
Abstract
Several factors must be considered to successfully integrate immunotherapy with radiation into clinical practice. One such factor is that concepts arising from preclinical work must be tested in combination with radiation in preclinical models to better understand how combination therapy will work in patients; examples include checkpoint inhibitors, tumor growth factor-beta (TGF-β) inhibitors, and natural killer (NK) cell therapy. Also, many radiation fields and fractionation schedules typically used in radiation therapy had been standardized before the introduction of advanced techniques for radiation planning and delivery that account for changes in tumor size, location, and motion during treatment, as well as uncertainties introduced by variations in patient setup between treatment fractions. As a result, radiation therapy may involve the use of large treatment volumes, often encompassing nodal regions that may not be irradiated with more conformal techniques. Traditional forms of radiation in particular pose challenges for combination trials with immunotherapy. This chapter explores these issues in more detail and provides insights as to how radiation therapy can be optimized to combine with immunotherapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Radiation was first used to treat cancer by Emil H. Grubbe in 1896. Since that time, it has become a major component of cancer treatment. More than half of all patients with cancer will be treated with radiation at some point during the course of the disease. Many forms of radiation therapy involve daily doses of relatively small fractions (e.g., 1.8–2 Gy), but advances in the technology used to plan and deliver radiation therapy allows the delivery of high radiation doses to tumors while sparing the nearby normal tissue. One such technique, stereotactic ablative radiation therapy (SABR), can produce local control rates of 98% and overall survival rates of 55% for patients with inoperable stage I lung cancer [1]; however, neither SABR nor other forms of radiation on its own can control distant disease (metastases) in patients with stage IV disease. Radiation can induce antitumor effects outside the radiation field (i.e., abscopal effects), but such reactions are rare and require a functioning immune system [2]. That radiation affects aspects of immune function is clear; the potential for inducing abscopal (systemic) effects with an essentially local form of therapy underscores the importance of clarifying more precisely how radiation modulates the immune system to identify which forms of immunotherapy will provide synergistic effects with radiation.
3.1 Checkpoints and Radiation Therapy
3.1.1 Checkpoint Inhibitors
Checkpoint inhibitors are a group of immune system molecules that act as negative regulators to help maintain a balance between autoimmunity and immune response. The most clinically relevant examples of checkpoint inhibitors include cytotoxic T lymphocyte associated protein 4 (CTLA4) and programmed cell death protein-1/programmed cell death ligand-1 (PD1/PDL1).
CTLA4 is a receptor found on mature, activated CD4+ and CD8+ T-cells and interacts with the B7-1(2)/CD80(86) ligand on antigen-presenting cells. It shares this ligand with its co-stimulatory molecule, CD28, which is also found on CD4+ and CD8+ T-cells. Once tumor antigens are recognized by antigen-presenting cells, they are presented via major histocompatibility complexes (MHCs) to T cells, which recognize the antigen by means of T-cell receptors (TCRs). Subsequently, CD28 binds its ligand B7-1(2)/CD80(86) and activates the T cells. Downstream effects include CD25 induction followed by activation of the interleukin-2 (IL-2) receptor, which ultimately induces differentiation and survival of Teffector cells. However, CTLA4 has a much higher binding affinity for the shared ligand, and it can outcompete CD28 for this position, causing the opposite effect. When activated, CTLA4 dampens T-cell activation and response by suppressing IL-2 production and T-cell proliferation. CTLA4 functions early during T-cell activation and can also attenuate signaling by kinases such as the PI3K/AKT pathway, which is originally activated by TCRs and CD28 [3]. Further, the CTLA4 receptor is constitutively expressed on regulatory T-cells (Tregs), and its activation can enhance their proliferation. These immunosuppressive cells function to directly and negatively control dendritic cell (DC) maturation as well as downregulating B7 expression on DCs to block the immunostimulatory signal of CD28:B7 [4, 5]. Ultimately, a lopsided balance in favor of Treg versus Thelper/Teffector cells results in T-cell anergy and tolerance.
In both preclinical and clinical models, radiation and CTLA4 blockade mediate synergistic effects that culminate in systemic clearance of tumor outside the radiation field (the “abscopal” or “bystander” effect [6,7,8]). The term abscopal comes from the Latin ab meaning [to position] “away from” and scopus referring to the target. Abscopal effects are consistent with increased release of tumor-associated antigens, resulting in tumor antigen-specific T-cells that infiltrate tumors both locally and distantly. The addition of CTLA4 blockade to radiation releases the inhibition on the immunostimulatory interaction of CD28 and its ligand B7-1, an interaction that improves T-cell activation and effector-cell generation. The combination of the two therapies creates a larger pool of effectively primed T-cells and creates an environment where they can proliferate without CTLA4 inhibition. The primed T-cells subsequently migrate, recognize, and attack tumor cells at distant sites, occasionally leading to eradication of systemic disease. This combination does not produce abscopal effects in all patients; however, additional studies are needed to determine which combinations of immunotherapies with radiation will result in systemic immunity. Because radiation therapy is traditionally considered a local therapy, the combination of immunotherapy and radiation therapy is in effect converting a local therapy modality into a systemic therapy [9, 10].
The optimal radiation dose for T-cell priming via tumor-associated antigens is still under investigation; however, one group suggested that hypofractionation, specifically three fractions of 8 Gy each, was the most effective at secondary tumor control compared with other fractionation schemes, such as one fraction of 20 Gy or five fractions of 6 Gy [6]. Another group, evaluating the effects of fractionation on local tumor control, showed that treatment with two fractions of 7.5 Gy each maintained lower splenocyte Treg levels than did other fractionation schemes such as five fractions of 3 Gy, three fractions of 5 Gy, and 1 fraction of 15 Gy. However, no significant differences in local control were found between any of the treatment conditions [11]. Another study supports the notion that single ablative doses may be better for local tumor control. In that study, a single fraction of 30 Gy was able to eradicate most of the tumors in the CT26 murine model. However, when the mice were given an additional 30 Gy in 10 fractions (3 Gy each, 60 Gy total), most mice experienced tumor regrowth and death [12]. This group further showed that the 30 Gy doses induced high CD8+ infiltration, whereas the additional fractionated 30 Gy caused recruitment of myeloid-derived suppressor cells to the tumor. This discrepancy in results may have been related to differences in the tumor models used in the various studies; however, they all suggest that SABR-like doses may be effective at invoking antitumor immunity. Notably, however, none of these studies tested conventional 2-Gy-per-fraction schemes. At this time, the abscopal effect remains elusive, but the hope is that such responses will become more common as the search continues for the optimal dosage and timing in terms of patient response and toxicity, and as more data become available [13].
