
Abstract
Patients with recurring or metastatic colorectal cancer (mCRC) have strikingly low long-term survival, while conventional treatments such as chemotherapeutic intervention and radiation therapy marginally improve longevity. Although many factors involving immunosurveillance and immunosuppression were recently validated as important for patient prognosis and care, a multitude of experimental immunotherapies designed to combat unresectable mCRC have, in few cases, successfully mobilized anti-tumor immune cells against malignancies nor conclusively or consistently granted protection, complete remission, and/or stable disease from immunotherapy, benefitting less than 10 % of those receiving therapy. After decades of progress, however, new insights into the mechanisms of immunosuppression, tolerance, and mutation profiling established novel therapies that circumvent these immunological barriers. This review underlines the most exciting methods to date that manipulate immune cells to curb mCRC, including adoptive cell therapy, dendritic cell vaccines, and checkpoint inhibitor antibodies, hinting at effective and enduring protection against disease progression and undetected micrometastases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) represents 10 % of malignancies worldwide and remains the fourth leading cause of cancer-related mortalities [1]. Although surgical resection of localized tumors greatly improves patient survival past 5 and 10 years [2], liver metastases will develop in over half of all CRC patients further lowering survival [3–5]. A minority of stage III CRC patients benefit from adjuvant chemotherapy (i.e., 5FU/oxaliplatin regimens), yet all endure toxicity [6]. Where chemotherapy and radiation fail, priming the immune system to inhibit metastatic spread may improve patient survival without severe toxicity.
Harnessing the immune system has successfully eradicated malignant cells in animal models, and even though human tumors are more complex, some phase III trials have been successful [7]. Immunotherapy introduces modulatory agents, such as adjuvants, antibodies, and/or immune cells, to initiate or amplify an existing anti-tumor response. Ipilimumab is an exciting example where immune cell-activating antibodies prolong the lives of melanoma patients [8].
The topic of CRC immunotherapy was explored elegantly in previous reviews; this review, however, mainly discusses the inherent immunosuppressive barriers in the local tumor environment that obstruct current immunotherapeutic intervention. Particularly, this review will (1) briefly overview the prognostic impact of common tumor-infiltrating immune cells in CRCs, (2) critique current and most promising therapies that orient the immune system against CRCs, and (3) suggest which immunosuppressive barriers can be targeted to improve immunotherapy efficacy.
Anti-tumor Immunity
Before the culmination of seminal studies performed by Freidman, Galon, and Pagès, who demonstrated that effector T cells infiltrate CRCs and benefit patient survival, Jass and colleagues provided direction for these investigators by connecting the mechanism of protective immunity to infiltrating lymphocytes in advanced rectal cancer [9]. Many studies concur with the supposition that immunosurveillance [10, 11] inhibits CRC tumorigenesis and progression [9, 12–15]. Anti-tumor activity is highly coordinated, requires specifically primed lymphocytes to eradicate aberrant cells, and is evidenced by high densities of tumor-infiltrating lymphocytes (TILs) in early-stage CRCs [16, 17].
Cytotoxic Immune Response
Rudimentary anti-tumor T cell responses are driven by anti-proliferative interferon-gamma (IFN-γ) [18]. This cytokine is secreted upon proteasomal degradation and subsequent presentation of antigens to T cell receptors (TCRs) by major histocompatibility complex (MHC) class I [19]. Cytotoxic CD8+ T lymphocytes (CTLs) are a major source of IFN-γ [20, 21] as well as apoptosis-inducing granzyme B [22] and require cooperative interactions from type-1 polarized CD4+ T (TH1) cells [23]. High densities of CD8+ effector memory T cells in CRCs associate with prolonged survival and lack of early signs of metastasis [13], suggesting TH1 polarization and associated CTLs quell metastasis after curative surgery [15].
CXCR3 Ligands
A complex network of chemokines and cognate receptors dictates the type and density of infiltrating T cells into tumors [24, 25]. The majority of CTLs and active TH1 cells express CXCR3, the receptor for a trio of IFN-γ-induced chemokines, CXCL9, CXCL10, and CXCL11 [26–29]. These chemokines home CXCR3-expressing cells towards inflammation.
