
Abstract
Purpose
Chidamide (CS055/HBI-8000) is a new benzamide class of histone deacetylase inhibitor with marked anti-tumor activity. This study reports the phase I results.
Methods
Patients with advanced solid tumors or lymphomas received oral doses of 5, 10, 17.5, 25, 32.5, or 50 mg chidamide either twice (BIW) or three times (TIW) per week for 4 consecutive weeks every 6 weeks. Safety, characteristics of pharmacokinetics (PK) and pharmacodynamics (PD), and preliminary efficacy were evaluated.
Results
A total of 31 patients were enrolled. No DLTs were identified in the BIW cohorts up to 50 mg. DLTs were grade 3 diarrhea and vomiting in two patients in the TIW cohort at 50 mg, respectively. PK analysis revealed t1/2 of 16.8–18.3 h, T max of 1–2 h in most cases, and a dose-related increase in C max and AUC. Significant induction of histone H3 acetylation in peripheral white blood cells was observed after a single dose of chidamide. Four patients with T-cell lymphomas and 1 patient with submandibular adenoid cystic carcinoma achieved a partial response.
Conclusions
Chidamide was generally well tolerated in patients with advanced solid tumors or lymphomas in the tested regimens. Favorable PK and PD profiles, as well as encouraging preliminary anti-tumor activity, were demonstrated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is increasingly apparent that tumor development depends not only on stable genetic alterations, but also on epigenetic changes, which can influence gene expression patterns critical for neoplastic development and progression [1]. Epigenetic changes contributing to malignancy can be produced by decrease in histone acetylation, a process controlled by the antagonistic actions of two large families of enzymes—histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs appear to act as transcription repressors via histone deacetylation–mediated chromatin condensation [2]. In humans, at least 18 HDACs have been identified, and they are classified into three classes based on their homology to yeast proteins [3, 4]. Class I includes HDAC1, 2, 3, and 8. HDAC4, 5, 6, 7, 9, and 10 belong to class II, and HDAC11 is placed in class IV. Although the precise biological role of individual HDACs is largely unknown, class I HDACs are over-expressed in tumors [5, 6], and accumulating data suggest that these HDACs are associated with cell cycle progression, metastasis, and apoptosis, making them promising targets for cancer therapy [1, 3, 4].
HDAC inhibitors (HDACi) have emerged as a novel therapeutic class of molecules with significant anticancer potential. HDACi represent a structurally diverse group of molecules, including hydroxamic acid derivatives, benzamides, cyclic peptides, and short-chain fatty acids [1, 3, 4]. Several oral or intravenous (IV) hydroxamic acids (SAHA/vorinostat, PXD-101/belinostat, and LBH589/panobinostat), one IV cyclic peptide (FK-228/romidepsin), and one oral benzamide (MGCD0103) have exhibited significant single-agent activity in clinical trials [7–10]. Vorinostat (ZOLINZA) and romidepsin (ISTODAX) were approved in the United States for the treatment of cutaneous T-cell lymphoma [11, 12], and recently romidepsin for peripheral T-cell lymphoma [13].
Despite numerous studies, HDACi have not exhibited significant single-agent clinical activity against solid tumors [14–16]. However, preliminary activity has been observed with non-small cell lung cancer and breast carcinoma in combination settings [17–19]. In addition to a requirement for combination with other anti-tumor agents [20], it is likely that optimal results in the solid tumor settings will require improved pharmacokinetic properties to enhance exposure and improved side effect profiles that minimize overlapping toxicities. Thus, a current focus of HDACi cancer research and development is the search for inhibitors with improved pharmaceutical and safety profiles, and the determination of optimal combination strategies with other anti-tumor agents. In this regard, the ability to produce and maintain target exposure continuously without significant fatigue or myelosuppression may provide a distinct advantage, particularly in combination settings.
