
Abstract
There are common mechanisms shared by genetically or pathologically distinct neurodegenerative diseases, such as excitotoxicity, mitochondrial deficits and oxidative stress, protein misfolding and translational dysfunction, autophagy and microglia activation. This indicates that although the original cause may differ in individual diseases or even subtypes of certain disorders, these disrupted common cell functions and signaling, together with aging, may lead to final execution of cell death through similar pathways. The variable neurodegenerative disease symptoms are probably caused by the type, location, and connection of the cell populations that suffer from dysfunction and loss. Besides apoptosis, necroptosis, and autophagy, an important form of death termed parthanatos plays a prominent role in stroke and several neurodegenerative diseases, which is due to PARP-1 overactivation, PAR accumulation, nuclear translocation of the mitochondria protein AIF, and large-scale DNA cleavage. Understanding the mechanisms and interactions of cell death signaling will not only help to develop neuroprotective strategies to halt neurodegeneration, but also provide biomarkers for monitoring disease progression and recovery.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Neurodegenerative diseases
- Cell death
- Oxidative stress
- Excitotoxicity
- Parthanatos
- Mitochondria
- Nitric oxide
- AIF
1 Introduction
Neurodegenerative diseases are caused by interactions of genetic factors (mutation of disease-associated genes) and environmental factors (including aging and life style). These disorders share common features, like excitotoxicity, synaptic dysfunction, misfolded protein aggregation, reactive oxidative stress (ROS), mitochondrial deficits, dysregulation of intracellular calcium, trophic factor loss, axonal transport deficits, transcription and translation disruption, and finally cell loss. Early cognitive and mood symptoms seen with neurodegenerative disease patients are mostly due to the impaired synaptic and other cellular function, and networking of affected cells by either gain-of-function or loss-of-function of the mutated disease protein. Disrupted cell functions together with aging-induced accumulation of DNA damage and oxidative stress gradually overwhelm the self-defense system including protein quality control system (for example, ubiquitin and autophagy) and others, leading to a shift in life-death balance culminating in programmed cell death (PCD) . It is likely that multiple cell death pathways are involved in the cell loss in neurodegenerative diseases and in experiment models of neurodegeneration. The fate (survival or death) and major path to death (how to die) of cells depend on the source, duration, and dose of different stressors, as well as the type, previous challenges, and metabolic conditions of the cell. Several types of cell death including apoptosis, necrosis, autophagy, and parthanatos may be involved in neurodegeneration [1]. We will briefly discuss apoptosis, necroptosis, autophagy and focus on parthanatos and their involvement and therapeutic potential in neurodegenerative diseases.
2 Cell Death in Neurodegeneration
2.1 Apoptosis
Apoptosis is the most studied and well-known form of PCD, which is found in both development and diseases. The generally accepted definition of apoptosis is an active form of cell death with cell plasma membrane and organelle integrity and mediation by caspases activation [2], which includes extrinsic apoptosis and caspase-dependent intrinsic apoptosis defined by Nomenclature Committee on Cell Death (NCCD) [3]. There are markers of apoptosis in postmortem tissues of patients, as well as in cell and animal models of neurodegenerative disorders, including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [2, 4]. It is worth noting that certain cell death-related phenomenon may accompany a cell death process while the final execution is carried out by a different cell death mechanism [3]. Reactive oxygen species, DNA damage, loss of mitochondria membrane potential, and aggregation of misfolded proteins are all common features associated with most neurodegenerative disorders and major forms of neuronal death. On the other hand, inhibiting cell death by Z-VAD-fmk , a pan caspases blocker, usually results in delayed death by autophagy or necrosis [1, 5]. Thus, extra caution needs to be taken when classifying a certain type of death as apoptosis.
2.2 Necroptosis
Necroptosis was first defined in 2005 as a regulated form of non-apoptotic cell death with necrotic cell morphology, triggered by TNFα receptor 1 (TNFR1) and can be inhibited by receptor-interacting protein kinase 1 (RIPK1) inhibitor necrostatin 1 (Nec-1) [3, 5, 6]. Besides death receptor activators, such as TNF and IFNγ, ROS have also been shown to promote necroptosis [5, 7]. Necroptosis has been shown to contribute to delayed mouse ischemic brain injury, which can be inhibited by necrostatin-1 [6]. Nec-1 reduced motor neuron death in coculture with astrocytes from ALS patients [8] and delayed motor deficits in the R6/2 transgenic mouse model of Huntington’s disease [9]. A group recently detected activation of RIPK1 and RIPK3 in multiple sclerosis (MS) patient cortical samples and also showed that RIPK1 inhibitor 7-Cl-O-Nec-1 reduced oligodendrocyte death in two MS animal models [10].
2.3 Autophagy
Autophagy (“self-eating”) is a process by which cells form intracellular autophagosomes (double-membrane structures) to “eat up” intracellular organelles and aggregated proteins and deliver them to lysosomes for degradation and nutrient recycling [11]. Several pathological mutations of proteins in AD, PD, HD, and ALS are linked to deficits of autophagy, such as substrate recognition, lysosome acidification, trafficking, and membrane permeabilization [12], which will not be discussed in detail here. Autophagy plays important role in cleaning up damaged mitochondria and aggregated proteins and thus contributes to neurodegenerative pathology. Indeed, upregulated autophagy can promote the clearance of pro-aggregate proteins, such as mutant huntingtin, and protect against neurodegeneration in both cell and animal models [13]. Defects in autophagy primarily play a role in neurodegeneration, but autophagy itself can also be involved in several forms of cell death.
