
Abstract
Background
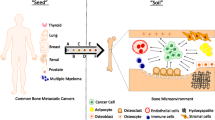
Bone is one of the most common sites for solid tumor metastasis. Bone metastasis of a malignant tumor seriously affects the quality of life and the overall survival of patients. Evidence has suggested that bone provides a favorable microenvironment that enables disseminated tumor cells to home, proliferate and colonize, leading to the formation of metastases. In the process of bone metastasis the bone microenvironment may be considered as an orchestra that plays a dissonant melody through blending (e.g. cross-talk between osteoclasts, osteoblasts and/or other cells), adding (e.g. a variety of biological factors) or taking away (e.g. blocking a specific pathway) players.
Conclusions
Here, we review the normal bone microenvironment, bone microenvironment-related factors that promote bone metastasis, as well as mechanisms underlying bone metastasis. In addition, we elude on directions for clinical bone metastasis management, focusing on potential therapeutic approaches to target bone microenvironment-related factors, including bisphosphonate, denosumab, CXCR4/CXCL12 antagonists and cathepsin K inhibitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Malignant cancers such as prostate, breast, lung, thyroid and kidney cancers tend to metastasize to the bone [1]. The bone microenvironment provides a favorable environment for cancer cells to colonize and reactivate proliferation within the bone matrix. Bone metastasis can lead to skeletal-related events (SREs) such as bone pain, pathological fractures and hypercalcemia. SREs are closely related to the overall survival and quality of life of patients with bone metastases. Studying the role of the bone microenvironment and its effect on the occurrence of bone metastasis may lead to the identification of new therapeutic targets and drugs for effectively inhibiting bone metastasis. Here, we review the role of the bone microenvironment in bone metastasis and elude on the identification of therapeutic targets.
2 The bone microenvironment
Bone is a dynamic organ that functions in structural support and movement, and as a reservoir of minerals and energy [2]. The bone microenvironment consists of cells and cytokines within the bone marrow and bone matrix. There are several different types of cells in the bone microenvironment, including osteoclasts (OCs), bone marrow endothelial cells, nerve cells, hematopoietic cells, macrophages and lymphocytes, as well as mesenchymal stem cells such as osteoblasts (OBs), chondroblasts and adipose cells, which provide a stable growth environment through cell-cell interactions to maintain bone stability [3]. OBs and OCs play major roles in bone remodeling, and the balance between OBs and OCs maintains the integrity of the bone structure. Among them, OBs represent monocytes differentiated from mesenchymal stem cells, which participate in bone remodeling by generating a new extracellular matrix (ECM) and calcium phosphate crystals that are deposited into the interstitial space of the matrix. OCs are multinuclear cells derived from monocyte/macrophage progenitors that participate in bone resorption by producing acid to solubilize calcium phosphate crystals and by secreting proteases to degrade exposed ECM [4]. The bone matrix provides structural support for bone cells and produces several growth factors. These growth factors are released during normal bone turnover and play important roles in maintaining the stability of the bone microenvironment.
3 The bone microenvironment and bone metastasis
Colonization of cancer cells within a metastatic organ involves interaction between cancer cells and cells from the microenvironment, or interaction between cancer cells and the ECM [5]. These interactions depend on factors involved in binding between cell surface receptors and ligands, intercellular adhesion, changes in cell morphology and changes in specific functions of the endothelium [6].
In 1889, Stephen Paget proposed that tumor cells tend to target certain organs. He first proposed the “seed and soil” hypothesis, saying that to facilitate the formation of metastases the “soil” (metastatic microenvironment) must be able to nourish “seeds” (disseminated cancer cells). The bone microenvironment may, for example, provide sufficient growth signals to promote bone metastasis of breast cancer cells and facilitate their proliferation, whereas the same signals may not promote bone metastasis of colon cancer cells. Changes in the bone “soil” can create a favorable growth environment for the metastasis of multiple types of malignant cancers. Metastatic tumor cells acquire characteristics through epithelial mesenchymal transition (EMT), which enhances the process by which metastatic tumor cells can enter the bone via blood vessels. The EMT process is characterized by downregulation of the epithelial adhesion molecule E-cadherin as also cytoskeletal changes, which result in loss of cell contacts, increased motility of tumor cells and increased invasiveness [7]. Although bone metastases are common in many solid cancers, they do not occur in all bone sites. The most common sites of bone metastases are the ends of vertebrae, ribs, pelvis and long bones [2]. The highly vascularized metaphysis is often the preferred site for cancer cell homing. The metaphysis is mainly composed of trabecular bone and red bone marrow with a rich blood supply [8]. Therefore, trabecular bones are common sites of bone metastasis. Cancer cells need to pass through the trabecular bone to reach the endosteum in order to enter the bone marrow. The diameter of the sinusoidal blood vessels on the surface of the trabecular bone is several times that of cancer cells. In addition, the blood flow rate is slow, which provides optimal conditions for contact between cancer cells and endothelial cells [8, 9]. Moreover, endothelial cells of the sinusoidal vessels express a variety of adhesion molecules, and many types of cells in the bone microenvironment can promote colonization and metastasis of cancer cells [9].
