
Abstract
Starting first from Paget’s “seed and soil” to the latest hypothesis about metastatic process involving the concept of a premetastatic niche, a large amount of data suggested the idea that metastatization is a multistep coordinated process with a high degree of efficiency. A specific subpopulation of cells with tumor-initiating and migratory capacity can selectively migrate toward sites that are able to promote survival, and/or proliferation of metastatic tumor cells through a microenvironment modification. Bone plays a pivotal role in this process, acting not only as a preferential site for cancer cells' homing and proliferation, due to a complex interplay between different cellular phenotypes such as osteoblasts and osteoclasts, but also as a source of bone marrow precursors that are able to facilitate the metastatic process in extra-skeletal disease. Moreover, bone microenvironment has the unique capacity to retain cancer stem cells in a quiescent status, acting as a reservoir that is able to cause a metastatic spread also many years after the resection of the primary tumor. To add a further level of complexity, these mechanisms are strictly regulated through the signalling through several soluble factors including PTH, vitamin D or calcium concentration. Understanding this complexity represents a major challenge in anti-cancer research and a mandatory step towards the development of new drugs potentially able not only to reduce the consequences of bone lesions but also to target the metastatization process from the “bone pre-neoplastic niche” to “visceral pre-neoplastic niches”.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Vascular Endothelial Growth Factor
- Bone Metastasis
- Cancer Stem Cell
- Prostate Cancer Cell
- Stem Cell Factor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Bone Premetastatic Niche
Paget’s “seed and soil” theory postulated that cancer cells “colonize” the organs whose microenvironment is advantageous. Starting from this theory, we can say that metastasis is a remarkably efficient, multistep process. The determinants of ‘successful metastatic growth in a given organ are poorly understood, but there is substantial evidence to suggest that tumor cells and host tissue both play important roles in metastasis. This hypothesis started from a new model of metastasis formation. This ‘early metastasis model’ suggests that tumor cells leave the primary site much earlier in tumourigenesis (Pardal et al. 2003). The model is based on experimental data about cancer stem-like cells (CSC), a population of cells, within tumor mass, able to navigate in the bloodstream and to localize new metastatic sites (Pardal et al. 2003; Al-Hajj et al. 2003).
Populations of cells with tumor-initiating capacity have been shown to exist in human acute myeloid leukaemia (Lapidot et al. 1994; Bonnet and Dick 1997) and in several solid malignancies, such as breast (Al-Hajj et al. 2003) and colon cancer (O’Brien et al. 2007). Accordingly, “premetastatic niches” can be defined as a localized microenvironment that is being formed in metastatic target organs, prior to the arrival of metastatic tumor cells. Moreover, premetastatic niches consist of a collection of specific proteins and Bone Marrow Derived Cells (BMDCs). In fact, during the early development of primary tumors, neovascularization is guaranteed by VEGF-receptor 1+ (VEGFR1+), hematopoietic progenitor cells (HPC) which support the recruitment and incorporation of bone marrow-derived VEGF receptor 2+ (VEGFR2+) endothelial progenitors cells (EPC). These bone marrow-derived cells are also able to form clusters of cells in the tissue parenchyma at common sites of metastasis before actual tumor cell seeding. At these sites, bone marrow-derived cells express VEGFR1, CD11b, c-kit and other markers of their progenitor cell status within the tissue parenchyma of the premetastatic niche. Then, in response to the primary tumor chemokine secretion and other events, the VEGFR1+ HPCs proliferate and circulate in the bloodstream, but also preferentially localize to areas of increased fibronectin, newly synthesized by resident fibroblasts and fibroblast-like cells. The VEGFR1+ HPCs express integrin VLA-4 (or α4β1), allowing them to adhere specifically to the newly synthesized fibronectin for the initiation of cellular cluster formation. Other mediators such as metalloprotease 9 (MMP-9) allow extravasion of VEGFR1+ HPC. The VEGFR1+ HPCs, along with fibronectin and associated stromal cells, cause modifications of the local microenvironment, which leads to the activation of other integrins and chemokines such as SDF-1. SDF-1 itself promotes attachment, survival and growth of tumor cells.
Therefore, premetastatic niches are thought to be fertile regions of tissue that facilitate the invasion, survival and/or proliferation of metastatic tumor cells, providing a highly novel mechanism for the promotion of metastasis (Kaplan et al. 2005). Furthermore, some tissues are more receptive to a given metastasizing tumor cell type, which can explain the tendency of tumor cells to metastasize to some organs more often than other organs in a way that cannot be explained by differences in blood flow. In addition to target organ-specific growth of metastases, metastatic tumor foci seem to grow preferentially in specific areas of some tissues. So, we can postulate that cross-talking between bone microenvironment and cancer cells facilitates bone tropism of cancer cells. Moreover, there is some evidence that a specific subpopulation of cancer cells forming primary tumor sites can circulate as stem cells do.
1.1 Starting From the Primary Tumor Site: The Cancer Stem Cells
1.1.1 Cancer Stem Cell Characteristics
Stem cells are defined by their ability for self-renewal, differentiation into adult tissue and migration. Cancer stem-like cells are defined as cells capable of giving rise to a new tumor, and are thought to be the root cause of cancer. While still controversial, the Cancer Stem-like Cell (CSC) hypothesis may be directly relevant to metastatic theory, as such cells are good candidates for the acquisition of migratory capabilities and propagation of heterogeneous tumor cell populations at distant sites. Cancer stem-like cells may show some similarities to normal stem cells, but there appear to be differences too. While normal stem cells are present in tissues in relatively low numbers, the proportion of cells with specific CSC surface markers residing in a given tumor seems to vary greatly, up to 24.5% in colon cancer or 12–60% in breast cancer. In addition, putative breast cancer stem-like cells expressing CD44 (an adhesion molecule that binds hyaluronate) and lacking CD24 (an adhesion molecule that binds P-selectin) have been shown to switch to a more differentiated CD24-positive phenotype in distant metastases with loss of CD44, which can even progress to initiate further metastases (Shipitsin et al. 2007). As tumors progress through clonal selection of more malignant and less differentiated cells, differentiation between putative highly successful cancer stem-like cells and the rest of the tumor cells is difficult (Shipitsin and Polyak 2008). Another CSCs surface marker is CD133, which has been shown to be expressed in several solid malignancies such as intestine, brain, lung and prostate (Sing 2003; Collins et al. 2005; Shmelkov et al. 2008; Bertolini et al. 2009). However, it was demonstrated that CD133 is expressed in differentiated epithelium cells. Moreover, both CD133-positive and CD133-negative metastatic cells were able to start tumorigenesis. These findings raised the question of whether the CD133-negative cells have represented a largely non-epithelial population of stromal and inflammatory cells.
1.1.2 The Acquisition of Self-Renewal and Migration Ability (The Epithelial Mesenchymal Transition)
Many signaling pathways involved in stem-cells renewal cause neoplastic proliferation and migration when dysregulated by mutations. The most studied pathways are WNT, sonic hedgehog (SHH), NOTCH and PTEN.
The features of these pathways in normal and neoplastic cells are summarized in Table 1.
1.1.2.1 WNT Pathway
WNTs are secreted proteins which bind Frizzled receptors. This link activates Dishevelled (DSH) which disrupts the complex of glycogen synthase kinase 3β (GSK3β), Casein Kynase 1 (CK1), axin and adenomatosis poliposis coli (APC). This disruption inhibits interaction with APC-β-catenin and allows β-catenin to accumulate and translocate to the nucleus, binding LEF\TCF transcription factors and activating the expression of target genes promoting cellular proliferation. Mutations of this pathway have been implicated in many types of cancers (Polakis 1999; Zhu and Watt 1999). Expression of stabilized β-catenin promotes the self-renewal of many types of stem cells. WNT signaling activates the same pathway in colorectal cancer cells (Chenn and Walsh 2002). Mutations that activate WNT signaling cause the hyperproliferation of crypt progenitors, generating benign polyps (Powell et al. 1992). So, tumorigenesis in the intestinal epithelium seems to be caused by the hyper-self-renewal of intestinal-crypt stem cells, followed by the accumulation of additional mutations (Kinzler and Vogelstein 1996).
1.1.2.2 PTEN Pathway
PTEN plays an important role not only in self-renewal and activation of hematopoietic stem cells but also in the prevention of leukemogenesis (Zhang et al. 2006). It is quite likely that PTEN also plays an important role in breast cancer stem cells by negatively regulating PI3K/mTOR/STAT3 signaling. Some in vitro studies showed that overexpression of PTEN decreased cancer cell tumorigenicity (Cheney et al. 1998; Zhou et al. 2007). PTEN/PI3K/mTOR/STAT3 signaling forms a complex signaling network that is able to maintain the cancer stem cell population within the whole cell population (Zhou et al. 2007).
1.1.2.3 Notch Signaling System
Notch plays a key role in the normal development of many tissues and cell types through diverse effects on stem cell renewal, cellular differentiation, survival and proliferation (Artavanis-Tsakonas et al. 1999). The Notch signaling system includes Notch ligands (Jagged 1), receptors, negative and positive modifiers and Notch target transcription factors. One of the most important functions of the Notch pathway is expansion of the hematopoietic stem cell compartment during bone development, and participation in osteoblast differentiation (Nobta et al. 2005). Moreover, this signaling system is aberrantly activated in a variety of human cancers, including T-cell acute lymphoblastic leukemia and carcinomas of the lung, colorectum, prostate and the breast (Radtke and Raj 2003; Kunnimalaiyaan and Chen 2007; Proweller et al. 2006). Notch upregulation is involved in tumor metastatization, and its inhibition impairs tumor spreading (Hughes 2009). Osteoblasts within the bone marrow are identified as the niche for supporting long-term hematopoietic stem cells, providing the Notch ligand Jagged1 and other factors under regulation by bone morphogenetic protein (BMP) and PTH/PTHrP signaling (Calvi et al. 2003). Increasing evidence suggests that the osteoblast niche inhibits drug-induced apoptosis and confers de novo drug resistance in myeloma cells (Nefedova et al. 2004). This type of paracrine Notch signaling in metastatic cancer cells could explain their predisposition to bone metastasis. Moreover, cross-talk occurs between TGF-β and the Notch pathway. TGF-β increases the expression of Hes-1, a direct target of Notch, in several cell types (Blokzijl et al. 2003). TGF-β induces the interaction of the intracellular domain of Notch1 with Smad3. TGF-β-induced EMT is blocked by RNA silencing of the Notch target gene Hey-1 and the Notch ligand Jagged1, and by chemical inactivation of Notch (Zavadil et al. 2004).
