
Abstract
At the clinical level, recent studies reveal the link between coagulation and other pathophysiological processes, including platelet activation, inflammation, cancer, the immune response, and/or infectious diseases. These links are likely to underpin the coagulopathy associated with risk factors for venous thromboembolic (VTE) and deep vein thrombosis (DVT). At the molecular level, the interactions between platelet-specific receptors and coagulation factors could help explain coagulopathy associated with aberrant platelet function, as well as revealing new approaches targeting platelet receptors in diagnosis or treatment of VTE or DVT. Glycoprotein (GP)Ibα, the major ligand-binding subunit of the platelet GPIb-IX-V complex, that binds the adhesive ligand, von Willebrand factor (VWF), is co-associated with the platelet-specific collagen receptor, GPVI. The GPIb-IX-V/GPVI adheso-signaling complex not only initiates platelet activation and aggregation (thrombus formation) in response to vascular injury or disease but GPIbα also regulates coagulation through a specific interaction with thrombin and other coagulation factors. Here, we discuss the structure and function of key platelet receptors involved in thrombus formation and coagulation in health and disease, with a particular focus on platelet GPIbα.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction: Coagulation and Platelets
Coagulation of human plasma is initiated by activation of the intrinsic (FXII-dependent) or extrinsic (tissue factor-dependent) pathways (Fig. 13.1a) [1]. In vivo, release of activated tissue factor at sites of damaged vasculature provides a triggering mechanism for initiating coagulation [2]. For the intrinsic pathway, however, while contact activation by negative surfaces is known to trigger coagulation in vitro, recent evidence suggests that collagen exposure, release of procoagulant polyphosphates from activated platelets [3], and possibly even pathological shear stress could provide a mechanism for activating factor XII (FXII) in vivo [4]. Other mechanisms could involve activation of prekallikrein that acts on FXII. Another intriguing possibility is that antibacterial leukocyte DNA-containing neutrophil extracellular traps (NETs) or associated proteins could activate intrinsic coagulation. Both intrinsic and extrinsic pathways activate the common coagulation pathway, involving activation of FX to FXa which converts prothrombin (FII) to active thrombin (FIIa; Fig. 13.1a).

Coagulation and platelet function. a Intrinsic (FXII-dependent) and extrinsic (tissue factor-dependent) coagulation pathways leading to the common pathway of thrombin (FIIa) generation. Thrombin induces clotting via conversion of fibrinogen to fibrin, a process accelerated by activated platelets, and b can bind to platelet GPIbα (of the GPIb-IX-V complex) and activate platelets via GPIbα signaling when GPV (a thrombin substrate) is removed, and G-protein-coupled protease-activated thrombin receptors, PAR-1 and PAR-4. Thrombin can thereby enhance thrombus formation following adhesion of circulating platelets to extracellular matrix or activated endothelium, or under shear stress, leading to secretion of ADP and procoagulant factors such as polyphosphates, increased expression of platelet surface phospholipids, and activation of integrin αIIbβ3 that binds fibrinogen or VWF and mediates platelet aggregation. Thrombin can be activated by vascular damage (releasing tissue factor) or by activation of FXII (intrinsic pathway) by collagen exposure, platelet secretion of procoagulant factors such as polyphosphates, collagen (that binds FXII under some conditions or activates prekallikrein under hyperglycemic conditions), or possibly by pathological shear stress a. F factor, GP glycoprotein, HK high molecular weight kininogen, PK prekallikrein, PL phospholipids, TSP thrombospondin, VWF von Willebrand factor
However coagulation is initiated, there is a clear role for platelets in spatial and temporal regulation of coagulation at prothrombotic sites [5]. Circulating platelets in the bloodstream are rapidly activated following vascular damage by exposure of collagen, von Willebrand factor (VWF), or other adhesive ligands in the subendothelial matrix or ruptured atherosclerotic plaque [6], VWF/P-selectin on activated endothelium, or VWF in stenotic vessels (Fig. 13.1b) [7]. Platelet glycoprotein (GP)Ibα of the GPIb-IX-V complex binds VWF [8] or thrombospondin (TSP), GPVI binds collagen or laminin (facilitated by activated platelet integrins, α2β1, or α5β1, respectively, facilitating adhesion or platelet activation via GPVI) [9]. Engagement of GPIb-IX-V/GPVI leads to activation of the integrin αIIbβ3, which binds fibrinogen or VWF and mediates platelet aggregation and fibrin formation [10]. Activated platelets secrete agonists such as ADP which acts on purinergic G-protein-coupled receptors [11], and secrete procoagulant factors such as polyphosphates [3] which promote coagulation and generation of active thrombin [12]; expression of phosphatidylserine or other procoagulant phospholipids on the surface of activated platelets also accelerates coagulation by localization and assembly of coagulation complexes (Fig. 13.1b). Thrombin activates platelets by using GPIbα as a cofactor in the activation of platelets via G-protein-coupled protease-activated receptors, PAR-1 or PAR-4, which in turn promote platelet activation and degranulation. Platelet activation is also associated with time-dependent metalloproteinase-mediated ectodomain shedding of platelet receptors, GPIbα (“glycocalicin”), GPV, and GPVI [13] (Fig. 13.1b). In this regard, elevated levels or plasma soluble GPVI (sGPVI) associated with disseminated intravascular coagulation (DIC) correlate with increased levels of coagulation markers [14]. Interestingly, GPV may be shed via cleavage at separate sites either by platelet sheddases or by thrombin [13, 15], with loss of surface GPV being associated with increased platelet activation by the interaction of thrombin with GPIbα.