4 Bone microenvironment-related factors that promote bone metastasis
4.1 Osteoclasts
Osteoclasts (OCs) play important roles in bone metastasis of malignant tumors. The establishment of osteolytic bone metastasis requires bone resorption mediated by activation of OCs from the bone marrow interface [10]. Mature OCs release cathepsin K, thereby creating an acidic environment and promoting degradation of the bone matrix [11]. This facilitates the entry of a large number of biological factors, such as cytokines and hormones, into the bone microenvironment. Among these factors, parathyroid hormone-related protein (PTHrP) and receptor activator of nuclear factor-κB ligand (RANKL) play important roles in osteolytic bone metastasis [6]. PTHrP is produced by cancer cells and binds to parathyroid hormone receptor 1 on the surface of osteoblasts (OBs) and other stromal cells (Fig. 1). This induces expression of RANKL in OBs and bone stromal cells and downregulates the expression of osteoprotegerin (OPG). Local increases in calcium levels after bone destruction may increase PTHrP production [7]. PTHrP may also promote the proliferation of cancer cells through autocrine stimulation and cancer angiogenesis [12]. RANKL is a member of the tumor necrosis factor (TNF) family [10, 13] and is mainly produced by OBs and bone stromal cells. RANKL binding to RANK, which is expressed by OCs, results in the formation and activation of OCs (Fig. 1). Cancer cells infiltrating the bone marrow cavity can directly promote OC-mediated bone resorption by secreting growth factors such as macrophage colony stimulating factor [14]. In addition, a feedback mechanism between cancer cells and OCs is involved in osteolytic bone metastasis (Fig. 1). PTHrP upregulates RANKL, which acts on OC precursors to promote their maturation and bone resorption. Bone resorption leads to a massive release of biological factors such as transforming growth factor (TGF)-β, epidermal growth factor (EGF) and insulin like growth factor 1 (IGF-1), which in turn stimulate the production of PTHrP [6, 15] (Fig. 1). TGF-β further increases PTHrP production through the Smad and p38 mitogen activated protein kinase signaling pathways [12, 16]. TGF-β can activate the Notch ligand Jagged-1, which is an important mediator of bone metastasis, by activating the Notch pathway in bone cells [12, 17]. In addition, TGF-β has been found to stimulate the expression of cyclooxygenase-2 (COX-2), resulting in the production of prostaglandin E2 (PGE2) in MDA-MB-231 breast cancer cells. Elevated PGE2 increases the production of RANKL by binding to the EP4 receptor on the surface of OBs, leading to the development of OCs [12]. In PC3 prostate cancer cells, modulation of COX-2 expression and activation by TGF-β has been found to be associated with the progression of bone metastases [18]. TGF-β also regulates other bone microenvironmental factors such as hypoxia to further promote the growth of cancer cells [12]. Treatment of MDA-MB-231 breast cancer cells with TGF-β has been found to induce vascular endothelial growth factor (VEGF) and CXCR4 expression through hypoxia inducible factor (HIF) under hypoxic conditions [19] (Fig. 1). In addition, c-Src plays an important role in the function of OCs. The c-Src family of non-receptor tyrosine kinases is involved in the control of signal transduction downstream of a variety of cell surface receptors, including integrins, cadherin E and tyrosine kinase receptors.

Tumor-osteoclast interactions. PTHrP is produced by tumor cells and binds to parathyroid hormone receptor 1 on the surface of osteoblasts and other stromal cells to induce the expression of the TNF family member RANKL. RANKL binds to RANK expressed by osteoclasts and mediates the formation and activation of osteoclasts. Subsequently, bone resorption leads to a massive release of biological factors such as TGF-β, EGF and IGF-1, which in turn stimulate the production of PTHrP. TGF-β further increases PTHrP production through SMAD and the p38 mitogen-activated protein kinase (MAPK) signaling pathway. TGF-β also activates the Notch ligand Jagged1, which activates the Notch pathway in osteocytes. TGF-β can also induce the expression of VEGF and CXCR4 by HIF. CXCR4 binds to CXCL12 secreted by cells such as osteoclasts and endothelial cells in the bone marrow, and activates multiple signaling pathways within cancer cells, such as those regulating cancer cell survival, gene transcription, integrin expression, angiogenesis and homing
4.2 Osteoblasts
Osteoblasts (OBs) are derived from bone stromal cells [2]. Along with the activation of OCs, a large number of cytokines is released, acting on bone stromal cells and activating osteogenic-related factors, eventually leading to the formation of osteogenic lesions. In osteogenic bone metastasis, the most important factors involved are bone morphogenetic proteins (BMPs) and endothelin-1 (ET-1) [6]. BMPs constitute a family of multifunctional cytokines of the TGF-β superfamily that can induce mesenchymal stem cells to differentiate into OBs by activating osteogenic-related transcription factors, such as runt-related transcription factor 2 (RUNX2), thereby protecting OBs from apoptosis induced by TNF or other death-inducing factors [8, 20] (Fig. 2). In bone tissue, BMPs are produced by osteoprogenitor cells, OBs, chondrocytes, platelets and endothelial cells. In addition, the ECM can temporarily store BMPs for use in bone repair and reconstruction [21]. BMP4 has been found to promote the osteogenic response of prostate cancer in bone by regulating BMP4-mediated paracrine signaling [12]. ET-1 is mainly secreted by endothelial cells and is a potent vasoactive peptide. ET-1 binds to the endothelin type A/B receptor on the surface of OBs through a G protein coupling mechanism. It can also promote the differentiation of OBs by inhibiting the negative regulator dickkopf-1 (DDK-1) [21]. ET-1 increases the mitotic effect of platelet-derived growth factor (PDGF), IGF-1, EGF and other growth factors [12] (Fig. 2). Both FGFs (FGF1 and FGF2) strongly stimulate bone formation. FGF2 stimulates the proliferation and differentiation of OBs by upregulating RUNX2 and BMP2 [10]. VEGF, which has been found to be produced by C4-2B prostate cancer cells, binds to VEGFR-1 and VEGFR-2 expressed by OBs, thereby stimulating the differentiation of OBs [16]. There also exists a vicious cycle between cancer cells and OBs (Fig. 2). Cancer cells stimulate OB formation by generating osteoblast stimulating factors such as BMPs, FGFs and PDGF. Cancer cells also activate ET-1 and down-regulate DDK-1, leading to new bone formation [7] (Fig. 2). Activated OBs, in turn, produce several factors such as interleukin-6 (IL-6), VEGF, monocyte chemotactic protein 1 (MCP-1) and macrophage inflammatory protein-2 (MIP-2). These factors promote the colonization and proliferation of cancer cells in the bone microenvironment [22] (Fig. 2).

Tumor-osteoblast interactions. Tumor cells stimulate osteoblast formation by generating osteoblastic stimulating factors such as BMPs, FGFs and PDGF. BMPs are multifunctional cytokines of the TGF-β superfamily that induce the differentiation and maturation of mesenchymal stem cells into osteoblasts by activating various osteogenic-related transcription factors (such as Runx2). Tumor cells also activate ET-1, leading to the formation of new bone. ET-1 is mainly secreted by endothelial cells and activates osteoblasts. It can also enhance the mitotic effect of IGF-1, PDGF, EGF and other growth factors. Activated osteoblasts, in turn, produce IL-6, MCP-1,VEGF and MIP-2, which help cancer cells to colonize and expand in the bone microenvironment
4.3 Endothelial cells
Endothelial cells are essential for the migration of cancer cells via the bloodstream to specific target organs [9]. After entering sinusoidal blood vessels the blood flow slows down, which is beneficial for contacts between cancer cells and endothelial cells, leading to adhesion, extravasation and colonization [9]. In experimental mice, cancer cells injected into the arterial circulation quickly fill the bones and then decrease in number, indicating that other factors are needed to support the survival of cancer cells in the bone microenvironment [2]. The continuous proliferation of cancer cells in bone involves multiple specific interactions between cancer cells and endothelial cells, such as cell adhesion, cell dormancy and angiogenesis [11]. Bone marrow endothelial cells express various cell adhesion factors necessary for homing of hematopoietic stem cells (HSCs), including P-selectin/CD62P, E-selectin/CD62E, intercellular adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM). These factors also promote the colonization of cancer cells in the bone marrow [23].