2.4 Parthanatos
Parthanatos , previously known as PARP-1-dependent cell death, is the second most studied form of cell death. When massive DNA damage is induced by ionizing radiation, prolonged toxic treatment of the DNA-alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) or N-methyl-d-aspartate (NMDA), oxygen glucose deprivation (OGD), hydrogen peroxide and other ROS, as well as ischemia and reperfusion, poly(ADP-ribose) polymerase-1 (PARP-1) overactivates and produces branched, long-chained poly (ADP-ribose) (PAR) polymers [14,15,16,17]. Ischemia and reperfusion causes massive release of the excitatory neurotransmitter glutamate, which excessively activates glutamate receptors, especially the NMDA receptor, and causes a large calcium influx, resulting in the activation of neuronal nitric oxide synthase (nNOS) and cell death, called “excitotoxicity ” [18,19,20]. nNOS plays an important role in excitotoxicity since nNOS deletion and NOS inhibitors protect primary brain cultures or mice against NMDA excitotoxicity, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and middle cerebral artery occlusion (MCAO)-induced neuronal injury [18, 19, 21,22,23]. Nitric oxide (NO) is able to diffuse freely through the plasma membrane and react with superoxide (O2.−) to form peroxynitrite (ONOO−), which induces DNA damage and PARP-1 activation [14, 24,25,26]. This “excitotoxicity” hypothesis is able to explain, at least in part, the cell death in acute ischemia and traumatic brain injury, as well as progressive neuron loss in neurodegenerative conditions such as HD, AD, PD, MS, and ALS [27,28,29].
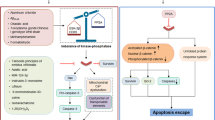
Under toxic stimuli and pathological conditions when PARP-1 is overactivated, the release of PAR polymers from the nucleus to cytosol and mitochondria induces translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, which leads to large-scale DNA fragmentation (approximately 50 kb) and chromatin condensation, followed by cell death, termed parthanatos (Fig. 16.1) [14,15,16,17]. This form of cell death named from “par” (for PAR polymer) and “Thanatos” (the personification of death in Greek mythology) is now officially recognized by the NCCD [3, 14, 17]. It has been thought previously that when PARP-1 overactivates, cells die due to cellular NAD+ and ATP depletion, in a way similar to necrosis. However, recent studies in primary cell cultures and mice stroke models indicate that energy depletion is not the primary cause of PARP-1 mediated cell death [30, 31]. In fact, mouse primary neurons are resistant to NMDA-induced cell death when cytosolic PAR was neutralized with PAR-specific antibodies [32]. On the other hand, introducing exogenous PAR polymer through lipid-based delivery results in neuron death, with higher dose and more complex PAR polymers inducing more death [32]. The other study [33] showing PAR polymer was able to induce AIF translocation from mitochondria to nucleus in cell-free in vitro assay further confirmed that PAR polymer is the mediator of death signal in parthanatos. The PAR-induced AIF translocation can be prevented by pretreatment of PAR degrading enzymes, but not the pan-caspase inhibitor z-VAD-fmk [33], indicating that the AIF release in parthanatos is distinct cellular event from that in apoptosis.

Comparison of the major signaling mediators in caspase-independent parthanatos and caspase-dependent apoptosis. Stressors such as ROS, NMDA, ischemia, alkylating agents (MNNG), radiation, UV can activate PARP-1 directly or indirectly through activation of nNOS, which produces NO to induce ROS and the subsequent DNA damage. PARP-1 overactivation produces free PAR by PARG-mediated hydrolysation, which serves as a death signal from the nucleus to mitochondria, where it induces the release of AIF. AIF then translocates together with a presumed nuclease PAAN to the nucleus where it induces large fragmentation of DNA. This form of cell death is called “parthanatos ,” which can be completely blocked by PARP-1 inhibitors, but not pan-caspase inhibitors including Z-VAD-fmk. Under some stress conditions with limited amount of DNA damage, cell may undergo apoptosis through p53 activation, which results in disrupted balance of BCL2 (anti-apoptotic) and BAK (pro-apoptotic) and the release of cytochrome c (Cyto c). Cytochrome c then associates with Apaf1 (not shown) and activates procaspase-9, which cleaves and activates caspase-3, results in protein degradation, DNA fragmentation, and cell death. Thus, this caspase-dependent apoptosis is distinct from parthanatos, and can be blocked by caspase inhibitors
Unlike caspase-dependent apoptosis, parthanatos cannot be rescued by pan-caspase inhibitors (such as Z-VAD-fmk), does not form apoptotic bodies, and causes large-scale instead of small-scale DNA fragmentation [17, 34]. Although both parthanatos and necrosis involve loss of cell membrane integrity, parthanatic cell death is distinct from necrosis, as it is marked with PARP-1 activation and AIF translocation and is not accompanied by cell swelling [17, 34]. The common triggers and signaling differences of caspase-dependent apoptosis and caspase-independent parthanatos are illustrated in Fig. 16.1.
The major forms of cell death found in neurodegenerative diseases are summarized in Table 16.1 (adapted from [3, 14, 17]). It is worth noting that the cell may favor one form of death over others depending on the nature, length, and degree of the stress as well as the pre-stimuli status of the cell. Thus, only cell death that can be completely blocked by inhibitors or deletion of PARP-1 should be considered as parthanatos primarily [14]. A more detailed summary of conditions that trigger parthanatos and the cellular events involved in parthanatos can be found in some recent reviews [14,15,16,17].
3 Components of Parthanatos
3.1 PARP-1
PARP-1 is the most abundant and best characterized member of the 17 mammalian PARP family proteins. PARP-1 is responsible for production of more than 90% of cellular PAR polymers [35, 36]. It is a nuclear protein with three major functional domains: (1) an N-terminal DNA-binding domain with three zinc finger motifs to sense DNA strand breaks and a nuclear localization sequence (NLS); (2) a central automodification domain for self (ADP-ribosyl)ation and a BRCA1 C-terminal (BRCT) motif for protein–protein interaction; (3) a C-terminal catalytic domain with NAD+-binding domain and conserved PARP signature motif (PSM) for PAR synthesis (Fig. 16.2) [35, 37]. PARP-1 is known to regulate DNA repair, transcription, cell differentiation, mitochondrial function, and cell death [35, 36, 38,39,40,41].