Chidamide (CS055/HBI-8000) is a new member of the benzamide class of HDACi rationally designed to block the catalytic pocket of class I HDACs and to exhibit enhanced metabolic stability relative to existing hydroxamic acid and benzamide class inhibitors [21, 22]. Enzyme inhibition profiling studies have demonstrated that chidamide inhibits class I HDACs 1, 2, 3, as well as class II HDAC 10, in the low-nanomolar concentration range [23]. Preclinical studies have shown that chidamide is orally bioavailable with broad-spectrum in vitro and in vivo anti-tumor activity [23]. In the current report, we present the results of a phase I study to evaluate tolerability and determine dose-limiting toxicities (DLTs) of chidamide in patients with advanced solid tumors or lymphomas. Other objectives included determination of pharmacokinetic (PK) and pharmacodynamic (PD) characteristics and evaluation of preliminary efficacy in patients treated with chidamide.
Patients and methods
Eligibility
Eligible patients must have had advanced solid tumors or lymphomas confirmed by histology or cytology, refractory or relapsed with respect to standard treatment, or for which no standard treatment existed. Other inclusion criteria were as follows: (1) age between 18 and 75; (2) ECOG performance status 0–2; (3) body weight between 47 and 87 kg for men and 35–75 kg for women; (4) life expectancy greater than 3 months; (5) had not been treated by chemotherapy, radiotherapy, targeted therapy, or endocrine therapy during the last 4 weeks before entry; and (6) adequate hepatic, renal, and hematologic functions: total bilirubin ≤1.5-fold upper limit of normal (ULN), ALT/AST ≤2.5-fold ULN, creatine ≤1.5-fold ULN, absolute neutrophil count ≥1.5 × 109/L, platelets ≥80 × 109/L, and hemoglobin ≥90 g/L.
Pregnant or lactating women, men, and women with reproductive potential without adequate contraception were excluded. Other exclusion criteria were as follows: history of Q-T prolongation, clinically significant VT, VF, heart block, MI within 1 year, CHF, clinically significant coronary artery disease, prolonged PT or PTT, or using anticoagulants, active infection, major surgery during previous 6 weeks, mental disorders, organ transplant recipients, history of drug abuse or alcoholism, uncontrolled brain metastasis with symptoms, or on another clinical trial of an investigational agent.
The trial was approved by the Chinese State Food and Drug Administration (2006L04595 and 2006L04596) and the institutional review board of the participating medical center. All patients provided written informed consent before enrollment.
Study design
Chidamide tablets (containing 2.5 or 5 mg of drug substance with the formulation using a solid dispersion of the drug substance in polyvinylpyrrolidone K30) were administered orally 30 min after the morning meal. The initial dosing schedule was twice per week (BIW; Mondays/Thursdays or Tuesdays/Fridays) for 4 consecutive weeks in a 6-week cycle, with the starting dose of 5 mg, which was roughly one-tenth the maximum tolerated dose (MTD) in rodent and non-rodent species with the same treatment schedule. Individual sequential cohorts received 5, 10, 17.5, 25, 32.5, or 50 mg according to a modified Fibonacci dose escalation scheme. Patients were administered with fixed dose of chidamide in each cohort, and the range of body weight was defined in the inclusion criteria as indicated above. Due to the lack of DLT observed at the highest level used (50 mg) in the BIW schedule, the protocol was amended to change the dosing frequency from TIW to three times per week (TIW; Mondays/Wednesdays/Fridays), and two dose levels (32.5 and 50 mg) were evaluated with this schedule.
At least three patents were enrolled in each cohort. If a patient in a particular cohort exposed to chidamide less than 50% of the total expected dose exposure in the first cycle due to various reasons, but not DLTs, one more patent should be enrolled in the same cohort. If one of the first three patients at a dose level experienced DLT, up to three additional patients (total up to six patients) were enrolled at that dose level. If more than two patients at a dose level experienced DLT, dose escalation was halted, and the dose level was determined to have exceeded maximum tolerated dose (MTD). Three additional patients were then entered on the next lower dose level. MTD was defined as the highest dose with an observed incidence of DLT in no more than one of six patients. Single-dose PK and PD studies were carried out in patients who received 25, 32.5, or 50 mg chidamide, regardless of dosing schedule. Multi-dose PK analysis was performed in patients who received 32.5 mg in the TIW schedule. Patients with measurable baseline disease were eligible for efficacy evaluation.
