
Abstract
The formation of platelet-rich thrombi, a critical step in the pathogenesis of atherothrombotic events, is a multistep process involving several components, among which von Willebrand Factor (VWF) plays a central role. Ruptured atherosclerotic plaques expose subendothelial matrix proteins which bind VWF that represents a bridge between the injured blood vessel and activated platelets, playing a crucial role in platelet adhesion and aggregation, especially in conditions of high-shear rate. Due to these peculiarities, the binding of VWF to GPIbα is an attractive drug target. Here we summarize the present knowledge on the different classes of drugs targeting the VWF–GPIb interaction and we give an account of their level of clinical development. In particular, the following compounds are discussed: AJW200, an IgG4 humanized monoclonal antibody against VWF-A1; 82D6A3, a monoclonal antibody against VWF-A3; ALX-0081 and ALX-0681, bivalent humanized nanobodies targeting the VWF-A1 domain; ARC1779 and its advanced formulation ARC15105, second-generation aptamers that bind the VWF-A1 domain; h6B4-Fab, a murine monoclonal antibody, and GPG-290, a recombinant chimeric protein, both directed against GPIbα.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glycoprotein Ib/IX/V
- von Willebrand factor
- Nanobody
- Aptamer
- Platelets
- Thrombosis
- Shear stress
- Stroke
- Acute coronary syndromes
- Antiplatelet
Platelet activation plays a critical role in the pathogenesis of atherothrombotic events, such as acute coronary syndromes (ACS) or stroke. The formation of arterial thrombi is a multistep process involving several components, and among them von Willebrand Factor (VWF) appears to be crucial, especially in the first phases of the process. Rupture of an atherosclerotic plaque exposes collagen to which VWF is bound, starting platelet adhesion followed by platelet activation (Nilsson et al. 1957). VWF plays a particularly important role in platelet adhesion and aggregation under high-shear conditions (Nilsson et al. 1957; Nillson et al. 1957). Moreover, plasma levels of VWF are raised in disease states associated with endothelial activation and may increase the thrombotic potential, and indeed VWF levels are predictive of adverse cardiac events, including vascular death (Spiel et al. 2008). In view of the above-summarized role of VWF in thrombogenesis, the interaction between VWF and its platelet receptor, GPIb/IX/V, has been considered as a promising new target for antiplatelet therapy (Spiel et al. 2008).
1 von Willebrand Factor
1.1 Structure and Functions
VWF is a large adhesive, multimeric glycoprotein that fulfills two crucial roles in primary hemostasis: it functions as a bridge between subendothelial matrix and circulating platelets, and it acts as a carrier for blood clotting factor VIII, protecting it from rapid inactivation. The gene for VWF is located to the short arm of chromosome 12 and it comprises 180 kb and 52 exons. VWF is mainly synthesized by endothelial cells, where it is stored in Weibel-Palade bodies and secreted either constitutively or by exocytosis, but also by megakaryocytes. VWF is synthesized as pre-pro-VWF, formed by a signal peptide of 22 aminoacids, a pro-peptide of 741 aminoacids and the mature subunit of 2,050 aminoacids, the latter consisting of four types of domains with specific functions (Fig. 1). The D′–D3 domain contains binding sites for FVIII and for P-selectin (Sadler 1998); P-selectin partially mediates the tethering of UL-VWF strings to the endothelial surface in flowing blood facilitating multimer degradation by ADAMTS-13 through the exposure of the cleavage site on VWF-A2 domain (Michaux et al. 2006); the A1 domain is the binding site for platelet GPIbα but it also contains binding sites for heparin, sulphated glycolipids and botrocetin, a lectin snake venom of Bothrops jararaca that elicits VWF binding to platelet GPIb leading to platelet agglutination (Sixma et al. 1991); the A3 domain is the major binding site for fibrillar collagen type I and III (Cruz et al. 1995); the A2 domain contains the cleavage site for the metalloproteinase ADAMTS-13; finally, the C1 domain is the binding site for GPIIb/IIIa (Beacham et al. 1992). In the endoplasmic reticulum proVWF dimerizes “tail-to-tail” through disulfide bonds between C-terminal cystine knot domains. Pro-VWF dimers are transported to the Golgi apparatus where a propeptide processing protease cleaves the propeptide (domains D1–D2); and additional disulfide bonds between D3 domains form “head to head” between D3 domains in the trans Golgi network resulting in multimers.

Structure and domains of VWF
Weibel-Palade bodies of endothelial cells are the main storage granules for VWF wherefrom it can be secreted either constitutively or via a pathway regulated by secretagogues, including estrogens, histamine, thrombin, or fibrin (Wagner 1989, 1990; Claus et al. 2010). VWF stored within Weibel-Palade bodies is composed of the largest multimeric species, the UL-VWF (up to 10,000 kDa), usually absent from blood of normal individuals (Wagner and Marder 1984). In physiologic conditions, UL-VWF are in fact cleaved by ADAMTS-13 and disappear from the circulation; they can be detected only transiently in the circulation of normal individuals following treatment with I-deamino-8-d-arginine vasopressin (DDAVP), that induces their secretion from endothelial storage granules (Ruggeri et al. 1982). On the contrary, in microangiopathic disorders, such as thrombotic thrombocytopenic purpura (TTP) or haemolytic uremic syndrome, UL-VWF accumulates in blood (Lowe and Werner 2005). Approximately 20% of VWF present in blood is stored in platelet α-granules (Harrison et al. 1993) and consists of UL-VWF multimers. Platelets activated at sites of vascular injury, mainly by thrombin, release their VWF content; therefore platelets and endothelial cells co-operate in the release of the most thrombogenic VWF multimers at a site of vascular wall damage. Moreover, when VWF is released acutely FVIII bound to it is detached by thrombin cleavage and made available for the clotting cascade (Nesheim et al. 1991).
In order to bind their ligands, the A1 and A3 domains of VWF are dependent on the fluid dynamic forces generated by blood flow. Shear stress, which expresses the forces produced by the sliding of different layers of blood inside vessels, is greater in arteries than in veins, and among different arteries it is higher in small arterioles of 10–50 μm, where it ranges from 500 to 5,000 s−1, and highest in diseased arteries, such as coronary arteries with stenosis (Mailhac et al. 1994).
Above a shear rate of 1,500 s−1 platelet adhesion to a damaged surface is strictly dependent on the interaction between VWF A1 domain and GPIbα. In fact, in conditions of elevated shear stress, VWF exposes the A1 domain normally hidden by the folding of the molecule, and induces a sustained binding to platelet GPIbα stopping them via tether formation. Subsequently, the binding of plasma VWF C1 domain to platelet GPIIb/IIIa (Savage et al. 1996) on one side and to the exposed subendothelial collagen through its A3 domain on the other (Ruggeri et al. 1983) contributes to thrombus growth. UL-VWF multimers contain a large number of binding sites for GPIbα giving rise to spontaneous platelet aggregation and thrombosis (Moake et al. 1982). For this reason, UL-VWF must be actively removed from plasma. When released, UL-VWF are anchored to the surface of endothelial cells through P-selectin and stretched by fluid shear stress to an open conformation that exposes the A2 domain to cleavage by ADAMTS-13 (Schneider et al. 2007; Ruggeri 2003).
The binding of soluble VWF to GPIb-IX-V can be artificially induced in vitro by ristocetin (Giannini et al. 2007), a vancomycin-like antibiotic from Nocardia lurida, that binds to the proline-rich sequence, Glu-700 to Asp-709, C-terminal to the Cys-509–Cys-695 disulfide bond of the A1 domain, or by botrocetin that binds to the A1 domain of VWF (Girma et al. 1990).
