
Abstract
Thrombotic thrombocytopenic purpura (TTP) is a disorder with characteristic von Willebrand factor (VWF)-rich microthrombi affecting the arterioles and capillaries of multiple organs. The disorder frequently leads to early death unless the patients are treated with plasma exchange or infusion. Studies in the last decade have provided ample evidence to support that TTP is caused by deficiency of a plasma metalloprotease, ADAMTS13. When exposed to high shear stress in the microcirculation, VWF and platelets are prone to form aggregates. This propensity of VWF and platelet to form microvascular thrombosis is mitigated by ADAMTS13, which cleaves VWF before it is activated by shear stress to cause platelet aggregation in the circulation. Deficiency of ADAMTS13, due to autoimmune inhibitors in patients with acquired TTP and mutations of the ADAMTS13 gene in hereditary cases, leads to VWF–platelet aggregation and microvascular thrombosis of TTP. In this review, we discuss the current knowledge on the pathogenesis, diagnosis and management of TTP, address the ongoing controversies, and indicate the directions of future investigations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Thrombotic thrombocytopenic purpura (TTP) refers to the disorder of widespread microvascular thrombosis involving the capillaries and arterioles of the brain and other organs. The patients typically present with thrombocytopenia, hemolysis with schistocytes on blood smears, and neurologic abnormalities such as headache, confusion, focal deficits, seizures or coma. Thrombocytopenia results from consumption of platelets in the thrombotic process, while erythrocyte fragmentation and hemolysis are believed to result from mechanical injury induced by abnormally high shear stress in the microvasculature.
Without treatment, TTP undertakes a rapid course of deterioration and death in most cases. Plasma exchange or infusion is the mainstay of treatment for TTP. Although quite effective, plasma therapy has defied many efforts to understand why it leads to remission of TTP.
Advances in recent years have transformed TTP from an intriguing mystery to a disease mostly explicable on the deficiency of a newly discovered metalloprotease, ADAMTS13. It leads to the appreciation that the spectrum of TTP presentation is much more variable than previously believed. Like monogenetic diseases, the phenotypic severity of TTP is affected by multiple epistatic genes and environmental modifiers.
1.1 TTP and thrombotic microangiopathy
The syndrome of thrombocytopenia and microangiopathic hemolysis, commonly referred as thrombotic microangiopathy, occurs when there is widespread microvascular thrombosis creating exceedingly high levels of shear stress. It has been associated with various diagnostic terms such as TTP, hemolytic uremic syndrome (HUS), typical HUS, diarrhea-associated HUS, or atypical HUS. Without definitive pathogenetic evidence to support a clear distinction among the various clinical entities, some investigators have classified these disorders under the generic term of TTP/HUS. Others have tried to distinguish TTP and HUS based on the presence or absence of neurological manifestations. TTP has also been defined on the basis of responses to plasma therapy. These classifications of thrombotic microangiopathy are based on assumptions that have never been validated.
In recent years, several molecular mechanisms have been identified for thrombotic microangiopathy. Defective regulation of VWF activity by a circulating metalloprotease, ADAMTS13, is found in most idiopathic cases presenting with thrombocytopenia, microangiopathic hemolysis and mental changes or focal neurological deficits. These patients characteristically have hematuria and proteinuria, but no or minimal renal clearance impairment. Separately, defective regulation of the complement system has been detected in many patients presenting with thrombocytopenia, microangiopathic hemolysis and renal failure. These observations, along with the well-defined disorders such as shiga toxins or neuraminidase-associated HUS, provide a new framework of classifying thrombotic microangiopathy according to molecular defects (Table 1). Cases without identifiable molecular defects are classified according to their etiologies or associated conditions.
A major message of the new disease classification is that different molecular defects may lead to microvascular thrombosis. These disorders share the common features of thrombocytopenia and microangiopathic hemolysis in the peripheral blood. Some of them even share the benefit of plasma infusion or exchange therapy. Nevertheless, to ensure appropriate management, it is critically important to distinguish these disorders based on the underlying pathogenetic mechanisms.
2 Pathology of TTP
Before plasma therapy was adopted in clinical practice, most of the patients presenting with acute TTP died. At autopsy, widespread hyaline thrombi, accompanied by variable fibroblastic infiltration and endothelial overlay, are found in the terminal arterioles and capillaries of multiple organs. The thrombi are found most extensively in the heart, brain, kidney, pancreas, spleen, mesentery and adrenal gland, and are composed primarily of platelets and von Willebrand factor (VWF) (Fig. 1) [1–3]. Small amount of fibrin may be present surrounding or sometimes penetrating the amorphous or granular materials. In older lesions, hyaline deposits may be seen in the sub-endothelial layers of capillaries and between the endothelium and muscular layers of arterioles. Pre-occlusive pseudoaneurysmal dilatation may also be present. Fibrinoid necrosis and vascular or perivascular inflammatory cell infiltration are characteristically absent or minimal.

Immunohistopathology of TTP. VWF-rich hyaline thrombi are present in the arterioles and capillaries but not the venules of multiple organs of a patient that died of TTP due to ADAMTS13 inhibitors. The endothelial cells are intact and contain abundant granules of VWF. No inflammatory cell infiltration is noted. The renal glomeruli contain foci of microthrombi but have well preserved architectures, consistent with the patient’s hematuria and minimal Cr elevation (1.2 mg/dL) at the time of death. VWF is not expressed in the sinusoidal endothelial cells of the liver and no thrombi are detected in the hepatic sinusoids
The distribution of pathological lesions is consistent with the clinical features of TTP: mental change, focal neurological deficits, electrocardiographic changes, abdominal pain with or without evidence of pancreatitis. Notably, the glomerular microthrombi are usually spotty; derangement of an entire glomerulus or cortical necrosis of the kidney is not seen in TTP. The renal pathology of TTP is consistent with the clinical findings of hematuria and proteinuria but no or minimal impairment of the renal clearance function.
3 ADAMTS13 is a VWF cleaving metalloprotease
In TTP, the thrombi consist primarily of VWF and platelets. Knowledge of the processes regulating the interaction between VWF and platelet is critical for understanding why microvascular thrombosis occurs in TTP.
3.1 The role of VWF in hemostasis
von Willebrand factor, whose deficiency leads to bleeding diathesis in patients with von Willebrand disease (VWD), is a plasma glycoprotein comprising a series of multimers with molecular weights ranging from approximately 1 × 106 to greater than 20 × 106 Da [4]. In addition to serving as the carrier of factor VIII, VWF is the ligand for mediating platelet adhesion and aggregation at sites of vessel injury. VWF binds to platelet receptors glycoproteins 1b/IX/V and αIIbβ3. Two intriguing features of VWF–platelet interaction are closely related to the pathogenesis of TTP: VWF–platelet interaction is enhanced by shear stress, and large multimers are hemostatically more effective than small multimers.
3.2 Endothelial VWF is different from plasma VWF multimers
von Willebrand factor is synthesized in vascular endothelial cells and megakaryocytes and released in the plasma and the subendothelium of some vessels. Secretion from endothelial cells accounts for more than 90% of plasma VWF.
Endothelial VWF is a homopolymer of the 2,050-residue VWF polypeptide [5]. The basic VWF polypeptide consists of a series of modular domains (D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK) (Fig. 2a) [4]. The epitope for binding platelet glycoprotein 1b is located in the A1 domain while the 2507RGDS2510 epitope for binding to platelet receptor GPαIIbβ3 is in the C1 domain near the C terminus.

The biosynthesis and the domain structure of VWF. a The domain structure of VWF. The location of the Y1605-M1606 scissile bond and the regions of the A2 domain and the D4-CK domains that are involved in binding with ADAMTS13 exocites (distintegrin-spacer domains [D-S] and TSR5-CUB domains, respectively) are indicated. Also shown are the regions of the VWF polypeptide that are involved in binding factor VIII, GP1b, collagen, heparin and GPαIIbβ3. b Formation of VWF polymers. In the endoplasmic reticulum (ER), two pro-VWF polypeptides are linked by disulfide bonds near the C terminus to form a dimer of pro-VWF. The dimers are bonded by disulfide bonds near the N terminus to form a polymer for secretion from endothelial cells. The process of polymerization starts in the trans Golgi network and proceeds to completion in the Weibel-Palade bodies. The propeptide is cleaved before the VWF polymer is secreted
The VWF polymer is formed in two steps [6, 7]. First, in the endoplasmic reticulum, two monomers of pro-VWF are joined via inter-monomeric disulfide bonds near the C terminus to form dimers (Fig. 2b). Next, a group of VWF dimers is joined successively via disulfide bonds in the D3 domain to form a high molecular weight (HMW) polymer. The process of VWF polymerization is believed to begin in the trans Golgi network and continues to completion in the elongated, endothelial cell specific Weibel-Palade bodies.
Secretion of VWF occurs primarily via the Weibel-Palade bodies. Exocytosis of VWF from the storage granules may be swiftly increased by exposing endothelial cells to secretagogues such as histamine, thrombin, phorbal myristate acetate, or calcium ionophore A23187.
VWF in the endothelial cell culture media may also contain a series of large multimers extending downward from the HMW endothelial VWF polymer. These multimers may result from incomplete polymerization, disulfide bond reduction or proteolysis of the VWF polymer by cellular enzymes during sample preparation for gel analysis.
In some endothelial cell cultures, a series of VWF oligomers are also detected. Treatment of the endothelial cells with cycloheximide, a protein synthesis inhibitor, promptly suppresses the secretion of these oligomers, indicating that the oligomers are constitutively secreted rather than secreted via the Weibel-Palade bodies. Constitutive secretion of VWF is observed in endothelial cells with incomplete sorting of VWF to the Weibel-Palade bodies but is believed to play little if any role in the release of VWF in vivo.
Although believed to be directly released from endothelial cells, the plasma multimers have never been demonstrated in endothelial cell cultures.