4.4 Adipocytes
Adipocytes, which originate from the same mesenchymal stem cell lineage as OBs [24], are important components of the bone microenvironment, accounting for 15% to 40% of the bone marrow space in early adulthood and increasing to 60% with age [2]. Bone marrow adipocytes can be subdivided into constitutive and regulatory adipocytes. Regulatory adipocytes, which are distributed in the hematopoietic area of the bone marrow, known as red marrow, increase with age. The areas in which the regulatory adipocytes infiltrate are the areas at which bone metastases are more likely to occur [2]. Bone marrow adipocytes interact with cancer cells, i.e., they secrete hormones, cytokines, fatty acids and other factors, thereby forming an environment suitable for the survival of cancer cells. On the other hand, cancer cells are attracted to bone marrow adipocytes, leading to increased mobility. Interactions between cancer cells and adipocytes may lead to chronic inflammatory environments that are prone to induce cancer cell growth [22]. Breast cancer cells have been found to be sensitive to the microenvironment of adipocytes in the bone marrow, and these adipocytes have been found to locally stimulate breast cancer cell proliferation, invasion and angiogenesis [25]. As a result, breast cancer cells are more likely to colonize marrow adipose tissue compartments. Bone marrow adipocytes mediate the transfer of stored lipids to metastatic tumor cells [18], and this process can improve the activity of cancer cells [26]. Bone marrow adipocytes can also promote the differentiation and activation of OCs by producing factors such as RANKL, CXCL1 and CXCL2, and inhibit the differentiation of OBs by suppressing BMP signals. Since both OBs and adipocytes are derived from mesenchymal stem cells, an OB-adipocyte progenitor cell with bidirectional differentiation potential must be present in the bone microenvironment. This progenitor cell differentiates into specific cell lineages by activation of the respective lipogenic or osteogenic factors [3]. Microenvironmental conditions that promote adipocyte differentiation indirectly reduce the number of OBs which, in turn, indirectly decreases bone formation [2]. Herroon et al. [26] showed that increases in the number of adipocytes in the bone marrow caused by a high-fat diet are related to accelerated growth of bone cancers in mice using a combination model of diet-induced bone marrow fat growth and tumor growth in the tibia. Bone marrow adipocytes may promote tumor growth in a FABP4-dependent manner [22, 25]. FABP4 is a lipid molecule that is mainly expressed in adipocytes, macrophages and endothelial cells. Herroon et al. proposed that inhibition of FABP4 may block the lipid transport between bone marrow adipocytes and cancer cells, thereby reducing tumor metastasis to the bone marrow. These studies revealed a role of adipocyte FABP4 in promoting bone metastasis. Adipocytes not only serve as an effective energy source, but also secrete a large number of bioactive substances such as IL-1, IL-6, leptin, adiponectin, VCAM-1, TNF and VEGF. These cytokines, adipokines and growth factors are important for bone homeostasis as well as for cancer cell behavior and survival. Increased production of IL-1 and leptin, for example, promotes interactions between breast cancer cells and bone marrow adipocytes. In melanoma, increases in IL-6 trigger increases in OC formation which, in turn, leads to tumor cell proliferation [3].
4.5 Platelets
Activated platelets are important sources of angiogenic factors, and the aggregation of these factors at cancer sites affects cancer growth. Megakaryocytes, the source of platelets, are located in the vascular sinus, and are thus the first cells to encounter cancer cells when entering the bone marrow environment [24]. Therefore, platelets and their megakaryocyte precursors play important roles in bone metastasis. Platelet adhesion to circulating cancer cells was originally thought to generate a “coat” for tumors to escape the innate immune system during metastasis. However, the role of platelets in cancer metastasis is clearly beyond the scope of immunosurveillance [10]. Platelet adhesion to MDA-MB-231 breast cancer cells has been found to induce the release of lysobisphosphatidic acid (LPA) [10]. LPA is a growth factor whose biological role is mediated by G protein-coupled receptor (GPCR) family members [6]. LPA promotes the proliferation of cancer cells and induces the release of interleukins [16, 27]. Nam et al. [28] found that LPA induces the expression of IL-8 and IL-11 in MDA-MB-231 breast cancer cells, and that IL-8 promotes the growth, migration, invasion and angiogenesis of breast cancer cells. IL-8 can also promote the formation of early malignant cell clones in the bone microenvironment [16], whereas IL-11 promotes breast cancer cell growth through various mechanisms. These cytokines, thus, play multiple roles in promoting the growth of malignant tumor cells by creating a favorable microenvironment for the occurrence of bone metastasis. LPA promotes bone resorption through a mechanism similar to that of PTHrP (by promoting the expression of RANKL and inhibiting the expression of OPG in OBs) [16, 28]. Interestingly, it has been reported that LPA1 receptor antagonists can significantly inhibit the growth of breast cancer cells and the occurrence of bone metastases [29].
4.6 Immune cells
Bone metastases induce inflammatory responses that attract infiltrating T cells and macrophages [8]. Data indicate that T cells regulate the growth of bone tumors independently of their interactions with bone cells [17]. Memory T cells are found in the bone marrow of breast cancer patients, suggesting that they may play a role in cancer immune surveillance. Activated T cells lead to the production of RANKL, which directly promotes the formation of OCs. In patients with bone metastasis, OC precursors differentiate into mature OCs in the presence of T cells without macrophage colony stimulating factor and RANKL, whereas OC formation does not occur in the presence of T cell depletion [17]. In addition, T cells promote the production of IL-7, which has both inhibitory and activating effects on OCs. IL-7 promotes OC formation by upregulating T cell-derived cytokines such as RANKL and TNF [17]. TH17 cells, a subgroup of helper T cells, secrete TGF and TNF which, in turn, activate OCs, promote bone resorption and enhance the vicious cycle of metastatic bone tumor growth [17]. Future studies are needed to reveal the roles of different T cell subsets in regulating bone metastasis.