The interaction of VWF A1 domain with platelet GPIbα is thought to play some role also under static conditions in vitro (Yamashita et al. 2004) or in venous-like blood flow conditions (Savage et al. 1992). Indeed, histology of thrombi from patients who died from pulmonary embolism revealed the presence of GPIIb/IIIa, VWF, and fibrin in close association (Takahashi et al. 2009).
2 GPIb and Platelet Activation
The GPIb/IX/V complex consists of leucine-rich repeat glycoproteins, GPIbα (130 kDa) and GPIbβ (22 kDa), that are disulfide-linked and non-covalently associated with GPIX (22 kDa) and GPV as a 2:2:2:1 complex. Under high-shear conditions (1,000–10,000 s−1) platelet–platelet interactions become progressively more VWF-dependent, with an important role of both GPIb and integrin αIIbβ3 in promoting the initial formation of platelet aggregates. Under pathologically high-shear conditions (>10,000 s−1), like those found at sites of severe vessel narrowing, platelet aggregation is exclusively mediated by VWF–GPIb adhesive bonds (Jackson et al. 2009). The interaction between VWF and GPIb may trigger platelet activation (Du 2007). The mechanism through which the VWF–GPIbα interaction signals and contributes to subsequent platelet activation has not been completely defined yet. The cytoplasmic region of GPIbα is associated with filamin (also named actin-binding protein), calmodulin and 14-3-3ζ which provide links to signaling proteins such as PI3K, FAK, Src-related tyrosine kinases, GTPase-activating protein and tyrosine phosphatases (PTP1β and SHPTP10) (Du 2007). Topographical association of the GPIb/IX/V complex with other membrane proteins, such as GPVI, FcR γ-chain, α2β1, FcγRIIA, most likely within specialized membrane microdomains known as lipid rafts, supports a crosslinking mechanism involved in GPIbα signaling (López et al. 2005). The engagement of GPIbα by immobilized VWF elicits activation signals, such as transient cytoplasmic Ca2+ elevations, protein phosphorylation (PLCγ2, ERK-1/2, Syk), TxA2 synthesis, ADP release and ultimately activation of αIIbβ3 (Rivera et al. 2009).
3 VWF/GPIb in Thrombotic Diseases
Plasma VWF is elevated in ACS, such as ST elevation myocardial infarction, and its early rise is predictive of a negative prognosis (Rivera et al. 2009; Montalescot et al. 1998; Ray et al. 2005). Reduced coronary flow was associated with high levels of VWF (Takahashi et al. 2009), and increased VWF levels, as well as low levels of ADAMTS-13, are associated with a high cardiovascular risk (Thompson et al. 1995; Bongers et al. 2009).
TTP is a rare (2–10 cases/million/year) microthrombotic disorder due to the accumulation in plasma of UL-VWF multimers generated by a deficiency, either inherited or acquired, of ADAMTS-13 (Remuzzi et al. 2002; Benz and Amann 2010; Levy et al. 2001; Tsai 2010).
Some polymorphisms of the gene coding for GPIbα have been associated with an increased risk of coronary artery disease in young individuals, like the threonine/methionine substitution at amino acid 145. The biological effect of the thr/met substitution remains to be determined, but it is thought to produce a change in the avidity of GPIb for VWF (Kunicki and Nugent 2002). Moreover, a T/C substitution in the region of the translation start site of the gene influences translation efficiency. The presence of the 5C allele (gene frequency of about 0.10 in various white populations) increases the mean level of GPIbα expressed on platelets (roughly, a 50% increase in homozygous individuals and a 33% increase in heterozygous individuals), and there are data supporting an association between −5C and the severity of negative outcomes following acute myocardial infarction in young individuals (≤62 years old) (Kunichi and Nugent 2002). Moreover, a synergistic effect between −5C and Met145 in increasing the risk of stroke in younger individuals has been documented (Sonoda et al. 2001).
4 VWF/GPIb in Haemorrhagic Disorders
4.1 von Willebrand Disease
VWD is the most common inherited bleeding disorder, with an estimated prevalence of ~1%, but the disorder is often asymptomatic, thus the prevalence of clinically significant cases is estimated to be around one in 10,000 (Sadler et al. 2006; Lillicrap 2007). VWD is divided into three subtypes: Type 1 (partially quantitative defect), Type 2 (qualitative defects), and Type 3 (virtually complete absence of VWF). Type 2 can be further divided into 2A (caused by dominant loss-of-function mutations, usually in the A2 domain, and occasionally in the A1 domain), 2B (caused by dominant gain-of-function mutations, usually missense, in the A1 domain), 2M (caused by a mutations in the A1 domain leading to defective VWF binding to GPIbα), and 2N (caused by recessive mutations in the D′ to D3 domains which inactivate the binding of VWF to FVIII) (Federici 2009).
VWD encompasses a wide spectrum of disease severity, ranging from rare and mild bleeding symptoms to severe hemorrhagic episodes that are similar to those of severe hemophilia. The most common bleeding symptoms in VWD reflect impaired platelet adhesion, and include mucosal bleeding, especially epistaxis and menorrhagia. Life-threatening bleeding may rarely occur, especially in VWD type 3 or certain cases of VWD type 2 (Sadler et al. 2000; Federici et al. 2006).
4.2 Bernard Soulier Syndrome
Bernard Soulier syndrome (BSS) is a rare autosomal recessive, inherited platelet function disorder, associated with mild to severe mucocutaneous bleeding, due to a defect of platelet GPIb/IX/V (Lopez et al. 1998; Ware et al. 2000). In a recent paper an attempt to correlate genotype and phenotype in BSS patients has been made (Savoia et al. 2011). Bleeding diathesis did not correlate with thrombocytopenia, which was always moderate, and platelet GPIbα expression which was always severely impaired (Savoia et al. 2011)
5 Strategies to Inhibit the von Willebrand Factor–GPIb/IX/V Interaction
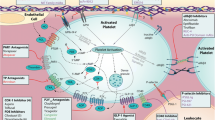
The VWF/GPIb interaction is a potentially attractive target for new antithrombotic agents. Indeed, since VWF has a role in platelet adhesion and activation essentially at high-shear stresses, it is expected that the blockade of VWF-mediated platelet activation would interfere more with platelet deposition in diseased arteries, such as stenosed coronary or cerebral arteries, than in healthy blood vessels, thus preventing thrombosis without affecting significantly physiologic haemostasis. Strategies aimed at interacting with the binding of VWF to GPIbα have involved the development of monoclonal antibodies, nanobodies or aptamers against VWF and GPIb (Fig. 2).

Strategies aimed at interacting with the binding of VWF to GPIbα
5.1 Monoclonal Antibodies Against VWF
5.1.1 AJW200
AJW200 is an IgG4 monoclonal antibody directed against the A1 domain of VWF, developed starting from the AJvW-2 murine monoclonal antibody, later humanized to minimize the immunological response when administered to humans (Fontayne et al. 2008). The precursor of AJW200, AJvW-2, was found to inhibit ristocetin- and botrocetin-induced aggregation, high-shear stress, but not low-shear stress-induced aggregation and adhesion to collagen type I of human platelets; moreover, photochemically induced thrombosis of the carotid artery of guinea pigs was inhibited by AJvW-2 without concomitant prolongation of the bleeding time (Kageyama et al. 1997).