1.1.2.4 SHH Pathway
Sonic hedgehog (SHH) ligands have an autocrine and paracrine action. When SHH reaches its target cell, it binds to the Patched-1 (PTCH1) and -2 (PTCH2) receptors. These in turn relieve the Smoothened (SMO) inhibition, leading to activation of the GLI (GLI 1, GLI 2, GLI 3) transcription factors. Abnormal activation of the pathway probably results in early carcinogenesis through transformation of adult stem cells into cancer stem cells (Dahmane et al. 1997; Ruiz i Altaba et al. 2002). In vitro models showed that cancer cells overexpressing SHH upregulated the expression of SHH-responsive target genes GLI1 and PTCH1 in pre-osteoblasts cells, leading to the induction of early phase osteoblast differentiation. Cancer cells that metastasize to bone are in close physical contact with bone stromal cells including bone cells and their osteoblast progenitors, fibroblasts, hematopoetic cells and multipotent mesenchymal stem cells, and the SHH pathway induces bone modification in order to create the conditions for the premetastatic niche. In fact, downstream mechanism through which SHH-signaling induces osteoblast differentiation is not fully understood. A recent study has demonstrated that SHH promotes osteoblast differentiation in multipotent mesenchymal cells by upregulating the expression and function of RUNX2 (Shimoyama et al. 2007; Spinella-Jaegle et al. 2001). Other data suggest that in cells that are already committed to the osteoblast lineage and express endogenous levels of RUNX2, the induction of osteoblast differentiation by SHH occurs through a mechanism that does not require further transcriptional upregulation of RUNX2 (Zunich et al. 2009).
These and other pathways enable cancer cells to acquire stem cell-like characteristics. Furthermore, the acquisition of mesenchymal markers such as fibronectin, and progressive loss of E-cadherin in tumor cells with nuclear β-catenin accumulation, suggest that they have undergone an epithelial–mesenchymal transition (EMT) or transdifferentiation. This process of EMT can be described by the chronological sequence of five morphogenetic events:
-
1.
Disassembly of tight junctions, which results in the redistribution of Zonula Occludens (ZO) proteins, claudins and occludins.
-
2.
Disruption of the polarity complex.
-
3.
Initiation of cytoskeleton reorganization (through actin reorganization).
-
4.
Metalloprotease upregulation.
-
5.
Increased deposition of extracellular matrix proteins.
In vitro and in vivo experiments showed that EMT is also promoted by Tumor Growth Factor β (TGF-β) (Miettinen et al. 1994; Piek et al. 1999; Derynck et al. 2001; Hugo et al. 2007). In fact, its related proteins cause transcription of different mesenchymal genes and repression of epithelial genes (Xu et al. 2009). These phenotypic changes finally promote cell mobility and migration to the premetastatic niche (bone microenvironment), and eventually cellular differentiation into distinct cell types (acquisition of the osteomimetic phenotype, osteomimicry). EMT is initiated by external signals, the extracellular matrix and soluble factors such as those of the TGF-β superfamily. These signaling pathways are thought to control the invasive behavior of solid cancers.
1.1.3 Tumor-Associated Macrophages
Solid tumors are not only composed of malignant cells, but they are also complex organ-like structures comprising many cell types, including a wide variety of migratory hematopoietic and resident stromal cells. Migration of these cell types into tumors has been interpreted as evidence for an immunological response of the host against any growing tumor. However, it is now acknowledged that tumors are largely recognized as self and lack efficient antigens. Instead, they appear to have been selected to escape the host immune system, to prevent rejection and facilitate tumor growth and spreading. This led to the proposal that infiltrates of hematopoietic cells have a causal role in carcinogenesis. Clinical data collected from a wide range of solid tumors underscore these findings, showing high densities of leukocytic infiltrations—mostly macrophages—correlating with a poor prognosis. Tumor-Associated Macrophages (TAM) originate from circulating monocytes and are activated macrophages of the polarized type II (M2 macrophages or activated macrophages), mainly induced by IL-4, IL-10, IL-13 and corticosteroids. Differential cytokine and chemokine production, and coordinated temporal and spatial activities of these cells in the tumor stroma are key features of polarized macrophages, which promote tumor angiogenesis and growth (Pollard 2009). These data suggest the new hypothesis that tumors can modify the behavior of macrophages from a potentially hostile antitumor phenotype to one that promotes malignancy. But what is the precise nature and function of these tumor-promoting macrophages? Can they participate in the building of a premetastatic niche? Increasing data support the identifying of a specific subpopulation of macrophages which:
-
1.
Express the endothelial-cell marker TIE2 receptor (also known as TEK); their importance is shown by their ablation that blocks angiogenesis in xenograft tumors (De Palma et al. 2007).
-
2.
Secrete VEGF through the HIF pathway (Murdoch et al. 2008).
-
3.
Facilitate tumor cell motility; moreover, intravasation of tumor cells also occurs next to clusters of macrophages on the vessel surface (Wyckoff 2007).
-
4.
Secrete MMP leading to ECM degradation (Hagemann 2005).
-
5.
Secrete TNF, and activates the wNT-β-catenin pathway (Pukrop et al. 2006).
Moreover, TAM produce TGF-β which is involved in the process of EMT (Mantovani et al. 2006; Pollard 2009; Kagan and Li 2003). By doing so, macrophages may participate in the building of a premetastatic niche. M2 macrophages are known as differentiated cells in response to parasitic infection, allergic conditions and during tissue repair. IL-13 and IL-4 are the most important cytokines supporting the process of EMT. In the tumor microenvironment however, macrophages develop in the presence of growth factors such as CSF1, or in response to molecules that signal through nuclear factor-κB (NF-κB), becoming non-immunogenic and trophic (Pollard 2009). Overall, these data suggest macrophages to substantially participate in the building of the premetastatic niche.
1.1.4 Lysyl Oxidase
Lysyl Oxidase (LOX) is produced by fibrogenic cells. The main activity of LOX is thought to be oxidation of specific lysine residues of collagen (Kagan and Li 2003). Increased expression of LOX has been found in metastatic and/or invasive breast cancer cell lines and is associated with higher stages of disease in patients with renal cell carcinoma (Kirschmann et al. 2002). Hypoxic tumor cells often have increased LOX expression and secretion, enabling cell movement to more oxygenated and nutrient-rich areas. Then, LOX increases cell invasion and migration through regulation of cell–matrix adhesion. In addition, matrix remodeling by invasive tumor cells provides a more ideal soil for any succeeding tumor cells, building a kind of “highway to metastasis”. LOX may be involved in tumor interactions with the cell–matrix required for intravasation and extravasation. Then, LOX is required for the formation of a mature ExtraCellular Matrix (ECM) at the secondary site, allowing tumor cell survival and possibly BMDC recruitment (Erler and Giaccia 2006). LOX secreted by hypoxic tumor cells accumulates at premetastatic sites, crosslinks collagen IV in the basement membrane and is essential for myeloid cell recruitment. Finally, tumor cells adhere to crosslinked collagen IV and produce matrix metalloproteinase-2, which cleaves collagen, enhancing the invasion and recruitment of BMDCs and metastasizing tumor cells (Erler et al. 2009).
1.2 The Long Way to the Bone: The Importance of Chemokines
Chemokines are small chemoattractant cytokines that bind to specific G-protein-coupled transmembrane receptors present on the plasma membranes of target cells. These molecules can guide circulating cancer cells to the bone.
1.2.1 CXCR4\SDF-1 Pathway
The chemokine receptor CXCR4 (or CD184) is an alpha-chemokine receptor for stromal-derived-factor-1 (SDF-1 or CXCL12) alpha or beta. SDF-1 and CXCR4 are a relatively ‘monogamous’ ligand-receptor pair (in contrast to other chemokines that bind several chemokine receptors in a more ‘promiscuous’ manner) (Arya et al. 2007). In the normal bone marrow, SDF-1 is produced by osteoblasts, fibroblasts and endothelial cells. Parathyroid hormone (PTH), PDGF (platelet-derived growth factor), interleukin-1 (IL-1), vascular endothelial growth factor (VEGF) and tumor necrosis factor alpha (TNF-α all induce SDF-1 production by osteoblasts (Jung et al. 2006). SDF-1 is important in hematopoietic stem cell homing to the bone marrow and in hematopoietic stem cell quiescence. It has been demonstrated that by blocking the CXCR4 receptor, hematopoietic stem cells mobilize into the bloodstream as peripheral blood stem cells. Moreover, it has been demonstrated that SDF-1 production can induce all of the following:
-
1.
Osteoclast precursor recruitment by promoting chemotaxis, proteinase activity and collagen transmigration (Yu et al. 2003a, b).
-
2.
Angiogenesis by recruiting endothelial progenitor cells (EPC) from the bone marrow via CXCR4-dependent mechanisms (Zheng et al. 2007).
- 3.
There are many experimental data concerning the importance of the CXCR4\SDF-1 pathway for neovascularisation and metastatic spreading to SDF-1-expressing tissues, especially the bones:
-
1.
Cancer cells in some organs such as lung, liver and bone produce large quantities of SDF-1 (Muller et al. 2001).
-
2.
SDF-1 mRNA expression is observed in the metaphysis of the long bones, near the endosteal surfaces covered by osteoblastics (Sun et al. 2005). In an experimental animal model of breast cancer bone metastasis, it has been demonstrated that single tumor cells are homing to the metaphyses of the long bones after systemic inoculation. Furthermore, they were mostly located in close proximity to osteoblasts and lining cells (Phadke et al. 2006).
-
3.
A gradient of chemokine expression was found between peripheral blood and bone marrow, with an increased expression of SDF-1 in the bone marrow along with lower concentrations of SDF-1 in serum, while increased expression of CXCR4 is found in peripheral blood with lower expression in bone marrow. This gradient promotes cancer cell migration into the bone marrow and prevents further trafficking of cancer cells (Hofbauer et al. 2008).
-
4.
There is some evidence that activation of the SDF-1/CXCR4 pathway not only regulates migration and homing of cancer cells to the bone but also regulates adhesion, invasion and cytoskeletal rearrangement of cancer cells (Gerritsen et al. 2002).
-
5.
Blocking CXCR4 with antibodies reduces the formation of experimental bone metastases induced by CXCR4-expressing breast or prostate cancer cells (Liang et al. 2004).
1.2.2 RANK\RANK-L Pathway
The RANK\RANK-L pathway is involved in tumor-induced osteoclastogenesis and osteolysis. RANK-L is physiologically produced by osteoblasts and stimulates osteoclast precursor recruitment and maturation. Then, mature osteoclasts cause bone reabsortion (Lacey et al. 1998). Normal glandular epithelial cells express RANK, and the RANK–RANKL pathway is involved in normal development of lactating mammary glands (Fata et al. 2000). It has been demonstrated that RANK expression facilitates cancer cell migration into the bones. Recent in vivo studies demonstrated the association between RANK expression and cancer cell osteotropism (Jones et al. 2006). In fact, it has been found that RANK is expressed by solid tumors, with a high concordance of the expression profile between bone metastases and corresponding primary tumors (Santini et al. 2010a, b). Moreover, RANK is clearly associated with early bone metastasis formation in breast cancer (Santini et al. 2010a, b) and, consistent with those data, it has been demonstrated that osteoprotegerin (OPG), a natural RANK-inhibitor, blocks cancer cell osteotropism (Dougall and Chaisson 2006).