Safety and efficacy measures
Toxicity was graded by the National Cancer Institute Common Toxicity Criteria (NCI-CTC, version 2.0). DLT was defined as first-cycle treatment-related adverse events ≥grade 3 non-hematologic or ≥grade 4 hematologic toxicity. Physical examination, ECG, and complete blood count (CBC) were performed at study entry and every week after dosing. At study entry and every 4 weeks during administration, laboratory studies for liver function (AST, ALT, total bilirubin, total protein, albumin), renal function (Cr, uric acid), blood electrolytes (Na, K, Cl), serum albumin (Alb), coagulation parameters (PTT or APTT), and myocardial enzyme (MBCK) were performed. Tumor response was evaluated by the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. At study entry and every 6 weeks during administration, patients with measurable tumor foci at baseline were evaluated.
Pharmacokinetic studies
Blood samples (3 mL) were collected in sodium heparin tubes at 0, 0.5, 1, 2, 6, 12, 24, 48, and 72 h after the first dose. For multi-dose PK studies, blood samples were also collected at the same time points after the last dose of the first cycle. Samples were immediately centrifuged at 3,000×g for 10 min at 4°C. Plasma was collected, aliquoted, and frozen at ≤−40°C until the time of analysis. Samples were analyzed for chidamide concentrations using a validated high-performance liquid chromatography/mass spectrometry method. The linear range of this method is 1–500 ng/mL, with a lower limit of quantitation of 0.2 ng/mL. Estimates of pharmacokinetic parameters for chidamide were derived from individual concentration–time data sets by non-compartmental analysis using the software package DAS 2.0 (Data Access System, Chinese Academy of Sciences, Beijing, China). The maximum concentration of drug in plasma (C max) and the time to reach C max (T max) were determined directly from the observed plasma concentration vs. time curves. The area under the plasma concentration versus time curve (AUC0−t) was calculated using the linear trapezoidal method from time zero to the time of the final quantifiable concentration. The AUC infinity (AUC0−∞) was estimated by the standard formula (AUC0−∞ = AUC0−t + Ct/λz). The corresponding half-life (t1/2) was calculated as ln2/λz using the terminal portion of the plasma concentration–time curve. The apparent oral clearance (CL/F) was obtained as the ratio of oral dose/AUC0−∞, and the apparent volume of distribution (Vd/F) was estimated as CL/λz.
Pharmacodynamic studies
From patients administered with different doses of chidamide, 3 mL of blood was collected in a sodium heparin tube at the time points of 0, 6, 24, 48, and 72 h. After centrifugation, the plasma was kept for the use of PK analysis, and the blood cells were resuspended in 7.6 mL of PBS containing 8.5% DMSO, 100 U/mL heparin sodium, and 1× protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN, USA) and stored at −20°C until use. Histone proteins from peripheral white blood cells (WBCs) were extracted and quantified, and the acetylation status of histone H3 was determined by a validated enzyme-linked immunosorbent assay (ELISA) method. The methods and procedures for the PD analysis are described in the supplementary Online Resource.
Statistical analysis
Quantitative indices included mean standard deviation, median, minimum value, and maximum value. Categorical parameters were described in terms of counts and percentages. Descriptive statistics were performed upon completion of the trial. Demographic data were included in this study with a statistical summary describing the number of patients in each cohort and the total number of patients.
Results
Patient demographics and treatment regimens
Thirty-one patients with advanced solid tumors or lymphomas were enrolled, and their baseline characteristics are presented in Table 1. Patients received chidamide orally in fixed doses for four consecutive weeks, followed by a two-week drug-free holiday. A complete treatment cycle was 6 weeks. Six dose levels ranging from 5 to 50 mg in a twice-per-week (BIW) schedule and two dose levels (32.5 and 50 mg) in a three-times-per-week (TIW) schedule were evaluated, respectively.
Dose-limiting toxicity
Two incidences of first-cycle dose-limiting toxicity (DLT) were observed in the first two patients enrolled in the 50 mg TIW cohort. A 56-year-old patient with ovarian cancer had grade 3 diarrhea on the second day after the 9th dose. The patient withdrew from the trial, and the symptom resolved within 2 days after anti-diarrheal treatment and fluid infusion. The other DLT was observed in a 60-year-old patient with lung cancer, who experienced grade 3 nausea and vomiting on the day of the 4th dose. The patient withdrew from the trial, and the symptoms resolved within 2 days after discontinuation of chidamide administration. Thus, GI side effects were determined to be DLTs in the 50 mg TIW cohort. No DLTs were observed in other cohorts for both BIW and TIW regimens.