PD1 is another immune checkpoint receptor that is expressed by T-cells, B cells, NK cells, DCs, and macrophages to help negatively regulate the immune response [14]. The PD1/PDL1 pathway acts differently from CTLA4, as it inhibits T-cell activity in the effector phase within peripheral tissues and tumors [3, 15]. Once the TCR recognizes antigen, the activated T-cell ultimately upregulates PD1 to dampen its own activation. PD1 has a multitude of immunosuppressive effects, including inhibition of T-cell proliferation, survival, and effector functions; cytokine release; cytotoxicity; induction of apoptosis of tumor-specific T-cells; and promotion of differentiation of CD4+ cells into Tregs [16, 17]. Therefore, upregulation of PD1 on immune cells and expression of PDL1 on tumors tends to promote tumor immune evasion.
Although radiation can modify the local tumor microenvironment, as discussed below, tumors can also exploit regulatory mechanisms to undermine T-cell responses. For example, tumor cells that express PDL1 can successfully undergo immunoevasion when that ligand interacts with its receptor PD1 on effector T-cells, which then mediates apoptosis of the infiltrating T-cells [18]. Interestingly, a negative feedback loop has been discovered in which effector T-cells in the tumor’s microenvironment produce interferon-gamma (IFN-y), which directly initiates PDL1 expression and then downregulates effector T-cells via the mechanism described above [19]. Several reports support this hypothesis, including a recent human melanoma study demonstrating strong correlations among intratumoral T-cell infiltration, PDL1, and IFN-γ [20]. Other studies have shown that fractionated radiation increases the expression of PDL1 by the tumor in response to effector CD8+ T-cell production of IFN-γ and that combining PD1/PDL1 inhibitors concurrently with radiation significantly improved durable systemic immune responses [21]. Another recent preclinical study examined the influence of PD1 expression on the abscopal effect after treatment with PD1/PDL1 blockade and radiation therapy [22]. That group found that the combination of conventional 2 Gy per fraction radiation and PD1/PDL1 blockade produced a curative rate of 66% in the CT26 model, and that concurrent administration of the drug with radiation produces the best effect. As the radiation reaches the tumor, type I IFNs (IFN-α and IFN-β) and IFN-ɣ modulate the microenvironment in ways that upregulate VCAM-1 (which allows T-cell trafficking and entry into the tumor), MHC-I, and chemokines to bring additional immune cells to local sites [23, 24].
3.2 Radiation and Immune Function
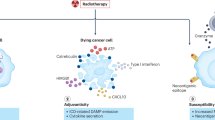
Ionizing radiation is known to release “danger-associated molecular patterns” such as upregulation of high mobility group box 1 protein (HMGB1), ATP, and calreticulin, all of which increase immune activation at the tumor site [25]. Also, release and uptake of tumor-associated antigens by antigen-presenting cells in the tumor microenvironment increase the probability of successful T-cell priming (Table 3.1) [36]. Curiously, the immune system seems to be unable to exploit these mechanisms to consistently produce specific, durable immune responses. One possible reason why is that even when T-cells are specifically activated in appropriate lymph nodes, homing and retention of Teffector cells into the tumor are limited because of the known ability of tumor cells to restrict the infiltration and activation of cytotoxic T-lymphocytes. This tumor-cell immunoevasion and ultimately immunosuppression is related to specific properties of tumor cells, some of which are highlighted below. General principles by which tumor cells evade the immune system include successful unchecked cell proliferation, de novo angiogenesis, suppression of T-cell penetration into tumor, increasing the ratio or concentration of immunosuppressive cells such as Tregs, and suppressing antigen presentation. Indeed, several of the ways by which tumor cells escape immune surveillance are identical to the immunosuppressive effects induced by radiation, particularly recruiting Tregs and producing TGF-β (Table 3.1) in addition to stimulating myeloid-derived suppressor cells and alternatively activated macrophages.
3.2.1 TGF-β
TGF-β is a multifunctional, polypeptide cytokine, and part of a cytokine superfamily that coordinates response to tissue injury and stress. It maintains immune self-tolerance while also assisting cancer cells in evading the host immune system. Of the three major isotypes of TGF-β (TGF-β1, TGF-β2, and TGF-β3), TGF-β1 is expressed mostly in the immune system. It is synthesized as an inactive pre-protein that is cleaved to form two homodimers joined by a disulfide bond. The homodimers interact with latency-associated protein and latent TGF-β-binding protein, which combine to form the so-called large latent complex. Release of free TGF-β requires first that the large latent complex be released, and then that the latency-associated protein be cleaved; this is accomplished in the extracellular matrix by metalloproteinases and thrombospondin. Changes or interactions with integrins can also activate latent TGF-β. Subsequently, free TGF-β binds to its transmembrane serine/threonine kinase heterodimeric receptor (TGFβRI/TGFβRII). Once bound, TGF-β has many downstream effects involved in tissue fibrosis, wound healing, carcinogenesis (e.g., proliferation, differentiation, migration, invasion, the epithelial–mesenchymal transition), and immune suppression [37, 38].