5.1.1.1 In Vitro Data
AJW200 selectively inhibits human (Kageyama et al. 2002a), canine (Kageyama et al. 2002b), and rabbit (Yamashita et al. 2003) platelet aggregation induced by ristocetin and botrocetin. AJW200 also suppresses high-shear stress (108 dyne/cm2)-induced platelet aggregation, as measured by the cone-and-plate viscometer, with an IC50 of 1.0 ± 0.1 μg/ml. In contrast, low-shear stress (12 dyne/cm2)-induced platelet aggregation is not affected by AJW200 up to a concentration of 80 μg/ml. Similarly, platelet adhesion to a type III collagen-coated surface was inhibited by AJW200 under high-shear stress conditions (1,500 s−1) (IC50 2.6 ± 0.2 μg/ml), but not at low-shear stress (360 s−1) (IC50 > 64 μg/ml). AJW200 also inhibited high-shear stress-induced thrombin generation (Kageyama et al. 2002a).
5.1.1.2 Preclinical Studies
An inhibition of ristocetin-induced platelet aggregation sustained over 24 h, 6 days, and 2 weeks was observed after a single bolus injection of, respectively, 0.3, 1, and 3 mg/kg of AJW200 in cynomolgus monkeys. AJW200 did not affect template skin bleeding time at the dose of 0.3 mg/kg, while it significantly prolonged it at 1 and 3 mg/kg, although moderately as compared with the anti-GPIIb/IIIa abciximab (Kageyama et al. 2002a).
AJW200 inhibited thrombus formation in stenosed coronary arteries of beagle dogs without prolonging the bleeding time, showing a safer profile as compared with abciximab (Kageyama et al. 2002a). At least 50% occupancy of VWF (corresponding to 0.7 μg/ml of AJW200 in plasma) was required for the inhibition of cyclic flow variations in vivo, while the bleeding time was extensively prolonged when VWF occupancy reached 80–100% (corresponding to ~20 μg/ml of AJW200 in plasma) (Kageyama et al. 2002a). In a model of balloon injury-induced thrombosis of the iliac arteries in rabbits, a bolus of AJW200 (3 mg/kg) significantly reduced fibrin-rich thrombus formation and the subsequent neointimal growth (Kageyama et al. 2002b) and prevented occlusive thrombus formation (Yamashita et al. 2003).
Finally, AJW200 reduced the number and weight of iliac vein thrombi and pulmonary thromboemboli generated by positioning a polyethylene tube in the iliac veins of rabbits (Takahashi et al. 2009).
5.1.1.3 Clinical Studies
In a randomized, double blind, Phase I study, placebo or three doses of AJW200 (0.01, 0.03 or 0.05 mg/kg) were infused i.v. in 24 healthy male subjects. No significant adverse events were recorded and there was no evidence of immunogenicity. The maximum VWF occupancy obtained was 19.4, 51.0 and 62.4%, respectively, and correlated with plasma AJW200 concentrations. AJW200 produced a dose-dependent inhibition of RiCof at 1 h post-infusion (58 ± 22 vs. 110 ± 25% and 34 ± 15 vs. 116 ± 41%, at 0.03 and 0.05 mg/kg, respectively) and a prolongation of the PFA-100 closure time, an effect that appeared to be related to the baseline VWF level, without concomitantly prolonging the skin bleeding time (Machin et al. 2003).
5.1.2 Monoclonal Antibody Against the A3 Domain of VWF
82D6A3 is a monoclonal antibody against the A3 domain of human VWF that inhibits the interaction of VWF with fibrillar collagen type I and III (Hoylaerts et al. 1997). 82D6A3 binds the A3-domain of VWF but not of denatured or reduced VWF, suggesting that 82D6A3 does not recognize a linear epitope.
5.1.2.1 In Vitro Data
82D6A3 inhibited platelet adhesion under flow conditions at different shear rates. The inhibitory effect increased with increasing shear stresses, with no significant effect at 650 s−1, a mild effect at 1,300 s−1, and an almost complete inhibition at 2,600 s−1, in agreement with the VWF dependence of the reaction (Vanhoorelbeke et al. 2003).
5.1.2.2 Preclinical Studies
The antithrombotic efficacy of 82D6A3 was evaluated in a modified Folts model in baboons. Baboons undergoing mechanical injury to the femoral artery were treated with an i.v. bolus of 82D6A3, at the doses of 100, 300, and 600 μg/kg, resulting in 58.3%, 100%, and 100% reduction, respectively, in cyclic blood flow variations. At the dose of 100 μg/kg 80% of the VWF-A3 domain was occupied, corresponding to a 30–36% ex vivo inhibition of VWF binding to collagen. 82D6A3 did not prolong the bleeding time up to 300 μg/kg mg/kg, even when 100% of VWF was occupied and 100% ex vivo inhibition of VWF-collagen binding was observed (Wu et al. 2002a). Under the same conditions, an anti-GPIIb/IIIa monoclonal antibody induced a strong prolongation of the bleeding time (Levy et al. 2001).
82D6A3 has been humanized by variable domain resurfacing and grafting on the constant region of a human IgG4 (Staelens et al. 2006) resulting in a h82D6A3 with an in vitro activity comparable to that of murine IgG, a step toward the use in humans. However, studies evaluating the antithrombotic activity of h82D6A3 in humans have not been reported yet.
5.2 Nanobodies
Despite significant clinical applications, monoclonal antibodies have several limitations that have opened the way to the development of smaller, more versatile antibodies. Nanobodies are naturally occurring antibodies devoid of light chains, initially discovered in the serum of dromedaries and then found in sera of all camelids; these antibodies, in addition to light chains, also lack the first constant domain of heavy chains (CH1). The variable domains of these antibodies, referred to as VnHs, represented the basis for the development of nanobodies, a new class of therapeutic proteins consisting of one or more single antigen-binding domains. Several nanobodies against various targets of potential therapeutic interest have been developed, including antithrombotic nanobodies (Siller-Matula et al. 2011).
5.2.1 ALX-0081
ALX-0081 is a first-in-class, bivalent humanized nanobody that binds with high avidity the A1 domain of VWF, thereby blocking the interaction between GPIbα and VWF under high-shear conditions (Van Bockenstaele et al. 2009). ALX-0081 consists of two identical building blocks, PMPaP2A2h1, bound to each other via a short linker. Bivalency is required to have a high affinity interaction with VWF A1 which translates in a potent inhibition of VWF binding to platelet GPIbα.
5.2.1.1 In Vitro Studies
ALX-0081 significantly inhibited ristocetin-induced binding of VWF to human platelets, as measured by ELISA, with an IC50 of 0.26 ± 0.04 nM (Markus et al. 2011). ALX-0081 (0.8 μg/ml) prevented platelet adhesion to collagen type III at high-shear rates (>1,500 s−1) but not at low-shear rates (<500 s−1). In the absence of VWF, ALX-0081 did not interfere with the platelet–collagen interaction (Ulrichts et al. 2011). The addition of ALX-0081 (0.1 to 3 μg/ml) to blood from patients with ACS treated with aspirin, clopidogrel, and unfractionated heparin, in which residual platelet activation was still observed, completely inhibited platelet adhesion under high-shear conditions (1,600 s−1). Moreover, ALX-0081 inhibited ristocetin-induced aggregation (RIPA) of platelets from healthy individuals, at the concentration of 0.4 μg/ml, and from CAD patients, at the concentration of 0.8 μg/ml. The fact that lower concentrations of ALX-0081 were required in healthy volunteers to inhibit platelet function as compared to those required in CAD patients is probably explained by the higher VWF-Ag levels observed in CAD patients (Van Loon et al. 2011). Indeed, the effective ALX-0081 dose resulting in complete suppression of platelet adhesion to collagen under high shear ranged from 0.2 to 0.8 μg/ml and correlated with the levels of VWF in the plasma of patients (Ulrichts et al. 2011). Finally, ALX-0081 dose-dependently prolonged the C/ADP closure time in the PFA-100 system (Van Loon et al. 2011).