1.2.3 The Integrin System
The study by Kaplan et al. (2005) showed that VLA4 or anti-VEGFR1 antibodies inhibit the proliferation and binding affinity of tumor cells to VEGFR1-positive HPCs, demonstrating their direct role in adhesion and growth of tumor cells. Moreover, other integrins are critical for cancer cell homing in other target organs. In fact, breast cancer cells expressing αvβ3-integrin and prostate cancer cells expressing αvβ2-integrin, which bind many bone matrix components, have a higher propensity for spreading to the bones (Pecheur et al. 2002). Additionally, proto-oncogenic tyrosine kinase c-SRC is similarly involved in the integrin’s pathway, and has an important role in cancer cell osteotropism. SRC binds to activated RANK, thereby recruiting TRAF6 and Grb2-associated binder 2 (Gab2), followed by phosphorylation of IκBα and JNK, which ultimately leads to activation of the transcription factors NF-κB and AP-1.
There are clinical and experimental data suggesting that SRC expression:
-
1.
Increases the survival of tumor cells in the bone microenvironment, leading to the establishment of bone metastases (latent phase of bone metastasis).
-
2.
Increases cell mobility (Zhang et al. 2009).
-
3.
Increases osteotropism of tumor cells (Rucci et al. 2006; Boyce et al. 2003).
Moreover, expression of SRC is involved in bone marrow seeding and sustains the outgrowth of indolent cancer cells in the bone marrow microenvironment. Activated SRC plays a critical role in the initiation and maintenance of a tumor cell response to bone-derived factors CXCL12/SDF and TRAIL. Latency status is maintained until overt progression to the phase of osteolytic outgrowth.
1.2.4 The BMP Receptor Axis
Bone morphogenetic proteins (BMP) are members of the TGF-β (transforming growth factor-β) family. So far, three BMP receptors have been characterized: BMP-R Ia, Ib and II (van Dijke et al. 1994). It has been shown that prostate, breast and lung cancer cells express BMP-2 mRNA and its protein. Moreover, BMP receptors are expressed in prostate cancer cell lines (Schwalbe et al. 2003). The most important functions of BMP-2 in cancer cells are:
-
1.
Promotion of cancer cell migration to the bones (Ite et al. 1997).
-
2.
Modulation of cancer cell migration through the integrins axis.
In fact, breast cancer cell lines upregulate bone sialoprotein (BSP) expression in pre-osteoblasts (Bunyaratavej et al. 2000), and in vivo inhibition of BMP in osteoinductive prostate cancer cells inhibits the osteoblastic response in bone (Schwaninger et al. 2007). Moreover, BMP has been shown to reverse TGF-β-induced EMT by decreasing vimentin expression and increasing E-cadherin expression in breast cancer cells and in normal mouse mammary epithelial cells (Zeisberg et al. 2003; Valcourt et al. 2005).
The role of the cited chemokine network is summarized in Table 2.
1.2.5 Other Adhesion Molecules
1.2.5.1 Osteopontin
Osteopontin (OPN) is a glycophosphoprotein and is one of the major components of non-collagenous bone matrix. OPN is expressed in osteoblasts and osteocyctes (bone-forming cells) as well as osteoclasts (bone-resorbing cells). Osteopontin promotes the adherence of osteoclasts and hematopoietic stem cells to the bone matrix (Asou et al. 2001). Hypocalcemia and hypophosphatemia both stimulate kidney proximal tubule cells to produce calcitriol (1α,25-dihydroxyvitamin D3) that stimulates OPN gene translation via the VDRE (vitamin D response element) in the OPN promoter region (Prince and Butler 1987; Yucha and Guthrie 2003). OPN expression is also regulated by exposure of cells to various factors and metabolic settings, including tumor necrosis factor α, infterleukin-1β, angiotensin II, transforming growth factor β (TGFβ) and parathyroid hormone (PTH), hyperglycemia and hypoxia (Noda and Rodan 1989; Hullinger et al. 2001). Finally, it has been shown that OPN is involved in bone metastatic spread of breast, prostate and lung cancer cells (Wai and Kuo 2004), and OPN expression confers migratory ability and invasive phenotype in human mammary cells (Tuck et al. 2003).
1.2.5.2 Sinusoidal Endothelial Cell Adhesion Molecules
Cancer cells usually settle in bone metaphysis that is rich in sinusoidal microvasculature, rather than bone diaphysis. This is mainly caused by:
-
1.
Hemodynamic properties of the sinusoidal vascular bed, with 90% of the blood circulating through metaphyseal sinusoids.
-
2.
Characteristics of the sinusoidal endothelia, allowing adhesion and transmigration of hematopoietic and lymphoid cells.
In a model studying the kinetics of metastatic breast cancer cell trafficking in bone, it has been shown that the majority of cancer cells tended to settle in the endosteal marrow, rather than in the centrum of the marrow and that primary tumor cells most often locate in close proximity to osteoblasts and bone lining cells.
However, migration to the sinusoids of the bone marrow is not sufficient to ensure colonization by cancer cells. Moreover, cancer cells migrate through the vasculature using a process of attachment–detachment through cell adhesion mediated by several adhesion molecules such as E-selectin, N-cadherin, intracellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule (VCAM-1) (Makuch et al. 2006). VCAM-1, a member of the immunoglobulin family of cell adhesion molecules, has ICAM-1 and VLA-4 as its main receptors, with the latter being constitutively activated in osteoclasts and are also found on many cancer cells. Inflammatory cytokines produced by osteoblasts in the presence of breast cancer cells may cause endothelial activation, expression of adhesion molecules and cancer cell invasion (Glinsky et al. 2001).
1.2.5.3 Endothelin 1 and ET Receptors
Endothelin-1 (ET-1) has been detected in osteocytes, osteoblasts, osteoclasts and vascular endothelial cells (Sasaki and Hong 1993). Endothelin-1 stimulates mitogenesis in osteoblasts (Stern et al. 1995). Moreover, ET-1 enhances the effect of other osteoblast-stimulatory factors, such as BMP-7, to induce bone formation (Nelson et al. 1995; Kitten et al. 1997).
Additionally, ET-1 stimulates the expression of osteopontin and osteocalcin in rat osteosarcoma cells (Shioide and Noda 1993), and mineralization of the bone matrix also depends on the ET-1 receptor pathway. Other studies of ET-1 null mice showed that ET-1 may regulate proliferation and migration of osteogenic cells rather than modulating the expression of bone matrix proteins (Kitano et al. 1998).
Nevertheless, the role of the ET-1 pathway in bone metastasis is not clear. Malignant tumors of the breast and prostate are typically associated with osteoblastic bone metastases, and both tumors express ET-1 and its receptors. Importantly, paracrine effects of ET-1 on bone cells may provide a favorable growth environment for tumor cells in bone. Endothelin-1 is found in normal prostate epithelium, throughout the entire gland. Addition of exogenous ET-1 increases the proliferation of prostate cancer cells and enhances the mitogenic effects of IGF-1, IGF-2, PDGF, epidermal growth factor (EGF) and FGF-2 on cancer cells. Moreover, ET-1 concentrations were significantly higher in men with advanced, hormone-refractory prostate cancer with established metastases to the bones as compared to patients with early-stage disease (Nelson et al. 1995). Increased ET-1 production has been described in prostate cancer cells through contact with bone (Chiao et al. 2000). Tumor-derived ET-1 stimulates new bone formation via ETA receptors on the surface of osteoblasts. Subsequently, growth factors produced by osteoblasts are incorporated into the new bone matrix as well as the local microenvironment. Interestingly, ET-1 mediates vasoconstriction of distal blood vessels, but not of those vessels directly supplying the tumor bed. This ‘vascular steal phenomenon’ improves local blood supply to the tumor and thereby improves oxygenation of tumor cells. Activation of endothelial cells by ET-1 stimulates the production of vascular endothelial growth factor (VEGF) by increasing the levels of hypoxia-inducible factor-1α (HIF-1α). Endothelin-1, acting through ETA, also induces the expression and activation of tumor-associated proteases, matrix metalloproteinases (MMPs) and urokinase plasminogen activator (uPA) (Spinella et al. 2003). Metalloproteinases degrade the local tissue matrix to permit local tumor invasion and formation of metastases. Urokinase plasminogen activator in turn converts plasminogen into plasmin, which degrades the tumor stroma, allowing the tumor to invade the surrounding tissue and prepare metastatic spread. Urokinase plasminogen activator might also activate MMP. Taken together, these factors have the potential to stimulate tumor growth as well as further increase tumor production of ET-1 (Guise et al. 2003).
1.3 Modification of the Bone Microenvironment: Premetastatic Niche Formation
Many data suggest the hypothesis that growth of macrometastases starts from the interaction between the target organ (e.g. bone) and the primary tumor that builds the premetastatic niche. In fact, studies in breast cancer demonstrated that although about 30% of patients may have micrometastatic disease in their bone marrow at the time of presentation, only 50% of these patients develop overt bone metastatic disease after 5 years. Furthermore, many patients with breast and prostate cancer do not develop bone metastases until many years after the surgical removal of the primary cancer. The Paget’s “seed and soil theory” hypothesizes that metastatic cells can enter into the bone marrow and remain in a quiescent status for many years. However, molecular mechanisms and signaling pathways that promote the switch to a premetastatic bone microenvironment are not well understood. We can postulate that cancer stem cells, like physiologic haematopoietic stem cells, can establish a relationship with bone marrow stroma in order to mantain their survival. Physiologically, two fundamental niches exist in the bone marrow:
-
1.
Endosteal Niche. Stem cells are closely associated with spindle-shaped N-cadherin positive osteoblasts (SNO). Moreover, these osteoblasts are involved in the maintenance of stem cell quiescence. The same osteoblasts might also bind cancer stem cells, thereby maintaining their dormancy (Yin et al. 2006).
-
2.
Vascular Niche. More differentiated cells are generally located in central parts of the bone marrow.
It is well understood that circulating cancer cells arrive in the endosteal niche and are kept in a dormancy status via the link with SNO and the release of inhibitory molecules by stromal cells (such as fibronectin). RANK\RANK-L is also involved in cancer cell reactivation via osteoclast activation and the consequent release of growth factors such as TGF-β, BMPs, PTH-related protein (Oh et al. 2004). These factors are typically involved in the process of epithelial–mesenchymal transition, and likely activate dormant metastatic cancer cells. Activated osteoclasts may directly induce hematopoietic stem cells, and probably metastatic cancer cells, to separate from the endosteal niche through increasing proteolytic activity of MMP-9 and cathepsin K, which clive and inactivate SDF-1, osteopontin and other niche factors (Kollett et al. 2006). Moreover, stromal cells secrete inactive metalloprotease-2, which is activated by cancer cells increasing their migratory capacity (Harada and Rodan 2003). At the time of bone marrow colonization, when pathologic bone lesions are still not evident, cancer cells acquire a bone-like phenotype, and this process is known as osteomimicry (Knerr et al. 2004).