3.2.2 TGF-β and the Immune System
Notably, TGF-β activity differs substantially in early versus late immune responses although the molecular event prompting the “switch” between early and late effects is unclear. Paradoxically, during the early stages of immune response, TGF-β suppresses malignancy directly by stopping tumor cell cycle progression, and indirectly through stromal effects (Table 3.2). However, later during tumorigenesis (i.e., the late stages of immune response), TGF-β contributes significantly to the immunosuppressive microenvironment in ways that encourage tumor progression, metastasis, resistance to therapy, and an overall poorer prognosis [39]. TGF-β is produced by both hematologic and solid tumor cells, such as breast, colon, liver, and lung and is also upregulated as a result of ionizing radiation. The latter mechanism directly implicates TGF-β in tissue fibrotic processes such as those that lead to radiation pneumonitis.
Another way in which TGF-β regulates a pro-invasive tumor microenvironment is by inducing the epithelial-to-mesenchymal transition. Cancer cells that have gone through this transition acquire stem-cell-like properties like self-renewal and resistance to chemotherapy or radiation [40]. Recent studies have linked suppression of CD8+ tumor-infiltrating lymphocytes with the epithelial-to-mesenchymal transition, which is modulated by a well-known transcription factor, zinc finger E-box binding homeobox 1 (ZEB1) [41]. That study further showed that microRNA-200 negatively targets PDL1 on tumor cells, but ZEB1 antagonizes this interaction by repressing miR-200, ultimately resulting in suppression of tumor-infiltrating lymphocytes and metastasis.
Radiation can also induce TGF-β production, which can have devastating consequences in delicate tissues such as the lung. TGF-β stimulates the formation of tissue collagen while reducing its degradation, which leads to tissue fibrosis. TGF-β has been linked with radiation pneumonitis, a potentially disabling condition that develops in up to 30% of patients who receive radiation therapy for thoracic malignancies. Usually appearing 1–6 months after radiation therapy, radiation pneumonitis manifests as shortness of breath, nonproductive cough, and fever [42], which can cause significant morbidity and, in severe cases, can be lethal. TGF-β may elevated in patients who develop RP; one recent study showed a seven-fold rise in TGF-β among patients who developed pneumonitis after 40 Gy of radiation, peaking at 3 months after therapy [43]. However, dosimetric variables did not predict who would develop pneumonitis in that study, prompting the authors to suggest stratifying patients at risk of developing pneumonitis based on their TGF-β levels before treatment. This topic is controversial, as other studies have shown weak or no correlation between serum TGF-β levels and RP after thoracic radiotherapy [44,45,46].
3.2.3 Anti-TGF-β
Preclinical efforts and some trials are now ongoing to evaluate whether blocking TGF-β expression or its downstream effects will promote immune surveillance and reverse the environment to a tumor-suppressing phenotype. Various methods of blocking this cytokine including TGF-β receptor kinase inhibitors (LY2109761, currently in preclinical testing for pancreatic cancer), antisense TGF-β oligonucleotides (Trabedersen/AP12009, currently in phase III trials for glioma, and in phase I trials for pancreatic, melanoma, and colorectal carcinoma), antibodies (fresolimumab/GC1008, now in preclinical and clinical evaluations), soluble receptors, and tumor cell vaccines (Lucanix/Belagen-pumatucel, being evaluated in a phase III for non-small cell lung cancer) [38].
3.3 NK Cells and the Tumor Microenvironment
Natural killer (NK) cells are key components of the innate immune system that specialize in eliminating targeT-cells via direct cytotoxicity and release of immunoregulatory cytokines. NK cells targeT-cells that express reduced numbers of MHC class I molecules, or, according to the “missing self” hypothesis, an incompatible or incomplete repertoire of MHC class I molecules [47, 48].
NK cells express numerous activating receptors that engage stress-induced ligands. NKG2D ligands have been found to be upregulated on murine tumor cells after stress-inducing events such as exposure to high doses of ionizing radiation, which can potentiate the antitumor cytotoxicity of both CD8+ T-cells and NK cells [49]. Interestingly, susceptibility to NK cell-mediated cytolysis seems to be enhanced after irradiation of KM12 and HeLa cells via upregulation of the NKG2D ligands MICB, ULBP1, and ULBP2 [50]. These findings suggest that combining radiation and NK cell-based therapy may have additive effects.
3.4 Radiation Dose: High or Low?
In certain settings, radiation can enhance the activity of the immune system against cancer. However, as mentioned previously, the optimal dose and fractionation of that radiation have yet to be clearly defined. Variations in both dose and fractionation have different biological effects that need to be understood to effectively use radiation to induce immune responses. A “one size fits all” solution is unlikely, but different radiation regimens may be needed according to the intrinsic radiosensitivity of a particular tumor, the composition of the tumor microenvironment, and the type of immunotherapy to be combined with the radiation.
Several lines of evidence indicate that low-dose radiation (10–50 cGy per fraction) is effective at enhancing an immune response. Epidemiologic findings have shown that individuals exposed to higher-than-normal levels of background radiation may actually have lower cancer mortality than those exposed to lower levels [51,52,53]. However, this remains controversial as other studies have shown either no effect or more incidence of cancer [54, 55]. One preclinical study showed that exposure of the whole body to low-dose radiation reduced lung metastasis and delayed tumor growth [56]. These findings were attributed to enhanced Th1-like cellular immunity [57,58,59,60], such as activated NK cells, DCs, macrophages, and T-cells and decreased numbers of Tregs [58, 59, 61]. On the contrary, low-dose radiation was also used as an immune suppressant to treat diseases like rheumatoid arthritis. Nevertheless, the applicability of low-dose radiation in clinical settings is limited.
High-dose radiation such as SABR has been shown to improve local disease control in addition to promoting abscopal regression when given with immunotherapeutic agents [7, 62, 63]. Preclinical studies have shown that ablative radiation can lead to the release of tumor-specific antigens, which then direct antigen-presenting cells to induce a T-cell-dependent response [2, 6, 62, 64, 65]. Moreover, induction of MHC class I expression on tumor cells may be radiation dose-dependent, with higher doses inducing more MHC-I expression [66].