Ligand Binding to Platelet GPIbα
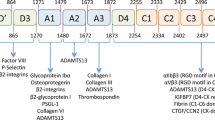
Coagulation factors including thrombin, FXII, FXI, and high molecular weight kininogen (HK) bind to the same ligand-binding domain of GPIbα involved in binding VWF, TSP, and other ligands [16, 17]. GPIbα is a multifunctional receptor which binds prothrombotic and procoagulant ligands within a versatile “shear-activated” ligand-binding region (Fig. 13.2). The absence or deficiency of GPIbα causes the inherited bleeding disorder, Bernard–Soulier Syndrome (BSS) [18] and along with the loss of high-shear- and VWF-dependent platelet-to-platelet interactions [19]; platelets from individuals with BSS are generally thought to have ablated procoagulant function [20]. GPIbα (~ 135 kDa) consists of an N-terminal globular domain (~ 40 kDa), a sialomucin core, an extracellular membrane-proximal tandem Cys sequence which forms disulfide bonds to 2 GPIbβ subunits (forming GPIb) [21], transmembrane domain, and cytoplasmic tail containing binding sites for intracellular signaling/cytoskeletal proteins. GPIb is noncovalently associated with GPIX and GPV, all members of the leucine-rich repeat family. The N-terminal globular domain of GPIbα (His1–Glu282) contains four important structural domains: the tandem leucine-rich repeats (~ 24 residues, each spanning the sequence 36–200), the N-terminal (residues 1–35) and C-terminal (201–268) disulfide-looped sequences, and an anionic sulfated tyrosine-rich sequence (269–282; Fig. 13.2) [22]. Using enzymes which specifically cleave at 282/283 (mocarhagin) [23] or inhibitory anti-GPIbα monoclonal antibodies mapped to specific structural regions [22, 24] as well as other approaches, it has been shown that GPIbα 1–282 contains discontiguous but overlapping binding sites for VWF [25], TSP [26], thrombin [27–29], FXII [30], FXI [31], HK [32], and counter receptors on activated endothelial cells (P-selectin) [33] or leukocytes (αMβ2; Mac-1) [34]. The sulfated tyrosine sequence also associates with the ectodomain of the immunoreceptor family protein, GPVI, on human platelets [35]. In addition, the procoagulant protein, recombinant FVIIa has been reported to bind to the sialomucin domain of GPIbα, downstream of Glu282 which could also localize thrombin generation to activated platelets.

N-terminal ligand-binding region of platelet GPIbα Structural regions based on primary sequence and crystal structure, and functional mapping of anti-GPIbα monoclonal antibodies which inhibit binding of one or more ligands. Antibodies against different epitopes, and the small-molecule allosteric inhibitor, the cyclic peptide OS1, interact with different regions within the 282-residue N-terminal domain illustrating the globular conformationally dependent binding sites for VWF and possibly other ligands. Coagulation factors of the intrinsic pathway (FXII, FXI, high molecular weight kininogen, with FXI being a substrate for both FXII and thrombin) also interact with the N-terminal domain of GPIbα
Extensive biochemical, crystallographic, and molecular simulation studies have analyzed binding of VWF to human GPIbα, revealing that the leucine-rich repeat sequence 60–128 (repeats “2–4”) is critical for interacting with the VWF A1 domain [28, 36–39]. Compared to the structure of the ligand-binding domain under resting conditions, as the shear stress increases from low to high physiological or pathological levels, the C-terminal disulfide-looped domain alters its conformation when complexed with VWF A1. A small molecular weight inhibitor, OS1, allosterically inhibits VWF binding to GPIbα by preventing the formation of the active conformation [40], while gain-of-function mutations within the C-terminal disulfide loop also increase binding to VWF-A1.