1.4 Osteomimicry
We still do not know whether cancer cells already possess the osteomimetic phenotype when they detach from their primary tumor, or whether these characteristics are acquired when they colonize the bone niche. There is some evidence though that at least some cancer cells do need a biological signature to invade osseous structures (Ramaswamy et al. 2003).
As shown in Table 3, many molecules are produced from cancer cells invading bone marrow.
1.4.1 PTHRP
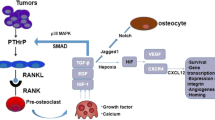
The parathyroid hormone-related protein is a homolog of PTH and has a direct action on PTH receptors, stimulating bone resorption and renal tubular calcium resorption (Yates et al. 1988). PTHRP is released by cancer cells of many solid tumors (Moseley et al. 1987; Burtis et al. 1987; Strewler et al. 1987), and contributes to metastatic spreading. It has been demonstrated that PTHRP is abundant in tumors with osteoclastic and osteoblastic bone metastases. In fact, breast carcinomas metastatic to the bones express PTHRP in >90% of the cases, compared with only 17% of metastases to extraosseous sites (Southby et al. 1990; Grill et al. 1991; Powell et al. 1991; Vargas et al. 1992). Furthermore, growth factors such as TGF-β or IGF, which are abundant in mineralized bone matrix (Hauschka et al. 1986), are released and activated by osteoclastic bone resorption (Pfeilschifter and Mundy 1987) and may enhance PTHRP secretion from cancer cells (Zakalik et al. 1992; Merryman et al. 1994). Finally, other studies have shown that PTHRP, secreted by prostate cancer cells, stimulates ostoblastogenesis and osteoblast differentiation (Liao et al. 2008). Accordingly, it was postulated that PTHRP causes bone microenvironment modification in order to facilitate the establishment of circulating cancer cells, the release of bone-derived growth factors and the formation of the premetastatic bone niche.
1.4.2 RANKL\OPG\TRAIL
RANKL is produced by osteoblasts and stromal cells for inducing osteoclast activation and bone resorption. It was demonstrated that many types of solid tumors produce RANKL, both at the primary site and in metastatic bone lesions (Brown et al. 2001a, b; Chen et al. 2006; Sasaki et al. 2007). OPG is a soluble decoy-receptor of RANKL, which is expressed by various cell types, including osteoblasts and tumor-associated stromal cells. OPG binds RANKL and prevents RANK–RANKL association, inhibiting osteoclast development (Cross et al. 2006). It was observed that many tumor cell lines produce OPG (Holen et al. 2005; Holen et al. 2002). High serum levels of OPG were found in patients with advanced-stage prostate cancer (Brown et al. 2001a, b). Furthermore, inhibition of RANKL results in inhibition of malignant bone lesions and tumor growth in bone (Canon et al. 2008; Kostenuik et al. 2009), while OPG inhibits cancer-induced osteoclastogenesis (Zhang et al. 2001) and increases bone density. Moreover, OPG shares some sequence homology with Endothelin-1 (ET-1), and was shown to stimulate bone formation through ET-A receptor activation (Nelson et al. 1999). OPG also binds tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) (Emery et al. 1998), the latter being an anticancer cytokine. Finally, based on high constitutive RANK expression in breast cancer cell lines, recent data actually indicate that RANK expression by cancer cells determines whether tumors predominantly migrate into bone, where the corresponding ligand RANKL is abundantly expressed (Jones et al. 2006). In murine animal models, the correlation between high expression of RANK and osteotropism has been demonstrated across different tumor types, including breast cancer and melanoma.
1.4.3 VEGF
Tumor induces angiogenesis through VEGF signaling (Ferrara 2009). Moreover, VEGF secretion attracts bone marrow-derived cells such as VEGFR1-positive HPC and VEGFR2-positive EPC to neoangiogenic sites in the tumor (Lyden et al. 1999). VEGF is closely associated with the early phases of bone remodeling and induces osteoblast chemotaxis and differentiation (Tombran-Tink and Barnstable 2004; Li et al. 2005). VEGF also upregulates RANK on endothelial cells, resulting in the amplification of the angiogenic response in the presence of RANKL (Min et al. 2003). In effect, VEGF mediates both a direct and an indirect effect on bone growth by activating osteoblasts and promoting angiogenesis in metastatic sites with high concentrations of RANKL (Street et al. 2002). For these reasons, the localized production of VEGF, such as that observed in metastatic tissue, is likely to contribute to osteolysis and local tumor progression (Aldridge et al. 2005).
1.4.4 C-KIT\SCF
C-kit (or CD 117) is a tyrosine kinase receptor of the Stem Cell Factor (SCF). In vitro models demonstrated that prostate cancer cells in bone express high levels of c-kit, while prostate cancer cells from extraosseous sites are c-kit negative. Additionally, SCF was found to be overexpressed in bone metastases from prostate cancer (Wiesner et al. 2008). In vivo models showed that prostate cancer cells preferentially metastasize to regions of the bone marrow where SCF was expressed by stromal cells (BMS) interacting with c-kit expressed on the surface of bone marrow progenitor cells (Heissig et al. 2002). There is an association between coexpression of SCF and c-kit in prostate cancer cells and bone metastases, suggesting that both ligand and receptor should be expressed by prostate cancer cells for intraosseous development, also suggesting some autocrine loops. The increased expression of c-kit in bone metastases from prostate cancer might be the result of a selective dissemination and/or growth into the bone of c-kit-positive cells already present in primary tumors of the prostate, which are known to be heterogeneous (Patrawala et al. 2006). These c-kit-positive cancer cells could also represent cancer stem cells which were reported to be present in some metastatic lesions from human carcinomas (Kleeberger et al. 2007; Wiesner et al. 2008).
1.4.5 IGF
The Insulin-Like Growth Factors (IGF) comprise 3 receptors, 3 ligands and 6 binding proteins (IGFBP). IGF-I and IGF-II are known to induce osteoblast differentiation, increase bone matrix apposition and decrease collagen degradation (Koch et al. 2005). IGF are abundant in the bone microenvironment, and in vitro studies showed that they increase prostate cells proliferation and chemotaxis. Moreover, the IGF-I pathway is upregulated in prostate cancer cells localized in the bones (Ritchie et al. 1997a, b; Rubin et al. 2004). However, IGF-I is neither necessary nor sufficient for an adequate osteoblast response to prostate cancer metastases (Rubin et al. 2004).
In addition, tumor cells invading the bone express several transcription factors that are involved in the acquisition of the osteomimetic phenotype, among other RUNX.
1.4.6 RUNX
RUNX is a family of transcription factors (RUNX2 and RUNX1 and RUNX3). It was demonstrated that RUNX2 is essential for the differentiation of osteoblasts and skeletal morphogenesis (Li et al. 2008). RUNX2 is overexpressed in metastatic breast cancers cells, promotes transcription of genes involved in the acquisition of migration and invasiveness, including MMP and VEGF among others. Importantly, inhibition of Runx2 function in metastatic breast cancer cells transplanted to bone results in prevention of tumorigenesis and osteolysis (Javed et al. 2005).
These data indicate that a multigenic program facilitates the acquisition of osteomimetic properties by certain cancer cells, improving their chance for survival, adaptation to the bone environment and the development of bone metastases.
1.4.7 The Role of the Calcium Sensing Receptor in Bone Marrow Homing
The calcium sensor (CaSR) is also expressed in several cell types in the kidney, osteoblasts, a variety of hematopoietic cells in the bone marrow, the gastrointestinal mucosa and squamous epithelial cells of the esophagus. At these sites, the CaSR has been implicated in the regulation of a number of cellular processes, such as ion and water transport, proliferation, differentiation and apoptosis. Moreover, human breast cancer cell lines express CaSR and its activation leads to increased PTHrP secretion from these cells. The secretion of PTHrP by tumoral cells drives osteoclast-mediated osteolysis, leading to the release of growth factors and calcium from the bone matrix, and further stimulation of cell proliferation (Coyle et al. 2006). Additionally, the loss of CaSR expression in the transition from normal colonic epithelial cells to malignant adenocarcinoma cells is associated with a low potential of colonic carcinomas to generate bone metastases (Rodland 2004).
1.5 Role of Calcium, Vitamin D and PTH in the Premetastatic Niche
1.5.1 Vitamin D
Serum concentrations of calcium are closely regulated by a close interplay between 25(OH)D levels and PTH levels. The precision of this integrated control is such that in normal individuals, serum ionized calcium fluctuates by no more than 0.1 mg/dl in either direction from its physiological set-point throughout the day. Interestingly, PTH gene expression is not only regulated through serum calcium concentrations by the CaSR but also independently through vitamin D metabolites, principally 25(OH)D, regardless of both 1,25(OH)D and calcium. (Pepe et al. 2006). 25(OH)D and 1,25 (OH)D control PTH gene expression, CaSR and VDR gene expression, and the proliferation of parathyroid cells. Therefore, low serum calcium concentrations and/or low vitamin D levels increase PTH secretion, in turn increasing distal tubular calcium reabsorption, intestinal 1,25(OH)D-mediated calcium absorption and osteoclast-medicated bone resorption to normalize serum calcium concentrations.