Some preclinical studies have found that hypofractionated radiotherapy (use of fractions >5 Gy) enhances the effectiveness of immunotherapy [66, 67] although more conventional fractionation can also have similar effects [21]. However, use of conventionally fractionated radiation over several weeks may continuously kill off infiltrating Teffector cells, as suggested by a study indicating that single-dose radiation was more effective than fractionated radiation or chemotherapy given with radiation [68]. Indeed, as mentioned earlier, another preclinical study involving the CT26 colon cancer model showed that an initial 30-Gy radiation dose resulted in CD8+ T-cell infiltration, but giving another 30 Gy in 10 fractions after initial treatment led to decreases in CD8+ T-cells and increases in myeloid-derived suppressor cells [12]. Although a single 30 Gy dose is not clinically relevant, these findings do suggest that higher radiation doses given in fewer fractions may result in better immune responses. On the other hand, another group found that fractionated and not single-dose radiation induced an immune-mediated abscopal effect in combination with an anti-CTLA4 antibody [6]. Notably, the fractionated doses in this study were ≥6 Gy, which is considered high-dose radiation; however, no low-dose fractions were tested.
In conclusion, radiation dose and fractionation are clearly important factors in activating an immune response, but the radiation regimens will necessarily differ depending on the tumor type. Additional investigations of which dose and fractionation schemes best enhance the immune system are needed if this type of therapy is to be effectively extended to patients.
3.5 Sequence of Radiation and Immunotherapy Combinations
Because radiation and immunotherapeutics have different mechanisms of action against cancer, their rational combination may well achieve excellent antitumor effects with few side effects. The question of which sequence is best is urgent, as it will dictate clinical practice when these therapies are extended to patients. However, because retrospective analyses of various combinations of radiation and ipilimumab have produced controversial and inadequate results (as noted below), prospective studies are needed to address this question.
In one retrospective analysis of 166 patients with metastatic melanoma given radiation therapy and ipilimumab, the median overall survival time was 9 months for patients who had concurrent radiotherapy and ipilimumab (radiation was given between first and fourth doses of ipilimumab) and 39 months in patients who received radiotherapy after the last dose of ipilimumab [69]. In a separate retrospective trial of 46 patients with melanoma brain metastases, patients who received stereotactic radiosurgery during or before ipilimumab had better overall survival and less regional recurrence than did patients who received stereotactic radiosurgery after ipilimumab [70]. Indeed, most of the trials investigating ipilimumab before, during, or after radiation to date have not revealed significant differences among treatment groups [71,72,73,74]; however, these trials were not designed to address the issue of sequencing. Another preclinical study also did not reveal significant differences between concurrent vs. sequential combinations of radiation and ipilimumab [75].
In contrast to ipilimumab, most preclinical studies that combine radiation with anti-PD1/PDL1 antibodies have given the two modalities concurrently; this approach has generated strong systemic antitumor effects [64, 76]. One preclinical study that did consider differences between concurrent versus sequential anti-PDL1 therapy and radiation found that anti-PD1 seemed to work better when given before the radiation is completed [21]. Because radiation induces PDL1 expression in the tumor microenvironment [64, 76], anti-PD1/PDL1 drugs given during the radiation should achieve the best synergistic effect.
Thus, although the doses and fractionation schedules of radiation seem to be important when radiation is to be combined with immunotherapy, few preclinical studies have compared various sequences of radiation doses or fractions to determine which doses and fractions achieve optimal antitumor immunity. This should be a focus in future preclinical investigation, as it will provide guidance for clinical trial designs.
3.6 Clinical Integration
Radiation therapy as currently used in the clinic has largely been optimized for local therapy, and historically it has been hampered by older imaging and radiation-delivery techniques that cannot precisely locate tumors, especially tumors that move, and rely on low-energy photon beams. Indeed, many of the techniques in current use for local control may not be optimal in terms of producing systemic immunologic responses. This section focuses on the implications of using different radiation field sizes with immunotherapy and how to identify the optimal sequencing of immunotherapy and radiation. A great many other important clinical issues also remain to be considered (e.g., appropriate radiation doses, use of induction chemotherapy, the number of sites to be irradiated, differences in T-cell priming based on the location of metastatic lesions [e.g., bone versus liver]), but these considerations are beyond the scope of this chapter.
3.6.1 Radiation Field Size
The highly conformal techniques used to plan and deliver SABR have several advantages when SABR is to be used in combination with immunotherapy. First, SABR treatment volumes are usually quite small, often directed to relatively small metastatic lesions that are typically safe distances from critical structures such as lung or heart. Because immunotherapy has its own forms of toxicity (e.g., colitis and pneumonitis), the ability to treat small volumes has inherent advantages in terms of nonadditive toxicity. Smaller volumes may also spare the draining lymphatics, which are important for T-cell education and priming. Also, the often-ablative doses used with SABR have unique immunologic effects on the tumor that may also be beneficial for eradicating the tumor stroma.
Despite these advantages, and the excellent local control possible with SABR, most patients with locally advanced disease require much larger treatment volumes that often encompass the tumor, involved lymphatics, and possibly other high-risk lymphatics. Although sterilizing draining lymphatics may be beneficial in terms of tumor control, it can also have detrimental effects on the host’s immunologic response against the tumor, because newly exposed tumor neoantigens are delivered to the lymph nodes, where T-cell priming takes place. Moreover, treating larger fields often requires prolonged courses of fractionated radiation, for example, giving 2-Gy fractions daily for 30–35 days. Thus, the antigen-presenting cells and the T-cells in the lymph node and in the tumor are essentially eradicated daily for 6–7 weeks. Further improvements in local control for locally advanced disease often require that radiation be combined with radiosensitizing agents. This poses further immunologic challenges because chemotherapy can cause myelosuppression and deplete lymphocytes [77]. Moreover, chemotherapy is often given with steroids to minimize the unpleasant side effects of chemotherapy; steroids can prevent T-cell priming, but they may not affect previously activated T-cells [78]. Finally, the large fields needed to treat locally advanced disease often approach the dose-volume limits necessary to minimize radiation-induced toxicity to normal tissues, and adding concurrent immunotherapy agents has a higher potential for toxicity, especially when the radiation involves organs with known inflammatory-mediated side effects from immunotherapy, such as the lung or bowel.