Specialized electrostatic “catch-slip” bonding facilitates high-affinity adhesion of VWF to receptor as shear rate increases, thereby enabling platelets to roll, skip, or firmly adhere to immobilized VWF in a shear-dependent manner [4, 38]. At high physiological or pathological shear rates such as encountered in a sclerotic or blocked artery, platelet adhesion becomes entirely GPIbα dependent [41]. However, examination of arterial thrombus formation in experimental models in vivo shows a significantly greater dependence on platelet GPIbα than VWF [42, 43], suggesting other ligands are also important. The extent to which conformational activation of GPIbα regulates interaction of ligands other than VWF is unknown, and precise binding sites for other ligands, including coagulation factors, are yet to be fully resolved. It is clear, however, that ligands such as TSP, P-selectin, and αMβ2 not only bind to the N-terminal domain of GPIbα under static conditions but also support GPIbα-dependent adhesion under flow conditions. The interaction of GPIbα with αMβ2 involves a domain of αM (“I-domain”) homologous to the GPIbα-binding A1 domain of VWF, although the binding sites for the two ligands are not identical. VWF A1 competes for binding of TSP, and some anti-GPIbα antibodies differentially block VWF or other ligands. It has been determined that the sulfated sequence (269–282) is critically involved in thrombin binding, with this interaction facilitating thrombin-dependent activation of platelet PAR-1. The extent to which co-localization of FXII, thrombin, and the common substrate FXI on a single receptor or adjacent copies of GPIbα within the GPIb-IX-V/GPVI complex is yet to be definitively established, although binding to GPIbα promotes activation of FXI by thrombin. Interestingly, regions of GPIbα beyond the 45-kDa N-terminal portion may be involved in platelet procoagulant function, as specific enzymatic removal of this region in murine washed platelets did not interfere with thrombin generation [44]. Targeted disruption of the cytoplasmic portion of GPIb-IX-V, for example, by site-directed mutagenesis, may help elucidate how the receptor complex modulates platelet procoagulant activity.
Functional Role of Interactions Involving GPIbα
Considering together the network of potential interactions of platelet GPIbα illustrates the potential for this receptor to co-localize, sequester, or otherwise regulate different components of platelet thrombus formation, coagulation, and platelet–leukocyte and platelet–endothelial cell interactions (Fig. 13.3). These interactions, rarely studied in combination, suggest how coagulation associated with platelet thrombus formation and inflammatory responses involving activated platelets and leukocytes in immune or infectious diseases could be coordinated by interactions involving GPIbα and adhesive or procoagulant ligands or counter-receptors under resting or activated conditions. GPIbα could provide a common regulatory receptor controlling time-dependent transition from initial platelet adhesion, activation and aggregation, to coagulation and inflammation in response to vascular injury or disease, for example, by progressively binding to VWF/TSP, thrombin/FXII/FXI/HK, or P-selectin/αMβ2, respectively. The capacity of platelets to rapidly adhere, become activated and degranulate in flowing blood mediated by GPIbα and other receptors would be a key property enabling the coordination of these pathophysiological processes. It is only recently that the role of platelets and platelet receptors has been investigated in detail in inflammation [45], coagulation [46], cancer [47–49], and infectious diseases [50, 51].

Interactions involving GPIbα In addition to other platelet surface receptors (black) with which it is co-associated (GPVI, GPV, GPIX, FcγRIIa), GPIbα interacts with at least ten other purported binding partners, including adhesive proteins (blue), coagulation factors (red), and receptors on either leukocytes (green) or activated endothelial cells (orange). Indirectly, the GPIbα-related network includes over 30 proteins. What this interaction map does not show is the spatial-temporal nature of these interactions or how these interactions are regulated under static or high-shear conditions (for example, when VWF binding is enhanced). These features are likely to control a coordinated, localized thrombotic, inflammatory, and coagulation response to injury, atheroma, immune disease, or infectious diseases
One potential link between coagulation factors and platelet receptors involves findings by Renne and colleagues [52], showing that while deficiency of FXII/FXI has minimal impact on bleeding times, there is marked inhibition of occlusive platelet thrombus formation at high shear in the arterial circulation in experimental models. It is unclear how FXII is activated under these conditions, but interaction with platelet GPIbα or GPIbα-dependent adhesion localizing platelet activation and secretion, and phosphatidylserine exposure could provide the means for stable occlusive thrombus formation in the presence of FXII. The role of HK in GPIbα binding and activation of FXII is also interesting in terms of possible GPIbα-mediated activation of FXII in vivo, and the link between coagulation and platelet–leukocyte adhesion as HK also engages the GPIbα counter-receptor on leukocytes, αMβ2 [53]. The HK binding site on αMβ2 overlaps a fibrinogen-binding site, and increased the capacity for binding GPIbα, providing a potential mechanism for HK-dependent enhancement of platelet–leukocyte adhesion [53]. The FXII activator, plasma kallikrein (PK), also interacts with collagen under hyperglycemic conditions [54], such that collagen exposure could lead to activation of PK/FXII as well as localizing platelet GPIb-IX-V/GPVI via interactions with collagen/VWF. Together, this would provide a mechanism for bridging platelets, leukocytes, and the subendothelial matrix leading to the activation of coagulation [55].