1.5.2 Antineoplastic Effect of Vitamin D
Vitamin D depletion (and secondary hyperparathyroidism) has been reported as a very common condition worldwide both in men and women, with many implications for general health conditions (Holick and Chen 2008). Very recently, it has been documented that low serum vitamin D levels are similarly prevalent in young individuals (Adami et al. 2009; Crew et al. 2009). Biological and epidemiological data suggest vitamin D levels to influence cancer development (IARC Working Group Reports 2008), but data are not consistent. For breast and prostate cancer, case-control studies suggest an inverse association between serum 25(OH)D concentration and the prevalence of these diseases, but this finding was not confirmed by prospective studies that analyzed 25(OH)D years before the diagnosis of cancer (Yin and Grandi 2010; Trump et al. 2009; Tretli et al. 2009; Yin et al. 2009; Chlebowski et al. 2008; Lappe et al. 2007). This might indicate that low 25(OH)D concentrations in serum are rather a consequence of the malignant disease than causing cancer. Also, studies on intake/supplementation of vitamin D in breast or prostate cancer patients showed conflicting results (Trump et al. 2006; Flaig and Barqawi 2006; Chan and Beer 2008; Attia et al. 2008). Many molecular pathways mediate the anticancer effects of calcitriol. The active form of vitamin D,1,25(OH)D has been established as an antiproliferative and pro-differentiation agent. More recent work showed calcitriol to be a proapoptotic agent and an inhibitor of cell migration and angiogenesis, supporting its potential in cancer prevention and cure (Peterlik and Grant 2009; Matthews et al. 2010). Among the breast cancer cell lines that do respond to 1,25(OH)D, a range of phenotype alterations have been reported, emphasizing that the mechanistic basis for the differentiating effects of 1,25(OH)D in cellular systems of breast cancer is very complex. (Gocek and Studzinski 2009). An interesting link to differentiation in 1,25(OH)D-treated breast cancer cells is the fact that vitamin D receptor (VDR) and Estrogen Receptor (ER) pathways converge to regulate BRCA-1, thus controlling the balance between cellular differentiation and proliferation (Campbell et al. 2000). In the prostate cancer cell line LNCaP, 1,25(OH)D up-regulates the expression of the insuline-like factor binding protein 3 (IGFBP-3) that functions to inhibit cell proliferation and up-regulates the expression and activity of the androgen receptor (AR) and the AR-mediated androgenic differentiation (Gocek and Studzinski 2009). The receptor of vitamin D (VDR) has been described in many types of cancer cells, including tumors of the breast, prostate, colon, bladder, skin, pancreas, leukaemia and lymphoma cells (Bouilon et al. 2006). In Caucasians, polymorphisms of VDR (VDR FokI and BsmI) migh modulate the risk of malignant tumors of the breast, skin and prostate, and possibly affect cancer risk also at other sites (Raimondi et al. 2009).
1.5.3 Inhibition of NFkB Activation, Angiogenesis, Invasion and Metastasis
Angiogenic factors such as IL8 and VEGF are crucial for the promotion of the premetastatic niche, and are similarly important for continued tumor growth and disease progression. NFkB plays a major role in the control of immune responses and inflammation, and promotes malignant behaviour by increasing the transcription of the antiapoptotic gene BCL-2, proteolytic enzymes such as matrix metalloproteinase 9 (MMP-9), urokinase-type plasminogen activator and angiogenic factors such as IL-8 and VEGF (Catz and Johnoston 2001). Calcitriol is known to directly modulate basal and cytokine-induced NFkB activity in many cells, including lymphocytes, monocytes, fibroblasts, osteoblasts and in cancer cells. In addition to the direct inhibition of NFkB, 1,25(OH)D indirectly inhibits NFkB-signaling by up-regulating the expression of other proteins that interfere with NFkB activation such as IGFBP-3 and Clusterin (CLU) (Folkman 1995). Early studies indicate that calcitriol is a potent inhibitor of tumor cell-induced angiogenesis by inhibiting VEGF-induced endothelial cell tube formation in vitro, decreasing tumor vascularization in mice and inhibiting angiogenesis through IL-8 in a NFkB-dependent manner (Bao et al. 2006). Furthermore, calcitriol directly inhibits the proliferation of endothelial cells (Chung et al. 2009). MMP in turn promote angiogenesis by mediating the degradation of the basement membrane of the vascular epithelium and the extracellular matrix. In human prostate cancer cells, calcitriol decreases the expression of MMP-9 by increasing the activity of TIMP-1 (tissue inhibitor of MMP-1) (Bao et al. 2006). Finally, it has been demonstrated that calcitriol reduces the invasive and metastatic potential of many malignant cells. In prostate cancer, calcitriol increase E-cadherin, a tumor suppressor gene, whose expression is inversely correlated with metastatic potential (Campbell et al. 1997).
1.5.4 Calcium
Inadequate calcium intake, low serum calcium (through CaSR) together with low vitamin D levels may directly or indirectly (through PTH) impact cell proliferation, differentiation and function. The CaSR, VDR and PTH-1R are expressed both in normal and malignant breast cells, and their expression is correlated with the occurrence of skeletal metastases. Interestingly, signaling pathways that are initiated via VDR and CaSR converge on the same downstream elements, e.g. the canonical Wnt pathway. Increasing extracellular calcium levels increases cellular differentiation in experimental models, decreases proliferation, induces apoptosis and down-modulates invasion, all of which seem to have tumor-protective effects (McGrath et al. 1984). Several studies have suggested an inverse association between dietary calcium intake and serum calcium levels with breast cancer risk in pre- and post-menopausal women (Almquist et al. 2007; Cui and Rohan 2006). However, the relationship between serum calcium concentrations and cancer risk is not consistent and complex, also because of the reciprocal effects of PTH and vitamin D levels. High serum calcium concentrations could indicate high bone turnover, which in turn suggests a bone microenvironment rich in chemotactic, adhesive and neoangiogenic factors that might promote the homing of cancer cells and the development of metastases (Schnieder et al. 2005). On the other hand, low serum calcium concentrations are generally associated with low 25(OH)D levels and high PTH levels that both promote cancer progression and bone metastases. Very likely, calcium concentrations play a crucial role in cellular signaling in bones in general and in the premetastatic niche in particular. The bone microenvironment is enriched in calcium during osteoclast-mediated bone resorption, reaching high local concentrations up to 40 mmol/L. Serum calcium homeostasis is closely regulated by the calcium sensing receptor (CaSR) expressed on parathyroid cells, modulating PTH secretion. However, CaSR is also expressed at high levels in breast cancer cells from patients with bone metastases and in prostate cancer cells (Mihai 2008). Activation of the CaSR by high calcium concentrations induces PTHrP expression, but may also attract breast and prostate cancer cells in areas of increased bone remodeling, thereby facilitating migration of cancer cells into the bones. Furthermore, high concentrations of calcium enhanced proliferation of prostate cancer cell lines and the proliferative response is associated with CaSR overexpression (Casimiro et al. 2009). Notably, CaSR is essential for stem cell migration and the settlement of HSC in the endosteal niche, suggesting preferential localization of these cells expressing CaSR in close proximity to calcium-releasing osteoclasts (Adams 2005).
1.5.5 PTH
1.5.5.1 PTH and Factors Involved in the Premetastatic Niche
Primary and secondary hyperparathyroidism is associated with a poor prognosis in patients with cancer (Schwartz 2008). PTH and PTHrP are immunologically distinct proteins that bind to the PTH-1R with equal affinity (Bryden et al. 2002). Many cancer cell types express PTHrP and its receptor PTH-1R, and PTHrP acts as an autocrine growth factor promoting proliferation, migration and disease progression (Deftos et al. 2005; Henderson et al. 2006). PTH could play a crucial role in promoting the homing of cancer cells in the bone environment, the persistence of the cancer stem cell niche, and the development of cancer metastases (Ritchie et al. 1997a, b). PTH and PTHrP both induce the activation of chemokines through PTH-1R, further inducing SDF-1 expression in the bone marrow. Major sources of SDF-1 in the marrow are cells of the osteoblastic lineage, mainly osteoblasts lining the bone endosteum (Ponomariov et al. 2000). The SDF-1/CXCR4 axis is known for regulating many aspects of stem cell functioning, including stem cell trafficking and development. It has been demonstrated that transgenic animals expressing constitutively active PTH/PTHrP receptors have increased numbers of hematopoietic stem cells recovered from the animals' bone marrow (Calvi et al. 2003). Interstingly, PTH increased the expression of SDF-1 in the local marrow environment in animal models, along with decreased SDF-1 in serum, creating a homing gradient for hematopoietic stem cells towards the bone marrow (Jung et al. 2006). There are many parallels between the metastasis of circulating carcinoma cells and the homing behavior of hematopoietic cells (Sun et al. 2003). Therefore, high circulating PTH levels in cancer patients could prime the marrow for metastatic spread by altering the SDF-1 axis. Physiologic bone remodeling takes place in specialized vascular structures called bone remodeling compartments (BRC), and PTH induces high bone turnover with subsequent expansion of the BRC space (Eriksen et al. 2007) Angiogenesis is closely associated with bone turnover, and angiogenic factors such as VEGF and endothelin are important regulators of both osteoclast and osteoblast activity. In addition, it has recently been demonstrated that PTH induces osteoclast formation in cooperation with RANKL, osteoclast activity through KDR/Flk-1 and/or Flt-1 receptors expressed in mature osteoclast, and survival involving beta-3-integrin-mediated attachment of osteoclasts to the extracellular matrix (Nakagawa et al. 2000). PTH/PTHrp induces PKC, ERK, MAPK and p38, with the MAPK pathway ultimately resulting in VEGF gene expression in osteoblasts and in epithelial cells of normal rat renal tubules (Esbrit et al. 2000; Alonso et al. 2008). VEGF expression was specifically observed in PTH1R-positive cancer cells after invasion of the bone marrow, using in vivo and in vitro models (Isowa et al. 2010). Finally, VEGF has been reported to increase SDF-1 expression in endothelial cells and in several prostate cancer cell lines (Dai et al. 2004).
1.5.5.2 PTH and the Hematopoietic Stem Cell Niche
The endosteal surface is rich in vasculature with close approximation of osteoblasts and vessel walls. Within the bone marrow, cells of the osteoblast lineage have unequivocally been shown to constitute a niche for Hematopoietic Stem Cells (HSC) (Xie et al. 2009). Cells derived from osteoprogenitors provide distinct niches for hematopoietic cells. Terminally differentiated osteoblasts along the endosteal surface could serve as a niche for HSC in their most quiescent stage, whereas the stromal reticular cell fraction including osteoprogenitors located in the bone marrow induce proliferation and differentiation of hematopoietic cells (Wu et al. 2009). It is widely known that stem cells are usually in the quiescent state or G0 phase, and this prevents stem cells from entering into the cell cycle and undergoing differentiation. CXCL12/CXCR4-mediated chemokine signaling plays an essential role in maintaining the quiescent pool of HSC (Sugiyama et al. 2006). Osteopontin (OP) and Angiopoietin-1 expressed by osteoblasts interact with Tie-2 expressed in HSC, activating N-cadherin and integrin (Arai et al. 2004). These interactions enhance the adhesion between the hematopoietic stem-cell niche and the stem cell, contributing to the maintenance of HCS quiescence. The bone morphogenetic protein (BMP) signaling pathway, which acts through BMP receptor type IA expressed in osteoblast, controls the number of HSC by regulating the size of the niche and is involved in maintaining quiescence and supressing proliferation. Different signaling pathways such as Wnt/cathenin and Notch/Jagged 1 promote self-renewal, proliferation and differentiation of HSC (Iwasaki and Suda 2009). Therefore, osteoblasts clearly have a role in the establishment and mainteinance of the HSC niche that could be modulated through PTH and other molecular mechanisms that are incompletely defined so far. Osteoblasts are the main targets of PTH/PTHrP, and PTH/PTHrp also has some modulating potential on HSC via osteoblasts. PTH may increase the proportion of bone marrow-derived stromal cells (BMC) that commit to the osteoblastic lineage both in vitro and in vivo, thus expanding the osteoblast pool. Furthermore, PTH induces osteoblasts to express BMP, SDF-1, VEGF, osteopontin and activate signaling pathways involved in the HSC niche. It has been demonstrated that PTH expands the HSC pool through activation of the PTH-1R on osteoblasts. The Jagged1/Notch signaling pathway is implicated in the control of stem cell self-renewal in several organs, and is necessary for PTH-dependent HCS expansion (Calvi 2006). PPR activation by the Notch ligand Jagged-1 in osteoblasts is associated with an increase in the number of HSC, and this increase can be stopped by administration of a secretase inhibitor. The same effects can be achieved by using exogenous PTH (Calvi 2006). Furthermore, PTH (as well as other stress conditions including inflammation, injury or chemotherapy) could result in a destabilization of HSC with induction of massive stem mobilization. Accordingly, osteoclast activity has been demonstrated to promote the proliferation and mobilization of hematopoietic progenitors from HCS. Bone resorbing osteoclasts secrete enzymes, including MMP-9 and cathepsin K that give them SDF-1 and osteopontin degradation capacity (Kollt et al. 2006). Therefore, PTH could impact both bone turnover and the bone marrow niche through modulation of osteoblast and osteoclast activity, resulting in improved engraftment both of HSC and cancer stem cells. It is notable that the majority of the signaling pathways involved in the interaction between normal stem cells and their niche are also involved in the interaction between cancer stem cells and their niche.