Radiation treatment volume also matters in terms of its hematologic effects; in one study [79], the cumulative incidence of grade 2 leukopenia among 27 men who received whole-pelvis irradiation (to 46 Gy) for prostate cancer was higher (15% vs. 2% without pelvic irradiation, P = 0.02), as was grade 2 anemia (8% vs. 0% without pelvic irradiation, P = 0.03). Because whole-pelvis radiation therapy may be more detrimental hematologically than prostate-only radiotherapy, and because neoadjuvant hormonal therapy reduces hemoglobin levels, one might speculate that negative effects on functioning immune system and tumor oxygenation could explain the disappointing results of RTOG 9413, which enrolled 1323 patients to compare whole-pelvis radiation versus prostate-only radiation therapy [80]. On the other hand, the immunostimulatory effects of radiotherapy (e.g., tumor cell death, changes in antigen availability and inflammatory signals) could have activated lymphocytes and DCs [81]. These and other questions remain to be answered in future trials designed specifically to examine the effects of radiation on the immune system.
3.6.2 Sequence
The optimal sequencing of immunotherapy and radiation therapy is another important consideration that will need to be worked out specifically for each type immunotherapy, as discussed previously. When this chapter was written, only two checkpoint inhibitors had been approved by the US Food and Drug Administration—anti-CTLA4 and anti-PD1. The need to determine the best sequencing will undoubtedly become increasingly challenging as more immunotherapies enter the clinic in the future because each new mechanism of action will need to be evaluated in the context of how to best combine each agent with radiation.
Perhaps the most extensive experience to date comes from the anti-CTLA4 agent ipilimumab, the first approved checkpoint inhibitor. The rationale for combining anti-CTLA4 with radiation is that radiation can increase the T-cell repertoire and diversity in a tumor, and blocking CTLA4 can promote the expansion of these newly activated T-cells [75]. This rationale also seems to favor the use of concurrent chemotherapy. Yet ipilimumab can also reduce inhibitor Treg populations, and thus pretreatment with ipilimumab could potentially “precondition” the tumor by increasing the CD8/Treg ratio, enabling a more robust T-cell response.
Several case reports have described abscopal responses when ipilimumab is used with radiation therapy; these reports may offer clues as to how this strategy works and the optimal sequencing of these two forms of treatment. In one report, a patient with metastatic non-small cell lung cancer received five fractions of 6 Gy each, starting concurrent ipilimumab on the day after the first fraction; this patient demonstrated an impressive abscopal response that lasted for several months [82]. However, another case report described a patient with metastatic melanoma that had progressed on ipilimumab; in that case, 28.5 Gy was delivered in three fractions to a paraspinal mass [7]. Several months later, that patient developed a systemic abscopal response in the liver, chest, and spleen. Thus giving radiation after disease has progressed on ipilimumab (which can deplete Tregs) may have distinct advantages and is the topic of a phase I/II trial testing ipilimumab with SABR for advanced solid tumors (NCT02239900). This trial will evaluate the timing of radiation during ipilimumab therapy (concurrent versus sequential) and will also evaluate different radiation doses and the effect of irradiating metastatic disease at different sites.
Experience with radiation plus anti-PD1/PDL1 therapies is becoming more prevalent as well. Several trials are underway to evaluate this combination, with most starting the anti-PD1 and the radiation at about the same times. Alternatively, other trials are evaluating checkpoint inhibition as adjuvant therapy after radiation for patients who require larger radiation fields (e.g., those with stage III non-small cell lung cancer or mesothelioma). This approach may prove to be safer by reducing the risk of pneumonitis. Radiation has also been shown to increase PDL1 expression in myeloid cells, which could further justify its use as an adjuvant to immunotherapy [76].
3.7 Clinical Trials of Radiation Plus Immunotherapy
3.7.1 Anti-CTLA4 Therapy
Historically, radiation was developed as means of providing local tumor control; immunotherapy, on the other hand, has been tested mainly for patients with metastatic disease, as a final option for tumors that have not responded to conventional treatments [83,84,85,86,87]. Clinical synergy between these two modalities has been shown in multiple studies of ipilimumab and concurrent radiation given for metastatic melanoma [7, 88]. Another group found mainly partial abscopal responses in 9 (43%) of 21 patients with melanoma, with 2 patients (10%) remaining stable [89]. Combining immune checkpoint blockade with radiation therapy has also shown promise in metastatic prostate cancer; in one phase I/II study of 50 men who received 4- to 10-mg/kg doses of ipilimumab plus 8-Gy fractions to each metastatic lesion [90], one patient had a complete response and six had stabilized disease; these results led to a randomized phase II trial (NCT01689974). Ipilimumab plus SABR is further being tested in several phase II trials for patients with stage IV melanoma and any number of metastases (NCT01970527, NCT02107755, NCT01565837).
In one nonrandomized phase I/II trial of 71 men with metastatic castration-resistant prostate cancer who had experienced disease progression after discontinuing antiandrogen therapy, ipilimumab as monotherapy (n = 29) was compared with ipilimumab plus a single radiation dose of 8 Gy (n = 41) per bone metastasis, given 24–48 h before the first ipilimumab dose (3 or 10 mg/kg) [90]. Of men with 28 evaluable tumors, ipilimumab at 10 mg/kg, with or without radiation, led to reductions in prostate-specific antigen levels of at least 50% in eight men, and another had a complete response. The phase III randomized, double-blind trial for men with metastatic castrate-resistant prostate cancer that had progressed after docetaxel, involved giving a bone-directed single 8-Gy dose of radiation followed by either ipilimumab 10 mg/kg (n = 399) or placebo (n = 400) every 3 weeks for up to 4 doses. Results from that trial showed a slightly longer median overall survival time of 11.2 months for men who received ipilimumab versus 10 months for the placebo group (P = 0.053); however, this trial did not reach a significant difference between the two cohorts [91]. Ipilimumab is also being tested with cetuximab and intensity-modulated radiation therapy in a phase Ib trial for patients with previously untreated stage III-IVB head and neck cancer (NCT01935921).