More recently, clear roles for leukocytes in the direct upregulation of platelet procoagulant function have emerged. While the majority of circulating microparticles in healthy individuals are platelet or megakaryocyte derived [46], leukocyte-derived microparticles originating from neutrophils, monocytes/macrophages, or lymphocytes as well as endothelial-derived microparticles are significantly upregulated in all stages of atherosclerosis and circulate at a high level in the bloodstream of patients with high atherothrombotic risk [56, 57]. Microparticles have been demonstrated to associate with resting platelets via CD36, lowering the required threshold concentration of agonist to activate platelets [58] and also via engagement of platelet GPIbα by active αMβ2 on microparticles derived from activated neutrophils [59]. In the second study, engagement of GPIbα by αMβ2–bearing microparticles triggered signaling pathways that led to surface expression of P-selectin and activation of αIIbβ3-mediating platelet aggregation. Both interactions provide a clear and distinct mechanistic link between platelet prothrombotic and leukocyte inflammatory states where microparticles from unstimulated versus activated neutrophils differentially facilitate interaction with either activated platelets (via PSGL-1/P-selectin) or resting platelets (via active αMβ2/GPIbα), respectively.
Platelet GPIbα also interacts with bacterial proteins, such as the Staphylococcal superantigen-like protein 5 (SSL5) via the sulfated-tyrosine sequence and carbohydrate moieties of GPIbα [60, 61]. SSL5 also interacts with extracellular immunoglobulin domains of GPVI [61]. These types of interactions could be involved in platelet activation associated not only with bacterial infection and increased thrombotic risk but also with the coagulopathy commonly associated with sepsis and other infections. Bacterial-induced activation of leukocytes also releases DNA-containing NETs, which may limit dispersal of bacterial, but are also associated with release of nuclear proteins such as histones [62]. NETs have been linked to the development of venous “red” (platelet-deficient) thrombus in experimental models of deep vein thrombosis (DVT) [63]; however, NETs and associated proteins such as VWF A1 domain-binding histones [64] could also promote platelet activation, secretion, and leukocyte recruitment in arterial “white” (platelet-rich) thrombus.
On the platelet surface, GPVI interacts with the sulfated region of GPIbα that binds thrombin [35], and could also influence coagulation in other ways. GPVI contains two extracellular immunoglobulin domains, and is co-associated with the accessory signaling receptor, FcRγ, required for GPVI surface expression [65–67]. The anti-GPIbα monoclonal antibody SZ2, inhibits collagen-dependent platelet activation via GPVI [68]. Masking GPVI also attenuates collagen-induced or tissue factor-dependent thrombin generation, thrombus formation [69], or pulmonary thromboembolism [70]. GPVI engagement could promote phosphatidylserine exposure on activated platelets [71–73], induce procoagulant platelet-derived microparticles [73], or activate platelets leading to secretion of procoagulant factors such as polyphosphates. However, GPVI blockade can also inhibit tissue factor-mediated coagulation in the absence of collagen or other known GPVI ligands [69], while GPVI ligands also induce dose-dependent increases in FXa and thrombin generation, regulated by a subpopulation of platelets with increased coagulation factor binding that is not related to increased phosphatidylserine exposure [71]. These mechanisms require further analysis in combination with interactions involving coagulation factors and GPIbα, which is co-associated with GPVI [35]. GPVI also binds to extracellular matrix metalloproteinase (MMP) inducer (EMMPRIN; CD147), like GPVI, a member of the immunoreceptor family expressed on activated platelets, monocytes, and tumor cells [74]. The GPVI–EMMPRIN interaction could contribute to platelet-mediated coagulation at sites of monocyte recruitment, for example, at atherosclerotic sites of the vasculature [75], or in the context of tumor growth.