CCR5 Ligand
CCR5 is co-expressed with CXCR3 on TH1 cells and CTLs in CRC invasive margins [30], however is absent from CRCs with lymphatic and liver metastasis and low densities of CD8+ T cells [31, 32]. Its ligand, CCL5 [33], is released from stored secretory compartments upon CD8+ T cell stimulation [34] to attract other T cells towards inflammation [35], and is strongly secreted from specific CRC subsets [36].
Natural Killer Cells in CRC
Among the arsenal of cytotoxic lymphocytes, natural killer (NK) cells and NK T cells recognize and eliminate MHC-negative tumor cells [37, 38]. However, their association with cancer progression is inconsistent across multiple cancer types. Recently, Sconocchia and colleagues demonstrated that certain cancer types harbor low densities of infiltrating NK-cells; breast cancers, melanomas, hepatocellular carcinomas, renal cell carcinomas, and CRCs all stain poorly for NK cell markers [39, 40]. These cells are not found in CRC-associated liver metastases [41] and have no prognostic relevance for CRC patients [42–44]. These studies suggest that evasion from NK cell detection and elimination is an obligate prerequisite for tumorigenesis [43]. The prognostic impact of TILs on CRC patient survival is summarized in Table 1.
Multidimensional Immunosuppression
Tumor eradication and host tolerance are dictated by the immune system. The type, polarity, and density of infiltrating immune cells can either reject or accept aberrant cell growth. Each CRC contains a unique composition of immune cells, including those that quench the tumor-fighting capacity of effector T cells described above. Other infiltrating immune cells produce proliferative signals and accelerate metastatic disease.
Immune-Regulating T Cells
In healthy tissues, regulatory FoxP3+CD4+CD25+ T cells (Tregs) [67, 68] suppress effector T cells and protect against autoimmune disease through an array of immune quenching factors that include TGF-β, IL-10, and cytotoxic T lymphocyte antigen 4 (CTLA-4) [69–71]. These factors silence memory CD8+ T cell activity and IFN-γ production [72]. For example, Tregs powerfully suppress T cells isolated from CRC patients [73], while depletion of Tregs via anti-CD25 antibody augments CRC rejection in mice [74].
Contradicting the studies mentioned above, Tregs may benefit CRC patients. FoxP3+ cell densities in CRCs positively correlate with patient survival [63, 64, 75] and absence of metastases [76]. These data question whether colon and rectum residing FoxP3+ cells truly represent Tregs. FoxP3 is transiently expressed by intra-tumoral effector T cells upon TCR stimulation [77], suggesting that FoxP3 poorly identifies immunosuppressive T cells. Alternatively, Tregs may protect against tumor progression in barrier organs by suppressing tumor-promoting inflammation initiated by foreign antigens [78]. Perhaps the true prognostic relevance of Tregs lies with their abundance to cytotoxic CD8+ T cells; tumor progression may be slowed or halted when CD8+ T cells far outnumber Tregs.
Considering all the studies mentioned above, only one factor supplied by Tregs is unlikely to exclusively inhibit immunosurveillance nor will only one cell type promote immunosuppressive environments. Therefore, effective immunotherapy likely requires blockage of other immunosuppressive cell types and their soluble factors. Without clear understanding, we risk overvaluing unreliable immunosuppressive markers and fail to personalize therapy. A better approach considers the holistic immune contexture of the tumor environment.
Current cancer immunotherapy research lacks mouse models that study multidimensional immunosuppression. Mice engineered to study one specific immune-modulating mechanism do not reflect the complexity of the human tumor environment.
Tumor-Promoting Myelogenous Cells
Macrophages residing in human tumors affect disease progression. These myelogenous cells polarize to promote either acute inflammation and adaptive T cell immunity or immunosuppression and tumor escape [79]. Therefore, the prognostic relevance of tumor-associated macrophages (TAMs) in CRCs is debatable.
Tumor-Associated Macrophages
TAMs represent major components of the colorectal intra-tumoral immune milieu and are classified into heterogeneous subgroups by an array of secreted cytokines [80, 81]. Angiogenesis- and metastasis-promoting cytokines link these cells to CRC progression and poor prognosis [82]. Immature TAMs, termed myeloid-derived suppressor cells (MDSCs), facilitate immunosuppression, inflammation, and angiogenesis [83–86] by secreting VEGF, T and NK cell-inhibiting (and Treg expanding) nitric oxide, reactive oxygen species, arginase, IL-10, and IL-6 [87, 88]. In contrary, the presence of TAMs at CRC invasive margins associate with improved survival [89]. Therefore, fully ascertaining the link between TAMs/MDSCs and patient survival will identify their mitigating effects on therapy.