3.3 ADAMTS13 cleaves endothelial VWF to plasma multimers
In the circulation, endothelial VWF polymer is converted to plasma multimers by ADAMTS13. ADAMTS13 cleaves VWF at the peptide bond Y1605-M1606 in the A2 domain, creating a 140 and a 176 kD fragments. Homodimers of the 140 and the 176 kD fragment represent the smallest fragments in plasma that are detectable with SDS PAGE and immunoblotting (Fig. 3). Cleavage of endothelial VWF by ADAMTS13 should also produce a monomeric form of the 140 kD fragment. This monomeric fragment has not yet been visualized, presumably because its concentration is too low for detection. Alternatively, cleavage of VWF at the end may be an infrequent event.

Generation of VWF multimers. At each brief exposure to high shear stress during the transit through the arteriole/capillary microcirculation, a VWF polymer or multimer is expected to be cleaved by ADAMTS13 at one of its Y1605-M1606 bonds at or near its center subunit (red, filled triangle). A series of multimers with progressively smaller sizes are generated when this process is repeated on the cleaved products. The smallest products of this proteolysis are homodimers of the 140 and the 176 kD fragments. The 140 kD monomer has not yet been detected in normal plasma. Only the VWF fragments would be generated if a VWF polymer were fully unfolded and cleaved simultaneously at all the scissile bonds
Proteolytic fragments of VWF are detectable in normal plasma. Nevertheless, no generation of VWF fragments occurs when normal plasma is incubated in a test tube, although it contains both VWF and ADAMTS13. This is because proteolysis of VWF occurs only when it is subjected to shear stress in the circulation [8]. The discovery that shear stress is essential for proteolysis of VWF underscores the basis of the current scheme of TTP pathogenesis. It also facilitated the development of assays for detecting VWF cleaving protease activity, eventually leading to the characterization, isolation, and cloning of ADAMTS13 and its association with TTP [9–19]. Mutations of the ADAMTS13 genes were detected in patients with the hereditary form of TTP [19]. The presence of ADAMTS13 inhibitors [20–28] or mutations [29–57] in patients with TTP has been repeatedly demonstrated in many studies from different parts of the world. However, some of these studies also raise controversy over whether ADAMTS13 deficiency defines the disorder of TTP. The reasons for the controversy will be addressed later in this review.
3.4 Structure of ADAMTS13
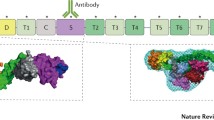
Human ADAMTS13 is a zinc metalloprotease consisting of 1427 amino acid residues and has a modular structure comprising a signal peptide, a short propeptide, a metalloproteinase domain (M), a disintegrin-like domain (D), a thrombospondin type 1 repeat (TSR), a cysteine-rich domain (C), a spacer domain (S), 7 additional TSRs (TSR 2–8) and two CUB (C1r/C1s, urinary epidermal growth factor, bone morphogenic protein) domains (Fig. 4a).

The structure and function of ADAMTS13. a ADAMTS13 has a modular structure comprising of a signal peptide (Sig), a propeptide (Pro), a metalloprotease domain (MP), a disintergrin-like domain (Dis), a thrombospondin type 1 repeat (TSR), a cysteine-rich region (Cys), a spacer domain (Spa), 7 additional TSRs (TSR 2-8), and two CUB domains. The 10 glycosylated N residues are indicated. The catalytic site (HEIGHSFGLEHD) is in the metalloprotease domain. The underline residues are conserved in the ADAMTS metalloprotease family. The sequence between the disintegrin and spacer domains interacts with regions of the VWF A2 domain downstream of the scissile bond. The sequence between TSR5 and CUB domains interacts with an epitope in the D4-CK region of VWF. This latter interaction has modest affinity and does not require shear stress. However, it may promote the other exocite interaction and subsequent Y1605-M1606 cleavage. b A comparison of ADAMTS13 with its 4 truncated variants for their VWF cleaving activity and binding and suppression by the IgG inhibitors of TTP patients
Plasma ADAMTS13 derives primarily from the stellate cells of the liver [58, 59]. ADAMTS13 may also be expressed, albeit at much lower levels, in other types of cells such as renal podocytes and tubular cells [60, 61], vascular endothelial cells [62, 63], and platelets [64, 65]. The plasma concentration of ADAMTS13 is approximately 1 μg/mL, or 5 nmol/L. The elimination half-life of ADAMTS13 is 1–2 days in the circulation. ADAMTS13 is present in the cryosupernatant fraction of plasma and is quite stable at either room temperature or 37°C for many hours but may be inactivated in pathological samples or by thrombin or plasmin of clotted plasma samples [66]. The cellular expression of ADAMTS13 may be affected by inflammatory cytokines [67].
The mature ADAMTS13 protein consists of 1353 amino acid residues but has a molecular weight of 190 kD as determined by either gel filtration or sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), substantially higher than the expected 145 kD. This large difference suggests that ADAMTS13 is highly glycosylated.
3.4.1 Both O- and N-glycosuylations are essential for secretion of active ADAMTS13
Human ADAMTS13 contains the disaccharide glucose-β1,3-fucose on most of its TSRs [68] and 10 N-linked glycans in the metalloprotease domain, in or surround the spacer domain and in the CUB domains [69]. Both N- and O-glycosylations are essential for correct protein folding and secretion. Conversion of oligomannose N-glycans to the complex type in the Golgi complex doubles the proteolytic activity of ADAMTS13. Interestingly, removal of the N-linked glycans does not affect the proteolytic activity of the mature ADAMTS13, suggesting that the N-glycans are needed for correct protein folding during biosynthesis but are not required for maintaining ADAMTS13 in its active form and not involved in the interaction of ADAMTS13 with VWF. In contrast, removal of N-glycans from VWF markedly increases the susceptibility of the substrate to cleavage by ADAMTS13 [70].
3.5 Cleavage of VWF by ADAMTS13
The interaction between ADAMTS13 and VWF is quite complex. The catalytic site of ADAMTS13 resides in the metalloprotease domain. The sequence downstream of the metalloprotease domain also interacts with VWF, thereby facilitating the attack of the VWF scissile bond by the ADAMTS13 catalytic site.
3.5.1 The sequence downstream of the spacer domain facilitates the cleavage of VWF multimers but hinders the cleavage of short VWF peptides
In expression studies, truncation of the sequence downstream of the spacer domain decreases the VWF cleaving activity of ADAMTS13 by 30–70% (Fig. 4b) [71, 72]. Gene transfer studies show the similarly truncated ADAMTS13 is proteolytically active in mouse circulation [73].
Further investigation has shown that the TSR5-CUB region of ADAMTS13 may bind with modest affinity to an epitope spanning residues 1874–2813 (corresponding to the D4-CK domains) of VWF (Figs. 1a, 4a) [74]. This binding may facilitate subsequent ADAMTS13-VWF interactions leading to the cleavage of VWF at the Y1605-M1606 bond.
ADAMTS13 variants truncated downstream of the spacer domain increases by two- to threefolds the activity of ADAMTS13 in cleaving abbreviated VWF peptides such as VWF73 (D1596-R1668), indicating that the distal part of ADAMTS13 enhances the cleavage of VWF multimers but hinders the cleavage of VWF73 [74].
3.5.2 Mouse strains with an IAP-retrotransposon in the ADAMTS13 gene
In some mouse strains, the ADAMTS13 gene contains a retrotransposon of the intracisternal A-type particle (IAP) in intron #23 [71, 75]. The presence of the IAP sequence leads to the synthesis of two truncated forms (IAPa, 1037 residues; and IAPb, 1028 residues) of ADAMTS13. In the mouse liver, the IAPa and IAPb variants account for approximately 75 and 25%, respectively, of the transcripts, while full length ADAMTS13 accounts for less than 10% [71].
Both ADAMTS13 variants lack the TSR1 #7 and #8 and the two CUB domains, and end with a novel IAP-derived 16-residue sequence (ALVWEAAPTFAVTRWR) not found in the full length mouse ADAMTS13. The minor form, IAPb, contains additional splicing variations in the spacer domain, which markedly decreases its secretion and proteolytic activity.
When compared to full length ADAMTS13, the IAPa variant is synthesized and secreted as efficiently as the full length ADAMTS13, but is only one-tenth as effective in cleaving VWF multimers [71]. Nevertheless, when tested against a short VWF peptide (VWF73, D1596-R1668) as the substrate, the IAPa variant is only marginally less effective [71]. This discrepancy explains why mouse strains with the IAP element in the ADAMTS13 gene have one-tenth plasma ADAMTS13 levels when tested against VWF multimers [71] but normal ADAMTS13 activity levels when tested against the VWF73 substrate [74]. Congenic mice generated by cross-breeding show that the IAPa variant of ADAMTS13 are proteolytically less effective in cleaving VWF in the animals [76].
The low proteolytic activity of the IAPa ADAMTS13 variant may result from the absence of the C-terminal domains. Nevertheless, the extraneous 16-residue sequence at the C terminus may further compromise the proteolytic activity of the IAP variant. This may explain why the IAP ADAMTS13 variant is much less active than similarly truncated native ADAMTS13 that does not contain the extraneous sequence.
3.5.3 The cysteine-rich and spacer domains are critical but not indispensible for the catalytic activity of ADAMTS13
Truncation of ADAMTS13 upstream of the cysteine-rich domain markedly decreases [71, 77], but does not abolish as initially proposed [78, 79], the activity of the protease in cleaving VWF multimers or VWF peptides (Fig. 4b). Further analysis shows that the ADAMTS13 sequence between the disintegrin and spacer domains contacts with VWF residues in the A2 domain downstream of the scissile bond (Figs. 2a, 4a) [77, 80, 81]. The ADAMTS13 exocites may orient the catalytic site of the protease to engage the Y1605-M1606 bond of VWF.
3.5.4 The VWF target sequence interacting with the ADAMTS13 spacer domain
The P1645-C1668 sequence in the VWF A2 domain is believed to interact with ADAMTS13 exocites between the disintegrin and spacer domains (Fig. 2a) [77, 80, 81]. Recombinant VWF fragment D1587-R1668 or D1596-R1668 is readily cleaved by ADAMTS13 [82]. Nevertheless, with further extension upstream, recombinant VWF fragment D1459-F1668 or E1554-D1668 is barely cleaved by ADAMTS13, suggesting that E1554-D1587 forms a shield of the Y1605-M1606 scissile bond.