Macrophages are derived from myeloid progenitor cells and are important components of the bone marrow. There are two major types of macrophages, M1 tumor suppressor macrophages and M2 tumor-associated macrophages (TAMs) [19, 30]. TAMs produce many pro-angiogenic factors such as uPA, TNF-α, IL-1, VEGF and NO, as well as growth factors and proteases such as MMP-7, MMP-9, FGF, HGF, EGF and PDGF. TAMs can also create hypoxic conditions, which are involved in cancer progression and metastasis [7, 31]. It has been found that, in addition to the OC-derived form, macrophage-derived cathepsin K (CTSK) may also play a key role in promoting the progression of prostate cancer bone metastasis [22, 32].
4.7 The CXCR4/CXCL12 signaling cascade
CXCL12, also known as stromal derived factor-1 [11], is secreted by OCs, endothelial cells and other cells in the bone marrow (Fig. 1). CXCR4 is highly conserved during evolution [33] and is expressed on hematopoietic cells, endothelial cells, stromal cells, neurons and various other cells [14]. The CXCL12/CXCR4 signaling cascade has been studied extensively. CXCL12 is a chemotactic factor in the bone matrix and interacts with CXCR4 on the surface of tumor cells, thereby promoting the migration of tumor cells into the bone matrix [34]. The CXCL12/CXCR4 signaling cascade has been found to play an important role in bone metastasis of several malignant cancers such as non-small cell lung cancer, breast cancer and prostate cancer [16, 35]. Binding of CXCL12 to CXCR4 activates multiple signaling pathways in cells, such as those involved in cancer cell survival, gene transcription and expression of integrins (such as αVβ3) on the surface of circulating tumor cells (CTCs) (Fig. 1). αVβ3 is a central molecule for OC function, and can regulate cell polarization, proliferation, actin ring formation and bone degradation [36]. Increased expression of αVβ3 has been found to promote the migration of prostate cancer cells into the bone microenvironment [11, 37]. The CXCL12-CXCR4 axis is important for solid tumor CTCs, and plays an important role in the generation of hematopoietic stem cells and the homing of cancer cells to the bone marrow (Fig. 1). In the bone microenvironment, CXCR4 binds to VCAM1 and upregulates the expression of the OC producing factor macrophage inflammatory protein 1α (MIP1α) [38]. The CXCL12-CXCR4 axis promotes the release of osteoclastogenic factors from cancer cells, thereby promoting bone metastasis. Targeted therapy against this signaling axis may have significant clinical implications for the treatment of bone metastases.
4.8 MicroRNAs (miRNAs)
miRNAs are involved in many aspects of bone metastasis, such as the detachment of cancer cells from the primary tumor, dissemination of cancer cells to bone, colonization of the bone marrow, proliferation of cancer cells in the bone marrow and interaction of cancer cells with the ECM in the microenvironment. miRNAs can regulate the expression of target genes and, thereby, initiate EMT and mesenchymal epithelial transition (MET) [1]. In the process of bone metastasis, miRNAs are released from cancer cells into the bone marrow microenvironment to promote or inhibit tumor growth [39]. Many miRNAs are involved in the promotion or inhibition of bone metastasis. miR-218, which activates the Wnt signaling pathway in breast cancer cells has, for example, been found to promote the proliferation of MDA-MB-231 breast cancer cells and to upregulate the expression of Wnt target genes in a Wnt-dependent manner. miR-218 has also been found to target Wnt inhibitors (SOST, DKK2 and SFRP2), indicating that it can enhance the activity of Wnt and promote abnormal expression of OB-associated genes [39]. miR-218 can also upregulate the expression of bone salivary protein (BSP), osteopontin (OPN) and CXCR4 in breast cancer cells [4]. In addition, miR-218 has been found to promote the secretion of PTHrP in cancer cells, which in turn activates RANKL in OBs, further promoting OC differentiation. Twist-1 and LPA can increase the invasiveness and growth of breast cancer cells by stimulating the expression of miR-10b and miR-21, respectively [16, 39]. miR-203 and miR-135, which are negative regulators of cancer cell bone metastasis, downregulate RUNX2 and, by doing so, inhibit the growth of malignant tumors in bone [40]. miR-34a inhibits OC differentiation, bone resorption and bone metastasis by suppressing TGF-β-induced factor 2 (Tgif2) [19, 22, 39]. In androgen sensitive PC3 prostate cancer cells, miR-143 and miR-145 inhibit EMT and reduce the tendency of the cancer cells to metastasize to bone [39]. miR-145 also attenuates EMT by inhibiting the oncogenic protein HEF1, which is a novel target of Wnt signaling [39]. Expression of miR-33a decreases the production of RANKL and the expression of PTHrP [4]. miR-335 downregulates tenascin C by inhibiting the transcription factor SOX4, thereby blocking the invasion of cancer cells into bone [16, 39]. Overexpression of miR-335 in small cell lung cancer has been found to downregulate the IGF1 receptor and RANKL, thereby inhibiting bone metastasis [22]. miR-30 acts as a negative regulator and inhibits the occurrence of bone metastasis [1, 4] by downregulating factors related to bone formation, such as αVβ3, RUNX2, cadherin 11, connexin 43 and connective tissue growth factor [39].
4.9 Bone matrix proteins
Osteopontin (OPN), an acidic protein secreted by stromal cells and cancer cells, plays an important role in tumor metastasis. Experiments in mice lacking or overexpressing OPN have shown that the level of OPN is related to the potential of bone metastasis [24]. Bone sialoprotein (BSP) is associated with the migration of cancer cells and the survival of cancer cells in the bone microenvironment. Different expression levels of OPN and BSP may be associated with two different processes involved in bone metastasis, i.e., osteolysis and osteosclerosis [41]. Periostin, the first bone marrow matrix protein identified to respond to tumors, has been found to promote the growth of metastatic tumors in experimental mouse models [24]. Other proteins in the bone matrix, including dentin sialophosphoprotein and dentin sialophosphoprotein 1 have been found to be related to the progression of breast and prostate cancer bone metastases [42]. By contrast, decorin has been found to inhibit breast cancer bone metastasis [24].