PARP-1 function domains. PARP-1 has three major function domains: the N-terminal DNA-binding domain, the auto-modification domain, and a C-terminal catalytic domain. The DNA-binding domain contains three zinc fingers that are important for DNA binding and DNA damage detection (FIII is dispensable) and a nuclear localization signal (NLS). The auto-modification domain has a BRCT motif for protein–protein interaction and glutamate and lysine residues as acceptors of ADP-ribose to regulate PARP-1 activity. The catalytic domain is composed of enzyme active site responsible for adding ADP-ribose from NAD+ to protein acceptors and a highly conserved sequence called the PARP signature motif (PSM) found in all members of PARP family proteins [15, 37]
Under physiological conditions, PARP-1 activity remains at a low level, but can increase up to 500-fold in a few seconds when sensing mild DNA damage [35]. Upon activation (Fig. 16.3), PARP-1 synthesizes negatively charged PAR by hydrolyzing oxidized nicotinamide adenine dinucleotide (NAD+) and linking them through glycosidic bonds (Fig. 16.3) with branching every 20–50 ADP-ribose units on average [42, 43]. PAR, averaging 50–200 units, are covalently attached to a variety of nuclear acceptor proteins by forming an ester bond with a glutamate, aspartate, or C-terminal lysine residues [41]. The nuclear acceptors of this post-translational modification, poly (ADP-ribosyl)ation, includes PARP-1 itself, histones, DNA ligase II, topoisomerases, DNA polymerases, transcription factors, and a host of other proteins [36]. Through this wide-ranged poly (ADP-ribosyl)ation and BRCT interaction, PARP-1 recruits DNA repair proteins to the DNA damage site to facilitate DNA repair [44]. Poly (ADP-ribosyl)ation of target proteins can alter its enzymatic activity, subcellular localization, and prevent protein–protein or protein–nucleic acid interactions. Possibly due to long branching and negative charge, PAR can modify the charge or mask the binding motif or functional domain of acceptor proteins [36, 41, 45].

Poly (ADP-ribosyl)ation metabolism . Several stressors activate PARP directly or indirectly through DNA damage, including the recently discovered PD-associated protein AIMP2. Activated PARP converts NAD+ to ADP-ribose, attaches it to a protein acceptor, and discharges nicotinamide (1, Initiation stage). The poly (ADP-ribose) long chain elongates (2, Elongation) and branches (3, Branching) until being dehydrolyzed by PARG (4, Breakdown). Mono-ADP-ribosyl-protein lyase then removes the final ADP-ribose on the protein acceptor (5) [15]
Given that structure and levels of PAR depend on the cell energy level and other factors of poly (ADP-ribosyl)ation, PARP-1 can promote both cell survival and death depending on the stress stimuli and status of the cell [37]. Mild or moderate stresses usually lead to PARP-1 activation that result in DNA repair and transcription regulation to restore genome stability, degrade oxidized or damaged proteins, and maintain homeostasis [37, 45,46,47]. On the other hand, hyperactivation of PARP-1 by severe or sustained stresses causes cell death programs, such as parthanatos [15, 48].
3.2 PARG
After PARP activation, poly (ADP-ribose) glycohydrolase (PARG) or the recently identified ADP-ribosylhydrolase 3 (ARH3) rapidly hydrolyze the glycosidic bonds to break down PAR from its modified proteins [15]. Therefore, poly (ADP-ribosyl)ation is an immediate, but transient (degrades within minutes), and strictly regulated post-translational modification [35, 46]. Full-length PARG localizes to the nucleus, but other splice variants with enzyme activity have been found in the cytosol and mitochondria [14, 49, 50].
Overexpression of PARG was found to protect against excitotoxicity, focal ischemia, H2O2, and PAR-mediated cell death [32, 51, 52]. On the other hand, reducing PARG increased the sensitivity of cells to DNA damaging agents, excitotoxicity, and ischemia. The PARG knockout (KO) mouse is embryonically lethal at around embryonic day 3.5 [32, 53]. Hanai and others have reported that mutant Drosophila lacking the PARG catalytic domain are lethal at the larval stages. Some mutants reached the adult stage at higher developmental temperature of 29 °C, but showed progressive neurodegeneration with reduced locomotor activity, extensive accumulation of PAR in central nervous system (CNS), and a short lifespan [54]. These results suggest that poly(ADP-ribose) metabolism is required for maintenance of the normal function of neuronal cells. In addition, PARG overexpression prevented PARP-1-dependent mitochondrial AIF release and cell death [33]. All this evidence suggests that PARG plays a critical role in the normal function of neuronal cells and parthanatos by regulating PAR levels [55].
3.3 PAR
PAR is a large, negatively charged polymer that can be either attached as a post-translational modification to a protein or as a free polymer produced by PARG hydrolysis (Fig. 16.3) [56]. Free or protein-bound PAR polymers can bind to other proteins non-covalently through their conserved PAR-binding motifs, including PAR-binding motifs (PBMs), macrodomains, WWE domains, and PAR-binding zinc finger (PBZF) domains [37]. These motifs usually overlap with other functional domains and thus the PAR polymer can alter the structure, function, and localization of its binding proteins [41, 56]. Free PAR polymer accumulation has been implicated as the major death signal in parthanatos, ischemic stroke, and some neurodegenerative diseases [14, 32, 33]. How the highly negatively charged large PAR polymer escapes from nucleus to mitochondria and induces parthanatos is not clear. It is presumed that a protein partner may bind PAR and help transport the death signal out of nucleus to mitochondria . Further study revealing the mechanism of PAR translocation will provide novel insight on how to prevent the death signal from escaping the nucleus.
3.4 AIF
AIF is a dual function molecule: it is a mitochondrial flavoprotein with important NADH oxidase activity, yet it is able to translocate to nucleus to induce large-scale DNA fragmentation upon toxic stimuli [57,58,59]. Studies using antibody neutralization, small interfering RNA (siRNA), and Harlequin mutant mice (express ~20% AIF compared to wild-type), all indicate that AIF is a death effector of parthanatos [33, 34, 60, 61]. It was originally thought that AIF resides in the mitochondrial intermembrane space [62]. However, a recent study using biochemical and electron microscopic analyses revealed that a small pool (30%) of AIF resides on the mouse outer mitochondrial membrane and is responsible for the rapid AIF release and neuron death following NMDA stimulation [63]. Unlike its name, AIF mediates caspase-independent cell death (parthanatos), while other death factors (such as cytochrome C) released from mitochondria participate in caspase-dependent cell death (Fig. 16.1). Some crosstalk between caspases and AIF have been proposed [64, 65].
