II clinical trials of oral, single-agent panobinostat, an extremely high incidence of grade 3–4 thrombocytopenia was seen with escalating doses beyond 30 mg three times a week, every other week [27]. With panobinostat 45 mg three times a week, every other week, the incidence of grade 3–4 thrombocytopenia in solid tumors and lymphomas was 57.2% and in hematologic malignancies was 81.8%. With the 60 mg three times a week, every other week regimen, the incidence of grade 3–4 thrombocytopenia in both hematologic and non-hematologic malignancies was 75%. Because of the already elevated risk of hemorrhage from bevacizumab and the increased incidence of thrombocytopenia with higher doses of panobinostat seen in prior studies, risk–benefit analysis strongly argued against escalating doses of panobinostat beyond 30 mg three times a week, every other week in our patient population. Therefore, although the final dose in our study did not meet the true definition of MTD, higher doses of panobinostat in combination with bevacizumab were not tested for safety purposes. Bevacizumab was supplied by Genentech (San Francisco, CA) and administered intravenously as 10 mg/kg on days 1 and 15 of every 28-day cycle.
Patients were evaluated every two weeks and as clinically indicated while on study. At each evaluation, patients were assessed for toxicity, complete blood count with differential, serum chemistries, liver function tests, and serial electrocardiograms were obtained.
We used a standard 3 + 3 dose finding design with the MTD defined as the dose at which less than one-third of patients experienced a DLT to the combination of panobinostat and bevacizumab. Toxicity was graded using the National Cancer Institute Common Toxicity Criteria (NCI-CTC) version 3.0. All patients who received therapy on study were considered evaluable for toxicity. DLTs were assessed during the first 30 days and defined as grade 3 thrombocytopenia; grade 4 neutropenia lasting >7 days; grade 4 anemia lasting >7 days despite transfusion or growth factors; febrile neutropenia if ANC < 0.5 × 109/L; a QT interval corrected for heart rate (QTc) of 500–515 ms that did not stabilize to <480 ms after one week; a second occurrence of QTc 500–515 ms; any QTc > 515 ms; any deep vein thrombosis (DVT) or pulmonary embolism (PE) while on fully therapeutic anticoagulation therapy; Grade 3 proteinuria lasting >14 days; or any other clinically significant Grade 3 toxicity despite maximal medical therapy lasting >7 days, any Grade 4 toxicity despite maximal medical therapy; or any Grade 3 or 4 toxicity resulting in study drug discontinuation (permanent removal from study treatment). Dose reductions were allowed within the first 30 days of treatment for any of the above toxicities.
Statistical analysis
Safety variables were summarized by descriptive statistics. Adverse events were reported for each dose level and described in terms of incidence and severity. Preliminary evidence of antitumor activity was evaluated using radiographic assessments based on the response assessment in neuro-oncology (RANO) criteria [28] as well as survival analysis based on Kaplan–Meier estimates.
Results
Patient characteristics
Twelve patients with recurrent GBM (10 patients), AA (1 patient), or AOA (1 patient) received intravenous bevacizumab 10 mg/kg every two weeks and panobinostat at the dose levels described in Table 1. Median age was 50 (range 30–71) and median KPS was 90 (range 70–100). Patient characteristics are further detailed in Table 2.
Toxicity
Potentially treatment-related grade 2 and 3 adverse events are summarized in Table 3. There were no grade 4 or 5 toxicities. In cohort 1 (20 mg three times per week, every week), one of the three patients developed grade 3 thrombocytopenia, the second patient developed prolonged grade 2 thrombocytopenia resulting in a treatment delay, and the third patient developed grade 2 thrombocytopenia resulting in a treatment hold but was able to restart at the same pre-hold dose. Due to concerns for thrombocytopenia at this weekly dose, the protocol was amended to examine an every other week regimen, a strategy that had been employed in other advanced solid tumors and hematologic malignancies [27]. The dose level was decreased to 20 mg three times per week, every other week (cohort 2). Since no DLTs were encountered in this cohort, the dose was escalated to 30 mg three times per week, every other week (cohort 3). There were no DLTs observed in the six patients treated in this last cohort. Based on data from studies of panobinostat in other cancers [27] and the likelihood that further dose escalation would result in thrombocytopenia, it was felt that the maximum feasible dose of panobinostat in combination with bevacizumab 10 mg/kg every other week was 30 mg three times a week, every other week.