Crystallography shows that the VWF A2 domain comprises a series of secondary structural elements in the order of β1–α1–β2–β3–α2–α3–β4–(α4 less loop)–β5–α5–β6–α6 [83]. The Y1605-M1606 scissile bond is buried at the center of the β4-sheet in the loosely packed hydrophobic core of the A2 domain. The P1645-C1668 sequence encompasses the β6 sheet and α6 helix elements. Interestingly, the sequence E1554-H1587 encompasses the α2 and α3 helices that form a ‘∩’ structure around the Y1605-M1606 scissile bond. The short sequence linking the α2–α3 structure to the β4 sheet forms the most flexible part of the A2 domain. It remains to be determined whether shear stress re-orients the α2–α3 structure away from the β4 sheet.
3.6 Shear stress promotes proteolytic cleavage of VWF
In the presence of VWF, shear stress promotes the platelet adhesion to injured blood vessel wall and the platelet–platelet aggregation. It was generally assumed that shear stress promoted VWF–platelet adhesion and aggregation by exerting its effect on platelets. Since shear stress promotes the cleavage of VWF [8], it is evident that shear stress affects the conformation of VWF, making it susceptible to cleavage by ADAMTS13. Further exposure to shear stress also increases its activity in mediating platelet aggregation.
A brief (<60 s) exposure to shear stress in a stainless steel capillary tubing with a wall shear rate of 3,000 s−1 (equivalent to 60 dynes/cm2 of shear stress, which is in the range encountered in the arterioles and capillaries) is sufficient to produce VWF cleavage by ADAMTS13 [8, 14]. Shear stress makes VWF susceptible to cleavage by ADAMTS13, presumably by exposing its scissile bonds and/or exocites to ADAMTS13.
The effect of shear stress is not specific for ADAMTS13; it also increases the cleavage of VWF by leukocyte proteases [10]. Interestingly, leukocyte proteases such as elastase, cathepsin G, proteinase 3 or matrix metalloprotease 9 also cleave VWF near the M1605-Y1606 bond. Crystal structure analysis has revealed that the cleavage sites are located in a β sheet that is loosely packed in the core of the A2 domain [83]. Since profound leucopenia is not accompanied by evidence of VWF size change, it is questionable that leukocyte proteases are involved in the regulation of VWF activity [84].
Although a direct comparison has not been performed, the available data suggest that shear stress makes VWF susceptible to cleavage before it is activated to support platelet attachment. This difference in response to shear stress may be critical for preventing platelet aggregation in the normal circulation.
Conformational unfolding of VWF from a compact, globular form to a stretched, filamentous form is observed using atomic force or fluorescent microscopy [85–87]. It is estimated that the shear stress necessary for inducing the conformational stretching may be as low as 35–90 dynes/cm2, depending on the devices used and the duration of exposure. The stretched form of VWF binds to collagen and causes platelet aggregation. Nevertheless, it is not clear whether complete conformational stretching is necessary for VWF to be cleaved by ADAMTS13.
Analysis using small angle neutron scattering and fluorescent probe binding measurement have shown that structural change of VWF at its globular sections is detectable at shear rates <3,000 s−1 [88], which is quite similar to the shear rates that cause VWF cleavage by ADAMTS13 in capillary tubes [14]. The conformational changes are most prominent at length scales <10 nm, suggesting that local structural rearrangements at the domain level are sufficient to make the VWF susceptible to proteolysis.
In addition to shear stress, chaotropic agents such as guanidine hydrochloride or urea also cause cleavage of VWF by ADAMTS13. Interestingly, treatment of VWF with guanidine hydrochloride causes local structural changes at the domain level (length scales <10 nm) without altering its overall size (i.e., radius of gyration) [89]. This observation further supports the view that stretching of the multimers to an elongated form is not necessary for cleavage by ADAMTS13 to occur.
In consistence with this view, stretching of a recombinant A2 domain with a laser tweezer leads to cleavage of the fragment by ADAMTS13 [90]. In force-probe simulations, the C-terminal part of the A2 domain exhibits stepwise unfolding when the A2 domain is stretched at both ends [91]. This unfolding may expose the VWF sequence in this region for binding to ADAMTS13 exocites and the Y1605-M1606 scissile bond to ADAMTS13 attack. The N-terminal part of the A2 domain is more resistant to stretching because the β1 strand is tightly embedded in the protein core, forming rupture-resistant interactions to adjacent β2 and β4.
3.7 Is cleavage of endothelium adherent VWF physiologically relevant?
In cell cultures and in ex vivo vascular preparations, it has been observed that some endothelial VWF polymers may remain attached to the cell surface after exocytosis [92]. The adherent VWF assumes an elongated form under conditions of shear stress as low as 2.5 dynes/cm2 and is readily cleaved by ADAMTS13. More recently, it is observed that the adherent VWF is also cleaved in the absence of shear stress [93]. This supports the current view that stretching of VWF to an elongated form is not essential for cleavage by ADAMTS13.
It has been postulated that cleavage of endothelial attached VWF produces the VWF multimers found in normal plasma. There are two reasons against this scheme. First, adherent VWF is observed primarily after endothelial cells are profoundly perturbed with histamine or other potent agonists. No adherent VWF has been observed as yet on unperturbed endothelial cells, which continually secrets the HMW VWF polymer.
Second, no studies have ever shown that the VWF detached from endothelial cells by ADAMTS13 consists of a series of multimers. In fact, generation of VWF multimers requires successive cleavages (Fig. 3). Cleavage of fully unfolded VWF attached to endothelial surface will generate VWF fragments, but not a series of multimers.
Non-adherent VWF in a tubular flow is only intermittently exposed to the peak shear near the wall-fluid boundary. Furthermore, only the shear stress in the arterioles and capillaries reaches the levels necessary for making VWF susceptible to cleavage by ADAMTS13. During each of the momentary bursts of tensile force in the arteriole/capillary, it is likely that only the central subunit of the VWF multimer is exposed to sufficient tensile force and cleaved by ADAMTS13 (Fig. 5). Successive cleavages occur as the VWF molecules repetitively traverse the microcirculation. This process leads to the generation of VWF multimers. In contrast, cleavage of endothelium adherent VWF by ADAMTS13 will not convert the VWF polymer to a series of multimers.

ADAMTS13 prevents VWF–platelet aggregation and thrombosis. a At sites of vessel injury under high shear stress conditions such as those in the arterioles and capillaries, the conformationally flexible VWF, upon attached to the extracellular matrix of the denuded subendothelium, is readily stretched to an extended form for mediating platelet adhesion and aggregation essential for hemostasis. The unfolded VWF may be protected by thrombospondin-1 from ADAMTS13 cleavage. Inactivation of ADAMTS13 by thrombin and plasmin generated at sites of vessel injury may also help minimize the cleavage of the unfolded VWF. b In the circulation, the stretching of VWF does not occur because ADAMTS13 cleaves the Y1605-M1606 bond of the A2 domain whenever it is exposed by shear stress. The cleavage occurs each time VWF traverses the microcirculation. The proposed scheme assumes that VWF is cleaved when its structural changes occur at the domain level, before it becomes stretched by shear stress to an elongated form. This process of proteolysis maintains the VWF in a compact form while it is converted to a series of multimers with progressively smaller sizes. c In the absence of ADAMTS13, VWF, after repetitive exposure to high levels of shear stress, is unfolded to an elongated active form that causes platelet aggregation in the circulation. Intra-vascular platelet thrombosis further increases the shear stress in the microcirculation, begetting more VWF unfolding and platelet aggregation. This self-perpetuating process may explain why TTP often deteriorates precipitously if it is not immediately treated with plasma therapy to replenish the missing ADAMTS13
3.8 Loss of large VWF multimers in patients with abnormal intravascular shear stress
Large VWF multimers are decreased in patients with HUS, aortic stenosis or pulmonary arterial hypertension [2, 94, 95]. In these conditions, ADAMTS13 levels are normal or only mildly decreased. It is believed that large multimers are decreased because high levels of shear stress in these disorders enhance proteolysis of VWF by ADAMTS13. Consequently, the proteolytic fragments are increased. The loss of large VWF multimers may contribute to bleeding diathesis of the patients. VWF multimer pattern normalizes when the high shear stress conditions resolve.
4 Severe ADAMTS13 deficiency causes TTP
4.1 Shear stress causes VWF–platelet aggregation
Exposure of VWF and platelets to shear stress leads to platelet aggregation [87, 96]. The levels of shear stress necessary for inducing platelet aggregation is in the range found in normal arteriolar and capillary circulation, but much higher that those in the venules or larger vessels. Therefore, VWF and platelet have a propensity to form aggregates in normal arterioles and capillaries. This propensity of VWF and platelets to form aggregates may be essential for the effective hemostasis at sites of arteriolar or capillary injury (Fig. 5a). However, it also creates a prothrombotic state in the microcirculation that needs to be constantly checked.
By cleaving VWF before it is activated by shear stress to cause platelet aggregation, ADAMTS13 mitigates the tendency of VWF and platelets to form aggregates in normal microcirculation (Fig. 5b). Thus, the physiological role of ADAMTS13 is to prevent intravascular platelet thrombosis. Deficiency of ADAMTS13 causes VWF–platelet aggregation in the arterioles and capillaries characteristic of TTP (Fig. 5c).
It is important to note that for proteolysis to occur, VWF does not need to be fully stretched to an elongated form. VWF is cleaved by ADAMTS13 when its structure is altered by shear stress in the domain level [88–90]. Thus, through repetitive cleavage in the absence of complete unfolding, VWF maintains its compact conformation while it is converted to a series of multimers with progressively smaller sizes. This scheme allows large VWF multimers to be present in the plasma and immediately available to sites of vessel injury without causing intravascular thrombosis.
The unfolded VWF at vessel injury sites may be protected by thrombospondin-1 [97] from cleavage by ADAMTS13. Inactivation of ADAMTS13 by thrombin and plasmin [66] generated at the vessel injury sites may provide additional protection of VWF from proteolysis.