5.2.1.2 Preclinical Studies
ALX-0081, fully active in humans, cross-reacts with VWF from primates and, partially, from guinea pigs, while it does not bind VWF from other rodents. Initial safety and efficacy studies in cynomologus monkeys showed that ALX-0081 (0.4 and 8 mg/kg, given by i.v. injection) inhibited ristocetin-induced platelet aggregation, for up to 48 h with the highest one (Ulrichts et al. 2011). The half life of a i.v. bolus of ALX-0081 ranged from 17 to 30 h. PK-PD analysis indicated that plasma levels exceeding 1 μg/ml of ALX-0081 resulted in complete RiCof inhibition.
The antithrombotic effect of ALX-0081 was tested in a Folt’s model in the femoral arteries of baboons. Plasma levels of ALX-0081 between 0.3 and 0.5 μg/ml induced full inhibition of cyclic flow reductions. The effect of ALX-0081, abciximab, and clopidogrel on bleeding was evaluated by a surgical bleeding method. Mean blood loss was higher in animals treated with clopidogrel (10 mg/kg) and to an even greater extent in those treated with abciximab (20–500 μg/kg) than in animals receiving ALX-0081 (3–300 μg/kg) (1.6- to sixfold lower as compared with clopidogrel and abciximab, respectively). Comparing the doses required to prevent cyclic flow reductions and those increasing surgical bleeding, ALX-0081 showed a larger therapeutic window as compared with abciximab and clopidogrel (Ulrichts et al. 2011).
In a model of middle cerebral artery (MCA) thrombosis induced by photochemical injury in guinea pigs, ALX-0081 was compared with tirofiban, a GPIIb/IIIa inhibitor, and rtPA, a thrombolytic agent. ALX-0081 (5 mg/kg), administered immediately after the total occlusion of the MCA, restored blood perfusion; tirofiban (10 mg/kg i.v. bolus followed by 20 mg/kg/min for 2 h infusion) and rtPA (0.1 mg/kg i.v. bolus plus 0.9 mg/kg/min for 30 min infusion) were also effective. However, while treatment with ALX-0081 prevented brain damage, assessed by vital staining 24 h after the induction of injury, both tirofiban and rtPA were ineffective. The antithrombotic effect of ALX-0081 was not associated with intracerebral hemorrhage while intracranial bleeding was observed in tirofiban and rtPA-treated animals (Momi et al. 2011).
5.2.1.3 Clinical Studies
A Phase I study tested ALX-0081, given by i.v. infusions (1 h) at doses ranging from 0.5 mg to 12 mg, in 40 male healthy volunteers. ALX-0081 was well tolerated and appeared to be safe, with no bleeding and no immunogenic response. ALX-0081 displayed non-linear pharmacokinetic properties, following a two compartment model. Ristocetin-induced platelet agglutination was fully inhibited at doses ≥2 mg, corresponding to plasma concentrations of 400 ng/mL, 1 h post-dosing, with a maximum duration of 12 h. A mild and transient reduction of FVIII and VWF in plasma was observed, fully reversible within 24 h.
A placebo-controlled, dose-escalating, Phase Ib study was carried out in 25 patients with stable angina undergoing PCI. Single escalating doses (2–9 mg) were followed by multiple dosing (four doses in 24 h for a total of 18 mg). A significant inhibition of ristocetin-induced platelet agglutination and ristocetin cofactor activity for 24 and 30 h was observed. Only mild and transient adverse events were reported and most of them seemed to be related to the PCI procedure; only minor bleedings were reported and these apparently did not differ between the treatment groups (Holz et al. 2009).
A Phase II, randomized, open-label study, designed to compare the safety, tolerability and biological effectiveness of ALX-0081 versus the GPIIb/IIIa inhibitor ReoPro in high risk PCI patients receiving standard treatment with acetylsalicylic acid plus clopidogrel and heparin is ongoing (http://clinicaltrials.gov/ct2/show/NCT01020383?term=ALX-0081&rank=1). Patients are randomly assigned to either ALX-0081 (four i.v. boluses, once every 6 h: the first of 6 mg, and the subsequent three doses of 4 mg) or ReoPro (0.25 mg/kg i.v. bolus followed by continuous i.v. infusion of 0.125 μg/kg/min for 12 h).
5.2.2 ALX-0081 Plus ALX-0681
ALX-0681 is a nanobody identical to ALX-0081, targeting the A1 domain of VWF but specifically formulated to be administered subcutaneously.
5.2.2.1 Clinical Studies
A Phase II, single-blind, randomized, placebo-controlled trial, involving 40 centers worldwide, designed to assess the efficacy and safety of anti-VWF nanobody as adjunctive treatment to plasma exchange in 110 patients with acquired TTP (TITAN Study) is ongoing (clinical trial identifier: NCT01151423). The dose regimen is 10 mg i.v. bolus of ALX-0081, prior to plasma exchange, followed by 10 mg ALX-0681 injected s.c. once or twice a day for 30 days; in the placebo-controlled group i.v. bolus injection prior to plasma exchange, followed by daily s.c. injection of placebo comparator, is administered.
Very preliminary results (5/110 patients) have been recently reported showing a significant shortening of the time to normalization of platelet count (primary end point of the study) as compared with plasma exchange alone, a reduction of UL-VWF in plasma and a sustained inhibition of ristocetin-induced platelet agglutination (Peyvandi et al. 2011). Final results of this trial are expected for 2013.
5.3 Aptamers
5.3.1 ARC 1779
Aptamers are nucleic acid molecules with high affinity and specificity for a selected target molecule, with an ability to fold into unique three-dimensional structures that promote target binding. Through conjugation with high molecular weight polyethylene glycol, aptamers are engineered to have some of the attributes of monoclonal antibodies and some of those of low molecular weight chemically synthesized drugs. ARC1779 is a nuclease-resistant aptamer conjugated to a 20 kDa polyethylene glycol at the 5′terminus. It binds with high affinity to the A1 domain of VWF and inhibits VWF-dependent platelet aggregation.
5.3.1.1 In Vitro Data
ARC1779 inhibits botrocetin-induced platelet aggregation with an IC50 of 344nM (Machin et al. 2003), VWF activity with an IC50 of 100nM, and shear dependent platelet function as assessed by the PFA-100 with an IC95 ∼400nM, in blood from healthy volunteers as well as from ACS patients (Diener et al. 2009). Moreover, ARC1779 dose-dependently inhibits platelet adhesion in a parallel plate perfusion chamber under high-shear rate (1,500 s−1), an effect completely absent at low-shear rate (Machin et al. 2003). In a model of human whole blood perfused at a high-shear rate (6,974 s−1) over de-endothelized porcine arteries, ARC1779 significantly reduced platelet accumulation (Machin et al. 2003).
5.3.1.2 Preclinical Studies
In a carotid artery thrombosis model induced by electrical injury in cynomolgus monkeys, ARC1779 (i.v. bolus + infusion) inhibited the formation of occlusive thrombi (Machin et al. 2003). At the plasma concentration of 700 nM (9.1 μg/ml), effective in preventing thrombosis, only a mild prolongation of the bleeding time was observed (Machin et al. 2003).
The antithrombotic effects of ARC1779 were also determined in an ex vivo model in which blood from patients on double antiplatelet therapy with aspirin and clopidogrel labeled with 111In was perfused over injured porcine aortic segments under high-shear rate. ARC1779 significantly reduced platelet adhesion at 75 and 250 nM (Diener et al. 2009).