3.7.2 Anti-PD1 Therapy
Several trials are ongoing to test anti-PD1 with radiation therapy for patients with melanoma or non-small cell lung cancer that did not respond to at least one regimen of systemic therapy or anti-PD-1 therapy. NCT02608385 is a phase I study of PD1 blockade with pembrolizumab and SABR for advanced solid tumors at the University of Chicago; NCT02303990 or “RADVAX” is a stratified phase I trial of pembrolizumab with hypofractionated radiation therapy for advanced and metastatic cancers at the Abramson Cancer Center of the University of Pennsylvania; and NCT02318771 is a trial of hypofractionated radiotherapy with pembrolizumab for recurrent/metastatic head and neck cancer, renal cell carcinoma, lung cancer, or melanoma.
Immunotherapy with radiation is also being tested to improve long-term local control of high-grade glial tumors. Ongoing trials include a multicenter, open-label nonrandomized phase II trial of MEDI4736 for patients with newly diagnosed, unmethylated MGMT glioblastoma in which patients are given anti-PDL1 (durvalumab) every 2 weeks with standard radiation therapy (NCT02336165); and a phase I trial of PD1 blockade with pembrolizumab and bevacizumab with 5 days of hypofractionated stereotactic radiotherapy for recurrent high-grade glial tumors has been initiated at the H. Lee Moffitt Cancer Center and Research Institute (NCT02313272).
The PD1 inhibitor pembrolizumab is currently being tested in a phase II trial for patients with surgically resectable squamous cell carcinoma of the head and neck; pembrolizumab is given intravenously approximately 2 or 3 weeks before surgery to be followed by risk-based intensity-modulated radiation therapy to 60 Gy (in daily 2-Gy fractions, NCT02296684); it is also being tested for patients getting reirradiation with inoperable locoregionally recurrent or second primary squamous cell carcinoma (NCT02289209). Another phase I single-arm, open-label trial is testing pembrolizumab in combination with cisplatin-based standard definitive chemoradiation therapy (to a total dose of 70 Gy, in daily 2-Gy fraction) for patients with stage III-IVB squamous cell carcinoma of the head and neck; the accrual goal of that trial is about 39 patients (NCT02586207).
Colorectal cancer, especially tumors of the rectum or anal cancer, has long been treated effectively by radiation therapy; whether immunotherapy could provide a stronger immune response to help destroy tumor cells is being investigated in a phase II trial of pembrolizumab plus radiotherapy or ablation for metastatic colorectal cancer (NCT02437071).
3.7.3 Cytokine Therapy
Intralesional injections of IFN-β (3–5 million units, 3 times/week, before radiation to a total dose of 40–60 Gy) has shown mixed results [92,93,94]. In one such trial, all 21 patients with metastatic melanoma showed either a complete remission (70%) or a partial remission, with a median survival time of 10 months [95]. In another phase I trial, SABR (one, two, or three doses of 20 Gy/fraction) is followed by high-dose IL-2 at 3 days after the last radiation fraction for metastatic melanoma or renal cell carcinoma. That regimen produced antitumor responses as defined by the Response Evaluation Criteria for Solid Tumors (one complete response and seven partial responses) in nonirradiated target lesions [96]. Ongoing trials combining concurrent high-dose IL-2 with radiation therapy for either renal cell carcinoma or melanoma are focusing on the immunological effects of this treatment [97, 98].
3.7.4 OX40 Therapy
Use of OX40 agents with radiation for breast cancer has been delayed somewhat given the prevalence of open trials of the monoclonal antibodies trastuzumab or pertuzumab for HER2-positive breast cancer. However, a phase I/II study is underway to test the anti-OX40 agent MEDI6469 in combination with SABR for metastatic liver or lung lesions in patients with progressive metastatic breast cancer (NCT01862900). Another phase I/II trial now ongoing is testing an OX40 agent, which is thought to induce proliferation of memory and effector T-cells, in combination with cyclophosphamide and radiation therapy for patients with progressive metastatic prostate cancer (NCT01303705).
3.8 Conclusions
The combination of immunotherapy plus radiation has great potential to extend the benefit of radiation beyond its current role of local control. As we go forward, we need to consider how our current radiation techniques are best combined with immunotherapy, and in some cases we will likely need to develop unique fields and dosing regimens to expand radiation benefit into new patient populations. Pretreatment assessment of a patient’s immune state will become important, as well as minimizing immunosuppressive agents such as steroids. Moving past checkpoint inhibitors we will need to personalize immunotherapy toward patient-specific mechanisms of resistance and XRT-specific immunotherapies. More preclinical studies are needed to evaluate these questions and establish the safety of combination therapy. By working to refine and perfect rational combinations of immunotherapy and radiation we have an opportunity to improve the lives of many patients.
References
Timmerman R, Paulus R, Galvin J, et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA. 2010;303:1070–6.
Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58:862–70.
Santarpia M, Gonzalez-Cao M, Viteri S, Karachaliou N, Altavilla G, Rosell R. Programmed cell death protein-1/programmed cell death ligand-1 pathway inhibition and predictive biomarkers: understanding transforming growth factor-beta role. Transl Lung Cancer Res. 2015;4:728–42.
Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5.
Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–3.
Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88.
Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925–31.
Stamell EF, Wolchok JD, Gnjatic S, Lee NY, Brownell I. The abscopal effect associated with a systemic anti-melanoma immune response. Int J Radiat Oncol Biol Phys. 2013;85:293–5.
Seyedin SN, Schoenhals JE, Lee DA, et al. Strategies for combining immunotherapy with radiation for anticancer therapy. Immunotherapy. 2015;7(9):967–80.
Tang C, Wang X, Soh H, et al. Combining radiation and immunotherapy: a new systemic therapy for solid tumors? Cancer Immunol Res. 2014;2:831–8.
Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys. 2012;83:1306–10.