Dendritic Cells
In the periphery, protective immunity requires antigen presentation by dendritic cells (DCs) [90]. These cells are equipped with molecular receptors that sample microbial components and program lymphocytes with the resulting information. Programming requires concurrent ligation of CD28 or OX40 on T cells to CD80/86 [91]. Like TAMs, DCs form heterogeneous populations that dictate immune responses either away or against tumor antigens [92]. DCs activated without maturation signals will promote tolerance and immunosuppression by inducing Tregs [93].
DC analysis in CRCs is limited to low-density immunohistochemistry and tissue microarrays. These methods analyze immune cells within small tissue fragments [94]. Immunohistochemistry is very useful for studying spatial arrangements of immune cells in solid tumors, however only identifies cells by a limited number of markers. Tissue microarrays utilize more markers, but require fragmenting tissues, thereby destroying spatial arrangements of immune cells. Markers such as CD83 [95], S-100 [96], and CD208 [81, 97] represent DCs in the stroma and invasive margins. However, no CRC study conclusively characterized the maturation status of DCs via cell surface markers or cytokine secretion using multidimensional approaches. As potential targets for immunotherapeutic intervention, much remains to be learned about the role of DCs in CRC progression. Common immunosuppressive mechanisms are summarized in Table 2.
Novel methods that improve immune infiltrate profiling and prognostic accuracy are encouraging; however, counting T cells, macrophages, and DCs alone does not optimally predict which patients will respond to therapy because the phenotype and activity of infiltrates are equally critical. We are very far from identifying all the immunosuppressive mechanisms, yet our currently knowledge will soon extend and improve the quality of patient lives.
The diversity of CRC environments requires research to develop multiple methods to directly manipulate phenotypes and activity of immune infiltrates. However, to eradicate tumors these methods must overcome three major obstacles. (1) Interrupting the immunosuppressive environment will strengthen the impact of existing anti-tumor responses. This can be done using simple molecules that target specific immune-modulating pathways. (2) Encouraging CTLs to migrate to the invasive margin will augment anti-tumor immunity. Tumor rejection may simply require overpowering the immunosuppressive environment by introducing an overwhelming number of T cells, cytokines, and cytotoxic mediators. And, (3) repolarizing myelogenous cells to secrete anti-tumor cytokines will suppress Tregs and encourage tumor antigen-specific immunity. Tumor-fighting DCs may vaccinate patients to produce tumor-fighting memory T cells. Immunosuppressive barriers that may inhibit immunotherapy are depicted in Fig. 1.

Common immunosuppressive barriers in CRC are potential targets for immunotherapy. MDSCs (brown) release Treg (orange)-promoting factors, IL-10, TGF-β, and IDO (gray, purple, and blue, respectively), and T cell-suppressing arginase and nitric oxide (olive and brown) (a). Tregs compete with other T cells for coreceptor ligation by expressing CTLA-4 and LAG-3. CTLA-4 binds to CD80/CD86 and LAG-3 binds to MHC class II, thereby blocking the transmission of stimulatory signals from DCs (purple) and other antigen-presenting cells (APCs) to effector T cells (b). Tregs and endogenous lymphocytes serve as sinks and limit the amount of homeostatic cytokines, IL-7 and IL-15 (light blue and red, respectively), that are available to effector T cells (c). Chronic T cell stimulation through viral challenge and cancer antigens perpetuates PD-1 expression leading to effector T cell exhaustion (d)
Directing the Immune System Against Metastatic CRC
The remainder of this review discusses methods that treat advanced CRC patients through immune manipulation and includes perspectives on how immunosuppression stymies these methods. In the context of cancer, immunotherapy broadly encompasses any immune manipulation that eliminates tumor cells. Immunotherapy promises to eradicate microscopic metastases more precisely than a surgeon’s scalpel and provides lasting protective immunological memory. The current arsenal used to prime the immune system against occult metastases is limited; however, major breakthroughs are propelling research to investigate alternative avenues.