Safety and tolerance
A total of seventy-five adverse events (AEs) that were at least possibly related to chidamide were reported in all 31 patients in the first treatment cycle. As shown in Table 2, treatment-related AEs were mostly grade 1 (72%), with 17% grade 2 and 11% grade 3. The most common AEs were fatigue (11 patients [35%]), thrombocytopenia (8 patients [26%]), anorexia (8 patients [26%]), leucopenia or neutropenia (7 patients [23%]), reduced hemoglobin (6 patients [19%]), nausea (5 patients [16%]), and diarrhea (5 patients [16%]) (Table 2). There were no grade 4 AEs in the trial, and fatigue was limited to grade 1. In general, the number and severity of AEs increased with exposure, particularly with respect to myelosuppression and GI events. Symptomatic pericarditis or pericardial effusion was not observed. In addition, first-cycle ECG examinations did not reveal any clinically significant changes from the baseline or NCI-CTC QTc prolongation.
In the TIW dosing schedule, two out of two patients developed grade 3 non-hematologic AEs (DLTs) in the 50 mg cohort, and four out of seven patients experienced grade 3 hematologic AEs at the 32.5-mg dose level. Therefore, 32.5 mg was determined to be the MTD for the TIW dosing schedule. No further dose escalation higher than 50 mg was performed in the BIW schedule, but based on a higher frequency of grade 3 hematologic AEs observed at the 50-mg dose level (Table 2), it was estimated that this dose level might be close to MTD for the BIW dosing schedule.
Anti-tumor activity
Out of the 25 evaluable patients, there were 5 patients with PR, 11 patients with SD, and 9 patients with PD. Four of the 5 patients with PRs were T-cell non-Hodgkin’s lymphoma (T-NHL) patients assigned to the 5, 32.5, and 50 mg BIW cohorts and the 32.5 TIW cohort. The other PR patient was enrolled with adenoid cystic carcinoma of the submandibular gland and was treated in the 32.5 mg BIW cohort (Table 3).
The 4 NHL patients with PRs represented 80% of the T-NHL patients included in the study. One patient (P1, Table 3) presented with stage IVa cutaneous T-cell lymphoma (CTCL), which had progressed rapidly after multiple chemotherapy treatments, total skin radiotherapy, and high-dose chemotherapy combined with stem cell transplantation. When enrolled in the study, the patient had large areas of damaged skin with measurable disease of 64 mm (sum LD). After completing one treatment cycle of 5 mg chidamide BIW, measurable lesions were eliminated and non-reference lesions were also significantly reduced. Upon exhibiting a PR at the end of the first cycle of treatment, P1 was subsequently administered 10 mg chidamide for two cycles followed by 17.5 mg for two cycles, with a total response duration of 133 days. A patient with subcutaneous panniculitis-like T-cell lymphoma in the 32.5 mg BIW cohort (P16, Table 3) exhibited a response lasting 126 days (four cycles of treatment).
Stable disease was observed in patients with B-cell NHL, adenoid cystic carcinoma of the submandibular gland, leiomyosarcoma of the retroperitoneum, alveolar soft part sarcoma, breast carcinoma, endometrial adenocarcinoma, thymic carcinoma, rectal adenocarcinoma, renal cell carcinoma, and lung adenocarcinoma. Six of the 11 patients with stable disease received more than two cycles of chidamide treatment, with disease stabilization lasting from 119 to 252 days.
Pharmacokinetic analysis
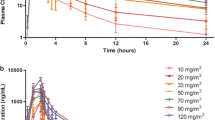
Single-dose PK studies were performed in patients who received 25, 32.5, and 50 mg chidamide, regardless of dosing schedule. The mean plasma drug concentrations at different time points after first dose and the individual values of T max and C max are plotted in Fig. 1a–c, respectively. Peak plasma concentrations for the majority of patients (13 out of 21, 62%) were observed within 0.5–2 h of drug administration and all, except one, within 12 h. Plasma drug concentrations generally returned to close to baseline level within 48 h, but remained quantifiable at 72 h after a single dose. Systemic exposures (C max and AUC) were generally dose dependent across the 25–50-mg dose range. However, substantial interpatient variability in those parameters, as well as CL/F and Vd/F, was apparent, implying varied systemic exposure to chidamide during drug treatment. The elimination half-life (t½) was similar among the different dose groups, with mean values ranging from 16.8 to 18.3 h. Single-dose PK parameters of chidamide are summarized in Table 4.