Filatenkov A, Baker J, Mueller AM, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. 2015;21:3727–39.
Reynders K, Illidge T, Siva S, Chang JY, De Ruysscher D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat Rev. 2015;41:503–10.
Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91.
Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29.
Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Lee SJ, Jang BC, Lee SW, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580:755–62.
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science Translational Medicine. 2012;4:127ra37.
Dovedi SJ, Adlard AL, Lipowska-Bhalla G, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74:5458–68.
Park SS, Dong H, Liu X, et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol Res. 2015;3:610–9.
Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180:3132–9.
Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One. 2012;7:e32542.
Burnette B, Fu YX, Weichselbaum RR. The confluence of radiotherapy and immunotherapy. Front Oncol. 2012;2:143.
Kwilas AR, Donahue RN, Bernstein MB, Hodge JW. In the field: exploiting the untapped potential of immunogenic modulation by radiation in combination with immunotherapy for the treatment of cancer. Front Oncol. 2012;2:104.
Matsumura S, Wang B, Kawashima N, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–107.
Melcher A, Gough M, Todryk S, Vile R. Apoptosis or necrosis for tumor immunotherapy: what’s in a name? J Mol Med (Berl). 1999;77:824–33.
Kotera Y, Shimizu K, Mule JJ. Comparative analysis of necrotic and apoptotic tumor cells as a source of antigen(s) in dendritic cell-based immunization. Cancer Res. 2001;61:8105–9.
McBride WH, Chiang CS, Olson JL, et al. A sense of danger from radiation. Radiat Res. 2004;162:1–19.
Friedman EJ. Immune modulation by ionizing radiation and its implications for cancer immunotherapy. Curr Pharm Des. 2002;8:1765–80.
Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Kachikwu EL, Iwamoto KS, Liao YP, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. 2011;81:1128–35.
Canney PA, Dean S. Transforming growth factor beta: a promotor of late connective tissue injury following radiotherapy? Br J Radiol. 1990;63:620–3.
Kaur P, Asea A. Radiation-induced effects and the immune system in cancer. Front Oncol. 2012;2:191.
Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–70.
Hawinkels LJ, Ten Dijke P. Exploring anti-TGF-beta therapies in cancer and fibrosis. Growth Factors. 2011;29:140–52.
Ivanovic V, Todorovic-Rakovic N, Demajo M, et al. Elevated plasma levels of transforming growth factor-beta 1 (TGF-beta 1) in patients with advanced breast cancer: association with disease progression. Eur J Cancer. 2003;39:454–61.
Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14:481–7.
Chen L, Gibbons DL, Goswami S, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241.
Chen Y, Williams J, Ding I, et al. Radiation pneumonitis and early circulatory cytokine markers. Semin Radiat Oncol. 2002;12:26–33.
Li J, Mu S, Mu L, Zhang X, Pang R, Gao S. Transforming growth factor-beta-1 is a serum biomarker of radiation-induced pneumonitis in esophageal cancer patients treated with thoracic radiotherapy: preliminary results of a prospective study. Onco Targets Ther. 2015;8:1129–36.
Evans ES, Kocak Z, Zhou SM, et al. Does transforming growth factor-beta1 predict for radiation-induced pneumonitis in patients treated for lung cancer? Cytokine. 2006;35:186–92.
Stenmark MH, Cai XW, Shedden K, et al. Combining physical and biologic parameters to predict radiation-induced lung toxicity in patients with non-small-cell lung cancer treated with definitive radiation therapy. Int J Radiat Oncol Biol Phys. 2012;84:e217–22.
Wang J, Qiao XY, Lu FH, et al. TGF-beta1 in serum and induced sputum for predicting radiation pneumonitis in patients with non-small cell lung cancer after radiotherapy. Chin J Cancer. 2010;29:325–9.
Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–8.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10.
Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–90.
Kim JY, Son YO, Park SW, et al. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp Mol Med. 2006;38:474–84.
Mifune M, Sobue T, Arimoto H, Komoto Y, Kondo S, Tanooka H. Cancer mortality survey in a spa area (Misasa, Japan) with a high radon background. Jpn J Cancer Res. 1992;83:1–5.
Wei LX, Zha YR, Tao ZF, He WH, Chen DQ, Yuan YL. Epidemiological investigation of radiological effects in high background radiation areas of Yangjiang, China. J Radiat Res. 1990;31:119–36.
Kendall GM, Muirhead CR, MacGibbon BH, et al. Mortality and occupational exposure to radiation: first analysis of the National Registry for Radiation Workers. BMJ. 1992;304:220–5.
Spycher BD, Lupatsch JE, Zwahlen M, et al. Background ionizing radiation and the risk of childhood cancer: a census-based nationwide cohort study. Environ Health Perspect. 2015;123:622–8.
Wall BF, Kendall GM, Edwards AA, Bouffler S, Muirhead CR, Meara JR. What are the risks from medical X-rays and other low dose radiation? Br J Radiol. 2006;79:285–94.
Hosoi Y, Ishii K, Yamada S, Ono T, Sakamoto K. Effect of combination treatment of 15 cGy total body irradiation and OK-432 on spontaneous lung metastasis and mitogenic response of splenocytes in mice. Radiat Oncol Investig. 1997;5:283–8.
Hosoi Y, Sakamoto K. Suppressive effect of low dose total body irradiation on lung metastasis: dose dependency and effective period. Radiother Oncol. 1993;26:177–9.
Ren H, Shen J, Tomiyama-Miyaji C, et al. Augmentation of innate immunity by low-dose irradiation. Cell Immunol. 2006;244:50–6.
Kojima S, Nakayama K, Ishida H. Low dose gamma-rays activate immune functions via induction of glutathione and delay tumor growth. J Radiat Res. 2004;45:33–9.
Liu SZ. Cancer control related to stimulation of immunity by low-dose radiation. Dose-Response. 2007;5:39–47.
Jin SZ, Pan XN, Wu N, Jin GH, Liu SZ. Whole-body low dose irradiation promotes the efficacy of conventional radiotherapy for cancer and possible mechanisms. Dose Response. 2007;5:349–58.