Plasma GPIbα and GPVI
Although the extracellular domain of platelet GPIbα is important for ligand binding, constitutive ectodomain shedding of GPIbα results in high levels of soluble GPIbα ectodomain (glycocalicin) in normal plasma (approximately two thirds of total GPIbα in blood) [76]. The functional consequences of glycocalicin-binding ligands are not addressed by existing studies, and it possibly has a regulatory role in some circumstances or is less efficacious than surface-expressed GPIbα within the GPIb-IX-V/GPVI adheso-signaling complex. Similarly, shedding of platelet GPVI liberating plasma soluble GPVI [77, 78] could downgrade the capacity for GPVI-dependent platelet activation or microparticle generation. Unlike GPIbα, levels of sGPVI in healthy plasma are relatively low (approximately one sixth of total blood GPVI) [79], but are elevated under prothrombotic [80–82] or procoagulant [14] conditions and may serve as a platelet-specific biomarker as an indicator of risk, for example, in the case of infectious diseases or immune disease [78]. In this regard, the platelet Fc receptor, FcγRIIa, utilises intracellular signaling pathways equivalent to GPVI/FcRγ, and engagement of FcγRIIa induces GPVI shedding [83]. Through this mechanism, antiplatelet autoantibodies, for example, targeting platelet factor 4/heparin complexes as seen in heparin-induced thrombocytopenia can activate platelets via FcγRIIa [84, 85], to release platelet-derived microparticles [86] and increase platelet thrombin generation [87].
Conclusions: Targeting Platelets in Human Disease
Whether selectively targeting platelet GPIb-IX-V/GPVI ligand binding, platelet activation or secretion to inhibit the impact of platelets on coagulation to aid in the therapeutic control of thrombosis [88, 89], or coagulopathy where platelets and platelet/leukocyte or platelet/endothelium interactions are implicated in procoagulant activity, warrants further investigation [90]. Further analysis, particularly centered on studies in human vascular systems of interactive sites, changes in binding under shear conditions, and the influence of other ligands under different conditions is required [91] to exploit these possibilities. It is also worth noting how multifunctional interactions of thrombin, FXa, and other factors beyond coagulation, broaden the range of interactions of platelet GPIb-IX-V/GPVI in human pathophysiology.
References
Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–34.
del Conde I Shrimpton CN Thiagarajan P López JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106(5):1604–11.
Müller F, Mutch NJ, Schenk WA, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139(6):1143–56.
Colace TV, Tormoen GW, McCarty OJ, Diamond SL. Microfluidics and coagulation biology. Ann Rev Biomed Eng. 2013;15:283–303.
Jackson SP, Nesbitt WS, Westein E. Dynamics of platelet thrombus formation. J Thromb Haemost. 2009;7(s1):17–20.
Winckers K, ten Cate H, Hackeng TM. The role of tissue factor pathway inhibitor in atherosclerosis and arterial thrombosis. Blood Rev. 2013;27(3):119–32.
Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327–58.
Chow TW, Hellums JD, Moake JL, Kroll MH. Shear stress-induced von Willebrand factor binding to platelet glycoprotein Ib initiates calcium influx associated with aggregation. Blood. 1992;80(1):113–20.
Andrews RK, Shen Y, Gardiner EE, Berndt MC. Platelet adhesion receptors and (patho)physiological thrombus formation. Histol Histopathol. 2001;16(3):969–80.
Cosemans JMEM, Schols SEM, Stefanini L, et al. Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood. 2011;117(2):651–60.
van der Meijden PEJ, Schoenwaelder SM, Feijge MAH, et al. Dual P2Y12 receptor signaling in thrombin-stimulated platelets-involvement of phosphoinositide 3-kinase β but not γ isoform in Ca2+ mobilization and procoagulant activity. FEBS J. 2008;275(2):371–85.
Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. 2011;118(26):6963–70.
Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J Thromb Haemost. 2007;5(7):1530–7.
Al-Tamimi M, Grigoriadis G, Tran H, et al. Coagulation-induced shedding of platelet glycoprotein VI mediated by Factor Xa. Blood. 2011;117(14):3912–20.
Rabie T, Strehl A, Ludwig A, Nieswandt B. Evidence for a role of ADAM17 (TACE) in the regulation of platelet glycoprotein V. J Biol Chem. 2005;280(15):14462–68.
Andrews RK, Gardiner EE, Shen Y, Whisstock JC, Berndt MC. Glycoprotein Ib-IX-V. Int J Biochem Cell Biol. 2003;35(8):1170–4.
Canobbio I, Balduini C, Torti M. Signalling through the platelet glycoprotein Ib-V-IX complex. Cell Signal. 2004;16(12):1329–44.
López JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndrome. Blood. 1998;91(12):4397–418.
Cranmer SL, Ashworth KJ, Yao Y, et al. High shear-dependent loss of membrane integrity and defective platelet adhesion following disruption of the GPIbα-filamin interaction. Blood. 2011;117(9):2718–27.
Dicker IB, Pedicord DL, Seiffert DA, Jamieson GA, Greco NJ. Both the high affinity thrombin receptor (GPIb-IX-V) and GPIIb/IIIa are implicated in expression of thrombin-induced platelet procoagulant activity. Thromb Haemost. 2001;86(4):1065–9.
Luo S-Z, Mo X, Afshar-Kharghan V, Srinivasan S, Lopez JA, Li R. Glycoprotein Ibα forms disulfide bonds with 2 glycoprotein Ibβ subunits in the resting platelet. Blood. 2007;109(2):603–9.
Andrews RK, Berndt MC, López JA. The glycoprotein Ib-IX-V complex. In Michelson AD, editor. Platelets. San Diego: Academic; 2006:145–63.
Ward CM, Andrews RK, Smith AI, Berndt MC. Mocarhagin, a novel cobra venom metalloproteinase, cleaves the platelet von Willebrand factor receptor glycoprotein Ibα. Identification of the sulfated tyrosine/anionic sequence Tyr-276-Glu-282 of glycoprotein Ibα as a binding site for von Willebrand factor and α-thrombin. Biochemistry. 1996;35(15):4929–38.
Cauwenberghs N, Vanhoorelbeke K, Vauterin S, et al. Epitope mapping of inhibitory antibodies against platelet glycoprotein Ibα reveals interaction between the leucine-rich repeat N-terminal and C-terminal flanking domains of glycoprotein Ibα. Blood. 2001;98(3):652–60.
Shen Y, Romo GM, Dong JF, et al. Requirement of leucine-rich repeats of glycoprotein (GP) Ibα for shear-dependent and static binding of von Willebrand factor to the platelet membrane GP Ib-IX-V complex. Blood. 2000;95(3):903–10.
Jurk K, Clemetson KJ, de Groot PG, et al. Thrombospondin-1 mediates platelet adhesion at high shear via glycoprotein Ib (GPIb): an alternative/backup mechanism to von Willebrand factor. FASEB J. 2003;17(11):1490–2.
Li CQ, Vindigni A, Sadler JE, Wardell MR. Platelet glycoprotein Ibα binds to thrombin anion-binding exosite II inducing allosteric changes in the activity of thrombin. J Biol Chem. 2001;276(9):6161–8.
Huizinga EG, Tsuji S, Romijn RAP, et al. Structures of glycoprotein Ibα and its complex with von Willebrand Factor A1 domain. Science. 2002;297(5584):1176–9.
Dumas JJ, Kumar R, Seehra J, Somers WS, Mosyak L. Crystal structure of the GPIbα-thrombin complex essential for platelet aggregation. Science.2003;301(5630): 222–6.
Bradford HN, Pixley RA, Colman RW. Human factor XII binding to the glycoprotein Ib-IX-V complex inhibits thrombin-induced platelet aggregation. J Biol Chem. 2000;275(30):22756–42.
Yun TH, Baglia FA, Myles T, et al. Thrombin activation of factor XI on activated platelets requires the interaction of factor XI and platelet glycoprotein Ibα with thrombin anion-binding exosites I and II, respectively. J Biol Chem. 2003;278(48):48112–9.
Joseph K, Nakazawa Y, Bahou WF, Ghebrehiwet B, Kaplan AP. Platelet glycoprotein Ib: a zinc-dependent binding protein for the heavy chain of high-molecular-weight kininogen. Mol Med. 1999;5(8):555–63.
Romo GM, Dong JF, Schade AJ, et al. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med. 1999;190(6):803–14.
Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 2000;192(2):193–204.
Arthur JF, Gardiner EE, Matzaris M, et al. Glycoprotein VI is associated with GPIb-IX-V on the membrane of resting and activated platelets. Thromb Haemost. 2005;93(4):716–23.
Cruz MA, Diacovo TG, Emsley J, Liddington R, Handin RI. Mapping the glycoprotein Ib-binding site in the von Willebrand factor A1 domain. J Biol Chem. 2000;275(25):19098–105.
Shen Y, Dong Jf JF, Romo GM, et al. Functional analysis of the C-terminal flanking sequence of platelet glycoprotein Ibα using canine-human chimeras. Blood. 2002;99(1):145–50.