4.2 What do we learn from the mouse models of ADAMTS13 deficiency
Inactivation of the ADAMTS13 gene in mice failed to generate the phenotype of TTP microvascular thrombosis, until the ADAMTS13 null allele was transferred to a wild derive mouse strain (CASA/Rk) [98, 99]. The CASA/Rk mice have substantially higher levels of VWF. Nevertheless, cross-breeding studies show that the development of TTP is independent of mouse plasma VWF levels.
In CASA/Rk mice with homozygous ADAMTS13 null alleles, spontaneous thrombosis and death occur postneonatally [98]. Administration of shiga toxin precipitates the onset of microvascular thrombosis. It is speculated that shiga toxins may induce thrombosis phenotype by causing a burst of VWF exocytosis from endothelial cells [100].
Clinically, there is no evidence of antecedent shiga toxin exposure in patients of TTP. Only a small fraction of TTP patients has elevated plasma VWF levels. Endothelial perturbation appears to be a consequence, rather than a prerequisite of TTP [101]. In general, there is no evidence of ADAMTS13 deficiency among patients with shiga toxin-associated HUS [2, 102]. Therefore, the relevance of the shiga toxin-ADAMTS13-deficient mouse model to either TTP or shiga toxin-associated HUS remains uncertain.
The lack of a thrombotic phenotype in some mouse strains with inactivated ADAMTS13 gene suggests that these strains contain modifiers that affect the response of VWF to shear stress.
4.3 Other pathogenetic mechanisms of TTP
Other mechanisms of TTP that have been investigated include defects in fibrinolytic activity [103] or prostacyclin homeostasis [104], increased circulating thrombomodulin, tPA or PAI-1 levels [105], anti-endothelial antibodies [106–108], immune complexes [109], platelet aggregating proteins [110], anti-CD36 [111], calcium-dependent cysteine proteases [112], or endothelial apoptotic factors [113]. One study also suggested that the presence of HLA DR53 might be protective against TTP [114]. These changes represent secondary changes or are not specific for TTP [115–119]. Immune complexes detected in patients with TTP may result from ADAMTS13 binding with its antibodies.
4.4 Causes of ADAMTS13 deficiency
Genetic mutations or autoimmune inhibitors cause ADAMTS13 deficiency in TTP. The role of low ADAMTS13 levels in sepsis or other disorders remains uncertain.
4.4.1 Genetic mutations of ADAMTS13
In patients with hereditary TTP, homozygous or compound heterozygous mutations of the ADAMTS13 gene lead to severe ADAMTS13 deficiency. More than 90 different ADAMTS13 mutations have been identified [29–57, 120]. The mutations include missense, non-sense, frame-shifting insertion or deletion and splicing site variations. The affected residues span across the entire spectrum of the ADAMTS13 gene. Impaired protein synthesis, secretion, or proteolytic activity has been described in association with the ADAMTS13 mutants in expression studies.
Heterozygous individuals have ADAMTS13 activity in the 40–70% of normal values. No phenotypic abnormalities have been attributed to partial deficiency of ADAMTS13.
It has been proposed that ADAMTS13 mutations alone are not sufficient to cause TTP. This is a mis-interpretation of the data in the literature. In fact, a TTP phenotype is present in more than 90% of the patients with double heterozygous or homozygous mutations. Furthermore, it is very likely that those few asymptomatic cases will become symptomatic later in life. Overall, the clinical data show that ADAMTS13 mutations cause TTP, which may nevertheless presents with variable phenotypic severity.
Only a few mutations have been described in more than one pedigree. A notable exception is 4143insA, which has been described in multiple pedigrees of Northern and Central Europe and in Turkey. Haplotype analysis suggests that many if not all of the 4143insA mutant alleles probably originated from a common ancestry [121]. Why this particular allele is much more frequent than other mutant alleles remains an intriguing question.
One ADAMTS13 variant allele, C1423T (P475S) is found in Japanese (5.1%), Korean (4%), and Chinese (0.5–1.7%) but has not been detected among Caucasians or African Americans [29, 122]. This polymorphism had raised considerable interest because in expression studies this mutation markedly compromises the activity of ADAMTS13 to approximately 10% of control, raising the speculation that partial deficiency of ADAMTS13 deficiency may be quite common among the Northeast Asians. Nevertheless, this prediction has not materialized. In carriers of the P475S polymorphism, the ADAMTS13 activity level is decreased by a clinically insignificant 10%. The previously reported low activity of the P475S variant resulted from the use of high urea concentration in the ADAMTS13 activity assay [123].
4.4.2 Autoimmune inhibitors of ADAMTS13
In patients with acquired TTP, deficiency of ADAMTS13 results from autoimmune inhibitors of the ADAMTS13 protease [20–28].
With the exception of patients who develop ADAMTS13 inhibitors following the institution of ticlopidine [124, 125], the etiologies of acquired TTP are unknown. ADAMTS13 inhibitors have not been described in other drug-associated thrombotic microangiopathy.
Many TTP patients exhibit positive autoimmune reactions to other antigens [25], suggesting that defective immune regulation may contribute to the development of TTP. HIV infection may also increase the risk of TTP, although this association has not been observed in all series [126].
The inhibitors are primarily IgG, although occasional cases with IgA and IgM antibodies have been described. The VH1-69 germline heavy chain gene appears to be used most frequently in producing the ADAMTS13 antibodies.
Epitope mapping studies shows that the spacer domain [127, 128], specifically residues T572-N579 and V657-G666 [129], comprise a common antigenic core region that is crucial for interaction with ADAMTS13 antibodies of TTP (Fig. 4b). The proteolytic activity of ADAMTS13 variants truncated upstream of the cysteine domain is not suppressible by the inhibitors of patients with TTP. Such non-suppressible ADAMTS13 products may be used to overcome the difficulty encountered in the management of patients with high inhibitor levels.
The levels of the ADAMTS13 inhibitors tend to be low (<10 U/mL) [26, 130], often receding to even lower or undetectable levels within weeks or months. Such characteristics of the ADAMTS13 inhibitors suggest that the immune response is induced by exposure to exogenous antigens with molecular mimicry to ADAMTS13.
The low level and self-limited course of ADAMTS13 inhibitors are critical for the success of plasma therapy in TTP. Persistently high levels of ADAMTS13 inhibitors may lead to treatment failure and death [131, 132], while persistent ADAMTS13 inhibitors may lead to refractory TTP requiring long-term plasma therapy [133]. A rising ADAMTS13 inhibitor level may be associated with switching of the IgG subclasses, suggesting cytokine dysregulation may underlie the escalating inhibitor levels observed in some of the TTP cases [132].
4.4.3 Other causes of ADAMTS13 deficiency
ADAMTS13 has been reported to be decreased in a variety of pathological conditions, including sepsis, disseminated intravascular coagulopathy, liver diseases, and plasmodium falciparum infection [134–140]. In most cases, the decrease of ADAMTS13 level is unlikely to contribute to thrombosis. Some of the observed decreases are controversial [141, 142]. ADAMTS13 is quite stable in normal plasma but may quickly deteriorate to very low levels in the plasma samples of patients with pathological conditions. Loss of ADAMTS13 activity in vitro may contribute to the profound decreases described in some studies. The pathophysiological significance of decreased ADAMTS13 levels in patients without autoimmune inhibitors or mutations remains uncertain.
4.5 VWF multimers in TTP
Abnormalities in VWF multimers are common in TTP [143]. The VWF multimer size often changes rapidly during acute TTP, reflecting the evolving balance between decreased VWF proteolysis, which increases VWF size, and increased VWF–platelet binding, which preferentially consumes the large multimers. In a typical case of TTP, large VWF multimers are often depleted at the time of initial acute presentation. The severity of depletion is affected by the severity and duration of thrombocytopenia. When the patient is placed on plasma exchange therapy, rising ADAMTS13 levels suppress VWF–platelet aggregation, leading to an increase in the platelet count and the appearance of ultra large VWF multimers. Ultra large multimers may not be detected if the ADAMTS13 rapidly increased to >20–30% of normal. Such serial changes suggest that ultra large VWF multimers are not the cause of thrombosis; they merely reflect inadequate VWF proteolysis in patients with low ADAMTS13 levels.
4.6 Modifiers of phenotypic expression in patients with ADAMTS13 deficiency
Variation in the severity of thrombotic phenotype is common in patients with either hereditary or acquired ADAMTS13 deficiency. In theory, the thrombotic propensity may be affected by four different processes: (1) plasma ADAMTS13 activity level, (2) endothelial release of VWF, (3) shear stress in the microcirculation, (4) platelet reactivity, and (5) modifiers of VWF response to shear stress. However, how each of these factors operates in individual patients remains poorly understood.
The ADAMTS13 assays generally have a sensitivity of 5–10%. The use of sensitive ADAMTS13 assays [144–146] may provide a more precise depiction of how plasma ADAMTS13 level evolves in patients with ADAMTS13 mutations or inhibitors. However, because of multiple modifiers of VWF–platelet interactions, it is unlikely that there will be a fixed threshold level of ADAMTS13 for causing platelet aggregation.
Both platelets and factor VIII have been shown in in vitro studies to promote the cleavage of VWF by ADAMTS13 [147, 148]. However, there is no evidence that VWF multimer size is shifted to larger forms in patients with severe hemophilia or thrombocytopenia. The roles of platelets or factor VIII in regulating VWF cleavage by ADAMTS13 remain uncertain.
In mouse ex vivo vascular models, thrombospodin-1 has been reported to retard the cleavage of VWF strands on endothelial cells [97]. However, inactivation of the thrombospondin 1 gene does not lead to increased proteolysis of VWF in mice. Whether thrombospondin 1 affects the cleavage of VWF in flowing blood remains unknown.
Overall, much remains to be learned on how the various potential modifiers affect the phenotype of ADAMTS13 deficiency. Advances in understanding the modifiers of TTP will improve the management of patients with TTP.