5 Treatment of bone metastases
5.1 Bisphosphonates
Bisphosphonates are pyrophosphate analogues that bind to calcium and are found primarily in vertebrate bones. When bisphosphonates are attached to bone, they can be absorbed by OCs, thereby inhibiting the binding of OCs to bone and inducing OC apoptosis [22]. Increased uptake of bisphosphonates at bone metastatic sites indicates increased cell activity and metastasis [10]. The mechanism of OC apoptosis depends on the type of bisphosphonates. Non-nitrogenous bisphosphonates are converted into ATP analogues containing methylidene in cells, which accumulate in macrophages and OCs leading to direct apoptosis [43]. Nitrogenous bisphosphonates cause OC apoptosis by inhibiting farnesyl pyrophosphate synthase and the sodium mevalerate pathway [4, 11]. Nitrogenous bisphosphonates specifically inhibit the activity of farnesyl pyrophosphate synthase, and farnesyl pyrophosphate synthase is a key enzyme in the sodium mevalerate pathway, which inhibits the propylation of small GTPases leading to the inactivation of OCs [36]. Bisphosphonates include zoledronic acid, ebandronic acid, pamidronic acid, alendronic acid and risedronic acid. Zoledronic acid is currently considered the most effective bisphosphonate [12]. Zoledronic acid significantly reduces skeletal complications of breast cancer bone metastases. Recent clinical trials show that the addition of zoledronic acid to adjuvant endocrine therapy may improve disease-free and overall survival of early breast cancer patients [12]. The addition of zoledronic acid to adjuvant endocrine therapy improves bone density and prevents osteoporosis caused by endocrine therapy. Zoledronic acid can also reduce the SREs of hormone refractory metastatic prostate cancer [12]. In addition, chlorophosphonates encapsulated by liposomes have been developed. Free chlorophosphonates cannot be phagocytized by macrophages and are, therefore, rapidly removed from the circulation. After liposome encapsulation, however, chlorophosphonates are released in cells and promote apoptosis [31].
5.2 Denosumab
Denosumab is a human IgG2 monoclonal antibody with a high affinity and specificity for RANKL. Its mechanism is similar to that of OPG, as it prevents RANKL binding to RANK [44], thus inhibiting the formation of OCs and reducing the incidence of osteolytic metastases [4]. Denosumab is more effective than zoledronic acid for preventing SREs in patients with solid tumor bone metastases, and since it is highly tolerated, it may be a new therapeutic option for bone metastasis [12, 45, 46]. Denosumab increases metastatic bone density [45] and may, therefore, have clinical benefits for inhibiting OC formation, enhancing bone density and inhibiting osteoporosis.
5.3 CXCR4/CXCL12 antagonists
Plerixafor/AMD3100 is a small molecule analogue of CXCR4 that can act as a CXCR4 antagonist. AMD3100 has been found to delay the metastasis and growth of breast cancer cells in mice [22]. CXCR4 also acts as a hematopoietic stem cell mobilizing agent, promoting the movement of cancer cells from the bone marrow into the blood, and increasing sensitivity to chemotherapy [24]. CTCE9908 is a small molecule analogue of CXCL12 that competes with CXCR4 for binding to CXCL12. In experimental studies, injection of CTCE9908 into the left ventricle of nude mice has been found to reduce the size of MDA-MB-231 breast cancer cell metastases [22].
5.4 Cathepsin K inhibitor
Bone metastases from breastr and prostate cancer exhibit higher levels of expression of the cysteine protease cathepsin K (CTSK) than their corresponding primary tumors [32]. Although in most prostate cancer patients bone metastases express OBs, these lesions are significantly associated with an increased OC activity [32]. Mature OCs secrete high levels CTSK. OC-specific cathepsin K inhibits bone resorption while maintaining the number of OCs, thereby generating sufficient activity to maintain bone formation [40]. AFG-495 is a cathepsin K inhibitor, and preclinical data show that it reduces bone destruction in animal breast cancer bone metastasis models [36]. This effect may be explained by the anti-reabsorption activity of AFG-495, which deprives the bone from growth factors necessary for breast cancer cell growth. This hypothesis was confirmed by the observation that AFG-495 exhibits no inhibitory effect on the growth of breast cancer cells in other body sites. Odanacatib is a highly active cathepsin K inhibitor with a similar effect on bone resorption as zoledronic acid [4].
5.5 Endothelin receptor antagonist
Endothelins (ET-1, ET-2 and ET-3) bind to two different membrane receptors, ETAR and ETBR. ETBR has a similar affinity for these three peptides, whereas ETAR preferentially binds to ET-1 and is considered the main regulator of the endothelin (ET) axis [47]. Atrasentan/ABT-627 is an ETAR antagonist, and the first ET receptor subtype antagonist identified for the treatment of prostate cancer [24]. It is an effective competitive inhibitor of ET-1, and its binding affinity for ETAR is 1800-fold greater than that for ETBR [47]. It can prevent OC bone metastasis and reduce the incidence of bone disease. Atrasentan has been actively studied as a single drug in castration-resistant prostate cancer (CRPC) patients and it was found that patients treated with Atrasentan had a significantly longer progression-free survival (PFS) [48]. Bosentan, a dual antagonist to ETAR and ETBR, has been associated with decreased osteolysis and cancer cell metastasis [49]. Zibotentan/ZD4054 is an oral ETAR-specific antagonist that specifically binds to ETAR but not ETBR. Zibotentan has been used in clinical trials in patients with bone metastasis, but as yet clinical data are limited [47, 48].
5.6 TGF-β pathway inhibitors
TGF-β plays a dual role in the development of cancer. TGF-β acts as a tumor suppressor by inhibiting cell growth in precancerous tissues. However, it also stimulates metastasis and angiogenesis in cancerous tissues [50]. BMP7 belongs to the TGF family of proteins and is secreted by bone marrow stromal cells. BMP7 binds to the BMPR2 receptor on the surface of tumor cells, inhibiting the ERK signaling pathway and activating p38 MAP kinase, leading to the dormancy of tumor cells [16, 51]. Preclinical trials have confirmed that BMP7 is an effective inducer that inhibits the formation of bone metastases [24]. Ki26894 is a TGF-β1 receptor inhibitor that has been found to block TGF signaling in MDA-MB-231 breast cancer cells by inhibiting Smad2 phosphorylation. In addition, in vitro experiments have shown that Ki26894 can reduce the movement and invasion of TGF-induced MDA-MB-231 breast cancer cells [52].