The most commonly reported grade ≥2 adverse events potentially related to panobinostat were thrombocytopenia (3 patients), neutropenia (3 patients), diarrhea (3 patients), and leukopenia (2 patients). The most commonly reported grade ≥2 adverse events potentially related to bevacizumab were neutropenia (2 patients) and thrombocytopenia (3 patients). There were no grade 4 or 5 toxicities attributed to panobinostat or bevacizumab. One patient developed a grade 3 deep venous thrombosis and one patient developed a grade 3 esophageal hemorrhage, both events occurring during the fifth cycle of treatment.
Two of the 12 patients required one dose-reduction each. A patient in cohort 1 was dose-reduced after two weeks on treatment due to grade 3 thrombocytopenia. A patient in cohort 3 was dose-reduced during the second cycle due to prolonged QTc > 500 ms. Another patient in cohort 3 required panobinostat to be held for QTc > 480, but treatment was able to be restarted without dose reduction.
Response and survival
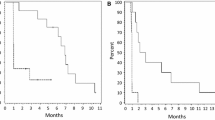
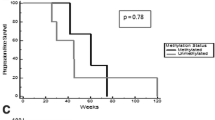
Three patients, all with GBM as their pathology, achieved partial responses (1 patient in cohort 2 and 2 patients in cohort 3). Two patients had progressive disease on their first restaging MRI (1 patient in cohort 1 and 1 patient in cohort 3). The remainder had stable disease. The PFS6 rate in all cohorts combined was 3/12 (25%); all of the patients who achieved PFS at 6 months received treatment at the maximum feasible dose. Three patients remain on treatment. Median PFS from the date of registration was 4.3 months (range 1.1–9.5+ months). Median OS from the date of registration was 8.2 months (range 4.8–12.1+ months); four patients are still alive.
Discussion
Although bevacizumab has demonstrated efficacy in phase II clinical trials of recurrent GBM and anaplastic glioma, responses are typically not durable. Preclinical data suggests that panobinostat has anti-angiogenic activity and may work synergistically with VEGF inhibitors. In addition, panobinostat depletes CXCR4 signaling, one of the putative mechanisms of VEGF resistance in GBM. These data suggest that the addition of panobinostat to bevacizumab may result in more durable responses. In this study, we found that the combination of panobinostat and bevacizumab in recurrent HGG is tolerable and safe. However, weekly dosing of panobinostat in combination with bevacizumab was limited by thrombocytopenia. This is consistent with pooled data from phase I and II studies in advanced hematologic malignancies or solid tumors suggesting that every other week dosing schedules of oral panobinostat was better tolerated that weekly dosing schedules with respect to thrombocytopenia [27]. The maximum feasible dose of panobinostat three times a week, every other week, in combination with bevacizumab from this study was 30 mg, similar to the MTD determined in a phase I study of single-agent panobinostat in patients with advanced solid tumors, non-Hodgkin’s lymphoma, or cutaneous T-cell lymphoma [29].
This study had several limitations. No pharmacokinetic (PK) or pharmacodynamic (PD) parameters were measured. Although collection of blood for PK studies was originally included in the study, these were subsequently removed when it was felt that the interaction between panobinostat and bevacizumab was likely to be minimal. Extensive PK and PD data is available with single agent panobinostat from other studies. Panobinostat is rapidly and extensively distributed in tissues and inter-patient variability in exposure is 60% with oral dosing, making it difficult to predict how an individual patient will respond to drug. In addition, because doses above 30 mg three times a week, every other week were not evaluated, we did not find the true MTD. As previously discussed, due to safety concerns for hemorrhage from bevacizumab and the high incidence of severe thrombocytopenia at higher doses of panobinostat from pooled analysis of other clinical trials [27], the 30 mg dose in combination with bevacizumab was deemed to represent the maximum feasible dose. Finally, although a few radiographic responses were seen, the sample size is too small to fully understand the impact of adding panobinostat to bevacizumab in recurrent HGG.
In conclusion, the combination of oral panobinostat and intravenous bevacizumab is feasible, with thrombocytopenia seen with more frequent dosing of panobinostat. Further clinical investigation of this regimen is underway in a multicenter phase II trial of bevacizumab and panobinostat in patients with recurrent HGG.