AIF is a high-affinity PAR-binding protein [66], and the binding of PAR with AIF is required for the release of AIF from the mitochondria to induce cell death, but it is not required for its NAD+ oxidase activity, or ability to induce DNA fragmentation [67]. PAR binding to AIF on the outer surface of mitochondria decreases its affinity to the mitochondria facilitating its release [67]. AIF translocation to the nucleus seems to be the no-return point for cell death in excitotoxicity, ischemia, and some neurodegenerative disorders [68, 69]. However, how PAR is released from the nucleus and what is the nuclease downstream of AIF are two missing links of parthanatos.
3.5 Iduna
Iduna , also known as ring finger protein (RNF) 146 , is an E3 ubquitin ligase named after the Norwegian goddess of protection and eternal youth. It ubiquitinates poly (ADP-ribosyl)ated substrates and targets them for degradation [70, 71]. Iduna was recently demonstrated as a novel endogenous inhibitor of parthanatos and protects neurons from excitotoxicity both in vitro and in vivo, as well as MCAO-induced stroke in mice [72]. Furthermore, the neuroprotective function of Iduna relies on its binding ability with PAR, since mutations of Iduna’s PBM motif result in attenuated protection [72]. Under mild toxic stimulations, Iduna can be induced and works as a guard in the cytosol to degrade PAR or PAR-binding proteins to prevent parthanatos, presumably by preventing PAR from reaching mitochondria and release AIF.
4 Parthanatos and Neurodegenerative Diseases
4.1 PARP-1 and Neurodegeneration
PARP-1 has emerged as a major death regulator in both acute neuronal injury and neurodegenerative disorders. Oxidative stress , one of the most important common features of neurodegenerative disorders and aging diseases, is able to activate PARP-1. Involvement of PARP-1 has been found in human patient brains, as well as cellular and rodent models of stroke [73], trauma [74], spinal cord injury [75], AD [76,77,78,79], PD [80,81,82,83,84], HD [85], MS [86], and ALS [87, 88] [14, 89]. Furthermore, PARP inhibitors and genetic deletion of PARP-1 are profoundly neuroprotective in NO-mediated toxicity [24], NMDA excitotoxicity [34, 90, 91], as well as experiment models of ischemic injury [73, 92], traumatic brain injury [74], AD [77], HD [93], and PD [82,83,84, 94].
4.1.1 Parkinson’s Disease
Parkinson’s disease is a complicated neurodegenerative movement disorder that results from the selective loss of dopaminergic neurons in the substantia nigra [95]. MPTP, a toxin known to induce PD-like symptoms, has been reported to increase PARP-1 activity in mice [69, 83, 96]. PARP-1 KO mice are highly resistant to MPTP-induced neurotoxicity compared to WT mice [83]. In another study, PARP-1 inhibitors ameliorate α-synuclein cytotoxicity, as well as primary dopaminergic neurons treated with 1-methyl-4-phenylpyridinium (MPP(+)) [84]. Both pretreatment and posttreatment with the PARP-1 inhibitor benzamide attenuated MPTP-induced neuronal damage in mice [94]. Moreover, in a 6-hydroxydopamine (6-OHDA) in vitro PD mice model, PARP-1 KO mice show reduced dopaminergic neurodegeneration and related PD symptoms, due to the suppression of AMP protein kinase (AMPK) activation and AIF nuclear translocation [81]. Mutations of parkin and loss of its ubiquitin E3 ligase function are associated with familial PD [97]. The parkin substrate aminoacyl-tRNA synthetase complex interacting multifunctional protein-2 (AIMP2) is found in Lewy body inclusions of PD substantia nigra and its accumulation leads to dopamine neuron death [98, 99]. A recent study [82] found that AIMP2 accumulation directly leads to PARP-1 overactivation and results in selective, age-dependent progressive loss of dopaminergic neurons, while the PARP-1 inhibitor AG-014699 or PARP-1 gene deletion protects AIMP2 transgenic mice against dopamine neuron loss and behavioral deficits.
4.1.2 Alzheimer’s Disease
Alzheimer’s disease is a dementia characterized by the accumulation of the β-amyloid peptide (Aβ) and hyperphosphorylation of the microtubule-associated protein tau within the brain [100]. The correlation between PARP-1 and AD was first reported in 1999, indicating higher levels of PARP-1 and PAR in AD postmortem human brains by immunostaining [77]. Early PARP-1 activation was detected in the entorhinal cortex and hippocampus of AD Swedish and Indiana double mutation mice at 3 months of age when initial amyloid deposition takes place. The increase of PARP-1 activity in Aβ-treated human SH-SY5Y cells can be prevented by PARP-1 inhibitor, MC2050 [79]. Moreover, Aβ injection increases PARP-1 levels in rat brains and nicotinamide, an endogenous PARP-1 inhibitor, downregulates PARP-1 and oxidative stress induced by Aβ [78]. Moreover, mice treated with the PARP-1 inhibitor PJ34 and PARP-1 KO mice have lower microglial activation after Aβ injections compared to controls [76]. In the same study, PARP-1 deletion attenuated the microglial activation and cognitive deficits in AD mice with Swedish and Indiana double mutations [76]. PARP-1 activation and accumulation of PAR have also been observed in astrocytes and microglia treated with different species of Aβ or transfected with human amyloid precursor protein (APP) [76, 79, 89, 101].