5.7 Src signaling pathway inhibitors
Src signaling plays an important role in the growth, invasion and metastasis of tumor cells. The Src kinase family includes nine non-receptor tyrosine kinases, of which Src is the most widely studied [53]. It is involved in cell proliferation, survival, movement and angiogenesis. The presence of high levels of Src in mature OCs suggests that Src plays an important role in maintaining the normal physiology of OCs [54]. Dasatinib, saracatinib and bosutinib are small molecule tyrosine kinase inhibitors that are currently subject to clinical research. Dasatinib has been found to inhibit osteoclastogenesis [4]. Animal experiments have shown that dasatinib can significantly reduce the growth of prostate tumors in the bone. When prostate cancer cells were injected into the tibia of mice, dasatinib increased bone density and reduced bone destruction [55]. In mice vaccinated with triple-negative breast cancer cells, dasatinib significantly prevented the formation of osteolytic metastases [56]. Saracatinib is a bispecific Src/Abl tyrosine kinase inhibitor [43] that has been found to prevent the migration of OC precursor cells from the OB layer to the bone surface, thereby inhibiting bone resorption [57].
5.8 Integrin inhibitors
Integrins are adhesion receptors on the surface of various cells, including cancer cells and OCs. Integrins are involved in cancer cell invasion and OC-mediated bone resorption. Integrin inhibitors can prevent bone metastasis in at least two ways: by inhibiting OC-mediated bone resorption and by directly targeting cancer cells [58]. ATN-161, a fibronectin-derived peptide that interferes with integrin α5β1 and αVβ3 binding, has been tested in preclinical animal models. It significantly reduced the incidence of bone metastases in nude mice injected with MDA-MB-231 breast cancer cells into the heart [22]. PSK-1404 is a non-peptide antagonist of αVβ3. Long-term PSK1404 therapy has been found to inhibit OC-mediated bone resorption in animal models [59] and to directly target αVβ3 expression in cancer cells, thereby decreasing bone colonization by cancer cells [58].
5.9 VEGF receptor tyrosine kinase inhibitors
VEGF is an important factor that stimulates tumor angiogenesis and plays a positive regulatory role in malignant tumor bone metastasis. PTK787 is a VEGF receptor tyrosine kinase inhibitor that has been found to reduce the ability of C4-2B prostate cancer cells to metastasize [44]. Cabozantinib, a dual inhibitor of VEGFR2 and the tyrosine kinase receptor MET, has been found to act as an effective inhibitor of bone metastasis [7].
5.10 Other inhibitors
Bortezomib is a proteasome inhibitor [22] that induces apoptosis of cancer cells and tumor-activated OCs, and promotes OB differentiation and bone formation [60]. In preclinical models, bortezomib has been found to inhibit osteolytic breast cancer metastasis [22]. Its anti-tumor effect mainly occurs in the bone microenvironment and it can promote bone formation [60]. Sunitinib is a multi-targeted tyrosine kinase inhibitor that targets bone marrow stromal cells and endothelial cells, leading to bone marrow vascular leakage, stromal cell apoptosis, as well as to inhibition of lung cancer cell colonization and osteolysis [24].
5.11 Other treatments
Radionuclides are currently used to treat bone pain in patients with multifocal osteoblastic metastases [4]. These include Sr-89, Rhe-186, Sm-153 and Ra-223, which are substitutes for bone hydroxyapatite and can be taken up by osteoblastic metastases. Calcilytics, which are antagonists of calcium-sensing receptors, have been studied for their role in the treatment of autosomal dominant hypocalcemia and have been shown to limit the growth of bone tumors [24]. Reveromycin is a novel antibiotic that inhibits OC activation and bone resorption. Its activity may be due to a specific transport of retroviral A into OCs at acidic pH. Histological analysis indicates that reveromycin significantly reduces bone damage caused by OCs, suggesting that it inhibits osteolytic bone metastasis.
6 Outlook
Although several cellular signaling pathways and molecules involved in the pathogenesis of bone metastasis have been identified, its precise mechanisms are not fully elucidated yet. Recent studies have confirmed that targeted therapies against different biological factors and cell signaling pathways can improve their efficacy and reduce toxic side effects. This may bring more benefits to patients with different types of bone metastasis. (Table 1) At present, some new drugs, including Src signaling pathway inhibitors, cathepsin K inhibitors and endothelin receptor antagonists are already being tested in clinical trials. As a novel emerging treatment strategy, immune checkpoint inhibitors may have therapeutic potential for inhibiting bone metastases. Although the application of nanotechnology to medicine is still in its infancy, our knowledge of the bone microenvironment may facilitate the use of nanoparticles targeting cancer cells within the bone. Epigenetic changes in tumor stromal cells may also play important roles in cancer biology and may be used as therapeutic intervention targets [24]. With the development of multidisciplinary treatment options for bone metastases, new treatment strategies may be developed as well as ideas for a personalized treatment of bone metastases.