Burnette B, Weichselbaum RR. The immunology of ablative radiation. Semin Radiat Oncol. 2015;25:40–5.
Okuma K, Yamashita H, Niibe Y, Hayakawa K, Nakagawa K. Abscopal effect of radiation on lung metastases of hepatocellular carcinoma: a case report. J Med Case Reports. 2011;5:111.
Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9.
Camphausen K, Moses MA, Menard C, et al. Radiation abscopal antitumor effect is mediated through p53. Cancer Res. 2003;63:1990–3.
Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–71.
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–23.
Lee Y, Auh SL, Wang Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–95.
Barker CA, Postow MA, Khan SA, et al. Concurrent radiotherapy and ipilimumab immunotherapy for patients with melanoma. Cancer Immunol Res. 2013;1:92–8.
Kiess AP, Wolchok JD, Barker CA, et al. Stereotactic radiosurgery for melanoma brain metastases in patients receiving ipilimumab: safety profile and efficacy of combined treatment. Int J Radiat Oncol Biol Phys. 2015;92:368–75.
Mathew M, Tam M, Ott PA, et al. Ipilimumab in melanoma with limited brain metastases treated with stereotactic radiosurgery. Melanoma Res. 2013;23:191–5.
Silk AW, Bassetti MF, West BT, Tsien CI, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2:899–906.
Gerber NK, Young RJ, Barker CA, et al. Ipilimumab and whole brain radiation therapy for melanoma brain metastases. J Neurooncol. 2015;121:159–65.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99.
Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7.
Deng L, Liang H, Burnette B, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–95.
Tang C, Liao Z, Gomez D, et al. Lymphopenia association with gross tumor volume and lung V5 and its effects on non-small cell lung cancer patient outcomes. Int J Radiat Oncol Biol Phys. 2014;89:1084–91.
Hinrichs CS, Palmer DC, Rosenberg SA, Restifo NP. Glucocorticoids do not inhibit antitumor activity of activated CD8+ T cells. J Immunother. 2005;28:517–24.
Pinkawa M, Djukic V, Klotz J, et al. Hematologic changes during prostate cancer radiation therapy are dependent on the treatment volume. Future Oncol. 2014;10:835–43.
Lawrence YR, Dicker AP. Radiation therapy and the immune system: learning to live together. Future Oncol. 2014;10:777–80.
Finkelstein SE, Salenius S, Mantz CA, et al. Combining immunotherapy and radiation for prostate cancer. Clin Genitourin Cancer. 2015;13:1–9.
Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res. 2013;1:365–72.
Zumwalt TJ, Goel A. Immunotherapy of metastatic colorectal cancer: prevailing challenges and new perspectives. Curr Colorectal Cancer Rep. 2015;11:125–40.
Zhang GQ, Zhao H, Wu JY, et al. Prolonged overall survival in gastric cancer patients after adoptive immunotherapy. World J Gastroenterol. 2015;21:2777–85.
Zhang GQ, Li F, Sun SJ, et al. Adoptive immunotherapy for small cell lung cancer by expanded activated autologous lymphocytes: a retrospective clinical analysis. Asian Pac J Cancer Prev. 2015;16:1487–94.
Wyluda EJ, Cheng J, Schell TD, et al. Durable complete responses off all treatment in patients with metastatic malignant melanoma after sequential immunotherapy followed by a finite course of BRAF inhibitor therapy. Cancer Biol Ther. 2015;16:662–70.
Massari F, Santoni M, Ciccarese C, et al. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer Treat Rev. 2015;41:114–21.
Hiniker SM, Chen DS, Reddy S, et al. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol. 2012;5:404–7.
Grimaldi AM, Simeone E, Giannarelli D, et al. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunol. 2014;3:e28780.
Slovin SF, Higano CS, Hamid O, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol. 2013;24:1813–21.
Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12.
Hazard LJ, Sause WT, Noyes RD. Combined adjuvant radiation and interferon-alpha 2B therapy in high-risk melanoma patients: the potential for increased radiation toxicity. Int J Radiat Oncol Biol Phys. 2002;52:796–800.
Nguyen NP, Levinson B, Dutta S, et al. Concurrent interferon-alpha and radiation for head and neck melanoma. Melanoma Res. 2003;13:67–71.
Conill C, Jorcano S, Domingo-Domenech J, et al. Toxicity of combined treatment of adjuvant irradiation and interferon alpha2b in high-risk melanoma patients. Melanoma Res. 2007;17:304–9.
Paul E, Muller I, Renner H, Bodeker RH, Cochran AJ. Treatment of locoregional metastases of malignant melanomas with radiotherapy and intralesional beta-interferon injection. Melanoma Res. 2003;13:611–7.
Seung SK, Curti BD, Crittenden M, et al. Phase 1 study of stereotactic body radiotherapy and interleukin-2--tumor and immunological responses. Sci Transl Med. 2012;4:137ra74.
Ridolfi L, De Rosa F, Granato AM, et al. Radiotherapy as an immunological booster in patients with metastatic melanoma or renal cell carcinoma treated with high-dose interleukin-2: interim analysis data. J Clin Oncol. 2015;33(suppl; abstr e14007).
Ridolfi L, de Rosa F, Ridolfi R, et al. Radiotherapy as an immunological booster in patients with metastatic melanoma or renal cell carcinoma treated with high-dose Interleukin-2: evaluation of biomarkers of immunologic and therapeutic response. J Transl Med. 2014;12:262.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Schoenhals, J.E., Skrepnik, T., Selek, U., Cortez, M.A., Li, A., Welsh, J.W. (2017). Optimizing Radiotherapy with Immunotherapeutic Approaches. In: Naing, A., Hajjar, J. (eds) Immunotherapy. Advances in Experimental Medicine and Biology, vol 995. Springer, Cham. https://doi.org/10.1007/978-3-319-53156-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-53156-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53155-7
Online ISBN: 978-3-319-53156-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)