4.1.3 Huntington’s Disease
Huntington’s disease is an autosomal-dominant, progressive neurodegenerative disorder due to expanded CAG repeat mutations that results from polyglutamine expansion at the N-terminus of the huntingtin protein [102]. Huntington’s disease manifests with ≥35 polyglutamine expansions. A strong neuronal and glial PARP expression and weak caspase-3 activation accompanied by massive cell death are detected in the caudate nucleus of severely affected HD brains, suggesting that caspase-mediated neuronal death has little contribution to HD pathology [85]. A recent study reported that R6/2 HD mice treated with the PARP-1 inhibitor INO-1001 survived longer and displayed ameliorated neurological dysfunction than the control mice, confirming the neuroprotective effect of PARP-1 inhibition in HD [93].
Involvement of PARP-1 in other neurodegenerative disorders will not be described in detail here. There are concerns about long-term use of PARP inhibitors for chronic neurodegenerative diseases due to the potential side effects including genome instability [103]. However, PARP-1 KO mice are viable and are resistant to numerous toxic stimuli as mentioned above, which suggests that PARP-2 or other PARP family members may also play a role in DNA repair and maintaining genome stability. Nevertheless, new potent selective PARP-1 inhibitors, which cross blood–brain barrier, such as AG-014699 and AG14361, could be beneficial in preventing or delaying neurodegenerative disease progression.
5 Involvement of Other Parthanatos Components in Neurodegeneration
In addition to PARP-1, AIF translocation has also been described in several neurodegenerative disorders and experiment models [57]. The first evidence for the involvement of AIF in neurodegenerative diseases is that cortical neurons of harlequin mice with reduced AIF expression were highly resistant to glutamate excitotoxicity [60]. AIF nuclear translocation from mitochondria has been reported in PC-12 cells [104], mouse dopaminergic cell lines [105, 106], and in vivo tyrosine-hydroxylase (TH)-positive neurons [69] treated with MPTP. Chu et al. has also found that MPP+ treatment results in nuclear translocation of AIF and cell death that cannot be prevented by caspase inhibitors but can be significantly reduced by short hairpin RNA against AIF [106]. Nuclear translocation of AIF has also been found in primary neurons of PD patients and in lipopolysaccharide (LPS)-injected mouse nigral dopaminergic neurons [107]. Interestingly, both Aβ and non-Aβ component of amyloid plaques induced PARP-1 activation and AIF translocation from mitochondria to nucleus in rat brain slices, but had no effect on caspase-3 activity [108]. Moreover, AIF release from the mitochondria and translocation into the nucleus were also observed in both in vitro and in vivo retinal degeneration models [109, 110] and the motor neurons of symptomatic superoxide dismutase 1 (SOD1) G93A transgenic mice, a model of ALS [111]. Despite the fact that harlequin mice are resistant to numerous forms of acute brain injury, they suffer from slow, progressive neurodegeneration, accompanied by markers of oxidative stress [112, 113], probably due to the loss of pro-survival mitochondrial function of AIF. Nevertheless, as a critical mediator of caspase-independent programmed cell death, future studies to reveal AIF isoforms functions, its protein function regions and pro-parthanatos nuclear partners, will lead to the development of more selective drugs to treat degenerative diseases and other neurological disorders involving AIF [114]. In fact, a nuclear effector of AIF-induced DNA cleavage and cell death has been identified and named as PAAN (parthanatos-dependent AIF-associated nuclease ) by our laboratory.
Iduna (RNF146/dactylidin) is upregulated early in highly vulnerable brain tissues of AD patients compared with aged controls [115], suggesting that Iduna may function as a molecular protector molecular early in the progression of neurodegenerative diseases. It will be interesting to investigate if Iduna function is impaired in neurodegenerative disorders, which could contribute to possible poly-(ADP-ribosyl)ated damaged or toxic protein accumulation.
6 Conclusions
In summary, parthanatos plays important roles in exitotoxicity, acute neurological diseases such as ischemic stroke and traumatic injury, and neurodegenerative disorders including PD, AD, HD, ALS, and MS. Although cell death itself is the endpoint event at the late stage of neurodegeneration, pharmacological inhibitors and genetic deletions of the mediators in cell death show beneficial effect in cellular and animal models of neurodegenerative diseases, indicating that preventing or reducing cell loss are still promising therapeutic interventions that could probably slow down the disease progress [1]. Further investigation of parthanatos-signaling mechanisms and roles of parthanatos mediators in different neurologic and neurodegenerative disorders are essential. We have highlighted some potential drug targets in the parthanatos-signaling pathways in Fig. 16.4.

Therapeutic targets of parthanatos. When excitotoxicity is involved, the beneficial effect of NMDAR toxic signaling blockers (such as Tat peptides interfering NR2B/PSD-95 and PSD-95/nNOS binding, [121,122,123]), NMDAR blocker (memantine, [124, 125]) and selective nNOS inhibitors [20] have been shown in experiment models and clinical trials in ischemia and some neurodegenerative disorders. Antioxidants can also reduce ROS and thus protect cells from parthanatos and other forms of death. The neuroprotective effect of PARP-1 inhibitors has been elaborated in the chapter. Other potential therapeutic targets downstream of PARP-1 activation includes but is not limited to: activation of endogenous PARP-1 blocker PARG and Iduna (such as preconditioning), prevention of death signal (PAR) translocation from nucleus to mitochondria, prevention of AIF release and translocation to nucleus with its partner PAAN, as well as disruption of critical mediator interactions and inhibition of PAAN nuclease function. Highlighted boxes with red outlines are therapeutic targets proposed for neurological and neurodegenerative disorders involving parthanatos
It will also be important to understand the interactions between parthanatos and other major forms of cell death in neurodegenerative diseases (Fig. 16.5). For example, PARP-1 can be cleaved by activated caspases, which reserves the energy needed for cell execution by apoptotic machinery [116]. While on the other hand, cells with PARP-1 overactivation suffer from cellular NAD+ and ATP depletion and undergo either necroptosis [117] or PAR polymer-mediated parthanatos [15], possibly accompanied by autophagy [118, 119]. It is worth mentioning that cell death factors such as PAR, AIF, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and tumor protein 53 (p53) are involved in various cell death programs and neurodegenerative diseases, and understanding their roles and interactions in these conditions will contribute to the development of therapies and biomarkers for neurodegenerative diseases [1, 14, 120].