References
N. Kamiya, H. Suzuk, T. Endo, M. Yano, M. Naoi, D. Nishimi, K. Kawamura, T. Imamoto, T. Ichikawa, Clinical usefulness of bone markers in prostate cancer with bone metastasis. Int. J. Urol. 19, 968–979 (2012)
J. Fornetti, A.L. Welm, S.A. Stewart, Understanding the bone in Cancer metastasis. J. Bone Miner. Res. 33, 2099–2113 (2018)
E.V. Morris, C.M. Edwards, Bone marrow adipose tissue: A new player in cancer metastasis to bone. Front. Endocrinol. (Lausanne) 7, 90 (2016)
U.H. Weidle, F. Birzele, G. Kollmorgen, R. Rüger, Molecular mechanisms of bone metastasis. Cancer Genomics Proteomics 13, 1–12 (2016)
P. Nilendu, S.C. Sarode, D. Jahagirdar, I. Tandon, S. Patil, G.S. Sarode, J.K. Pal, N.K. Sharma, Mutual concessions and compromises between stromal cells and cancer cells: Driving tumor development and drug resistance. Cell. Oncol. 41, 353–367 (2018)
A. Schmid-Alliana, H. Schmid-Antomarchi, R. Al-Sahlanee, P. Lagadec, J.C. Scimeca, E. Verron, Understanding the progression of bone metastases to identify novel therapeutic targets. Int. J. Mol. Sci. 19, 148 (2018)
M. van Driel, J.P. van Leeuwen, Cancer and bone: A complex complex. Arch. Biochem. Biophys. 561, 159–166 (2014)
A.M. Mastro, C.V. Gay, D.R. Welch, The skeleton as a unique environment for breast cancer cells. Clin. Exp. Metastasis 20, 275–284 (2003)
Tu Q, Zhang J, Fix A, Brewer E, Li Y. P, Zhang Z. Y, Chen J. Targeted overexpression of BSP in osteoclasts promotes bone metastasis of breast Cancer cells. J. Cell. Physiol. 218, 135–145 (2009)
L.J. Suva, C. Washam, R.W. Nicholas, R.J. Griffin, Bone metastasis: Mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 7, 208–218 (2011)
A.S. Gdowski, A. Ranjan, J.K. Vishwanatha, Current concepts in bone metastasis, contemporary therapeutic strategies and ongoing clinical trials. J. Exp. Clin. Cancer Res. 36, 108 (2017)
J. Fang, Q. Xu, Differences of osteoblastic bone metastases and osteolytic bone metastases in clinical features and molecular characteristic. Clin. Transl. Oncol. 17, 173–179 (2015)
D. Cruceriu, O. Baldasici, O. Balacescu, I. Berindan-Neagoe, The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: Molecular insights and therapeutic approaches. Cell. Oncol. 43, 1–18 (2020)
A.C. Hirbe, E.A. Morgan, K.N. Weilbaecher, The CXCR4/SDF-1 chemokine axis: A potential therapeutic target for bone metastases? Curr. Pharm. Des. 16, 1284–1290 (2010)
G.R. Mundy, Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2, 584–593 (2002)
P. Clezardin, Pathophysiology of bone metastases from solid malignancies. Joint Bone Spine 84, 677–684 (2017)
L. D'Amico, I. Roato, The impact of immune system in regulating bone metastasis formation by Osteotropic tumors. J. Immunol. Res. 2015, 143526 (2015)
A.L. Hardaway, M.K. Herroon, E. Rajagurubandara, I. Podgorski, Bone marrow fat: Linking adipocyte induced inflammation with skeletal metastases. Cancer Metastasis Rev. 33, 527–543 (2014)
Y. Shiozawa, M.R. Eber, J.E. Berry, R.S. Taichman, Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep. 4, 689 (2015)
A.C. Carreira, F.H. Lojudice, E. Halcsik, R.D. Navarro, M.C. Sogayar, J.M. Granjeiro, Bone morphogenetic proteins: Facts, challenges, and future perspectives. J. Dent. Res. 93, 335–345 (2014)
L. Rosano, F. Spinella, A. Bagnato, Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 13, 637–651 (2013)
J.Y. Krzeszinski, Y. Wan, New therapeutic targets for cancer bone metastasis. Trends Pharmacol. Sci. 36, 360–373 (2015)
K.M. Bussard, C.V. Gay, A.M. Mastro, The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 27, 41–55 (2008)
K.N. Weilbaecher, T.A. Guise, L.K. McCauley, Cancer to bone: A fatal attraction. Nat. Rev. Cancer 11, 411–425 (2011)
Z.S. Templeton, W.R. Lie, W. Wang, Y. Rosenberg-Hasson, R.V. Alluri, J.S. Tamaresis, M.H. Bachmann, K. Lee, W.J. Maloney, C.H. Contag, B.L. King, Breast Cancer cell colonization of the human bone marrow adipose tissue niche. Neoplasia 17, 849–861 (2015)
M.K. Herroon, E. Rajagurubandara, A.L. Hardaway, K. Powell, A. Turchick, D. Feldmann, I. Podgorski, Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 4, 2108–2123 (2013)
Y.S. Hwang, P.F. Lindholm, Constitutive and inducible expression of invasion-related factors in PC-3 prostate cancer cells. J. Cancer Prev. 20, 121–128 (2015)
J.S. Nam, A.R. Sharma, L.T. Nguyen, S. Jagga, Y.H. Lee, G. Sharma, S.S. Lee, Lysophosphatidic acid enhances breast cancer cells-mediated. Korean J. Physiol. Pharmacol. 22, 503–511 (2018)
M. David, J. Ribeiro, F. Descotes, C.M. Serre, M. Barbier, M. Murone, P. Clézardin, O. Peyruchaud, Targeting lysophosphatidic acid receptor type 1 with Debio 0719 inhibits spontaneous metastasis dissemination of breast cancer cells independently of cell proliferation and angiogenesis. Int. J. Oncol. 40, 1133–1141 (2012)
A. Salmaninejad, S.F. Valilou, A. Soltani, S. Ahmadi, Y.J. Abarghan, R.J. Rosengren, A. Sahebkar, Tumor-associated macrophages: Role in cancer development and therapeutic implications. Cell. Oncol. 42, 591–608 (2019)
K. Hiraoka, M. Zenmyo, K. Watari, H. Iguchi, A. Fotovati, Y.N. Kimura, F. Hosoi, T. Shoda, K. Nagata, H. Osada, M. Ono, M. Kuwano, Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/ macrophages. Cancer Sci. 99, 1595–1602 (2008)
M.K. Herroon, E. Rajagurubandara, D.L. Rudy, A. Chalasani, A.L. Hardaway, I. Podgorski, Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene 32, 1580–1593 (2013)
S.J. Coniglio, Role of tumor-derived chemokines in osteolytic bone metastasis. Front. Endocrinol. (Lausanne) 9, 313 (2018)
J. Wang, Y. Shiozawa, Y. Wang, Y. Jung, K.J. Pienta, R. Mehra, R. Loberg, R.S. Taichman, The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 283, 4283–4294 (2008)
O. Wald, CXCR4 based therapeutics for Non-Small Cell Lung Cancer (NSCLC). J. Clin. Med. 7, 303 (2018)