Shen Y, Cranmer SL, Aprico A, et al. Leucine-rich repeats 2-4 (Leu60-Glu128) of platelet glycoprotein Ibα regulate shear-dependent cell adhesion to von Willebrand factor. J Biol Chem. 2006; 281 (36): 26419–26423.
Dumas JJ, Kumar R, McDonagh T, et al. Crystal structure of the wild-type von Willebrand Factor A1-glycoprotein Iba complex reveals conformation differences with a complex bearing von Willebrand Disease mutations. J Biol Chem. 2004;279(22):23327–34.
McEwan PA, Andrews RK, Emsley J. Glycoprotein Ibα inhibitor complex structure reveals a combined steric and allosteric mechanism of von Willebrand factor antagonism. Blood. 2009;114(23):4883–5.
Yago T, Lou J, Wu T, et al. Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest. 2008;118(9):3195–207.
Bergmeier W, Piffath CL, Goerge T, et al. The role of platelet adhesion receptor GPIbα far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci U S A. 2006;103(45):16900–5.
Bergmeier W, Chauhan AK, Wagner DD. Glycoprotein Ibα and von Willebrand factor in primary platelet adhesion and thrombus formation: lessons from mutant mice. Thromb Haemost. 2008;99(2):264–70.
Ravanat C, Strassel C, Hechler B, et al. A central role of GPIb-IX in the procoagulant function of platelets that is independent of the 45-kDa GPIbα N-terminal extracellular domain. Blood. 2010;116(7):1157–64.
Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908–16.
Owens AP, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108(10):1284–97.
Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123–34.
Jain S, Harris J, Ware J. Platelets: linking hemostasis and cancer. Arterioscler Thromb Vasc Biol. 2010;30(12):2362–7.
Goubran HA, Burnouf T, Radosevic M, El-Ekiaby M. The platelet-cancer loop. Eur J Internal Med. 2013 24(5):393-400.
Semple JW, Italiano JE, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–74.
Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–9.
Renne T, Pozgajova M, Gruner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–81.
Chavakis T, Santoso S, Clemetson KJ, et al. High molecular weight kininogen regulates platelet-leukocyte interactions by bridging Mac-1 and glycoprotein Ib. J Biol Chem. 2003;278(46):45375–81.
Liu J, Gao B-B, Clermont AC, et al. Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nat Med. 2011;17(2):206–10.
Morrissey JH, Choi SH, Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood. 2012;119(25):5972–9.
Shantsila E, Kamphuisen PW, Lip GYH. Circulating microparticles in cardiovascular disease: implications for atherogenesis and atherothrombosis. J Thromb Haemost. 2010;8(11):2358–68.
Morel O, Toti F, Hugel B, et al. Procoagulant microparticles. Arterioscler Thromb Vasc Biol. 2006;26(12):2594–604.
Ghosh A, Li W, Febbraio M, et al. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest. 2008;118(5):1934–43.
Pluskota E, Woody NM, Szpak D, et al. Expression, activation, and function of integrin αMβ2 (Mac-1) on neutrophil-derived microparticles. Blood. 2008;112(6):2327–35.
De Haas CJC Weeterings C Vughs MM De Groot PG Van Strijp JA Lisman T. Staphylococcal superantigen-like 5 activates platelets and supports platelet adhesion under flow conditions, which involves glycoprotein Ibα and αIIbβ3. J Thromb Haemost. 2009;7(11):1867–74.
Hu H, Armstrong PCJ, Khalil E, et al. GPVI and GPIbα mediate Staphylococcal Superantigen-like protein 5 (SSL5) induced platelet activation and direct toward glycans as potential inhibitors. PLoS ONE. 2011;6(4):e19190.
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10(1):136–44.
Ward CM, Tetaz TJ, Andrews RK, Berndt MC. Binding of the von Willebrand factor A1 domain to histone. Thromb Res. 1997;86(6):469–77.
Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor γ chain complex on human platelets. J Exp Med. 1998;188(2):267–76.
Jandrot-Perrus M, Busfield S, Lagrue A-H, et al. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: a platelet-specific collagen receptor from the immunoglobulin superfamily. Blood. 2000;96(5):1798–807.
Suzuki-Inoue K, Tulasne D, Shen Y, et al. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J Biol Chem. 2002;277(24):21561–6.
Ruan CG, Du XP, Xi XD, Castaldi PA, Berndt MC. A murine antiglycoprotein Ib complex monoclonal antibody, SZ2, inhibits platelet aggregation induced by both ristocetin and collagen. Blood. 1987;69(2):570–7.