Pharmacokinetic analysis of chidamide. a Mean plasma drug concentrations at various time points with different doses. b Individual T max values versus dose. c Individual C max values versus dose
Preliminary multi-dose PK analysis was conducted in patients in the 32.5 mg TIW cohort. Three out of seven patients received all 12 doses, and their individual PK parameters from the first and last dose are shown in Table 4. The overall systemic exposures expressed as AUC appeared to be greater after administration of the last dose of chidamide. The C max was greater after the 12th dose in two of three patients. Meanwhile, decreased CL/F and Vd/F values after the 12th dose were shown in all the three patients. The data suggest that increased systemic exposure may occur after multiple dose administration of chidamide on a three-times-per-week dosing schedule.
Pharmacodynamic analysis
PD analysis was carried out by examining histone H3 acetylation in peripheral WBCs from 19 patients who received a single dose of 25 mg (3 patients), 32.5 mg (10 patients), or 50 mg chidamide (6 patients), regardless of dosing schedule. The first dose of 25, 32.5, and 50 mg chidamide induced a 1.3- to 5-fold increase in histone H3 acetylation in peripheral WBCs from 13 out of 18 patients with detectable H3 acetylation at baseline (induction could not be assessed in one patient, due to undetectable histone acetylation at baseline). In general, peak induction of H3 acetylation was observed between 24 and 48 h after treatment, with increased acetylation persisting for up to >72 h after a single dose of chidamide (Fig. 2). Although there was a trend in dose and response of the largest induction and longest duration, substantial variability was apparent, which might be related to varied systemic exposure to the drug and a limited number of patients analyzed.

Mean induction increase in histone H3 acetylation in peripheral white blood cells at various time points with different doses. Procedures are referred to the supplementary Online Resource
Discussion
The most common DLTs associated with all chemical classes of HDACi are anorexia, diarrhea, fatigue, nausea, thrombocytopenia, and vomiting. Additional grade 3/4 adverse events have included anemia, asthenia, atrial fibrillation, dehydration, electrocardiogram (ECG) changes including QTc prolongation, electrolyte disturbances, fever, hyperbilirubinemia, hyperglycemia, hypoalbuminemia, hypocalcemia, hyponatremia, hypophosphatemia, hypotension, neutropenia, pulmonary embolism, pyrexia, and transaminase elevation [24, 25].
This phase 1 study demonstrated that chidamide was generally well tolerated in patients with advanced solid tumors or lymphomas when administered orally on twice-per-week (BIW) and three-times-per-week (TIW) schedules for 4 consecutive weeks in a 6-week cycle. Grade 3 diarrhea and grade 3 vomiting were reported as DLTs in the 50 mg TIW cohort, which resolved within 2 days after discontinuation of the drug. There were no grade 4 AEs in this trial, and myelosuppression was limited to ≤grade 2 at weekly chidamide exposures up to 65 mg and ≤grade 3 at weekly exposures up to 150 mg.
Favorable PK properties of chidamide have been demonstrated in this patient population. In general, chidamide was rapidly absorbed after oral administration and exhibited an elimination half-life in plasma of 17–18 h. Elimination half-lives reported for other HDACi generally range from 2 to 9 h [24, 25], with an extreme exception of MS-275 (entinostat) for about 50–150 h [26, 27]. A reasonable longer half-life may represent an advantage for chidamide, as preclinical studies have demonstrated that induction of apoptosis in tumor cells requires continuous exposure to HDACi for at least 16 h [28]. Systemic exposures were generally dose dependent across the 25- to 50-mg dose range. In three patients administered 12 consecutive doses of 32.5 mg of chidamide TIW, there was an increase in systemic exposure following the last dose, most likely due to changes in elimination and/or metabolism. However, significant variability precludes a definitive conclusion, and a greater number of patients would be needed to confirm the apparent changes in CL/F and Vd/F with repeated-dose administration of chidamide.