References
Adami S, Bertoldo F, Braga S et al (2009) 25-hydroxy vitamin D levels in healthy premenopausal women: association with bone turnover markers and bone mineral density. Bone 45:423–426
Adams GB (2005) Stem cell engraftment at the endosteal niche is specified by the Calcium sensing receptor. Nature 439:599–603
Aldridge SE, Lennard TW, Williams JR et al (2005) Vascular endothelial growth factor acts as an osteolytic factor in breast cancer metastases to bone. Br J Cancer 92:1531–1537
Al-Hajj M, Wicha MS, Benito-Hernandez A et al (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100:3983–3988
Almquist M, Manjer J, Bondenson L et al (2007) Serum calcium and breast cancer risk: results from a prospective cohort study of 7, 847 women. Cancer Cause Control 18:595–602
Alonso V, deGortazar AR, Ardura JA (2008) Parathyroid hormone related protein increases human osteoblastic cell survival by activation of vascular endothelial factor receptor-2. J Cell Physiol 217:717–727
Arai F, Hirao A, Ohmura M et al (2004) Tie2/angiopoietin -1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 18:149–161
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284:770–776
Arya M, Ahmed H, Silhi N et al (2007) Clinical importance and therapeutic implications of the pivotal CXCL12-CXCR4 (chemokine ligand-receptor) interaction in cancer cell migration. Tumour Biol 28(3):123–131
Asou Y, Rittling SR, Yoshitake H et al (2001) Osteopontin facilitates angiogenesis, accumulation of osteoclasts, and resorption in ectopic bone. Endocrinology 142:969–973
Attia S, Eickhoff J, Wilding G et al (2008) Randomized double blinded phase II evaluation of docetaxel with or without doxecalciferol in patients with metastatic androgen independent prostate cancer. Clin Canc Res 14:2437–2443
Bao BY, Yao J, Lee YF (2006) Alpha 25hydroxyvitamin D3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis 247:122–129
Bertolini G, Roz L, Perego P (2009) Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA 106(38):16281–16286
Bleul CC, Fuhlbrigge RC, Casasnovas JM (1996) A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med 184(3):1101–1109
Blokzijl A, Dahlqvist C, Reissmann E (2003) Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol 163(4):723–728
Bonnet D, Dick JE (1997) Human acute myeloid leukaemia is organised as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730–737
Bouilon E et al (2006) Vitamin D and cancer. J Steroid Biochem Mol Biol 102:156–162
Boyce BF, Xing L, Shakespeare W et al (2003) Regulation of bone remodelling and emerging breakthrough drugs for osteoporosis and osteolytic bone metastases. Kidney Int Suppl 85:S2–S5
Brown J, Corey E, Lee Z et al (2001a) Osteoprotegerin and rank ligand expression in prostate cancer. Urology 57:611–616
Brown JM, Vessella RL, Kostenuik PJ et al (2001b) Serum osteoprotegerin levels are increased in patients with advanced prostate cancer. Clin Cancer Res 7(10):2977–2983
Bryden AAG, Hoyland JA, Freemong AJ et al (2002) Parathyroid hormone relate peptide and receptor expression in paired primary prostate cancer and bone metastases. Br J Cancer 86:322–325
Bunyaratavej P, Hullinger TG, Somerman MJ (2000) Bone morphogenetic proteins secreted by breast cancer cells upregulate bone sialoprotein expression in preosteoblast cells. Exp Cell Res 260:324–333
Burtis WJ, Wu T, Bunch C, Wysolmerski J et al (1987) Identification of a novel 17,000-dalton parathyroid hormone-like adenylate cyclase-stimulating protein from a tumor associated with humoral hypercalcemia of malignancy. J Biol Chem 262(15):7151–7156
Calvi L (2006) Osteoblastic activation in the hematopoietic stem cell niche. Ann NY Acad 1068:477–488
Calvi LM, Adams GB, Weibrecht KW et al (2003) Osteoblastic cells regulate the hematopoietic stem cell niche. Nature 425:841–846
Campbell MJ, Elstner E, Holden S et al (1997) Inhibition of proliferation of prostate cancer cells by a 19-norhexafluoride vitamin D 3 analogue involves the induction of p21 waf1p27 kip 1 and E-cadherin. J Mol Endocrinol 19:15–27
Campbell MJ, Gombart AF, Kwok SH et al (2000) The antiproliferative effects of 1alpha 25(OH)D on breast and prostate cancer cell are associated with induction of BRCA1 gene expression. Oncogene 19:5091–5097
Canon JR, Roudier M, Bryant R et al (2008) Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin Exp Metastasis 25(2):119–129
Casimiro S, Guise A, Chirgwin J (2009) The critical role of bone microenvironment in cancer metastasis. Mol Cell Endocrinol 310:71–81
Catz SD, Johnoston JL (2001) Transcriptional regulation of BCL-2 by nuclear factor kappa and its significance in prostate cancer. Oncogene 41:3283–3294
Chan JS, Beer TM, Quin DI et al (2008) A phase II study of high dose calcitriol combined with mitoxantrone and prednisone for androgen.indipendent prostate cancer. BJU Int 102:1601–1606
Chen G, Sircar K, Aprikian A et al (2006) Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 107:289–298
Cheney IW, Johnson DE, Vaillancourt MT et al (1998) Suppression of tumorigenicity of glioblastoma cells by adenovirus-mediated MMAC1/PTEN gene transfer. Cancer Res 58(11):2331–2334
Chenn A, Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297:365–369
Chiao JW, Moonga BS, Yang YM et al (2000) Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer 83:360–365
Chlebowski RT, Jhonson KC, Kooperberger C et al (2008) Calcium plus vitamin D supplementation and the risk of breast cancer. J Natl Cancer Inst 100:1581–1591
Chung I, Han G, Seshadri M et al (2009) Role of vitamin D receptor in the proliferative effects of cacitriol in tumor derived endothelial cells and tumor angiogenesis in vivo. Cancer Res 69:967–975
Collins AT, Berry PA, Hyde C et al (2005) Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 65:10946–10951
Coyle D, McNeill RE, Hennessy E (2006) Calcium sensing receptor gene expression and breast to bone metastases. Br J Surg 93:898
Crew KD, Shane E, Cremers S et al (2009) High prevalence of vitamin D deficiency despite supplementation in premenopausal women with breast cancer undergoing adjuvant chemotherapy. J Clin Oncol 27:2151–2156
Cross SS, Harrison RF, Balasubramanian SP et al (2006) Expression of receptor activator of nuclear factor kappabeta ligand (RANKL) and tumour necrosis factor related, apoptosis inducing ligand (TRAIL) in breast cancer, and their relations with osteoprotegerin, oestrogen receptor, and clinicopathological variables. J Clin Pathol 59(7):716–720
Cui Y, Rohan TE (2006) Vitamin D, calcium and breast cancer risk: a review. Canc Epidemiol Biomarkers Prev 15:1427–1437
Dahmane N, Lee J, Robins P et al (1997) Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 389(6653):876–881
Dai J, Kitagawa Y, Zhang J et al (2004) Vascular endothelial growth factors contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogentic protein. Cancer Res 64:994–999
De Palma M, Murdoch C, Venneri MA et al (2007) Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol 28:519–524
Deftos LJ, Barken I, Burton DW (2005) Direct evidences that PTHrp expression promotes prostate cancer progression in bone. Biochem Biophys Res Commun 327:468–472
Derynck R, Akhurst RJ, Balmain A (2001) TGF-b signaling in tumor suppression and cancer progression. Nat Genet 29:117–129
Dougall WC, Chaisson M (2006) The RANK/RANKL/OPG triad in cancer-induced bone diseases. Cancer Metastasis Rev 25:541–549
Emery JG, McDonnell P, Burke MB et al (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273:14363–14367
Eriksen EF, Eghbali-Fatourechi GZ, Klosha S (2007) Remodeling and vascular space in bone. J Bone Miner Res 22:1–6
Erler JT, Giaccia AJ (2006) Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res 66(21):10238–10241
Erler JT, Bennewith KL, Cox TR et al (2009) Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15:35–44
Esbrit P, Alvarez-Arroyo MV, DeMiguel F et al (2000) C-terminal parathyroid hormone-related protein increases vascular endothelial growth factor in human osteoblastci cell. J Am Soc Nephrol 11:1085–1092
Fata JE, Kong YY, Li J et al (2000) The osteoclast differentiation factor osteoprotegerin ligand is essential for mammary gland development. Cell 103:41–50
Ferrara N (2009) Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 29(6):789–791
Flaig TW, Barqawi A, Miller G et al (2006) A phase II trail of dexamethasone, vitamin D, and carboplatin in patients with hormone refractory prostate cancer. Cancer 107:266–274
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27–31
Gerritsen ME, Peale FV, Wu T (2002) Gene expression profiling in silico: relative expression of candidate angiogenesis associated genes in renal cell carcinomas. Exp Nephrol 10:114–119
Glinsky VV, Glinsky GV, Rittenhouse-Olson K et al (2001) The role Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res 61:4851–4857
Gocek E, Studzinski GP (2009) Vitamin D and differentiation in cancer. Crit Rev Clin Lab Sci 46:190. doi:10.1080/10408360902982128
Grill V, Ho P, Body JJ et al (1991) Parathyroid hormone-relate protein: elevated levels in both humoral hypercalcemia and hypercalcemia complicating metastatic breast cancer. J Clin Endocrinol Metab 73:1309–1315
Guise TA, Yin JJ, Mohammad KS (2003) Role of endothelin-1 in osteoblastic bone metastases. Cancer 97(3):779–784
Hagemann T (2005) Macrophages induce invasiveness of epithelial cancer cells via NF-κB and JNK. J Immunol 175:1197–1205
Harada S, Rodan GA (2003) Control of osteoblast function and regulation of bone mass. Nature 423:349–355
Hauschka PV, Mavrakos AE, Iafrati MD et al (1986) Growth factors in bone matrix. J Biol Chem 261:12665–12674
Heissig B, Hattori K, Dias S et al (2002) Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109:625–637
Henderson MA, Danks JA, Slavin JL et al (2006) Parathyroid hormone-related protein localization in breast cancers predict improved prognosis. Cancer Res 66:7402–7411
Hofbauer L, Rachner T, Singh S (2008) Fatal attraction: why breast cancer cells home to bone. Breast Cancer Res 10:101