Ohlmann P, Hechler B, Ravanat C, et al. Ex vivo inhibition of thrombus formation by an anti-glycoprotein VI Fab fragment in non-human primates without modification of glycoprotein VI expression. J Thromb Haemost. 2008;6(6):1003–11.
Schulte V, Reusch HP, Pozgajova M, Varga-Szabo D, Gachet C, Nieswandt B. Two-phase antithrombotic protection after anti-glycoprotein VI treatment in mice. Arterioscler Thromb Vasc Biol. 2006; 26 (7): 1640–1647.
Gilio K, van Kruchten R, Braun A, et al. Roles of platelet STIM1 and Orai1 in glycoprotein VI- and thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem. 2010;285(31):23629–38.
Heemskerk JW, Siljander P, Vuist WM, et al. Function of glycoprotein VI and integrin α2β1 in the procoagulant response of single, collagen-adherent platelets. Thromb Haemost. 1999;81(5):782–92.
Siljander P, Farndale RW, Feijge MAH, et al. Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: Involvement of p38 MAP kinase and calpain. Arterioscler Thromb Vasc Biol. 2001;21(4):618–27.
Seizer P, Borst O, Langer HF, et al. EMMPRIN (CD147) is a novel receptor for platelet GPVI and mediates platelet rolling via GPVI-EMMPRIN interaction. Thromb Haemost. 2009;101(4):682–6.
Schulz C, von Bruhl ML, Barocke V, et al. EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thromb Haemost. 2011;9(5):1007–19.
Coller BS, Kalomiris E, Steinberg M, Scudder LE. Evidence that glycocalicin circulates in normal plasma. J Clin Invest. 1984;73(3):794–9.
Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood. 2004;104(12):3611–7.
Al-Tamimi M, Arthur JF, Gardiner EE, Andrews RK. Focusing on plasma glycoprotein VI. Thromb Haemost. 2012;107(4):648–55.
Al-Tamimi M, Mu FT, Moroi M, Gardiner EE, Berndt MC, Andrews RK. Measuring soluble platelet glycoprotein VI in human plasma by ELISA. Platelets. 2009;20(3):143–9.
Al-Tamimi M, Gardiner EE, Thom JY, et al. Soluble glycoprotein VI is raised in the plasma of patients with acute ischemic stroke. Stroke. 2011;42(2):498–500.
Bigalke B, Potz O, Kremmer E, et al. Sandwich immunoassay for soluble glycoprotein VI in patients with symptomatic coronary artery disease. Clin Chem. 2011;57(6):898–904.
Al-Tamimi M, Tan C, Qiao J, et al. Pathological shear triggers shedding of vascular receptors: a novel mechanism for downregulation of platelet glycoprotein (GP)VI in stenosed coronary vessels. Blood. 2012;119(18):4311–20.
Gardiner EE, Karunakaran D, Arthur JF, et al. Dual ITAM-mediated proteolytic pathways for irreversible inactivation of platelet receptors: De-ITAM-izing FcγRIIa. Blood. 2008;111(1):165–74.
Gardiner EE, Al-Tamimi M, Mu FT, et al. Compromised ITAM-based platelet receptor function in a patient with immune thrombocytopenic purpura. J Thromb Haemost. 2008;6(7):1175–82.
Reilly MP, Taylor SM, Hartman NK, et al. Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcγRIIA. Blood. 2001;98(8):2442–7.
Warkentin TE, Hayward CP, Boshkov LK, et al. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparin-induced thrombocytopenia. Blood. 1994;84(11):3691–9.
Tardy-Poncet B, Piot M, Chapelle C, et al. Thrombin generation and heparin-induced thrombocytopenia. J Thromb Haemostas. 2009;7(9):1474–81.
Andrews RK, Du X, Berndt MC. The 14-3-3ζ-GPIb-IX-V complex as an antiplatelet target. Drug News Perspect. 2007;20(5):285–92.
Clemetson KJ, Clemetson JM. Platelet GPIb complex as a target for anti-thrombotic drug development. Thromb Haemost. 2008;99(3):473–9.
Jackson SP. Arterial thrombosis-insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–36.
Wei AH, Schoenwaelder SM, Andrews RK, Jackson SP. New insights into the haemostatic function of platelets. Br J Haematol. 2009;147(4):415–30.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gardiner, E., Andrews, R. (2014). Structure and Function of Platelet Receptors Initiating Blood Clotting. In: Corey, S., Kimmel, M., Leonard, J. (eds) A Systems Biology Approach to Blood. Advances in Experimental Medicine and Biology, vol 844. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2095-2_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2095-2_13
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2094-5
Online ISBN: 978-1-4939-2095-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)