5 Clinical implications and controversies
5.1 ADAMTA13 analysis in the diagnosis and management of TTP
Many patients of TTP present with the triad of thrombocytopenia, microangiopathic hemolysis and neurological abnormalities. Some of them may also have fever and renal abnormalities (pentad). However, it is critical to note that neither the triad nor the pentad of presentation can be relied upon for diagnosis of TTP. In practice, a constellation of thrombocytopenia and microangiopathic hemolysis should always raise the suspicion of TTP. On the other hand, it is increasingly recognized that a patient of TTP may be asymptomatic, has isolated thrombocytopenia requiring distinction from idiopathic thrombocytopenic purpura, or even presents with a transient ischemic attack (TIA) or stroke without hematological changes.
5.1.1 ADAMTS13 analysis for diagnosis of TTP
The plasma ADAMTS13 activity level is less than 10% (or 5%, depending on the assays) in acute TTP. The deficiency is caused by inhibitors of ADAMTS13 or mutations of the ADAMTS13 gene. An ADAMTS13 level >10% excludes the diagnosis of TTP in a patient presenting with acute thrombocytopenia. For patients who are receiving plasma therapy for suspected TTP, a plasma ADAMTS13 activity greater than 10% excludes the diagnosis of TTP only if the platelet count is stable or decreasing at the time of the assay; it cannot be used to exclude the diagnosis of TTP if the platelet count is rising or normal.
During remission of TTP, measurements of ADAMTS13 levels may also be quite informative: an ADAMTS13 level below 5% (or 10%) is associated with high risk of relapse in the near future [149].
Inhibitors of ADAMTS13 are detectable in approximately 80–90% of patients with severe ADAMTS13 deficiency at the time of acute presentation. In an acquired TTP patient with no detectable inhibitors by plasma mixing studies, IgG isolated from the patient’s plasma will often exhibit inhibitory activity against ADAMTS13. Alternatively, the presence of ADAMTS13 inhibitors may be inferred from persistently low ADAMTS13 levels after the patient receives plasma exchange therapy.
Inhibitors are not detectable in patients with hereditary TTP. As mentioned above, because of in vitro inactivation, ADAMTS13 values may be quite low in a variety of pathological conditions without concurrent inhibitors of ADAMTS13.
Different designs have been developed for the analysis of ADAMTS13 activity. The assays are affected differentially by a variety of conditions, such plasma bilirubin [150] or hemoglobin [40]. Assays using overnight incubations tend to yield falsely low ADAMTS13 levels in pathological samples. High plasma VWF levels yield falsely low ADAMTS13 levels for assays that do not correct for the plasma VWF concentrations. In some cases, the factors affecting the assays are not known [35]. Interpretation of ADAMTS13 assay results requires correlation with the patient’s clinical status. When in doubt, the measurement should be repeated, preferably using a different assay.
5.1.2 Distinction of TTP from other types of thrombotic microangiopathy
A clinical distinction between TTP and atypical HUS or other microangiopathic disorders is difficult to make without ADAMTS13 assays. ADAMTS13 assays are now available for most clinical practices. In contrast, the tests for the diagnosis of atypical HUS are performed primarily in research laboratories. Furthermore, the molecular defects of atypical HUS remain unknown in many cases. Therefore, for patients presenting with thrombocytopenia and microangiopathic hemolysis, ADAMTS13 analysis is currently the most informative diagnostic test.
5.1.3 TTP without ADAMTS13 deficiency?
It has been claimed in some clinical series that approximately one-third or more of the patients with the diagnosis of TTP do not have severe ADAMTS13 deficiency. In those studies, TTP is diagnosed on the basis of thrombocytopenia and microangiopathic hemolysis. This definition of TTP will include patients with complement dysregulation or other molecular mechanisms of thrombotic microangiopathy. Disease classification based on clinical features are notoriously unreliable. Some patients with complement regulation defects were unnecessarily subjected to rituximab, azathioprine or even splenectomy because they were incorrectly given the diagnosis of TTP.
5.2 TTP presenting as TIA or stroke
TIA or stroke is a well-recognized complication of TTP. However, it was not known until recently that TTP may present with TIA or stroke without concurrent thrombocytopenia or microangiopathic hemolysis [151, 152]. It is likely that ADAMTS13 deficiency accounts for a small fraction of the cases presenting with TIA or stroke without concurrent thrombocytopenia or microangiopathic hemolysis. Nevertheless, since such patients respond to plasma therapy or rituximab but not to anticoagulation therapy, it is important to include ADAMTS13 deficiency in the differential diagnosis of TIA/stroke.
5.3 Distinction between the disease and phenotype of TTP
The absence of thrombocytopenia or microangiopathic hemolysis in some patients with severe ADAMTS13 deficiency has led some investigators to question whether ADAMTS13 deficiency per se is sufficient to cause TTP. This is analogous to questioning the diagnosis of sickle cell disease in a patient with homozygous hemoglobin S but no vaso-occlusive crisis.
5.3.1 Sub-clinical TTP
It refers to patients who have profound ADAMTS13 deficiency (plasma levels <5% or 10% depending on the assays) but no clinical manifestations of thrombocytopenia, microangiopathic hemolysis, neurologic deficits, or other thrombotic complications. The deficiency may be due to mutations of the ADAMTS13 gene, or persistent autoantibodies of the ADAMTS13 protease.
In some patients, the ADAMTS13 may be below the detection limit of the assay but above the threshold level of platelet aggregation. In others, there may be subclinical platelet aggregation which is masked by bone marrow compensation.
Both types of patients have extremely high risk of thrombotic complications when they are exposed to various physiological or pathological conditions such as pregnancy, surgery, trauma, fever, or infection. Prophylactic measures to raise the ADAMTS13 level is critical for preventing thrombotic complications in such cases.
Some cases of severe ADAMTS13 deficiency remain asymptomatic, but develop chronic renal failure, presumably due to repeated renal insult by subclinical microthrombi. Urinalysis may uncover the presence of microhematuria or proteinuria before the onset of chronic renal failure. A trial of plasma therapy is warranted for such cases to determine whether it improves or stabilize the renal abnormalities.
Occasionally, after many years of an uneventful course, a patient with severe ADAMTS13 deficiency may present with acute thrombotic complications such as strokes without obvious precipitating events. For hereditary cases that have experienced unprovoked thrombotic complications of TTP, prophylactic plasma therapy should be continued to prevent recurrence of thrombosis. Immunosuppressive therapy should be considered for acquired TTP patients to decrease the autoimmune response and raise the ADAMTS13 level. Further studies are needed to delineate the indications of primary long-term prophylaxis for asymptomatic ADAMTS13 deficient patients before they experience thrombotic complications.
5.4 Clinical improvement upon plasma therapy is not specific for the diagnosis of TTP
It is important to recognize that improvement upon plasma therapy is common in patients with complement regulation defects due to deficiency in factor H or factor I. Spontaneous recovery may also occur [153, 154]. Because the laboratory tests for complement regulation defects are not generally available in clinical practice, such patients are included in TTP series that do not exclude cases without severe ADAMTS13 deficiency.
Patients with factor H or I deficiency often require maintenance plasma therapy to prevent continuing deterioration of the renal function, which may occur without hematological relapses. Therefore, such patients may develop chronic renal failure if they are treated only during periods of hematological exacerbation.
Plasma therapy is not expected to be effective for patients with hyperactive factor B mutations or defects in membrane bound proteins such as membrane cofactor protein (CD46) or thrombomodulin. Nevertheless, spontaneous recovery is not uncommon for these atypical HUS patients. Therefore, improvement upon plasma therapy is not a basis for establishing the diagnosis of TTP.
5.5 Immunosuppressive therapy of TTP
Plasma exchange is the mainstay treatment of TTP. However, plasma therapy does not address the autoimmune cause of the disease. Most patients eventually achieve clinical remission because the ADAMTS13 inhibitor levels decline over time. Nevertheless, the immune response does not disappear completely. Fluctuation of the autoimmune response to ADAMTS13 is the reason that relapses of TTP occur in the majority of patients. Splenectomy, vincristine, cyclophosphamide, and azathioprine have been used with variable efficacy in TTP. More recently, rituximab and cyclosporine A have been used to treat the disease [151, 155].
The efficacy of rituximab has been described in case series, mostly of patients with refractory TTP due to persistently low ADAMTS13 levels. In a recent review of 73 cases treated with rituximab, the complete hematological remission rate is 95%, achieved at a median of 15 (range 5–75) days [156]. The median duration of relapse free survival is 10 months (range 1–49 months). Infectious complications developed in 4% of the cases. Because of reporting bias, the published data may not accurately reflect the efficacy of rituximab for TTP.
5.6 Does pregnancy increases the risk of TTP?
During pregnancy, the syndrome of microangiopathic hemolysis and thrombocytopenia may result from preeclampsia, disseminated intravascular coagulopathy, hemolysis with elevated liver enzyme and low platelet (HELLP) syndrome, or atypical HUS, with or without evidence of complement dysregulation. These disorders have different pathophysiology and require different therapeutic considerations.
Occurrence of TTP affecting pregnant women raises the speculation that pregnancy is associated with higher risk of TTP [157]. However, the evidence in support of this association remains inconclusive. In the Oklahoma Registry database, 14 (7%) of the 202 cases of TTP/HUS were pregnant. Nevertheless, only two of the 13 cases with ADAMTS13 measurements had severe ADAMTS13 deficiency. Thus, most of the pregnant cases had other types of thrombotic microangiopathy, and an association between pregnancy and TTP cannot be established based on that series. Since TTP is more likely to affect women of reproductive ages, TTP and pregnancy may occur coincidentally. On the other hand, pregnancy may cause acute exacerbations in patients with hereditary or acquired TTP.
5.6.1 ADAMTS13 activity during pregnancy
Serial measurement of ADAMTS13 activity levels has shown the normal pregnancy is accompanied by progressive decrease of ADAMTS13 during gestation. The ADAMTS13 activity level is decreased to a mean of 52% (range 22–89%) of normal plasma by the end of pregnancy [158]. In pregnancies complicated with HELLP syndrome, the ADAMTS13 activity is further decreased to 31% (12–43%) [159]. Thus, in patients with TTP, pregnancy may precipitate exacerbation of thrombotic complications by decreasing the ADAMTS13 activity level below the threshold necessary for preventing VWF–platelet thrombosis. Speculatively, other hemostatic changes such as increased VWF activity and platelet reactivity may also contribute to the exacerbation of TTP during pregnancy.