5.3.1.3 Clinical Studies
A first in man phase I, randomized, double blind, placebo controlled, dose-escalating study was carried out in healthy volunteers testing the pharmacodynamic profile of ARC1779 administered either as an i.v. bolus or as an i.v. bolus followed by a 4-h infusion (Spiel et al. 2009). ARC1779 induced complete inhibition of the PFA-100 C/ADP closure time with an EC50 of 2–3 μg/ml. These plasma concentrations of ARC1779 were achieved at C max with doses as low as 0.1 mg/kg and were sustained after slow i.v. bolus administration of 1.0 mg/kg for at least 6 h. No bleeding manifestations were observed (Spiel et al. 2009).
In a phase II, randomized, cross-over, double-dummy, pilot study carried out in type 2B VWD patients, ARC1779 0.23 mg/kg plus a 4-hour continuous infusion of 0.001 mg/kg/min, that results in a steady state concentration of 4–5 μg/ml) completely blocked the VWF A1 domain, enhanced desmopressin-induced RiCo and FVIII activity and prevented the rapid consumption of VWF multimers together with agglutinated platelets that occurs in response to desmopressin in these patients (Jilma et al. 2010).
A prospective, open-label clinical trial with a partial cross-over design has been carried out to test the efficacy and safety of ARC1779, added to plasma exchange, in patients with TTP. Three different administration regimens were used: subcutaneous injections of 50 mg of ARC1779 on seven consecutive days, a low-dose infusion of ARC1779 (0.002 mg/kg/min) for 24–72 h, and a high-dose infusion (0.004–0.006 mg/kg/min) for up to 72 h. ARC1779 was well tolerated without any bleeding at concentrations spanning over three orders of magnitude.
Infusion of ARC1779 dose-dependently inhibited VWF-dependent platelet function and increased or stabilized platelet counts in congenital TTP. However, the tested doses, particularly the daily s.c. injections that did not reach therapeutically effective plasma concentrations, did not correct all clinical or laboratory features of TTP (Jilma-Stohlawetz et al. 2011).
More recently, the effect of treatment with ARC1779 on a surrogate marker of cerebral embolism, i.e., embolic signals assessed by trans-cranial Doppler Ultrasound, was evaluated in patients undergoing carotid endarterectomy. Subjects planned for carotid endarterectomy for symptomatic or asymptomatic carotid stenosis were randomized to placebo or ARC1779. The dose regimen for ARC1779 was 0.00015 mg/kg/min for 20 min, 0.003 mg/kg/min for 20 min, 0.006 mg/kg for 20 min followed by a continuous infusion at 0.0006 mg/kg/min. The administration of the study drug was begun 1 h before induction of anesthesia and continued for 3.5 h after skin closure. ARC1779 resulted in a rapid reduction in the frequency and mean intensity of embolic signals. Anemia was reported in the ARC1779 arm of the study (Markus et al. 2011).
5.3.2 ARC15105
ARC15105 is a second-generation VWF A1 domain-inhibitory aptamer; more specifically it is a 21 nucleotide all 2′OMe aptamer, conjugated to 40 kDa polyethylene glycol, with potency and pharmacokinetic characteristics suitable for chronic s.c. treatment. ARC15105 binds VWF with a KD of ~1 nM, is highly stable in human, monkey, and rat serum, with 87–99% of the intact aptamer remaining after 72 h. The 40 kDa PEGylated aptamer had a t 1/2 of 18 h in rats, ~66 h in monkeys with a bioavailability of almost 98%: allometric scaling estimates the human t 1/2 in approximately 217 h (Siller-Matula et al. 2011).
5.3.2.1 In Vitro Studies
ARC15105 inhibited platelet adhesion to collagen under arterial shear flow conditions in a perfusion chamber more effectively than ARC1779 (IC50: 18.5 vs. 175 nM, p < 0.001). ARC15105 40nM inhibited by 93% platelet adhesion to denuded porcine aortas perfused under high-shear conditions with blood from healthy volunteers. Moreover, ARC15105 1 μM completely inhibited ristocetin-induced platelet agglutination but also reduced to some extent collagen, ADP, arachidonic acid, and TRAP-induced platelet aggregation (Siller-Matula et al. 2011), a finding not commented upon.
Finally, ARC15105 completely suppressed VWF activity, as measured by ELISA, in samples collected from patients with myocardial infarction (IC50: 27 nM) (Siller-Matula et al. 2011).
5.4 GPIb Receptor Antagonists
GPIbα is the central component of the receptor complex formed by glycoproteins GPIbα, GPIbβ, GPV, and IX. Human platelets contain approximately 25,000 copies of the GPIb/IX/V complex. GPIbα anchors the complex to the cytoskeleton and harbors the VWF-binding function in its ~290 NH2-terminal residues (Huizinga et al. 2002).
A number of potent anti-GPIb inhibitory antibodies have been produced and extensively tested with respect to their in vitro effects on platelets under both static and flow conditions (Huizinga et al. 2002; Miller et al. 1991; Uff et al. 2002; Cauwenberghs et al. 2001).
5.4.1 h6B4-Fab
The murine monoclonal antibody 6B4 was raised against purified human GPIb. In particular, it recognizes the epitope mapped to the C-terminal flanking region of GPIbα (His1-Val289) (Cauwenberghs et al. 2001). A fully recombinant and humanized version of 6B4-Fab fragment (h6B4-Fab) was recently developed (Fontayne et al. 2006).
5.4.1.1 In Vitro Studies
The intact 6B4 IgG monoclonal antibody blocks dose-dependently the binding of GPIb to VWF, it inhibits ristocetin-induced platelet agglutination and platelet adhesion to human collagen type I, in a parallel plate perfusion chamber at a shear rate of 2,600 s−1 (Cauwenberghs et al. 2001). The MoAb 6B4 Fab fragment blocked ristocetin- and botrocetin-induced platelet aggregation with an IC50 of 1.2 ± 0.3 μg/ml and 2.0 ± 0.5 μg/ml, respectively (Cauwenberghs et al. 2000). Moreover, it inhibited more effectively than intact 6B4 platelet adhesion under shear rates of 650, 1,300, and 2,600 s−1 at the doses of 3.5 μg/ml, 1.1 μg/ml, and 0.5 μg/ml, respectively (Cauwenberghs et al. 2000).
5.4.1.2 Preclinical Studies
The injection of 100 μg/kg of intact 6B4 into baboons caused a rapid drop of the platelet count (<30 × 109/l) within 10 min after injection, with a slow increase observed after 48 h. The same dose of the 6B4-Fab fragment induced a rapid decrease of platelet count to ~120–150 × 109/l but after 24 h the number of circulating platelets was completely normalized (Cauwenberghs et al. 2001).
Pre-treatment of baboons with the 6B4 Fab fragment (80 and 160 μg/kg) reduced platelet deposition on a thrombogenic device (a polytetrafluoroethylene-silicon rubber arteriovenous shunt), by 43 and 65%, measured 15 min after treatment. No complete inhibition of platelet adhesion was observed, even at high doses, probably due to the medium shear rate used in these experiments (700 and 1,000 s−1). Injection of 6B4 Fab fragment (110 μg/kg) in baboons 6 min after a thrombus was allowed to form did not affect platelet deposition indicating that, at least in this model, GPIb did not play a major role in platelet–platelet interactions (Cauwenberghs et al. 2001).