Holen I, Croucher PI, Hamdy FC et al (2002) Osteoprotegerin is a survival factor for human prostate cancer cells. Cancer Res 62:1619–1623
Holen I, Cross SS, Neville-Webbe HL et al (2005) Osteoprotegerin expression by breast cancer cells in vitro and breast tumours in vivo—a role in tumour cell survival? Breast Cancer Res Treat 92:207–215
Holick MF, Chen TC (2008) Vitamin D deficiency: a world wide problem with health consequences. Am J Clin Nutr 87:10805–10865
Hughes DP (2009) How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat Res 152:479–496
Hugo H, Ackland ML, Blick T et al (2007) Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol 213:374–383
Hullinger TG, Pan Q, Viswanathan HL (2001) TGFbeta and BMP-2 activation of the OPN promoter: roles of smad- and hox-binding elements. Exp Cell Res 262(1):69–74
Isowa A, Shimo T, Ibaragi S et al (2010) PTHrP regulates angiogenesis and bone resorption via VEGF expression. Anticancer Res 30:2755–2768
Ite H, Yoshida T, Matsumoto N, Aoki K, Osada Y, Sugimura T, Terada M (1997) Growth regulation of human prostate cancer cells by bone morphogenetic protein-2. Cancer Res 57:5022–5027
Iwasaki H, Suda T (2009) Cancer stem cell and their niche. Cancer Sci 100:1166–1172
Javed A, Barnes GL, Pratap J et al (2005) Impaired intranuclear trafficking of Runx2 (AML3/CBFA1) transcription factors in breast cancer cells inhibits osteolysis in vivo. Proc Natl Acad Sci USA 102:1454–1459
Jones DH, Nakashima T, Sanchez OH et al (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440:692–696
Jung Y, Wang J, Schneider A (2006) Regulation of SDF-1 (CXCL12) production by osteoblasts a possible mechanism for stem cell homing. Bone 38:497
Kagan HM, Li W (2003) Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem 88:660–672
Kaplan RN, Riba RD, Zacharoulis S et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438(7069):820–827
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87:159–170
Kirschmann DA, Seftor EA, Fong SF et al (2002) A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res 62:4478–4483
Kitano Y, Kurihara H, Kurihara Y et al (1998) Gene expression of bone matrix proteins and endothelin receptors in endothelin-1-deficient mice revealed by in situ hybridization. J Bone Miner Res 13:237–244
Kitten AM, Harvey SA, Criscimagna N et al (1997) Osteogenic protein-1 downregulates endothelin A receptors in primary rat osteoblasts. Am J Physiol 272((6 Pt 1)):E967–E975
Kleeberger W, Bova GS, Nielsen ME, Epstein JI, Berman DM et al (2007) Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res 67:9199–9206
Knerr K, Ackermann K, Neidhart T et al (2004) Bone metastasis: osteoblasts affect growth and adhesion regulons in prostate tumor cells and provoke osteomimicry. Int J Cancer 11:152–159
Koch H, Jadlowiec JA, Campbell PG (2005) Insulin-like growth factor-I induces early osteoblast gene expression in human mesenchymal stem cells. Stem Cells Dev 14:621–631
Kollett O, Dar A, Schvtiel S et al (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12:657–664
Kollt O, Dar A, Shivtiel S et al (2006) Osteoclasts degradate endosteal components and promote mobilization of hematopoietic progenitor cells. Nature Med 12:657–664
Kostenuik PJ, Nguyen HQ, McCabe J et al (2009) Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res 24:182–195
Kunnimalaiyaan M, Chen H (2007) Tumor suppressor role of Notch-1 signaling in neuroendocrine tumors. Oncologist 12:535–542
Lacey DL, Timms E, Tan HL et al (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176
Lapidot T, Sirard C, Vormoor J et al (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367:645–648
Lappe JM, Travers-Gustafson D, Davies KM et al (2007) Vitamin D and calcium supplementation reduces cancer risk:results of a randomized trial. Am J Clin Nutr 85:1586–1591
Li G, Cui Y, McIlmmuray L et al (2005) rhBMP2, rhVEGF (165), rhPTN and thrombin related peptide, TP 508 induce chemotaxis of human osteoblasts and microvascular endothelial cells. J Ortop Res 23:680–685
Li X, Huang M, Zheng H, Wang Y (2008) CHIP promotes Runx2 degradation and negatively regulates osteoblast differentiation. J Cell Biol 181(6):959–972
Liang Z, Wu T, Lou H et al (2004) Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res 64:4302–4308
Liao J, Li X, Koh AJ et al (2008) Tumor expressed PTHrP facilitates prostate cancer-induced osteoblastic lesions. Int J Cancer 123(10):2267–2278
Lyden D, Young AZ, Zagzag D et al (1999) Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature 401:670–677
Ma Q, Jones D, Borghesani PR et al (1998) Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA 95(16):9448–9453
Makuch LA, Sosnoski DM, Gay CV (2006) Osteoblast-conditioned media influence the expression of E-selectin on bone-derived vascular endothelial cells. J Cell Biochem 98:1221–1229
Mantovani A, Schioppa T, Porta C et al (2006) Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev 25(3):315–322
Matthews D, LaPorta E, Zinser GM et al (2010) Genomic vitamin D signalling in breast cancer: insights from animal models and human cells. J Steroid Biochem Mol Biol 121:36–367
McGrath CM, Soule HD et al (1984) Calcium regulation of normal human mammary epithelial cell growth in culture. In Vitro 20:625–662
Merryman JI, DeWille JW, Werkmeister JR et al (1994) Effects of transforming growth factor-β on parathyroid hormone-related protein production and ribonucleic acid expression by a squamous carcinoma cell line in vitro. Endocrinology 134:2424–2430
Miettinen PJ, Ebner R, Lopez AR, Derynck R (1994) TGF-b induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 127:2021–2036
Mihai M (2008) The calcium sensing receptor: from understanding parathyroid calcium homeostasis to bone metastasis. Ann R Coll Sur Engl 90:271–277
Min JK, Kim JM et al (2003) Vascular endothelial growth factor up-regulates expression of receptor activator of NF-kappaB (RANK) in endothelial cells. Concomitant increase of angiogenic response to RANK ligand. J Biol Chem 278:39548–39557
Moseley JM, Kubota M, Diefenbach-Jagger H et al (1987) Parathyroid hormone-related protein purified from a human lung cancer cell line. Proc Natl Acad Sci USA 84:5048–5052
Muller A, Homey B, Soto H et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56
Murdoch C, Muthana M, Coffelt SB et al (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nature Rev Cancer 8:618–631
Nakagawa M, Kaneda T, Arakawa T et al (2000) Vascular endothelial growth factor (VEF^GF) directly enhances osteoclastic bone resorption and survival of mature osteoclast. FEBS Lett 473:161–164
Nefedova Y, Cheng P, Alsina M (2004) Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood 103(9):3503–3510
Nelson JB, Hedican SP, George DJ et al (1995) Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med 1:944–949
Nelson JB, Nguyen SH, Wu-Wong JR et al (1999) New bone formation in an osteoblastic tumor model is increased by endothelin-1 overexpression and decreased by endothelin A receptor blockade. Urology 53(5):1063–1069
Nobta M, Tsukazaki T, Shibata Y et al (2005) Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated notch1 signaling. J Biol Chem 280(16):15842–15848
Noda M, Rodan GA (1989) Transcriptional regulation of osteopontin production in rat osteoblast-like cells by parathyroid hormone. J Cell Biol 08(1):713–718
O’Brien CA, Pollet A, Gallinger S et al (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110
Oh HS, Moharita A, Potian JG et al (2004) Bone marrow stroma influences tranforming growth factor-beta production production in breast cancer cells to regulate c-myc activation of the preprotachykinin-I gene in breast cancer cells. Cancer Res 64:6327–6336
Pardal R, Clark MF, Morrison SJ (2003) Applying the principals of stem-cell biology to cancer. Nat Rev Cancer 3:895–902
Patrawala L, Calhoun T, Schneider-Broussard R et al (2006) Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 25:1696–1708
Pecheur I, Peyruchaud O, Serre CM et al (2002) Integrin ανβ3 expression confers on tumor cells a greater propensity to metastasixe to bone. FASEB J 16:1266–1268
Pepe J, Romagnoli E, Nofroni I et al (2006) Vitamin D status as the major factor determining the circulating levels of parathyroid hormone: a study in normal subjects. Osteopor Int 16:805–812
Peterlik M, Grant WB, Cross HS (2009) Calcium, vitamin D and cancer. Anticancer Res 29:3687–3698
Pfeilschifter J, Mundy GR (1987) Modulation of transforming growth factor β activity in bone cultures by osteotropic hormones. Proc Natl Acad Sci USA 84:2024–2028
Phadke PA, Mercer R, Harms JF (2006) Kinetics of metastatic breast cancer cell trafficking bone. Clin Cancer Res 12(5):1431–1440
Piek E, Moustakas A, Kurisaki A et al (1999) TGF-b type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 112:4557–4568
Polakis P (1999) The oncogenic activation of β-catenin. Curr Opin Genet Dev 9:15–21
Pollard JW (2009) Trophic macrophages in development and disease. Nat Rev Immunol 9(4):259–270
Ponomariov T, Peled A, Petit I et al (2000) Induction of the chemokines stromal-derived factor-1 following DNA damage improves human stem cell functions. J Clin Invest 106:1331–1339
Powell GJ, Southby J, Danks JA et al (1991) Localization of parathyroid hormone-related protein in breast cancer metastasis: increased incidence in bone compared with other sites. Cancer Res 51:3059–3061
Powell SM, Zilz N, Beazer-Barclay Y et al (1992) APC mutations occur early during colorectal tumorigenesis. Nature 359:235–237
Prince CW, Butler WT (1987) 1, 25-Dihydroxyvitamin D3 regulates the biosyntheis of osteopontin, a bone-derived cell attachment protein, in clonal osteoblast-like osteosarcoma cells. Coll Relat Res 7:305–313
Proweller A, Tu L, Lepore JJ et al (2006) Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res 66:7438–7444
Pukrop T, Klemm F, Hagemann TH et al (2006) Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc Natl Acad Sci USA 103:5454–5459
Radtke F, Raj K (2003) The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer 3:756–767
Raimondi S, Johansson H, Maisonneuve P et al (2009) Review and meta-analysis on vitamin D receptor polymorphisms and cancer risk. Carcinogenesis 30:1170–1180