P. Clézardin, Therapeutic targets for bone metastases in breast cancer. Breast Cancer Res. 13, 207 (2011)
Y.X. Sun, M. Fang, J. Wang, C.R. Cooper, K.J. Pienta, R.S. Taichman, Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate 67, 61–73 (2007)
P.I. Croucher, M.M. McDonald, T.J. Martin, Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 16, 373–386 (2016)
M. Croset, C. Kan, P. Clézardin, Tumour-derived miRNAs and bone metastasis. Bonekey Rep. 4, 688 (2015)
H. Taipaleenmäki, G. Browne, J. Akech, J. Zustin, A.J. van Wijnen, J.L. Stein, E. Hesse, G.S. Stein, J.B. Lian, Targeting of Runx2 by miR-135 and miR-203 impairs progression of breast cancer and metastatic bone disease. Cancer Res. 75, 1433–1444 (2015)
G. Carlinfante, D. Vassiliou, O. Svensson, M. Wendel, D. Heinegard, G. Andersson, Differential expression of osteopontin and bone sialoprotein in bone metastasis of breast and prostate carcinoma. Clin. Exp. Metastasis 20, 437–444 (2003)
A. Bellahcene, V. Castronovo, K.U. Ogbureke, L.W. Fisher, N.S. Fedarko, Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer 8, 212–226 (2008)
B.T. Li, M.H. Wong, N. Pavlakis, Treatment and prevention of bone metastases from breast cancer: A comprehensive review of evidence for clinical practice. J. Clin. Med. 3, 1–24 (2014)
P. Clezardin, A. Teti, Bone metastasis: Pathogenesis and therapeutic implications. Clin. Exp. Metastasis 24, 599–608 (2007)
D.L. Lacey, W.J. Boyle, W.S. Simonet, P.J. Kostenuik, W.C. Dougall, J.K. Sullivan, J. San Martin, R. Dansey, Bench to bedside: Elucidation of the OPG–RANK–RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 11, 401–419 (2012)
R.E. Coleman, Bone cancer in 2011: Prevention and treatment of bone metastases. Nat. Rev. Clin. Oncol. 9, 76–78 (2012)
M.A. Carducci, A. Jimeno, Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin. Cancer Res. 12, 6296s–6300s (2006)
J.B. Nelson, K. Fizazi, K. Miller, C. Higano, J.W. Moul, H. Akaza, T. Morris, S. McIntosh, K. Pemberton, M. Gleave, Phase 3, randomized, placebo-controlled study of zibotentan (ZD4054) in patients with castration-resistant prostate cancer metastatic to bone. Cancer 118, 5709–5718 (2011)
D. Dreau, A. Karaa, C. Culberson, H. Wyan, I.H. McKillop, M.G. Clemens, Bosentan inhibits tumor vascularization and bone metastasis in an immunocompetent skin-fold chamber model of breast carcinoma cell metastasis. Clin. Exp. Metastasis 23, 41–53 (2006)
M. Petrillo, G. Scambia, G. Ferrandina, Novel targets for VEGF-independent antiangiogenic drugs. Expert Opin. Investig. Drugs 21, 451–472 (2012)
F. Rossari, C. Zucchinetti, G. Buda, E. Orciuolo, Tumor dormancy as an alternative step in the development of chemoresistance and metastasis - clinical implications. Cell. Oncol. 43, 155–176 (2020)
S. Ehata, A. Hanyu, M. Fujime, Y. Katsuno, E. Fukunaga, K. Goto, Y. Ishikawa, K. Nomura, H. Yokoo, T. Shimizu, E. Ogata, K. Miyazono, K. Shimizu, T. Imamura, Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 98, 127–133 (2007)
T. Koreckij, H. Nguyen, L.G. Brown, E.Y. Yu, R.L. Vessella, E. Corey, Dasatinib inhibits the growth of prostate cancer in bone and provides additional protection from osteolysis. Br. J. Cancer 101, 263–268 (2009)
F. Saad, A. Lipton, SRC kinase inhibition: Targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat. Rev. 36, 177–184 (2010)
L.J. Lombardo, F.Y. Lee, P. Chen, D. Norris, J.C. Barrish, K. Behnia, S. Castaneda, L.A. Cornelius, J. Das, A.M. Doweyko, C. Fairchild, J.T. Hunt, I. Inigo, K. Johnston, A. Kamath, D. Kan, H. Klei, P. Marathe, S. Pang, R. Peterson, S. Pitt, G.L. Schieven, R.J. Schmidt, J. Tokarski, M.L. Wen, J. Wityak, R.M. Borzilleri, Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino) thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical. J. Med. Chem. 47, 6658–6661 (2004)
X.H. Zhang, Q. Wang, W. Gerald, C.A. Hudis, L. Norton, M. Smid, J.A. Foekens, J. Massagué, Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16, 67–78 (2009)
T.J. de Vries, M.G. Mullender, M.A. van Duin, C.M. Semeins, N. James, T.P. Green, V. Everts, J. Klein-Nulend, The Src inhibitor AZD0530 reversibly inhibits the formation and activity of human osteoclasts. Mol. Cancer Res. 7, 476–488 (2009)
P. Clëzardin, Integrins in bone metastasis formation and potential therapeutic implications. Curr. Cancer Drug Targets 9, 801–806 (2009)
Y. Zhao, R. Bachelier, I. Treilleux, P. Pujuguet, O. Peyruchaud, R. Baron, P. Clément-Lacroix, P. Clezardin, Tumor alphavbeta integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 67, 5821–5830 (2007)
M.D. Jones, J.C. Liu, T.K. Barthel, S. Hussain, E. Lovria, D. Cheng, J.A. Schoonmaker, S. Mulay, D.C. Ayers, M.L. Bouxsein, G.S. Stein, S. Mukherjee, J.B. Lian, A proteasome inhibitor, bortezomib, inhibits breast cancer growth and reduces osteolysis by downregulating metastatic genes. Clin. Cancer Res. 16, 4978–4989 (2010)
Funding
This work was supported by the National Natural Science Foundation of China (Grant No.81774221), The Capital Health Research and Development of Special (No.2018–2-1113), the Basic-Clinical Cooperation Program from Capital Medical University (No.17JL14), the Research Foundation of Beijing Friendship Hospital, Capital Medical University (No.yyqdkt2016–4) and the Beijing Municipal 215 High-level Health Person Foundation Project (No.2014-3-004).
Author information
Authors and Affiliations
Contributions
Xiaoting Ma wrote the manuscript, collected materials, and consulted the literature; Jing Yu revised the paper and provided funding.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, X., Yu, J. Role of the bone microenvironment in bone metastasis of malignant tumors - therapeutic implications. Cell Oncol. 43, 751–761 (2020). https://doi.org/10.1007/s13402-020-00512-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-020-00512-w