Moreover, the i.v. injection of a single 0.5 mg/kg bolus of h6B4-Fab significantly reduced, while two subsequent administrations, resulting in the cumulative doses of 1.5 and 2.5 mg/kg, completely abolished cyclic flow reductions of a stenosed femoral artery in baboons (Yamashita et al. 2003). Intravenous administration of 0.5 mg/kg of h6B4-Fab resulted in a plasma concentration of 6.3 ± 1.1 μg/ml, with a t 1/2 of 15.5 min. Plasma concentrations raised to 25.9 ± 3.5 μg/ml after an additive dose of 1 mg/kg. A plasma concentration of 10 μg/ml fully inhibited ristocetin-induced platelet agglutination (Fontayne et al. 2008).
The antithrombotic effect of h6B4-Fab was accompanied by an only mild prolongation of the skin template bleeding time and of bleeding loss from a standardized incision. No thrombocytopenia was observed (Fontayne et al. 2008).
Another study evaluated the anti-thrombotic effects of several doses of the 6B4-Fab fragments in combination with the anti-GPIIb/IIIa antibody MA-16N7C2 in baboons. Pre-treatment of baboons with a combination of 1.5 μg/ml of 6B4-Fab and 0.5 μg/mL of MA-16N7C2 inhibited ex vivo collagen-coated surface coverage by platelets by 76%, whereas 88% inhibition was achieved with 2.25 μg/ml 6B4-Fab and 0.75 μg/ml of MA-16N7C2 as measured by a parallel plate perfusion chamber at the shear rate of 1,500 s−1. 6B4-Fab (0.6 mg/kg) did not affect skin bleeding time in baboons while MA-16N7C2 (0.3 mg/kg) significantly prolonged it. The combination of the two antibodies (0.6 mg/kg of 6B4 plus 0.1 mg/kg of MA-16N7C2) completely abolished ristocetin-, ADP- and collagen-induced platelet aggregation and significantly reduced cyclic flow variations but, interestingly, did not further prolong the bleeding time as compared with the single drugs (Fontayne et al. 2008).
5.4.2 GPG-290
GPG-290 is a recombinant, chimeric protein containing the 290 amino-terminal amino acids of GPIba linked via a proline to a human IgG1 Fc produced and purified from Chinese hamster ovary (CHO) cells. GPG-290 is highly pure, stable, and well tolerated in animals and has a half-life of approximately 1.5 days.
5.4.2.1 Preclinical Studies
The antithrombotic effect of GPG-290, alone or in combination with clopidogrel, was evaluated in a canine model of electrolytic injury-induced thrombosis of the left circumflex coronary artery (Hennan et al. 2006).
GPG-290 (50, 100, and 500 μg/kg i.v.) dose-dependently prolonged the time to coronary artery occlusion. Template tongue bleeding time was unchanged after GPG-290 50 and 100 μg/kg while it was prolonged (>2.5-fold) after the administration of the highest dose, but only at the 1 h time point. Clopidogrel was administered orally in two dose regimens: a therapeutic dosing regimen of 4.3 mg/kg on day 2 followed by 1.1 mg/kg for the subsequent two days (the last dose was administered 1 h before the surgical procedure) and a loading dose regimen of 4.3 mg/kg 3 h before the procedure. Clopidogrel was effective in prolonging the time to thrombotic occlusion, but significant bleeding was observed after the loading dose (Hennan et al. 2006).
The combination of clopidogrel (therapeutic dosing regimen) with GPG-290 100 μg/kg further prolonged, although slightly, the time to artery occlusion as compared with the single drugs, and improved blood flow (Hennan et al. 2006). The combination of GPG-290 100 μg/kg with the loading dose of clopidogrel (4.3 mg/kg 3 h before the procedure) provided incremental protection against thrombosis, prolonging the occlusion time and reducing the number of occluded arteries, with no additional prolongation of the bleeding time as compared with clopidogrel alone (Wu et al. 2002b).
The antithrombotic effect of GPG-290 was also assessed in a Folt’s model of stenosed coronary artery in dogs. GPG-290, at doses ranging from 25 to 100 μg/kg which correspond to plasma concentrations of 0.6–2.0 μg/ml, completely abolished cyclic flow variations in 67–100% of treated dogs, without prolonging the bleeding time. Moreover, GPG-290 had no-effect on plasma VWF antigen and VWF–collagen binding activity. Interestingly, the moderate prolongation of the bleeding time (three to fourfold increase) induced by GPG-290 at the highest dose tested of 500 μg/kg (10 times the efficacious antithrombotic dose) was normalized by DDAVP (0.3 μg/kg over 5 min) (Wadanoli et al. 2007).
6 Conclusions
Given the pivotal role of VWF in mediating platelet adhesion under high-shear stress conditions, the inhibition of the GPIb–VWF axis is a potentially promising new strategy to widen the therapeutic window of antiplatelet therapy. The antithrombotic potential of drugs interfering in different ways with the GPIb–VWF interaction has been documented in animal models and in preliminary clinical studies in humans. Aptamers, monoclonal antibodies or nanobodies directed against the A1 domain of VWF, the A3 domain of VWF or against GPIb are expected to be tested soon for their therapeutic potential in acute coronary syndromes and in acute ischemic stroke. Whether the reduced bleeding risk associated with the inhibition of GPIb–VWF interaction, and the pre-eminent activity in conditions of elevated shear stress shown in several animal models and in preliminary clinical trials, will translate in enhanced clinical benefit remains to be established by large, prospective, multicenter clinical trials.
Knowledge Gaps
-
A better definition of the bleeding/antithrombotic balance of inhibitors of the VWF/GPIb axis in vivo is needed.
-
A precise definition of the degree of VWF blockade or GPIb occupancy required to obtain an antithrombotic effect but not inducing bleeding needs to be established.
-
Combination studies with other antiplatelet/anticoagulant drugs (including the new oral anticoagulants) are warranted.
-
For blockers of the VWF–collagen interaction (A3 domain inhibitors) further studies to exclude possible negative effects, due to the crucial physiologic role this interaction may have in platelet adhesion, are warranted.
-
The potential variation of the effective concentration of VWF–GPIb blockade depending on the plasma VWF levels (e.g. higher in ACS patients as compared to healthy people), and its possible impact on the therapeutic effectiveness of this novel therapeutic approach, needs to be fully evaluated.
-
Whether chronic inhibition of the VWF–GPIb interaction may have an effect on atherosclerosis progression remains to be established.
Key Messages
-
VWF acts as a bridging element between damaged endothelial sites and the GPIb receptor on platelets.
-
VWF plays a key role in platelet adhesion and aggregation, especially under high-shear conditions.
-
In inflammatory and atherosclerotic conditions, chronically elevated levels of VWF are observed, that may contribute to an increased thrombotic tendency.
-
In patients with TTP, ultra-large VWF multimers are present in plasma owing to a deficiency of the VWF-cleaving protease ADAMTS13.
-
Inhibitors of the VWF/GPIb axis have been developed, including aptamers, nanobodies, Monoclonal antibodies.
-
Several preliminary phase II trials have tested inhibitors of VWF or GPIb in TTP with promising results in terms of reduction of platelet activation, especially in condition of high-shear rate, and safety.
-
Prospective clinical trials, evaluating the safety and efficacy of novel VWF inhibitors in cardiovascular disease, are ongoing.