5.6.2 Hereditary TTP and pregnancy
Pregnancy often leads to exacerbation of thrombotic complications in patients with hereditary TTP. Some women of hereditary TTP are asymptomatic or only mildly thrombocytopenic but develop the full-blown manifestation of TTP for the first time during their pregnancies. Untreated, hereditary TTP is associated with a high risk of serious maternal and fetal complications [57]. Nevertheless, congenital TTP is not a contraindication for pregnancy. With meticulous plasma infusion therapy to prevent maternal thrombocytopenia and thrombosis, successful pregnancy outcome is expected.
5.6.3 Acquired TTP and pregnancy
Pregnancy counseling for women with a history of acquired TTP continues to be a challenge. The ADAMTS13 level remains decreased in many patients of acquired TTP during remission. A further decrease of ADAMTS13 during pregnancy may be sufficient to precipitate exacerbation of TTP. However, the ADAMTS13 activity level tends to fluctuate in patients with acquired TTP, and the impact of pregnancy on the inhibitor titers is unknown. Thus, it is difficult to predict the risk of relapse in individual cases. Until more definitive data become available, pregnancy should be provisionally considered to have a high risk of TTP relapse if the patient’s baseline ADAMTS13 levels are less than 10–30% of normal control.
6 Future directions
Following the esoteric discovery that normal plasma contains a VWF cleaving protease, the past decade has witnessed the most exciting advances in the history of TTP. However, many challenges lie ahead. Development of sensitive and reliable ADAMTS13 assays with rapid turn-around time will greatly improve the management of TTP. The courses of both hereditary and acquired TTP and the nature of modifiers of TTP phenotype in patients with the disease require further delineation. Systemic studies are needed to define the role of immunosuppressive therapies in acquired TTP. The development of recombinant ADAMTS13 proteins may obviate the use of fresh frozen plasma. Bioengineered ADAMTS13 that is non-suppressible by TTP antibodies may circumvent the difficulties that recombinant ADAMTS13 replacement therapies may encounter in patients with acquired TTP. Basic research to elucidate the interaction between VWF and ADAMTS13 and the molecular mechanisms of ADAMTS13 inhibitors may provide new directions toward improving the diagnosis and management of TTP.
References
Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–79.
Tsai HM, Chandler WL, Sarode R, et al. von Willebrand factor and von Willebrand factor-cleaving metalloprotease activity in Escherichia coli O157:H7-associated hemolytic uremic syndrome. Pediatr Res. 2001;49:653–9.
Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127:834–9.
Sadler JE. New concepts in von Willebrand disease. Annu Rev Med. 2005;56:173–91.
Tsai HM, Nagel RL, Hatcher VB, Sussman II. Multimeric composition of endothelial cell-derived von Willebrand factor. Blood. 1989;73:2074–6.
Wagner DD, Lawrence SO, Ohlsson-Wilhelm BM, Fay PJ, Marder VJ. Topology and order of formation of interchain disulfide bonds in von Willebrand factor. Blood. 1987;69:27–32.
Handin RI, Wagner DD. Molecular and cellular biology of von Willebrand factor. Prog Hemost Thromb. 1989;9:233–59.
Tsai HM, Sussman II, Nagel RL. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood. 1994;83:2171–9.
Furlan M, Robles R, Lamie B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87:4223–34.
Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–44.
Furlan M, Robles R, Solenthaler M, et al. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–103.
Tsai HM, Sussman II, Ginsburg D, et al. Proteolytic cleavage of recombinant type 2A von Willebrand factor mutants R834W and R834Q: inhibition by doxycycline and by monoclonal antibody VP-1. Blood. 1997;89:1954–62.
Furlan M, Robles R, Solenthaler M, Lammle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91:2839–46.
Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–94.
Furlan M, Robles R, Galbusera M, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339:1578–84.
Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood. 2001;98:1662–6.
Gerritsen HE, Robles R, Lammle B, Furlan M. Partial amino acid sequence of purified von Willebrand factor-cleaving protease. Blood. 2001;98:1654–61.
Soejima K, Mimura N, Hirashima M, et al. A novel human metalloprotease synthesized in the liver and secreted into the blood: possibly, the von Willebrand factor-cleaving protease? J Biochem (Tokyo). 2001;130:475–80.
Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–94.
Rick ME, Moll S, Taylor MA, et al. Clinical use of a rapid collagen binding assay for von Willebrand factor cleaving protease in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2002;88:598–604.
Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98:1765–72.
Remuzzi G, Galbusera M, Noris M, et al. von Willebrand factor cleaving protease (ADAMTS13) is deficient in recurrent and familial thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Blood. 2002;100:778–85.
Bohm M, Vigh T, Scharrer I. Evaluation and clinical application of a new method for measuring activity of von Willebrand factor-cleaving metalloprotease (ADAMTS13). Ann Hematol. 2002;81:430–5.
Vesely SK, George JN, Lammle B, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood. 2003;102:60–8.
Coppo P, Bengoufa D, Veyradier A, et al. Severe ADAMTS13 deficiency in adult idiopathic thrombotic microangiopathies defines a subset of patients characterized by various autoimmune manifestations, lower platelet count, and mild renal involvement. Medicine (Baltimore). 2004;83:233–44.
Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103:4043–9.
Studt JD, Kremer Hovinga JA, Alberio L, Bianchi V, Lammle B. Von Willebrand factor-cleaving protease (ADAMTS-13) activity in thrombotic microangiopathies: diagnostic experience 2001/2002 of a single research laboratory. Swiss Med Wkly. 2003;133:325–32.
Peyvandi F, Ferrari S, Lavoretano S, Canciani MT, Mannucci PM. von Willebrand factor cleaving protease (ADAMTS-13) and ADAMTS-13 neutralizing autoantibodies in 100 patients with thrombotic thrombocytopenic purpura. Br J Haematol. 2004;127:433–9.
Kokame K, Matsumoto M, Soejima K, et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci USA. 2002;99:11902–7.
Kentouche K, Budde U, Furlan M, et al. Remission of thrombotic thrombocytopenic purpura in a patient with compound heterozygous deficiency of von Willebrand factor-cleaving protease by infusion of solvent/detergent plasma. Acta Paediatr. 2002;91:1056–9.
Bestetti G, Stellari A, Lattuada A, et al. ADAMTS 13 genotype and vWF protease activity in an Italian family with TTP. Thromb Haemost. 2003;90:955–6.
Assink K, Schiphorst R, Allford S, et al. Mutation analysis and clinical implications of von Willebrand factor-cleaving protease deficiency. Kidney Int. 2003;63:1995–9.
Antoine G, Zimmermann K, Plaimauer B, et al. ADAMTS13 gene defects in two brothers with constitutional thrombotic thrombocytopenic purpura and normalization of von Willebrand factor-cleaving protease activity by recombinant human ADAMTS13. Br J Haematol. 2003;120:821–4.
Schneppenheim R, Budde U, Oyen F, et al. von Willebrand factor cleaving protease and ADAMTS13 mutations in childhood TTP. Blood. 2003;101:1845–50.
Savasan S, Lee SK, Ginsburg D, Tsai HM. ADAMTS13 gene mutation in congenital thrombotic thrombocytopenic purpura with previously reported normal VWF cleaving protease activity. Blood. 2003;101:4449–51.
Matsumoto M, Kokame K, Soejima K, et al. Molecular characterization of ADAMTS13 gene mutations in Japanese patients with Upshaw-Schulman syndrome. Blood. 2004;103:1305–10.
Pimanda JE, Maekawa A, Wind T, et al. Congenital thrombotic thrombocytopenic purpura in association with a mutation in the second CUB domain of ADAMTS13. Blood. 2004;103:627–9.
Uchida T, Wada H, Mizutani M, et al. Identification of novel mutations in ADAMTS13 in an adult patient with congenital thrombotic thrombocytopenic purpura. Blood. 2004;104:2081–3.
Veyradier A, Lavergne JM, Ribba AS, et al. Ten candidate ADAMTS13 mutations in six French families with congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome). J Thromb Haemost. 2004;2:424–9.
Studt JD, Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105:542–4.
Licht C, Stapenhorst L, Simon T, et al. Two novel ADAMTS13 gene mutations in thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome (TTP/HUS). Kidney Int. 2004;66:955–8.
Snider CE, Moore JC, Warkentin TE, et al. Dissociation between the level of von Willebrand factor-cleaving protease activity and disease in a patient with congenital thrombotic thrombocytopenic purpura. Am J Hematol. 2004;77:387–90.
Camilleri RS, Cohen H, MacKie IJ, et al. Prevalence of the ADAMTS-13 missense mutation R1060 W in late onset adult thrombotic thrombocytopenic purpura. J Thromb Haemost. 2008;6:331–8.
Noris M, Bucchioni S, Galbusera M, et al. Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol. 2005;16:1177–83.
Liu F, Jin J, Dong NZ, Wang YG, Ruan CG. Identification of two novel mutations in ADAMTS13 gene in a patient with hereditary thrombotic thrombocytopenic purpura. Zhonghua Xue.Ye.Xue.Za Zhi. 2005;26:521–4.
Donadelli R, Banterla F, Galbusera M, et al. In vitro and in vivo consequences of mutations in the von Willebrand factor cleaving protease ADAMTS13 in thrombotic thrombocytopenic purpura. Thromb Haemost. 2006;96:454–64.
Peyvandi F, Lavoretano S, Palla R, et al. Mechanisms of the interaction between two ADAMTS13 gene mutations leading to severe deficiency of enzymatic activity. Hum Mutat. 2006;27:330–6.
Plaimauer B, Fuhrmann J, Mohr G, et al. Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and a missense mutation. Blood. 2006;107:118–25.