Interactions of cell death programs in neurodegenerative disease context. The main interconnected cell death programs in neurodegenerative disorders (parthanatos: purple, apoptosis: green, necroptosis: blue, and autophagy: red). Mitochondrial dysfunction, ROS, and associated DNA damage are the most common source of trigger of all four types of death, which happen to be the most affected cell targets in neurodegenerative diseases and aging. Some secondary deficits including protein misfolding, organelle damage, microtubule disorganization can also promote several forms of cell death. Some of the interplays between different types of death are also shown here (adapted from [126])
Abbreviations
- 3-MA:
-
3-Methyladenine
- 6-OHDA:
-
6-Hydroxydopamine
- Aβ:
-
β-Amyloid peptide
- AD:
-
Alzheimer’s disease
- AIF:
-
Apoptosis-inducing factor
- AIMP2:
-
Aminoacyl-tRNA synthetase complex interacting multifunctional protein-2
- ALS:
-
Amyotrophic lateral sclerosis
- AMPK:
-
AMP-activated protein kinase
- APP:
-
Amyloid precursor protein
- ARH3:
-
ADP-ribosylhydrolase 3
- BRCT:
-
BRCA1 C-terminal
- CNS:
-
Central nervous system
- DPQ:
-
3,4-Dihydro-5-(4-(1-piperidinyl)butoxyl)-1(2H)-isoquinolinone
- HD:
-
Huntington’s disease
- IFNγ:
-
Interferon gamma
- KO:
-
Knock out
- LPS:
-
Lipopolysaccharide
- MCAO:
-
Middle cerebral artery occlusion
- MNNG:
-
N-Methyl-N′-nitro-N-nitrosoguanidine
- MPP+:
-
1-Methyl-4-phenylpyridinium
- MPT:
-
Mitochondrial permeability transition
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MS:
-
Multiple sclerosis
- mTOR:
-
Mechanistic target of rapamycin
- NAD+:
-
Oxidized nicotinamide adenine dinucleotide
- Nec-1:
-
Necrostatin-1
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLS:
-
Nuclear localization sequence
- NMDA:
-
N-Methyl-d-aspartate
- nNOS:
-
Neuronal nitric oxide synthase
- NO:
-
Nitric oxide
- OGD:
-
Oxygen glucose deprivation
- P53:
-
Tumor protein 53
- PAAN:
-
Parthanatos-dependent AIF-associated nuclease
- PAR:
-
Poly(ADP-ribose)
- PARP1:
-
Poly(ADP-ribose) polymerase 1
- PARG:
-
Poly(ADP-ribose) glycohydrolase
- PBM:
-
PAR-binding motif
- PBZF:
-
PAR-binding zinc finger
- PD:
-
Parkinson’s disease
- PI3K:
-
Phosphoinositide3-kinase
- PSM:
-
PARP signature motif
- ROS:
-
Reactive oxygen species
- siRNA:
-
Small interfering RNA
- SOD1:
-
Superoxide dismutase 1
- TH:
-
Tyrosine-hydroxylase
- TNF:
-
Tumor necrosis factor
- TNFR1:
-
TNFα receptor 1
- RIPK1:
-
Receptor-interacting protein kinase 1
- RIPK3:
-
Receptor-interacting protein kinase 3
- Z-VAD-fmk:
-
N-Benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
References
Bredesen DE, Rao RV, Mehlen P (2006) Cell death in the nervous system. Nature 443(7113):796–802
Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1(2):120–129
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al (2012) Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ 19(1):107–120
Nijhawan D, Honarpour N, Wang X (2000) Apoptosis in neural development and disease. Annu Rev Neurosci 23:73–87
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1(2):112–119
Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W (1992) Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 267(8):5317–5323
Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S et al (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81(5):1001–1008
Zhu S, Zhang Y, Bai G, Li H (2011) Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease. Cell Death Dis 2:e115
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP et al (2015) Activation of necroptosis in multiple sclerosis. Cell Rep 10(11):1836–1849
Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16(6):345–357
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997
Harris H, Rubinsztein DC (2012) Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 8(2):108–117
Fatokun AA, Dawson VL, Dawson TM (2014) Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 171(8):2000–2016
David KK, Andrabi SA, Dawson TM, Dawson VL (2009) Parthanatos, a messenger of death. Front Biosci 14:1116–1128
Andrabi SA, Dawson TM, Dawson VL (2008) Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci 1147:233–241
Wang Y, Dawson VL, Dawson TM (2009) Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol 218(2):193–202
Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH (1991) Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A 88(14):6368–6371
Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH (1993) Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci Off J Soc Neurosci 13(6):2651–2661
Dawson VL, Dawson TM (1998) Nitric oxide in neurodegeneration. Prog Brain Res 118:215–229
Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA (1994) Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 265(5180):1883–1885
Schulz JB, Matthews RT, Muqit MM, Browne SE, Beal MF (1995) Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem 64(2):936–939
Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ et al (1997) Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci Off J Soc Neurosci 17(18):6908–6917
Zhang J, Dawson VL, Dawson TM, Snyder SH (1994) Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science 263(5147):687–689
Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL (1996) Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A 93(13):6770–6774
Gonzalez-Zulueta M, Ensz LM, Mukhina G, Lebovitz RM, Zwacka RM, Engelhardt JF et al (1998) Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J Neurosci Off J Soc Neurosci 18(6):2040–2055
Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL (2013) Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol 698(1–3):6–18
Mattson MP (2003) Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. NeuroMolecular Med 3(2):65–94
Ikonomidou C, Turski L (1995) Excitotoxicity and neurodegenerative diseases. Curr Opin Neurol 8(6):487–497
Fossati S, Cipriani G, Moroni F, Chiarugi A (2007) Neither energy collapse nor transcription underlie in vitro neurotoxicity of poly(ADP-ribose) polymerase hyper-activation. Neurochem Int 50(1):203–210
Goto S, Xue R, Sugo N, Sawada M, Blizzard KK, Poitras MF et al (2002) Poly(ADP-ribose) polymerase impairs early and long-term experimental stroke recovery. Stroke 33(4):1101–1106
Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M et al (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103(48):18308–18313
Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM et al (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A 103(48):18314–18319
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297(5579):259–263
D'Amours D, Desnoyers S, D'Silva I, Poirier GG (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342(Pt 2):249–268
Hong SJ, Dawson TM, Dawson VL (2004) Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 25(5):259–264