Analysis of histone acetylation showed that an induction of histone H3 acetylation in peripheral WBCs was detected within 6 h after dosing, with a peak at 24–48 h. The duration of enhanced histone acetylation lasted 24–72 h, despite the finding that drug plasma concentrations generally peaked within the first 12 h and returned to close to baseline level within 48 h after dosing. It is possible that prolonged enhanced histone H3 acetylation may be explained by a slow-on/slow-off or tight binding mechanism of inhibition, as demonstrated for other benzamide-based inhibitors [29, 30].
Previous clinical studies have demonstrated that HDACi produce significant single-agent anti-tumor activity in lymphomas, particularly cutaneous T-cell lymphomas [31, 32]. Consistent with this known class feature, chidamide produced 4 PRs out of 5 patients enrolled with T-cell lymphoma. Furthermore, one patient with adenoid cystic carcinoma of the submandibular gland also exhibited a PR. The anti-tumor mechanisms of action of HDACi have not been fully elucidated, particularly at the clinical level. Preclinical studies have established several potential mechanisms, including growth inhibition, cell cycle arrest, apoptosis, angiogenesis inhibition, and enhancement of anti-tumor immune responses [3]. HDACi may sensitize tumor cells to natural killer (NK) cell–mediated cytotoxicity, for example, by inducing expression of MHC class I-related proteins and NKG2D ligand on tumor cells [33, 34]. Our previous reported results demonstrate that chidamide alters expression of a number of genes involved in immune cell–mediated tumor cell cytotoxicity in peripheral WBCs from T-cell lymphoma patients who responded to chidamide treatment. NK cell activity and expression of functionally related proteins, such as CD16, NKG2D, and GZMA, were also increased in PBMCs from healthy donors after in vitro treatment with chidamide at nanomolar concentrations [23]. Taken together, these data imply that chidamide, and probably other HDACi as well, may produce indirect immune system-mediated anti-tumor effects, in addition to direct anti-tumor and anti-angiogenic activities.
In summary, oral chidamide (CS055/HBI-8000) was well tolerated in patients with advanced solid tumors or lymphomas at doses up to 50 mg BIW and 32.5 mg TIW for 4 consecutive weeks in a 6-week cycle. Chidamide exhibited a relatively long half-life and a long-lasting histone H3 acetylation response. The compound also produced significant preliminary anti-tumor activity at well-tolerated doses. The favorable PK, PD, and side effect profiles and preliminary activity support further investigation of chidamide in hematologic and solid tumor settings. In particular, the PK and side effect profiles elucidated in this study suggest that chidamide is a promising candidate in combination with a variety of marketed anti-tumor agents.
References
Mai A, Altucci L (2009) Epi-drugs to fight cancer: from chemistry to cancer treatment, the road ahead. Int J Biochem Cell Biol 41:199–213
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42
Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6:38–51
Rasheed W, Bishton M, Johnstone RW, Prince HM (2008) Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther 8:413–432
Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M (2007) Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep 18:769–774
Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, Kristiansen G (2008) Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer 98:604–610
Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K (2006) A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res 12:4628–4635
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M (2007) Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25:3109–3115
Crump M, Andreadis C, Assouline S, Rizzieri D, Wedgwood A, McLaughlin P, Laille E, Li Z, Martell RE, Younes A (2008) Treatment of relapsed or refractory non-Hodgkin lymphoma with the oral isotype-selective histone deacetylase inhibitor MGCD0103: interim results from a Phase II study. J Clin Oncol 26 (Abstract 8528)
Gimsing P, Hansen M, Knudsen LM, Knoblauch P, Christensen IJ, Ooi CE, Buhl-Jensen P (2008) A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol 81:170–176
FDA (2006) US Food and Drug Administration, Center for Drug Evaluation and Research, Office of Oncology Drug Products (OODP). Approval letter for ZOLINZA™, NDA 21-991, Merck & Co. Inc
FDA (2009) US Food and Drug Administration, Center for Drug Evaluation and Research, Office of Oncology Drug Products (OODP). Approval letter for ISTODAX, NDA 022393, Gloucester Pharmaceuticals