Antibody Therapy
The monoclonal antibody therapy anti-CTLA-4, or ipilimumab, is prominently used to improve metastatic melanoma patient survival [101]. As a checkpoint blocker, anti-CTLA-4 prevents CTLA-4 from outcompeting the T cell co-stimulation receptor, CD28. Its effectiveness requires existing immunosuppressed intra-tumoral T cells, making it the obvious choice for treating highly infiltrated CRCs. However, a pilot study reported no objective responses in 3 colon cancer patients [102], and a larger study using tremelimumab—another anti-CTLA-4 antibody—could not report any conclusive findings other than increased incidence of immune enterocolitis in 45 treatment-refractory CRC patients [103]. Other checkpoint proteins may be more appropriate for CRC therapy.
Programmed Death Receptor-1/Ligand-1 Blockade
Programmed death receptor-1/ligand-1 (PD-1/PD-1L) are members of the CD28 and B7 families and induce tolerance by regulating T cells. Activated CD8+ T cells express PD-1; however, PD-L1/L2-expressing tumor cells, circulating MDSCs [84], and DCs can impart unresponsiveness and exhaustion of immune-directed T cell activities [98]. An initial study reported only marginal responses to anti-PD-1 antibody where one CRC patient demonstrated complete response [104]. This study speculated that anti-PD-1 encourages lymphocyte migration into tumors and tissues [104]. Two larger studies included 19 patients with advanced CRC treated with anti-PD-1 [105] and 18 treated with anti-PD-L1 [106] neither observed equitable clinical responses when comparing to other cancers. Considering other tumor types, the first study witnessed objective responses in 9 of the 25 patients with PD-L1-positive tumors and none with PD-L1-negative tumors.
A recent study demonstrated that defective mismatch repair CRCs strongly upregulate PD-1 and CTLA-4, as well as other immune checkpoints: PD-L1, lymphocyte activation gene-3 (LAG-3), and indoleamine 2,3-dioxygenase (IDO) [107••]. While these tumors rarely metastasize, these data provide evidence that multidimensional immunosuppression is important in more aggressive tumors. Two interesting notes: approximately 30 % of intra-tumoral CD4+ FoxP3− T cells expressed LAG-3 [99•, 108, 109], and IDO expression in CRCs associates with liver metastasis, tumor progression, and decreased overall survival [100, 110]. These data suggest that strong anti-tumor T cell responses are concomitantly surmounted by multiple mechanisms of immunosuppression.
Questions remain as to why patients with metastatic melanoma and other cancers benefit from checkpoint blockade while CRC patients do not. The simple answer is that previous CRC studies did not reach statistical significance; fortunately, larger clinical trials are underway. The complex answer is that checkpoint blockers may not provide durable or prolonged protection. For example, ipilimumab allows Treg numbers to rebound after treatment and fails to increase CRC-specific T cell abundance in peripheral blood [102], tremelimumab treatment fostered misdirected/non-specific responses with mostly ineffective or undesirable consequences [103], anti-PD-1/PD-L1 antibodies may only be effective against PD-1/PD-L1-positive tumors [105], and finally, blockading only one axis of immunosuppression will lower efficacy in every CRC clinical trial [105, 106].
Adoptive T Cell Therapy
Adoptive cell transfer (ACT) introduces overwhelming numbers of tumor-targeting effector T cells to circumvent the need to reverse tolerance to tumor antigens. Therapy requires the transfer of autologous T cells—specific to tumor antigens or transfected to encode tumor-directing T cell receptors (TCRs)—directly to the patient. However, recruiting T cells to tumor beds is challenging because T cell infiltration is mitigated by immunosuppressive pressures.
Early attempts to elicit protective immunity against CRCs through adoptive transfer have been effective. Six out of 19 patients exhibited complete or partial response after their lymphocytes were isolated, propagated, sensitized to autologous tumor antigen, and then stimulated with a streptococcal immunopotentiator before transferred back [111]. Fourteen CRC patients received reinfusion of IL-2-stimulated TILs, however did not demonstrate meaningful clinical results other than preserved TCR ζ- and ε-chain expression [112]. Three out of 14 gastric and colon cancer patients achieved clinical response with IL-2-stimulated T cells. These patients stably expressed TCR ζ-chain on peripheral blood T cells, which is normally downregulated with disease progression and immunosuppression [113].