Luo X, Kraus WL (2012) On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 26(5):417–432
Ji Y, Tulin AV (2010) The roles of PARP1 in gene control and cell differentiation. Curr Opin Genet Dev 20(5):512–518
Kraus WL (2008) Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol 20(3):294–302
Kraus WL, Lis JT (2003) PARP goes transcription. Cell 113(6):677–683
Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7(7):517–528
Alvarez-Gonzalez R, Jacobson MK (1987) Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry 26(11):3218–3224
Rouleau M, Aubin RA, Poirier GG (2004) Poly(ADP-ribosyl)ated chromatin domains: access granted. J Cell Sci 117(Pt 6):815–825
Li M, Lu LY, Yang CY, Wang S, Yu X (2013) The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev 27(16):1752–1768
Hassa PO, Hottiger MO (2008) The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci 13:3046–3082
Gagne JP, Hendzel MJ, Droit A, Poirier GG (2006) The expanding role of poly(ADP-ribose) metabolism: current challenges and new perspectives. Curr Opin Cell Biol 18(2):145–151
Kim MY, Zhang T, Kraus WL (2005) Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev 19(17):1951–1967
Koh DW, Dawson TM, Dawson VL (2005) Poly(ADP-ribosyl)ation regulation of life and death in the nervous system. Cell Mol Life Sci 62(7–8):760–768
Meyer RG, Meyer-Ficca ML, Whatcott CJ, Jacobson EL, Jacobson MK (2007) Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-ribose) glycohydrolase (PARG) activity. Exp Cell Res 313(13):2920–2936
Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK (2004) Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res 297(2):521–532
Blenn C, Althaus FR, Malanga M (2006) Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem J 396(3):419–429
Cozzi A, Cipriani G, Fossati S, Faraco G, Formentini L, Min W et al (2006) Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J Cereb Blood Flow Metab 26(5):684–695
Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC et al (2004) Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A 101(51):17699–17704
Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H et al (2004) Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A 101(1):82–86
Zhou Y, Feng X, Koh DW (2011) Activation of cell death mediated by apoptosis-inducing factor due to the absence of poly(ADP-ribose) glycohydrolase. Biochemistry 50(14):2850–2859
Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39(1):8–24
Krantic S, Mechawar N, Reix S, Quirion R (2007) Apoptosis-inducing factor: a matter of neuron life and death. Prog Neurobiol 81(3):179–196
Modjtahedi N, Giordanetto F, Madeo F, Kroemer G (2006) Apoptosis-inducing factor: vital and lethal. Trends Cell Biol 16(5):264–272
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM et al (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397(6718):441–446
Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC et al (2005) Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci Off J Soc Neurosci 25(6):1324–1334
Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M et al (2005) Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci Off J Soc Neurosci 25(44):10262–10272
Cregan SP, Dawson VL, Slack RS (2004) Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 23(16):2785–2796
Yu SW, Wang Y, Frydenlund DS, Ottersen OP, Dawson VL, Dawson TM (2009) Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro 1(5):e00021
Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol 159(6):923–929
Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N et al (2000) Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J 14(5):729–739
Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL et al (2008) Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res 36(22):6959–6976
Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA et al (2011) Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci Signal 4(167):ra20
Dawson VL, Dawson TM (2004) Deadly conversations: nuclear-mitochondrial cross-talk. J Bioenerg Biomembr 36(4):287–294
Wang H, Shimoji M, Yu SW, Dawson TM, Dawson VL (2003) Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann N Y Acad Sci 991:132–139
DaRosa PA, Wang Z, Jiang X, Pruneda JN, Cong F, Klevit RE et al (2015) Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 517(7533):223–226
Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP et al (2011) Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc Natl Acad Sci U S A 108(34):14103–14108
Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z et al (2011) Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med 17(6):692–699
Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J et al (1997) Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3(10):1089–1095
LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM et al (2001) Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma 18(4):369–376
Maier C, Scheuerle A, Hauser B, Schelzig H, Szabo C, Radermacher P et al (2007) The selective poly(ADP)ribose-polymerase 1 inhibitor INO1001 reduces spinal cord injury during porcine aortic cross-clamping-induced ischemia/reperfusion injury. Intensive Care Med 33(5):845–850
Kauppinen TM, Suh SW, Higashi Y, Berman AE, Escartin C, Won SJ et al (2011) Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J Neuroinflammation 8:152
Love S, Barber R, Wilcock GK (1999) Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain 122(Pt 2):247–253
Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A (2014) Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1-42)-induced rat model of Alzheimer's disease. Free Radic Res 48(2):146–158
Martire S, Fuso A, Rotili D, Tempera I, Giordano C, De Zottis I et al (2013) PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS One 8(9):e72169
Infante J, Sanchez-Juan P, Mateo I, Rodriguez-Rodriguez E, Sanchez-Quintana C, Llorca J et al (2007) Poly (ADP-ribose) polymerase-1 (PARP-1) genetic variants are protective against Parkinson’s disease. J Neurol Sci 256(1–2):68–70
Kim TW, Cho HM, Choi SY, Suguira Y, Hayasaka T, Setou M et al (2013) (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis 4:e919
Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D et al (2013) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci 16(10):1392–1400
Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME et al (1999) Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A 96(10):5774–5779
Outeiro TF, Grammatopoulos TN, Altmann S, Amore A, Standaert DG, Hyman BT et al (2007) Pharmacological inhibition of PARP-1 reduces alpha-synuclein- and MPP+-induced cytotoxicity in Parkinson's disease in vitro models. Biochem Biophys Res Commun 357(3):596–602
Vis JC, Schipper E, de Boer-van Huizen RT, Verbeek MM, de Waal RM, Wesseling P et al (2005) Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol 109(3):321–328