Schneppenheim R, Kremer Hovinga JA, Becker T, et al. A common origin of the 4143insA ADAMTS13 mutation. Thromb Haemost. 2006;96:3–6.
Shibagaki Y, Matsumoto M, Kokame K, et al. Novel compound heterozygote mutations (H234Q/R1206X) of the ADAMTS13 gene in an adult patient with Upshaw-Schulman syndrome showing predominant episodes of repeated acute renal failure. Nephrol Dial Transplant. 2006;21:1289–92.
Tao Z, Anthony K, Peng Y, et al. Novel ADAMTS-13 mutations in an adult with delayed onset thrombotic thrombocytopenic purpura. J Thromb Haemost. 2006;4:1931–5.
Hommais A, Rayes J, Houllier A, et al. Molecular characterization of four ADAMTS13 mutations responsible for congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome). Thromb Haemost. 2007;98:593–9.
Meyer SC, Jeddi R, Meddeb B, et al. A first case of congenital TTP on the African continent due to a new homozygous mutation in the catalytic domain of ADAMTS13. Ann Hematol. 2008;87:663–6.
Palla R, Lavoretano S, Lombardi R, et al. The first deletion mutation in the TSP1-6 repeat domain of ADAMTS13 in a family with inherited thrombotic thrombocytopenic purpura. Haematologica. 2009;94:289–93.
Kokame K, Aoyama Y, Matsumoto M, Fujimura Y, Miyata T. Inherited and de novo mutations of ADAMTS13 in a patient with Upshaw-Schulman syndrome. J Thromb Haemost. 2008;6:213–5.
Garagiola I, Valsecchi C, Lavoretano S, et al. Nonsense-mediated mRNA decay in the ADAMTS13 gene caused by a 29-nucleotide deletion. Haematologica. 2008;93:1678–85.
Fujimura Y, Matsumoto M, Kokame K, et al. Pregnancy-induced thrombocytopenia and TTP, and the risk of fetal death, in Upshaw-Schulman syndrome: a series of 15 pregnancies in 9 genotyped patients. Br J Haematol. 2009;144:742–54.
Zhou W, Inada M, Lee TP, et al. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005;85:780–8.
Uemura M, Tatsumi K, Matsumoto M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106:922–4.
Manea M, Tati R, Karlsson J, Bekassy ZD, Karpman D. Biologically active ADAMTS13 is expressed in renal tubular epithelial cells. Pediatr. Nephrol. 2009 [Epub ahead of print].
Manea M, Kristoffersson A, Schneppenheim R, et al. Podocytes express ADAMTS13 in normal renal cortex and in patients with thrombotic thrombocytopenic purpura. Br J Haematol. 2007;138:651–62.
Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS-13. J Thromb Haemost. 2006;4:1396–404.
Shang D, Zheng XW, Niiya M, Zheng XL. Apical sorting of ADAMTS13 in vascular endothelial cells and Madin-Darby canine kidney cells depends on the CUB domains and their association with lipid rafts. Blood. 2006;108:2207–15.
Suzuki M, Murata M, Matsubara Y, et al. Detection of von Willebrand factor-cleaving protease (ADAMTS-13) in human platelets. Biochem Biophys Res Commun. 2004;313:212–6.
Liu L, Choi H, Bernardo A, et al. Platelet-derived VWF-cleaving metalloprotease ADAMTS-13. J Thromb Haemost. 2005;3:2536–44.
Crawley JT, Lam JK, Rance JB, et al. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. Blood. 2005;105:1085–93.
Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost. 2008;6:1233–5.
Ricketts LM, Dlugosz M, Luther KB, Haltiwanger RS, Majerus EM. O-fucosylation is required for ADAMTS13 secretion. J. Biol. Chem. 2007;282:17014–23.
Zhou W, Tsai HM. N-Glycans of ADAMTS13 modulate its secretion and von Willebrand factor cleaving activity. Blood. 2009;113:929–35.
McKinnon TA, Chion AC, Millington AJ, Lane DA, Laffan MA. N-linked glycosylation of VWF modulates its interaction with ADAMTS13. Blood. 2008;111:3042–9.
Zhou W, Bouhassira EE, Tsai HM. An IAP retrotransposon in the mouse ADAMTS13 gene creates ADAMTS13 variant proteins that are less effective in cleaving von Willebrand factor multimers. Blood. 2007;110:886–93.
Tao Z, Peng Y, Nolasco L, et al. Recombinant CUB-1 domain polypeptide inhibits the cleavage of ULVWF strings by ADAMTS13 under flow conditions. Blood. 2005;106:4139–45.
Niiya M, Endo M, Shang D, et al. Correction of ADAMTS13 deficiency by in utero gene transfer of lentiviral vector encoding ADAMTS13 genes. Mol Ther. 2009;17:34–41.
Zanardelli S, Chion AC, Groot E, et al. A novel binding site for ADAMTS13 constitutively exposed on the surface of globular VWF. Blood. 2009.
Banno F, Kaminaka K, Soejima K, Kokame K, Miyata T. Identification of strain-specific variants of mouse Adamts13 gene encoding von Willebrand factor-cleaving protease. J Biol. Chem. 2004;279:30896–903.
Banno F, Chauhan AK, Kokame K, et al. The distal carboxyl-terminal domains of ADAMTS13 are required for regulation of in vivo thrombus formation. Blood. 2009;113:5323–9.
Gao W, Anderson PJ, Majerus EM, Tuley EA, Sadler JE. Exosite interactions contribute to tension-induced cleavage of von Willebrand factor by the antithrombotic ADAMTS13 metalloprotease. Proc Natl Acad Sci USA. 2006;103:19099–104.
Soejima K, Matsumoto M, Kokame K, et al. ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood. 2003;102:3232–7.
Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278:30136–41.
Wu JJ, Fujikawa K, McMullen BA, Chung DW. Characterization of a core binding site for ADAMTS-13 in the A2 domain of von Willebrand factor. Proc Natl Acad Sci USA. 2006;103:18470–4.
de Groot R, Bardhan A, Ramroop N, Lane DA, Crawley JT. Essential role of the disintegrin-like domain in ADAMTS13 function. Blood. 2009;113:5609–16.
Kokame K, Matsumoto M, Fujimura Y, Miyata T. VWF73, a region from D1596 to R1668 of von Willebrand factor, provides a minimal substrate for ADAMTS-13. Blood. 2004;103:607–12.
Zhang Q, Zhou YF, Zhang CZ, et al. Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc Natl Acad Sci USA. 2009;106:9226–31.
Phillips MD, Vu C, Nolasco L, Moake JL. Granulocyte proteases do not process endothelial cell-derived unusually large von Willebrand factor multimers to plasma VWF in vivo. Am J Hematol. 1991;37:80–3.
Siedlecki CA, Lestini BJ, Kottke-Marchant KK, et al. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 1996;88:2939–50.
Reininger AJ, Heijnen HF, Schumann H, et al. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood. 2006;107:3537–45.
Schneider SW, Nuschele S, Wixforth A, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci USA. 2007;104:7899–903.
Singh I, Themistou E, Porcar L, Neelamegham S. Fluid shear induces conformation change in human blood protein von Willebrand factor in solution. Biophys J. 2009;96:2313–20.
Singh I, Shankaran H, Beauharnois ME, et al. Solution structure of human von Willebrand factor studied using small angle neutron scattering. J. Biol. Chem. 2006;281:38266–75.
Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330–4.
Schneppenheim R, Budde U, Obser T, et al. Expression and characterization of von Willebrand factor dimerization defects in different types of von Willebrand disease. Blood. 2001;97:2059–66.
Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–9.
Turner N, Nolasco L, Dong JF, Moake J. ADAMTS-13 cleaves long von Willebrand factor multimeric strings anchored to endothelial cells in the absence of flow, platelets or conformation-altering chemicals. J Thromb Haemost. 2009;7:229–32.
Vincentelli A, Susen S, Le TT, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349:343–9.
Veyradier A, Nishikubo T, Humbert M, et al. Improvement of von Willebrand factor proteolysis after prostacyclin infusion in severe pulmonary arterial hypertension. Circulation. 2000;102:2460–2.
Donadelli R, Orje JN, Capoferri C, Remuzzi G, Ruggeri ZM. Size regulation of von Willebrand factor-mediated platelet thrombi by ADAMTS13 in flowing blood. Blood. 2006;107:1943–50.
Bonnefoy A, Daenens K, Feys HB, et al. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. 2006;107:955–64.
Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115:2752–61.
Banno F, Kokame K, Okuda T, et al. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood. 2006;107:3161–6.
Nolasco LH, Turner NA, Bernardo A, et al. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106:4199–209.
Romani DW, Fijnheer R, Brinkman HJ, et al. Endothelial cell activation in thrombotic thrombocytopenic purpura (TTP): a prospective analysis. Br J Haematol. 2003;123:522–7.
Hunt BJ, Lammle B, Nevard CH, Haycock GB, Furlan M. von Willebrand factor-cleaving protease in childhood diarrhoea-associated haemolytic uraemic syndrome. Thromb Haemost. 2001;85:975–8.
Kwaan HC. The role of fibrinolysis in disease processes. Semin Thromb Hemost. 1984;10:71–9.
Tsai AL, Manner CE, Rudersdorf T, Wu KK. Quantitation of serum prostacyclin-binding in thrombotic thrombocytopenic purpura. Thromb Res. 1988;51:583–92.
Wada H, Kaneko T, Ohiwa M, et al. Increased levels of vascular endothelial cell markers in thrombotic thrombocytopenic purpura. Am J Hematol. 1993;44:101–5.
Burns ER, Zucker-Franklin D. Pathologic effects of plasma from patients with thrombotic thrombocytopenic purpura on platelets and cultured vascular endothelial cells. Blood. 1982;60:1030–7.
Leung DY, Moake JL, Havens PL, Kim M, Pober JS. Lytic anti-endothelial cell antibodies in haemolytic-uraemic syndrome. Lancet. 1988;2:183–6.
Praprotnik S, Blank M, Levy Y, et al. Anti-endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int Immunol. 2001;13:203–10.