These data suggest that effective adoptive transfer of IL-2-stimulated T cells must avoid immunosuppressive preprogramming. To achieve this, sentinel lymph node-derived CD4+ TH1 cells can be expanded instead of TILs. Intriguingly, four out of nine stage IV CRC patients achieved complete remission [114]. This study was successful because lymphocytes were acquired from lymph nodes that first received drainage from tumors and therefore were segregated from an immunosuppressive environment [114]. T cells residing in sentinel lymph nodes have greater proliferative potential upon stimulation [115] and should be the standard mode of therapy. To criticize, however, harvesting sentinel lymph nodes complicates surgery and may not yield enough T cells.
Chimeric Antigen Receptors
The presumption that TILs are defective through immunosuppressive preprogramming calls for novel approaches that refocus adoptive T cell therapy. Engineering T cells to express chimeric antigen receptors (CARs) specific for carcinoembryonic antigen (CEA)—commonly expressed by colorectal tumors—benefits some advanced-stage CRC patients. CARs are antigen-specific heavy and light chain antibodies genetically attached to cytoplasmic signaling molecules, such as 4-1BB, OX40, Luk, and TCR ζ-chain [116], and designed to relay activation signals to T cells without peptide presentation. CAR therapy is advantageous when MHC class I is downregulated [117].
However, an unfortunate case study describes treatment failure for a patient with ERBB2 overexpressing colon cancer. T cells recognized erb-b2 receptor tyrosine kinase 2 (ERBB2) on normal lung cells and triggered high levels of tumor necrosis factor-alpha (TNF-α) and IFN-γ, pulmonary toxicity, and death [118]. Another study transferred CEA-specific T cells retrovirally transduced to express murine TCRs; although one patient demonstrated regression of lung and liver metastases, all patients demonstrated severe transient inflammatory colitis [119].
These studies highlight how ubiquitously expressed self-antigens in the lungs and throughout the gastrointestinal tract are risky targets for immunotherapy. Future efforts need to consider the perils of targeting self-antigens or at least weigh clinical benefit against the consequences of treatment.
Lymphodepletion
Suppressor cells, as well as endogenous lymphocytes, effectively add another layer of immunosuppression by serving as sinks for T cell-promoting cytokines IL-7 and IL-15 [120]. Therefore, increased access to homeostatic cytokines may be critical for successful ACT. Lymphodepleting chemotherapy prior to immunotherapy reduces the number of circulating Tregs, promotes successful engraftment of adoptively transferred T cells, and improves the overall survival of melanoma patients [121]. Cyclophosphamide decreased peripheral FoxP3+ Tregs in all six metastatic CRC patients and increased IFN-γ-producing T cells after 22 days [122]. Local ablation of immune cells using fludarabine, cyclophosphamide, and/or temozomide may dramatically increase the durability of ACT by reducing immunosuppressive pressures and widening a niche for tumor-specific T cells. This treatment may best suit tumors devoid of beneficial CD8+ T cells, and not mismatch repair defective CRCs, which are highly infiltrated by CD8+ T cells.
Allogeneic CD34+ Stem Cell Transplantation
Allogeneic CD34+ stem cell transplantation (allo-SCT) is common for treating blood-related diseases; however, early attempts against CRCs have achieved mild success [123, 124]. A preparative regimen of lymphodepleting drugs is administered before matched donor hematopoietic stem cells (HSCs) are transferred to cancer patients. The ensuing curative graft-versus-tumor (GvT) effect is driven by donor cytotoxic T cells that proliferate in vivo and target malignancies [125]. Injecting 111-indium-labeled lymphocytes via the hepatic portal artery confirmed that donor CD3+, CD19+, and CD56+ lymphocytes home to liver metastases [126].