Ramaswamy S, Ross KN, Lander ES et al (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33:49–54
IARC Working Group Reports (2008) IARC vitamin D and cancer, vol 5. International Agency for Research on Cancer
Ritchie CK, Andrews LR, Thomas KG et al (1997a) The effects of growth factors associated with osteoblasts on prostate carcinoma proliferation and chemotaxis: implications for the development of metastatic disease. Endocrinology 138:1145–1150
Ritchie CK, Thomas KG, Andrews LR et al (1997b) Effects of the calciotrophic peptides calcitonin and PTH on prostate growth and chemotaxis. Prostate 30:183–187
Rodland KD (2004) The role of the calcium-sensing receptor in cancer. Cell Calcium 35(3):291–295
Rubin J, Chung LW, Fan X et al (2004) Prostate carcinoma cells that have resided in bone have an upregulated IGF-I axis. Prostate 58:41–49
Rucci N, Recchia I, Angelucci A et al (2006) Inhibition of protein kinase c-Src reduces the incidence of breast cancer metastases and increases survival in mice: implication for therapy. J Pharmacol Exp Ther 318:161–172
Ruiz i Altaba A, Sánchez P, Dahmane N (2002) Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2(5):361–372
Santini D, Perrone G, Roato I et al (2010a) Expression pattern of receptor activator of NFκB (RANK) in a series of primary solid tumors and related bone metastases. J Cell Physiol 226:780–784
Santini D, Vincenzi B, Russo A et al. (2010b) Association of receptor activator of NF-kb (RANK) expression with bone metastasis in breast carcinomas. ASCO 2010 abstract 1053
Sasaki T, Hong MH (1993) Endothelin-1 localization in bone cells and vascular endothelial cells in rat bone marrow. Anat Rec 237:332–337
Sasaki A, Ishikawa K, Haraguchi N et al (2007) Receptor activator of nuclear factor-kappaB ligand (RANKL) expression in hepatocellular carcinoma with bone metastasis. Ann Surg Oncol 14(3):1191–1199
Schnieder A, Kalikin LM, Mattos AC (2005) Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology 146:1727–1736
Schwalbe M, Sanger J, Eggers R et al (2003) Differential expression and regulation of bone morphogenetic protein 7 in breast cancer. Int J Oncol 23:89–95
Schwaninger R, Rentsch CA, Wetterwald A, van der Horst G, van Bezooijen RL, van der Pluijm G, Lowik CW, Ackermann K, Pyerin W, Hamdy FC et al (2007) Lack of noggin expression by cancer cells is a determinant of the osteoblast response in bone metastases. Am J Pathol 170:160–175
Schwartz GG (2008) Prostate cancer, serum parathyroid hormone and the progression of skeletal metastases. Canc Epidemiol Biomarkers Prev 17:478–483
Shimoyama A, Wada M, Ikeda F et al (2007) Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol Biol Cell 18:2411–2418
Shioide M, Noda M (1993) Endothelin modulates osteopontin and osteocalcin messenger ribonucleic acid expression in rat osteoblastic osteosarcoma cells. J Cell Biochem 53:176–180
Shipitsin M, Polyak K (2008) The cancer stem cell hypothesis: in search of definitions, markers and relevance. Lab Invest 88:459–463
Shipitsin M, Campbell LL, Argani P et al (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell 11:69–82
Shmelkov SV, Butler JM, Hooper AT et al (2008) CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J Clin Invest 118:2111–2120
Sing SK (2003) Identification of a cancer stem cell in human brain tumours. Cancer Res 63:5821–5828
Southby J, Kissin MW, Danks JA et al (1990) Immunohistochemical localization of parathyroid hormone-related protein in breast cancer. Cancer Res 50:7710–7716
Spinella F, Rosanò L, Di Castro V et al (2003) Endothelin-1 decreases gap junctional intercellular communication by inducing phosphorylation of connexin 43 in human ovarian carcinoma cells. J Biol Chem 278(42):41294–41301
Spinella-Jaegle S, Rawadi G, Kawai S et al (2001) Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J Cell Sci 114:2085–2094
Stern PH, Tatrai A, Semler DE et al (1995) Endothelin receptors, second messengers, and actions in bone. J Nutr 125(Suppl):2028S–2032S
Street J, Bao M, deGuzman L et al (2002) Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci USA 99:9656–9661
Strewler GJ, Stern P, Jacobs J, Eveloff J, Klein RF, Leung SC et al (1987) Parathyroid hormone-like protein from human renal carcinoma cells structural and functional homology with parathyroid hormone. J Clin Invest 80:1803–1807
Sugiyama T, Kohara H, Noda M et al (2006) Maintainance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signalling in bone marrow stromal cell niches. Immunity 25:977–988
Sun YX, Shelburne CE, Lopatin DE et al (2003) The expression of CXCR4 and CXCL12 (SDF-1) in human prostate camncers in vivo. J Cell Biochem 89:462–473
Sun YX, Schneider A, Jung Y et al (2005) Skeletal localization and neutralization of the SDF1 (CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseus sites in vivo. J Bone Miner Res 20:318–329
Tombran-Tink J, Barnstable CJ (2004) Osteoblasts and osteoclasts express PEDF, VEGF-A isoforms, and VEGF receptors: possible mediators of angiogenesis and matrix remodelling in the bone. Biochem Biophys Res Commun 316:573–579
Tretli S, Hernes E, Berg JP (2009) Association between serum 25(OH)D and death from prostate cancer. Br J Canc 100:450–454
Trump DL, Potter DM, Muindi J et al (2006) Phase II trial of high dose intermittent calcitriol and dexamethasone in androgen independent prostate cancer. Cancer 106:2136–2142
Trump DL, Chadha MK, Sunga AY et al (2009) Vitamin D deficiency and insufficiency among patients with prostate cancer. BJU Int 104:909–914
Tuck AB, Hota C, Wilson SM et al (2003) Osteopontin-induced migration of human mammary epithelial cells involves activation of EGF receptor and multiple signal transduction pathways. Oncogene 22:1198–1205
Valcourt U, Kowanetz M, Niimi H et al (2005) TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 16:1987–2002
van Dijke P, Yamashita H, Sampath TK, Reddi AH, Estevex M, Riddle DL, Ichijo H, Heldin CH, Miyazono K (1994) Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem 269:16985–16988
Vargas SJ, Gillespie MT, Powell GJ et al (1992) Localization of parathyroid hormone-related protein mRNA expression and metastatic lesions by in situ hybridization. J Bone Miner Res 7(8):971–980
Wai PY, Kuo PC (2004) The role of osteopontin in tumor metastasis. J Surg Res 121:228–241
Wiesner C, Nabha SM, Dos Santos EB et al (2008) C-kit and its ligand stem cell factor: potential contribution to prostate cancer bone metastasis. Neoplasia 10:996–1003
Wu JY, Scadden DT, Kronenberg M (2009) Role of the osteoblast lineage in the bone marrow hematopoietic niches. J Bone Min Res 24:759–763
Wyckoff JB (2007) Direct visualization of macrophageassisted tumor cell intravasation in mammary tumors. Cancer Res 67:2649–2656
Xie Y, Yin T, Wiegraebe W et al (2009) Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 457:97–101
Xu J, Lamouille S, Derynck R (2009) TGF-beta-induced epithelial to mesenchymal transition. Cell Res 19(2):156–172
Yates AJ, Gutierrez GE, Smolens P et al (1988) Effects of a synthetic peptide of a parathyroid hormone-related protein on calcium homeostasis, renal tubular calcium reabsorption, and bone metabolism in vivo and in vitro in rodents. J Clin Invest 81:932–938
Yin T, Li L et al (2006) The stem cell niche in bone. J Clin Invest 116:1195–1201
Yin L, Raum E, Haug U et al (2009) Meta-analysis of longitudinal studies: serum vitamin D and prostate cancer risk. Cancer Epidem 33:435–445
Yin L, Grandi N, Raum E et al (2010) Meta-analysis: serum vitamin D and breast cancer risk. Eu J Cancer 46:2196–2205
Yu XF, Collin-Osdoby P, Osdoby P (2003a) SDF-1 increases recruitment of osteoclast precursors by upregulation of matrix metalloproteinase 9 activity. Connect Tissue Res 44:79–84
Yu XF, Huang YF, Collin-Osdoby P et al (2003b) Stromal cell derived factor 1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase 9 (MMP9) activity, and collagen transmigration. J Bone Miner Res 18(8):1404–1418
Yucha C, Guthrie D (2003) Renal homeostasis of calcium. Nephrol Nurs J 30:755–764
Zakalik D, Diep D, Hooks MA et al (1992) Transforming growth factor β increases stability of parathyroid hormone-related protein messenger RNA. J Bone Miner Res 7(Suppl 1):104A, S118
Zavadil J, Cermak L, Soto-Nieveset N et al (2004) Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J 23(5):1155–1165
Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R (2003) BMP-7 counteracts TGF-b1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9:964–968
Zhang J, Dai J, Qi Y, Lin DL et al (2001) Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest 107(10):1235–1244
Zhang J, Grindley JC, Yin T et al (2006) PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441:518–522
Zhang XH, Wang Q, Gerald W et al (2009) Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16:67–78
Zheng H, Fu G, Dai T, Huang H (2007) Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1alpha/CXCR4 via PI3 K/Akt/eNOS signal transduction pathway. J Cardiovasc Pharmacol 50(3):274–280
Zhou J, Wulfkuhle J, Zhang H et al (2007) Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci USA 104(41):16158–16163
Zhu AJ, Watt FM (1999) β-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development 126:2285–2298
Zunich SM, Douglas T, Valdovinos M, Chang T, Bushman W, Walterhouse D, Iannaccone P, Lamm ML (2009) Paracrine sonic hedgehog signalling by prostate cancer cells induces osteoblast differentiation. Mol Cancer 8:12
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Santini, D., Pantano, F., Vincenzi, B., Tonini, G., Bertoldo, F. (2012). The Role of Bone Microenvironment, Vitamin D and Calcium. In: Joerger, M., Gnant, M. (eds) Prevention of Bone Metastases. Recent Results in Cancer Research, vol 192. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-21892-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-642-21892-7_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-21891-0
Online ISBN: 978-3-642-21892-7
eBook Packages: MedicineMedicine (R0)