References
Beacham DA, Wise RJ, Turci SM et al (1992) Selective inactivation of the Arg-Gly-Asp-Ser (RGDS) binding site in von Willebrand factor by site-directed mutagenesis. J Biol Chem 267:3409–3415
Benz K, Amann K (2010) Thrombotic microangiopathy: new insights. Curr Opin Nephrol Hypertens 19:242–247
Bongers TN, de Bruijne EL, Dipped DW et al (2009) Lower levels of ADAMTS13 are associated with cardiovascular disease in young patients. Atherosclerosis 207:250–254
Cauwenberghs N, Meiring M, Vauterin S et al (2000) Antithrombotic effect of platelet glycoprotein Ib-blocking monoclonal antibody Fab fragments in nonhuman primates. Arterioscler Thromb Vasc Biol 20:1347–1353
Cauwenberghs N, Vanhoorelbeke K, Vauterin S et al (2001) Epitope mapping of inhibitory antibodies against platelet glycoprotein Iba reveals interaction between the leucine-rich repeat N-terminal and C-terminal flanking region domains of glycoprotein Ibα. Blood 98:652–660
Claus RA, Bockmeyer CL, Sossdorf M et al (2010) The balance between von-Willebrand factor and its cleaving protease ADAMTS13: biomarker in systemic inflammation and development of organ failure? Curr Mol Med 10:236–248
Cruz MA, Yuan H, Lee JR et al (1995) Interaction of the von Willebrand factor (VWF) with collagen. Localization of the primary collagen-binding site by analysis of recombinant VWF A domain polypeptides. J Biol Chem 270:10822–10827
Diener JL, Danile Lagasse HA, Duerchmied D et al (2009) Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost 7:1155–1166
Du X (2007) Signaling and regulation of the glycoprotein Ib/IX/V complex. Curr Opin Hematol 14:262–269
Federici AB (2009) Classification of inherited von Willebrand Disease and implications in clinical practice. Thromb Res 124:S2–S6
Federici AB, Castaman G, Thompson A, Berntorp E (2006) Von Willebrand’s disease: clinical management. Haemophilia 12:152–158
Fontayne A, Vanhoorelbeke K, Pareyn I et al (2006) Rational humanization of the powerful antithrombotic anti GPIba antibody: 6B4. Thromb Haemost 96:671–684
Fontayne A, Meiring M, Lamprecht S et al (2008) The humanized anti-glycoprotein Ib monoclonal antibody h6B4-Fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb Haemost 100:670–677
Giannini S, Mezzasoma AM, Leone M, Gresele P (2007) Laboratory diagnosis and monitoring of desmopressin treatment of von Willebrand’s disease by flow cytometry. Haematologica 92:1647–1654
Girma JP, Takahashi Y, Yoshioka A, Diaz J, Meyer D (1990) Ristocetin and botrocetin involve two distinct domains of von Willebrand factor for binding to platelet membrane glycoprotein Ib. Thromb Haemost 64:326–332
Harrison P, Cramer EM (1993) Platelet alpha-granules. Blood Rev 7:52–62
Hennan JK, Swillo RE, Morgan GA et al (2006) Pharmacologic inhibition of platelet VWF-GPIbα interaction prevents coronary artery thrombosis. Thromb Haemost 95:469–475
Holz J, Bartunek J, Barbato E et al (2009) ALX-0081 a novel anti-thrombotic: first results of a multiple dose phase 1 study in patients with stable angina undergoing PCI. J Thromb Haemost 7:PP-WE-416
Hoylaerts M, Yamamoto H, Nuyts K et al (1997) von Willebrand factor binds to native collagen VI primarily via its A1 domain. Biochem J 324:185–191
Huizinga EG, Tsuji S, Romijn RAP et al (2002) Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297:1176–1179
Jackson P, Nesbitt WS, Westein E (2009) Dynamics of platelet thrombus formation. J Thromb Haemost 7:17–20
Jilma B, Paulinska P, Jilma-Stohlawetz P et al (2010) A randomized pilot trial of the anti-von Willebrand factor aptamer ARC1779 in patients with type 2B von Willebrand disease. Thromb Haemost 104:563–570
Jilma-Stohlawetz P, Gilbert JC, Gorczyca ME, Knöbl P, Jilma B (2011) A dose ranging phase I/II trial of the von Willebrand factor inhibiting aptamer ARC1779 in patients with congenital thrombotic thrombocytopenic purpura. Thromb Haemost 106:539–547
Kageyama S, Yamamoto H, Nagano M, Arisaka H, Kayahara T, Yoshimoto R (1997) Anti-thrombotic effects and bleeding risk of AJvW-2, a monoclonal antibody against human von Willebrand factor. Br J Pharmacol 122:165–171
Kageyama S, Yamamoto H, Nakazawa J et al (2002a) Pharmacokinetics and pharmacodynamics of AJW200, a humanized monoclonal antibody to von Willebrand factor, in monkey. Arterioscler Thromb Vasc Biol 22:187–192
Kageyama S, Matsushita J, Yamamoto H (2002b) Effect of a humanized monoclonal antibody to von Willebrand factor in a canine model of coronary arterial thrombosis. Eur J Pharmacol 443:143–149
Kunicki T, Nugent D (2002) The influence of platelet glycoprotein polymorphisms on receptor function and risk for thrombosis. Vox Sang 83(Suppl 1):85–90
Levy GG, Nichols WC, Lian EC et al (2001) Mutations in a member of the ADAMTS13 gene family cause thrombotic thrombocytopenic purpura. Nature 413:488–494
Lillicrap D (2007) Von Willebrand disease-phenotype versus genotype: deficiency versus disease. Thromb Res 120:S11–S16
Lopez JA, Andrews RK, Afshar-Kharghan V et al (1998) Bernard-Soulier syndrome. Blood 91:4397–4418
Lopez JA, del Conde I, Shrimpton CN (2005) Receptors, rafts, and microvesicles in thrombosis and inflammation. J Thromb Haemost 3:1737–1744
Lowe EJ, Werner EJ (2005) Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome in children and adolescent. Semin Thromb Hemost 31:717–730
Machin S, Clarke C, Ikemura O et al (2003) A humanized monoclonal antibody against VWF A1 domain inhibits VWF:RiCof activity and platelet adhesion in human volunteers. J Thromb Haemost (Suppl 1):OC328
Mailhac A, Badimon JJ, Fallon JT et al (1994) Effect of an eccentric severe stenosis on fibrin(ogen) deposition on severely damaged vessel wall in arterial thrombosis. Relative contribution of fibrinogen and platelets. Circulation 90:988–996
Markus HS, McCollum C, Imray C et al (2011) The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy – a randomized trial. Stroke 42:2149–2153
Michaux G, Pullen TJ, Haberichter SL et al (2006) P-selectin binds to the D’-D3 domains of von Willebrand factor in Weibel-Palade bodies. Blood 107:3922–3924
Miller JL, Thiam-Cisse M, Drouet LO (1991) Reduction in thrombus formation by PG-1 F(ab’)2, an anti-guinea pig platelet glycoprotein Ib monoclonal antibody. Arterioscler Thromb Vasc Biol 11:1231–1236
Moake JL, Rudy CK, Troll JH et al (1982) Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med 307:1432–1435
Momi S, Tantucci M, Van Roy M et al (2011) Selective blockade of the A1 domain of von Willebrand factor (VWF), prevents ischemic stroke in the guinea pig: comparison with the thrombolytic rtPA. J Thromb Haemost 9(Suppl 2):O-TU-098
Montalescot G, Philippe F, Ankri A et al (1998) Early increase of von Willebrand factor predicts adverse outcome in unstable coronary artery disease: beneficial effects of enoxaparin. French Investigators of the ESSENCE Trial. Circulation 98:294–299
Nesheim M, Pittman DD, Giles AR et al (1991) The effect of plasma von Willebrand factor on the binding of human factor VIII to thrombin-activated human platelets. J Biol Chem 266:17815–17820
Nillson IM, Blomback M, Jorpes E et al (1957) Von Willebrand’s disease and its correction with human plasma fraction 1-0. Acta Med Scand 159:179–188