Neame PB, Hirsh J. Circulating immune complexes in thrombotic thrombocytopenic purpura (TTP). Blood. 1978;51:559–60.
Siddiqui FA, Lian EC. Characterization of platelet agglutinating protein p37 purified from the plasma of a patient with thrombotic thrombocytopenic purpura. Biochem Mol Biol Int. 1993;30:385–95.
Tandon NN, Rock G, Jamieson GA. Anti-CD36 antibodies in thrombotic thrombocytopenic purpura. Br J Haematol. 1994;88:816–25.
Kelton JG, Moore JC, Murphy WG. The platelet aggregating factor(s) of thrombotic thrombocytopenic purpura. Prog Clin Biol Res. 1990;337:141–9.
Mauro M, Kim J, Costello C, Laurence J. Role of transforming growth factor beta1 in microvascular endothelial cell apoptosis associated with thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome. Am J Hematol. 2001;66:12–22.
Joseph G, Smith KJ, Hadley TJ, et al. HLA-DR53 protects against thrombotic thrombocytopenic purpura/adult hemolytic uremic syndrome. Am J Hematol. 1994;47:189–93.
Zeigler Z, Kelton J, Moore J, et al. Calpain activity in bone marrow transplant-associated thrombotic thrombocytopenic purpura. Bone Marrow Transplant. 1999;24:641–5.
Rock G, Chauhan K, Jamieson GA, Tandon NN. Anti-CD36 antibodies in patients with lupus anticoagulant and thrombotic complications. Br J Haematol. 1994;88:878–80.
Schultz DR, Arnold PI, Jy W, et al. Anti-CD36 autoantibodies in thrombotic thrombocytopenic purpura and other thrombotic disorders: identification of an 85 kD form of CD36 as a target antigen. Br J Haematol. 1998;103:849–57.
Rock G, Clark W, Sternbach M, Kolajova M, McLaine P. Haemolytic uraemic syndrome is an immune-mediated disease: role of anti-CD36 antibodies. Br J Haematol. 2005;131:247–52.
Hori Y, Wada H, Mori Y, et al. Plasma sFas and sFas ligand levels in patients with thrombotic thrombocytopenic purpura and in those with disseminated intravascular coagulation. Am J Hematol. 1999;61:21–5.
Manea M, Kristoffersson A, Tsai HM, et al. ADAMTS13 phenotype in plasma from normal individuals and patients with thrombotic thrombocytopenic purpura. Eur J Pediatr. 2007;166:249–57.
Schneppenheim R, Kremer Hovinga JA, Becker T, et al. A common origin of the 4143insA ADAMTS13 mutation. Thromb Haemost. 2006;96:3–6.
Jang MJ, Kim NK, Chong SY, et al. Frequency of Pro475Ser polymorphism of ADAMTS13 gene and its association with ADAMTS-13 activity in the Korean population. Yonsei Med J. 2008;49:405–8.
Akiyama M, Kokame K, Miyata T. ADAMTS13 P475S polymorphism causes a lowered enzymatic activity and urea lability in vitro. J Thromb Haemost. 2008;6:1830–2.
Tsai HM, Rice L, Sarode R, Chow TW, Moake JL. Antibody inhibitors to von Willebrand factor metalloproteinase and increased binding of von Willebrand factor to platelets in ticlopidine-associated thrombotic thrombocytopenic purpura. Ann Intern Med. 2000;132:794–9.
Sugio Y, Okamura T, Shimoda K, et al. Ticlopidine-Associated thrombotic thrombocytopenic purpura with an IgG-type inhibitor to von Willebrand factor-cleaving protease activity. Int J Hematol. 2001;74:347–51.
Terrell DR, Williams LA, Vesely SK, et al. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost. 2005;3:1432–6.
Zhou W, Dong L, Ginsburg D, Bouhassira EE, Tsai HM. Enzymatically active ADAMTS13 variants are not inhibited by anti-ADAMTS13 autoantibodies: a novel therapeutic strategy? J Biol. Chem. 2005;280:39934–41.
Luken BM, Turenhout EA, Hulstein JJ, et al. The spacer domain of ADAMTS13 contains a major binding site for antibodies in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2005;93:267–74.
Luken BM, Turenhout EA, Kaijen PH, et al. Amino acid regions 572–579 and 657–666 of the spacer domain of ADAMTS13 provide a common antigenic core required for binding of antibodies in patients with acquired TTP. Thromb Haemost. 2006;96:295–301.
Tsai HM, Li A, Rock G. Inhibitors of von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura. Clin. Lab. 2001;47:387–92.
Tsai HM. High titers of inhibitors of von Willebrand factor-cleaving metalloproteinase in a fatal case of acute thrombotic thrombocytopenic purpura. Am J Hematol. 2000;65:251–5.
Dong L, Chandrasekaran V, Zhou W, Tsai HM. Evolution of ADAMTS13 antibodies in a fatal case of thrombotic thrombocytopenic purpura. Am J Hematol. 2008;83:815–7.
Yomtovian R, Niklinski W, Silver B, Sarode R, Tsai HM. Rituximab for chronic recurring thrombotic thrombocytopenic purpura: a case report and review of the literature. Br J Haematol. 2004;124:787–95.
Mannucci PM, Canciani MT, Forza I, et al. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2002;98:2730–5.
Park YD, Yoshioka A, Kawa K, et al. Impaired activity of plasma von Willebrand factor-cleaving protease may predict the occurrence of hepatic veno-occlusive disease after stem cell transplantation. Bone Marrow Transplant. 2002;29:789–94.
Uemura M, Matsuyama T, Ishikawa M, et al. Decreased activity of plasma ADAMTS13 may contribute to the development of liver disturbance and multiorgan failure in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2005;29:264S–71S.
Ono T, Mimuro J, Madoiwa S, et al. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006;107:528–34.
Nguyen TC, Liu A, Liu L, et al. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica. 2007;92:121–4.
Kremer Hovinga JA, Zeerleder S, Kessler P, et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. 2007;5:2284–90.
Martin K, Borgel D, Lerolle N, et al. Decreased ADAMTS-13 (A disintegrin-like and metalloprotease with thrombospondin type 1 repeats) is associated with a poor prognosis in sepsis-induced organ failure. Crit Care Med. 2007;35:2375–82.
Bianchi V, Robles R, Alberio L, Furlan M, Lammle B. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100:710–3.
Lisman T, Bongers TN, Adelmeijer J, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44:53–61.
Moake JL, McPherson PD. Abnormalities of von Willebrand factor multimers in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. Am J Med. 1989;87:9N–15N.
Jin M, Cataland S, Bissell M, Wu HM. A rapid test for the diagnosis of thrombotic thrombocytopenic purpura using surface enhanced laser desorption/ionization time-of-flight (SELDI-TOF)-mass spectrometry. J Thromb Haemost. 2006;4:333–8.
Wu JJ, Fujikawa K, Lian EC, et al. A rapid enzyme-linked assay for ADAMTS-13. J Thromb Haemost. 2006;4:129–36.
Kato S, Matsumoto M, Matsuyama T, et al. Novel monoclonal antibody-based enzyme immunoassay for determining plasma levels of ADAMTS13 activity. Transfusion. 2006;46:1444–52.
Cao W, Krishnaswamy S, Camire RM, Lenting PJ, Zheng XL. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc Natl Acad Sci USA. 2008;105:7416–21.
Shim K, Anderson PJ, Tuley EA, Wiswall E, Sadler JE. Platelet–VWF complexes are preferred substrates of ADAMTS13 under fluid shear stress. Blood. 2008;111:651–7.
Jin M, Casper TC, Cataland SR, et al. Relationship between ADAMTS13 activity in clinical remission and the risk of TTP relapse. Br J Haematol. 2008;141:651–8.
Eckmann CM, De Laaf RT, Van Keulen JM, van Mourik JA, De Laat B. Bilirubin oxidase as a solution for the interference of hyperbilirubinemia with ADAMTS-13 activity measurement by FRETS-VWF73 assay. J Thromb Haemost. 2007;5:1330–1.
Gutterman LA, Kloster B, Tsai HM. Rituximab therapy for refractory thrombotic thrombocytopenic purpura. Blood Cells Mol Dis. 2002;28:385–91.
Downes KA, Yomtovian R, Tsai HM, et al. Relapsed thrombotic thrombocytopenic purpura presenting as an acute cerebrovascular accident. J Clin Apheresis. 2004;19:86–9.
Jokiranta TS, Zipfel PF, Fremeaux-Bacchi V, et al. Where next with atypical hemolytic uremic syndrome? Mol Immunol. 2007;44:3889–900.
Delvaeye M, Noris M, De VA, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–57.
Cataland SR, Jin M, Lin S, et al. Effect of prophylactic cyclosporine therapy on ADAMTS13 biomarkers in patients with idiopathic thrombotic thrombocytopenic purpura. Am J Hematol. 2008;83:911–5.
Elliott MA, Heit JA, Pruthi RK, et al. Rituximab for refractory and or relapsing thrombotic thrombocytopenic purpura related to immune-mediated severe ADAMTS13-deficiency: a report of four cases and a systematic review of the literature. Eur J Haematol. 2009.
Vesely SK, Li X, McMinn JR, Terrell DR, George JN. Pregnancy outcomes after recovery from thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Transfusion. 2004;44:1149–58.
Sanchez-Luceros A, Farias CE, Amaral MM, et al. von Willebrand factor-cleaving protease (ADAMTS13) activity in normal non-pregnant women, pregnant and post-delivery women. Thromb Haemost. 2004;92:1320–6.
Lattuada A, Rossi E, Calzarossa C, Candolfi R, Mannucci PM. Mild to moderate reduction of a von Willebrand factor cleaving protease (ADAMTS-13) in pregnant women with HELLP microangiopathic syndrome. Haematologica. 2003;88:1029–34.
Acknowledgments
This work is supported in part by a grant (R01 HL62136) from the National Heart Lung and Blood Institute of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Tsai, HM. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol 91, 1–19 (2010). https://doi.org/10.1007/s12185-009-0476-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-009-0476-1