FDA (2011) US Food and Drug Administration, Center for Drug Evaluation and Research, Office of Oncology Drug Products (OODP). Approval letter for ISTODAX, NDA 022393/S-004, Celgene Corporation
Blumenschein GR Jr, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C, Frankel SR (2008) Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs 26:81–87
Modesitt SC, Sill M, Hoffman JS, Bender DP, Gynecologic Oncology Group (2008) A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 109:182–186
Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schoffski P (2008) Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs 26:483–488
Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ, Belani CP (2007) Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res 13:3605–3610
Munster PN, Lacevic M, Thomas S, Christian R, Ismail-Khan R, Melisko M, Rugo H, Minton SE (2009) Phase II trial of the histone deacetylase inhibitor, vorinostat, to restore hormone sensitivity to the antiestrogen tamoxifen in patients with advanced breast cancer who progressed on prior hormone therapy. J Clin Oncol 27(Suppl) (Abstract 1075)
Ramalingam SS, Maitland M, Frankel P, Argiris AE, Koczywas M, Gitlitz B, Espinoza-Delgado I, Vokes EE, Gandara DR, Belani CP (2009) Randomized, double-blind, placebo-controlled Phase II study of carboplatin and paclitaxel with or without vorinostat, a histone deacetylase inhibitor (HDAC), for first-line therapy of advanced non-small cell lung cancer (NCI 7863). J Clin Oncol 27 (Suppl) (Abstract 8004)
Nolan L, Johnson PW, Ganesan A, Packham G, Crabb SJ (2008) Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential? Br J Cancer 99:689–694
Xie AH, Liao CZ, Li ZB, Ning ZQ, Hu W, Lu XP, Shi LM, Zhou J (2004) Quantitative structure-activity relationship study of histone deacetylase inhibitors. Curr Med Chem Anticancer Agents 4:273–299
Yin ZH, Wu ZW, Lan YK, Liao CZ, Shan S, Li ZL, Ning ZQ, Lu XP, Li ZB (2004) Synthesis of chidamide, a new histone deacetylase (HDAC) inhibitor. Chin J New Drugs 13:536–538
Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X, Lu XP (2011) Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharm. doi: 10.1007/s00280-011-1766-x
Glaser KB (2007) HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 74:659–671
Rasheed W, Bishton M, Johnstone RW, Prince HM (2008) Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther 8:413–432
Kummar S, Gutierrez M, Gardner ER, Donovan E, Hwang K, Chung EJ, Lee MJ, Maynard K, Kalnitskiy M, Chen A, Melillo G, Ryan QC, Conley B, Figg WD, Trepel JB, Zwiebel J, Doroshow JH, Murgo AJ (2007) Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin Cancer Res 13:5411–5417
Gore L, Rothenberg ML, O’Bryant CL, Schultz MK, Sandler AB, Coffin D, McCoy C, Schott A, Scholz C, Eckhardt SG (2008) A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res 14:4517–4525
Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J 409:581–589
Bonfils C, Kalita A, Dubay M, Siu LL, Carducci MA, Reid G, Martell RE, Besterman JM, Li Z (2008) Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin Cancer Res 14:3441–3449
Chou CJ, Herman D, Gottesfeld JM (2008) Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem 283:35402–35409
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M (2007) Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25:3109–3115
Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH (2010) Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol 28:4485–4491
Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N (2005) Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res 65:11136–11145
Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, Steinle A, Salih HR (2005) Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res 65:6321–6329
Acknowledgments
The authors thank Xiao-Rong Liu, Song Shan, and Jing-Zhong Zhu from Chipscreen Biosciences for analysis of histone acetylation. We also thank Dr. Michael J. Newman from HUYA Bioscience International for the critical review of the manuscript. This work was partially supported by grants from the Chinese National “863” Project (2008AA02Z303), National Prize for Small- and Middle-sized enterprises (04C26214420752), and the Significant Project in Biotech Field from Guangdong Province (2003A10903) and Shenzhen City (2005-K2-009).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Dong, M., Ning, ZQ., Xing, PY. et al. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother Pharmacol 69, 1413–1422 (2012). https://doi.org/10.1007/s00280-012-1847-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-012-1847-5