Nilsson IM, Blomback M, Von FI (1957) On an inherited autosomal hemorrhagic diathesis with antihemophilic globulin (AHG) deficiency and prolonged bleeding time. Acta Med Scand 159:35–57
Peyvandi F, Breems DA, Knoebl P et al (2011) First results of the Phase II TITAN trial: anti-von Willebrand factor Nanobody®as adjunctive treatment for patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost 9(Suppl 2):SY-TH-027
Ray KK, Morrow DA, Gibson GM et al (2005) Predictors of the rise in VWF after ST elevation myocardial infarction: implications for treatment strategies and clinical outcome: an ENTIRE-TIMI 23 substudy. Eur Heart J 26:440–446
Remuzzi G, Galbusera M, Noris M et al (2002) Von Willebrand cleaving protease (ADAMTS13) is deficient in recurrent and familial thrombotic thrombocytopenic purpura and haemolytic uremic syndrome. Blood 100:778–785
Rivera J, Lozano ML, Navarro-Núñez L, Vicente V (2009) Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 94:700–711
Ruggeri ZM (2003) Von Willebrand factor: a matrix protein that tries to be soluble. Blood 101:2450
Ruggeri ZM, Mannucci PM, Lombardi R et al (1982) Multimeric composition of factor VIII/von Willebrand factor following administration of DDAVP: implications for pathophysiology and therapy of von Willebrand’s disease subtypes. Blood 59:1272–1278
Ruggeri ZM, De Marco L, Gatti L et al (1983) Platelets have more than one binding site for von Willebrand factor. J Clin Invest 72:1–12
Sadler JE (1998) Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem 67:395–424
Sadler JE, Mannucci PM, Berntorp E, et al. (2000) Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 84:160–174
Sadler JE, Budde U, Eikelboom JC et al (2006) Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 4:2103–2114
Savage B, Shattil SJ, Ruggeri ZM (1992) Modulation of platelet function through adhesion receptors. A dual role for glycoprotein IIb-IIIa (integrin alpha IIb beta 3) mediated by fibrinogen and glycoprotein Ib-von Willebrand factor. J Biol Chem 267:11300–11306
Savage B, Saldivar E, Ruggeri ZM (1996) Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84:289–297
Savoia A, Pastore A, De Rocco D, et al. (2011) Clinical and genetic aspects of Bernard-Soulier syndrome: searching for genotype/phenotype correlations. Haematologica 96:417–423
Schneider SW, Nuschele S, Wixforth A et al (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci USA 104:7899–7903
Siller-Matula JM, Mehri Y, Tanguay JF et al (2011) ARC15105 a potent antagonist of von Willebrand factor (VWF) platelet activation and adhesion. J Thromb Haemost 9(Suppl 2):O-MO-027
Sixma JJ, Schiphorst ME, Verweij CL et al (1991) Effect of deletion of the A1 domain of von Willebrand factor on its binding to heparin, collagen and platelets in the presence of ristocetin. Eur J Biochem 196:369–375
Sonoda A, Murata M, Ikeda Y, Fukuchi Y, Watanabe K (2001) Stroke and platelet glycoprotein Ib alpha polymorphisms. Thromb Haemost 85:573–574
Spiel AO, Gilbert JC, Jilma B (2008) von Willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation 117:1449–1459
Spiel AO, Mayr FB, Ladani N et al (2009) The aptamer ARC1779 is a potent and specific inhibitor of von Willebrand factor mediated ex vivo platelet function in acute myocardial infarction. Platelets 20:334–340
Staelens S, Hadders MA, Vauterin S, Platteau C et al (2006) Paratope determination of the antithrombotic antibody 82D6A3 based on the crystal structure of its complex with the von Willebrand factor A3-domain. J Biol Chem 281:2225–2231
Takahashi M, Yamashita A, Moriguchi-Goto S, et al. (2009) Critical role of von Willebrand factor and platelet interaction in venous thromboembolism. Histol Histopathol 24:1391–1398
Thompson SG, Kienast J, Pyke SD et al (1995) Hemostatic factors and risk of myocardial infarction or sudden death in patients with angina pectoris. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. N Engl J Med 332:635–641
Tsai HM (2010) Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol 91:1–19
Uff S, Clemeston JM, Harrison T et al (2002) Crystal structure of the platelet glycoprotein Ibα N-terminal domain reveals an unmasking mechanism for receptor activation. J Biol Chem 38:35657–35663
Ulrichts H, Silence K, Schoolmester A et al (2011) Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared to currently marketed antiplatelet drugs. Blood 118:757–765
Van Bockenstaele F, Holz JB, Revets H (2009) The development of nanobodies for therapeutic applications. Curr Opin Invest Drugs 10:1212–1224
Van Loon JE, de Jaegere PPT, Ulrichts H et al (2011) The in vivo effect of the new antithrombotic drug candidate ALX-0081 on blood samples of patients undergoing percutaneous coronary intervention. Thromb Haemost 106:165–171
Vanhoorelbeke K, Depraetere H, Romijn RA et al (2003) A consensus tetrapeptide selected by phage display adopts the conformation of a dominant discontinuous epitope of a monoclonal anti-VWF antibody that inhibits the von Willebrand factor-collagen interaction. J Biol Chem 278:37815–37821
Wadanoli M, Sako D, Shaw GD et al (2007) The von Willebrand factor antagonist (GPG-290) prevents coronary thrombosis without prolongation of bleeding time. Thromb Haemost 98:397–405
Wagner DD (1989) Storage and secretion of von Willebrand factor. In: Zimmerman TS, Ruggeri ZM (eds) Coagulation and bleeding disorders. The role of factor VIII and von Willebrand factor. Dekker, New York, pp 161–180
Wagner DD (1990) Cell biology of von Willebrand factor. Annu Rev Cell Biol 6:217–246
Wagner DD, Marder VJ (1984) Biosynthesis of von Willebrand protein by human endothelial cells: processing steps and their intracellular localization. J Cell Biol 99:2123–2130
Ware J, Russell S, Ruggeri ZM (2000) Generation and rescue of a murine model of platelet dysfunction: the Bernard-Soulier syndrome. Proc Natl Acad Sci USA 97:2803–2808
Wu D, Vanhoorelbeke K, Cauwenberghs N et al (2002a) Inhibition of the von Willebrand (VWF)-collagen interaction by an antihuman VWF monoclonal antibody results in abolition of in vivo arterial platelet thrombus formation in baboons. Blood 99:3623–3628
Wu D, Meiring M, Kotze HF et al (2002b) Inhibition of platelet glycoprotein Ib, glycoprotein IIb/IIIa, or both by monoclonal antibodies prevents arterial thrombosis in baboons. Arterioscler Thromb Vasc Biol 22:323–328
Yamashita A, Asasda Y, Sugimura H et al (2003) Contribution of von Willebrand factor to thrombus formation on neointima of rabbits stenotic iliac artery under high blood-flow velocity. Arterioscler Thromb Vasc Biol 23:1105–1110
Yamashita A, Furukoji E, Marutsuka K et al (2004) Increased vascular wall thrombogenicity combined with reduced blood flow promotes occlusive thrombus formation in rabbit femoral artery. Arterioscler Thromb Vasc Biol 24:2420–2424
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Gresele, P., Momi, S. (2012). Inhibitors of the Interaction Between von Willebrand Factor and Platelet GPIb/IX/V. In: Gresele, P., Born, G., Patrono, C., Page, C. (eds) Antiplatelet Agents. Handbook of Experimental Pharmacology, vol 210. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-29423-5_12
Download citation
DOI: https://doi.org/10.1007/978-3-642-29423-5_12
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-29422-8
Online ISBN: 978-3-642-29423-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)