Kauppinen TM, Suh SW, Genain CP, Swanson RA (2005) Poly(ADP-ribose) polymerase-1 activation in a primate model of multiple sclerosis. J Neurosci Res 81(2):190–198
Kim SH, Engelhardt JI, Henkel JS, Siklos L, Soos J, Goodman C et al (2004) Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology 62(2):319–322
Kim SH, Henkel JS, Beers DR, Sengun IS, Simpson EP, Goodman JC et al (2003) PARP expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. J Neuropathol Exp Neurol 62(1):88–103
Martire S, Mosca L, d'Erme M (2015) PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev 146-148:53–64
Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A et al (2000) NMDA but not non-NMDA excitotoxicity is mediated by poly(ADP-ribose) polymerase. J Neurosci Off J Soc Neurosci 20(21):8005–8011
Pieper AA, Verma A, Zhang J, Snyder SH (1999) Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 20(4):171–181
Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA (1997) Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab 17(11):1143–1151
Cardinale A, Paldino E, Giampa C, Bernardi G, Fusco FR (2015) PARP-1 inhibition is Neuroprotective in the R6/2 mouse model of Huntington's disease. PLoS One 10(8):e0134482
Yokoyama H, Yano R, Kuroiwa H, Tsukada T, Uchida H, Kato H et al (2010) Therapeutic effect of a novel anti-parkinsonian agent zonisamide against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) neurotoxicity in mice. Metab Brain Dis 25(2):135–143
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87
Cosi C, Colpaert F, Koek W, Degryse A, Marien M (1996) Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res 729(2):264–269
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608
Corti O, Hampe C, Koutnikova H, Darios F, Jacquier S, Prigent A et al (2003) The p38 subunit of the aminoacyl-tRNA synthetase complex is a Parkin substrate: linking protein biosynthesis and neurodegeneration. Hum Mol Genet 12(12):1427–1437
Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O et al (2005) Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci Off J Soc Neurosci 25(35):7968–7978
O'Brien RJ, Wong PC (2011) Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34:185–204
Abeti R, Abramov AY, Duchen MR (2011) Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 134(Pt 6):1658–1672
Walker FO (2007) Huntington's disease. Lancet 369(9557):218–228
Graziani G, Szabo C (2005) Clinical perspectives of PARP inhibitors. Pharmacol Res 52(1):109–118
Liou AK, Zhou Z, Pei W, Lim TM, Yin XM, Chen J (2005) BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J 19(10):1350–1352
Chee JL, Guan XL, Lee JY, Dong B, Leong SM, Ong EH et al (2005) Compensatory caspase activation in MPP+-induced cell death in dopaminergic neurons. Cell Mol Life Sci 62(2):227–238
Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J (2005) Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem 94(6):1685–1695
Burguillos MA, Hajji N, Englund E, Persson A, Cenci AM, Machado A et al (2011) Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: evidence in Parkinson’s disease patients. Neurobiol Dis 41(1):177–188
Adamczyk A, Czapski GA, Jesko H, Strosznajder RP (2005) Non A beta component of Alzheimer’s disease amyloid and amyloid beta peptides evoked poly(ADP-ribose) polymerase-dependent release of apoptosis-inducing factor from rat brain mitochondria. J Physiol Pharmacol 56(Suppl 2):5–13
Paquet-Durand F, Silva J, Talukdar T, Johnson LE, Azadi S, van Veen T et al (2007) Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J Neurosci Off J Soc Neurosci 27(38):10311–10319
Sanges D, Comitato A, Tammaro R, Marigo V (2006) Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci U S A 103(46):17366–17371
Oh YK, Shin KS, Kang SJ (2006) AIF translocates to the nucleus in the spinal motor neurons in a mouse model of ALS. Neurosci Lett 406(3):205–210
El Ghouzzi V, Csaba Z, Olivier P, Lelouvier B, Schwendimann L, Dournaud P et al (2007) Apoptosis-inducing factor deficiency induces early mitochondrial degeneration in brain followed by progressive multifocal neuropathology. J Neuropathol Exp Neurol 66(9):838–847
Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN et al (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419(6905):367–374
Lorenzo HK, Susin SA (2007) Therapeutic potential of AIF-mediated caspase-independent programmed cell death. Drug Resist Updat 10(6):235–255
von Rotz RC, Kins S, Hipfel R, von der Kammer H, Nitsch RM (2005) The novel cytosolic RING finger protein dactylidin is up-regulated in brains of patients with Alzheimer’s disease. Eur J Neurosci 21(5):1289–1298
Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME (1998) Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J Biol Chem 273(22):13703–13712
Ha HC, Snyder SH (1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 96(24):13978–13982
Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodriguez MI et al (2012) ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 22(7):1181–1198
Virag L, Robaszkiewicz A, Rodriguez-Vargas JM, Oliver FJ (2013) Poly(ADP-ribose) signaling in cell death. Mol Asp Med 34(6):1153–1167
Vila M, Przedborski S (2003) Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 4(5):365–375
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW et al (2002) Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 298(5594):846–850
Cook DJ, Teves L, Tymianski M (2012) Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 483(7388):213–217
Zhou L, Li F, Xu HB, Luo CX, Wu HY, Zhu MM et al (2010) Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med 16(12):1439–1443
Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE et al (2009) Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15(12):1407–1413
Witt A, Macdonald N, Kirkpatrick P (2004) Memantine hydrochloride. Nat Rev Drug Discov 3(2):109–110
Basello DA, Scovassi AI (2015) Poly(ADP-ribosylation) and neurodegenerative disorders. Mitochondrion 24:56–63
Acknowledgments
This work was supported by grants from MSCRFII-0429 and MSCRFII-0125 to V.L.D. 2013-MSCRF-0028 to J.F. and NIH/NINDS NS67525 to T.M.D. and V.L.D. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We thank I-Hsun Wu for drawing the figures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Fan, J., Dawson, T.M., Dawson, V.L. (2017). Cell Death Mechanisms of Neurodegeneration. In: Beart, P., Robinson, M., Rattray, M., Maragakis, N. (eds) Neurodegenerative Diseases. Advances in Neurobiology, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-57193-5_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-57193-5_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57191-1
Online ISBN: 978-3-319-57193-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)