References
CBTRUS (2009) CBTRUS. Statistical report: primary brain tumors in the United States, 2000-2004. http://www.cbtrus.org/reports/2009-NPCR-04-05/CBTRUS-NPCR2004-2005-Report-.pdf. Accessed October 2009
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359(5):492–507
Friedman HS, Prados MD, Wen PY et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27(28):4733–4740
Kreisl TN, Kim L, Moore K et al (2009) Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27(5):740–745
Desjardins A, Reardon DA et al (2008) Bevacizumab plus irinotecan in recurrent WHO grade 3 malignant gliomas. Clin Cancer Res 14(21):7068–7073
Vredenburgh JJ, Desjardins A, Herndon JE 2nd et al (2007) Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13(4):1253–1259
Norden AD, Drappatz J, Wen PY (2008) Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol 7(12):1152–1160
Quant EC, Norden AD, Drappatz J et al (2009) Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol 11(5):550–555
Atadja P (2009) Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett 280(2):233–241
Gensert JM, Baranova OV, Weinstein DE, Ratan RR (2007) CD81, a cell cycle regulator, is a novel target for histone deacetylase inhibition in glioma cells. Neurobiol Dis 26(3):671–680
Ugur HC, Ramakrishna N, Bello L et al (2007) Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J Neurooncol 83(3):267–275
Yin D, Ong JM, Hu J et al (2007) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin Cancer Res 13(3):1045–1052
Sharma V, Koul N, Joseph C, Dixit D, Ghosh S, Sen E (2010) HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity. J Cell Mol Med 14(8):2151–2161
Eyupoglu IY, Hahnen E, Trankle C et al (2006) Experimental therapy of malignant gliomas using the inhibitor of histone deacetylase MS-275. Mol Cancer Ther 5(5):1248–1255
Wetzel M, Premkumar DR, Arnold B, Pollack IF (2005) Effect of trichostatin A, a histone deacetylase inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J Neurosurg 103(6 Suppl):549–556
Eyupoglu IY, Hahnen E, Buslei R et al (2005) Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J Neurochem 93(4):992–999
Sawa H, Murakami H, Kumagai M et al (2004) Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol 107(6):523–531
Galanis E, Jaeckle KA, Maurer MJ et al (2009) Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 27(12):2052–2058
Deroanne CF, Bonjean K, Servotte S et al (2002) Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21(3):427–436
Qian DZ, Kato Y, Shabbeer S et al (2006) Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res 12(2):634–642
Qian DZ, Wang X, Kachhap SK et al (2004) The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res 64(18):6626–6634
Sawa H, Murakami H, Ohshima Y et al (2002) Histone deacetylase inhibitors such as sodium butyrate and trichostatin A inhibit vascular endothelial growth factor (VEGF) secretion from human glioblastoma cells. Brain Tumor Pathol 19(2):77–81
Batchelor TT, Sorensen AG, di Tomaso E et al (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11(1):83–95
Mandawat A, Fiskus W, Buckley KM et al (2010) Pan-histone deacetylase inhibitor panobinostat depletes CXCR4 levels and signaling and exerts synergistic antimyeloid activity in combination with CXCR4 antagonists. Blood 116(24):5306–5315
Kioi M, Vogel H, Schultz G et al (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120(3):694–705
Rubin JB, Kung AL, Klein RS et al (2003) A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci USA 100(23):13513–13518
Lin R, Hu J, Paul S, et al. (2009) Characteristics of thrombocytopenia in patients treated with oral panobinostat (LBH589). Blood (ASH Annual Meeting Abstracts) 114: Abstract 2740
Wen PY, Macdonald DR, Reardon DA et al (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28(11):1963–1972
Novartis (2011) LBH 589 (Panobinostat) Investigator’s Brochure 7th edn, Brochure. Basel, Switzerland
Acknowledgments
E.Q. Lee has served on the advisory board of Novartis and Genentech/Roche. T. T. Batchelor has served on the advisory board for Genentech/Roche. R. Beroukhim has received research support from Novartis. S.M. Snodgrass is an employee of Novartis. J.J. Raizer has received research support from Novartis and Genentech/Roche, served on the advisory boards of both Novartis and Genentech/Roche and is on the speaker’s bureau of Genentech/Roche. P.Y. Wen has received research support from Novartis and Genentech/Roche and served on the advisory boards of both Novartis and Genentech/Roche.
Conflict of interest
All other authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
First three authors contributed equally to this work
Rights and permissions
About this article
Cite this article
Drappatz, J., Lee, E.Q., Hammond, S. et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol 107, 133–138 (2012). https://doi.org/10.1007/s11060-011-0717-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-011-0717-z