Successful allo-SCT requires local cytokine production. Neutralization of TNF-α and IL-1β in target epithelium inhibits acute GvT effects in mice [127], suggesting cytokine-mediated cytotoxicity obviates multiple layers of immunosuppression. One allo-SCT patient with CRC demonstrated increased tumoral expression of HLA-class I-associated β2-microglobulin molecule—an indicator of CD8+ T cell activity—but not severe enough for any meaningful clinical benefit. Donor cytotoxic T cells, however, inadvertently targeted a recipient’s lymphoid system [128]. Three out of 15 metastatic colon cancer patients receiving allo-SCT experienced disease stabilization or partial remission. Responders harbored intra-tumoral CEA-specific CD8+ T cells [129]. Interestingly, patients receiving HSCs from unrelated donors engrafted CD3+ cells faster than those receiving HLA-identical HSCs [130], suggesting non-perfect matching may induce more aggressive GvT effects.
This approach has many obstacles. First, complete T cell engraftment lags behind myeloid and B cells and can take over 60 days. Fortunately, patients who fail engraftment can receive infusion of CD3+ T cells [131]. Second, an overwhelming majority of patients who benefit from engraftment eventually succumb to disease, suggesting allo-SCT fades or becomes immunosuppressed by the tumor environment. Third, a study unrelated to gastrointestinal cancer observed striking upregulation of IDO activity in colon tissues of patients receiving allo-SCT. This is expected because subsequent tryptophan depletion hallmarks intense gut inflammation [132]. Therefore, therapeutic inhibition of IDO activity during allo-SCT must be explored. And fourth, the number of T cells generated after transplant is limited; large and advanced tumors may require an absurd number of generated T cells.
Anti-tumor Vaccines
The ideal vaccine is easy to administer, offers prolonged protection, and induces relatively low toxicity; however, no vaccine has induced reproducible clinically relevant regression of metastatic colorectal cancer (mCRC). Induction of memory T cells activates the anti-tumor cascade and provides prolonged protection against existing micrometastases [133]. Cancer vaccines must effectively break immunological tolerance and induce or amplify antigen-directed T cell assaults.
Initial efforts to break tolerance to CRC antigens were directed against CEA. When delivered by DNA vaccine, or “naked” plasmid DNA, CEA is presented by MHC class I/CTL pathways [134]. A clinical trial did not detect relevant CEA-specific antibody responses in all 17 metastatic CRC patients, yet 4 patients demonstrated proliferation of peripheral lymphocytes [134]. This proliferation, however, was most likely triggered by CEA expressed by normal tissues. Overcoming tolerance to self-antigens is challenging, especially in profusely immunosuppressive environments, and requires adjuvants to intensify vaccination. Directing an immunological attack against self-antigens while ignoring healthy tissues is fraught with many unidentified obstacles.
Pathogen-Derived Adjuvants
Bacterial or viral immunopotentiators can break inherent tolerance to self-antigens. Diphtheria toxin (DT) conjugated to beta-human chorionic gonadotropin (β-hCG) peptide—often expressed by CRCs—induced humoral immune responses in 73 % of IV CRC patients. Higher antibody responses to β-hCG associated with increased survival; however, patients who mounted stronger antibody responses to DT antigen did not benefit from treatment [135]. Another study treated 161 patients with DT conjugated to gastrin-17 (G-17), a growth factor that contributes to gastrointestinal tumor growth. Three percent of patients achieved partial responses while 32 % achieved stable disease, and those who generated antibodies to G-17 survive longer [136]. A promising study utilized adenovirus serotype-5 (Ad5)—known to trigger robust T cell responses—to deliver CEA. This vaccine induced cell-mediated T cell responses in 61 % of advanced CRC patients; however, the small study size could not conclude any survival advantage [137].
Alternatively, immune responses can be directed against tumor antigens by combining irradiated autologous tumor cells with pathogen-derived adjuvants. Colon cancer patients survived longer when given autologous tumor cell-bacillus Calmette-Guérin (BCG) vaccines [138]. A trial consisting of 254 colon cancer patients reported that benefits were limited to stage II disease [139]. Two other studies used Newcastle disease virus (NDV) as an adjuvant because viral challenge induces production of interferons and CCL5 and encourages chemotaxis of cytotoxic lymphocytes. Both studies boasted delayed disease reoccurrence and improved survival for colon cancer patients [140, 141]. No explanation was given to why rectal cancer patients did not benefit from either BCG or NDV. These studies strongly suggest that both the route of antigen presentation and the condition of immunostimulation dictate the efficacy of anti-cancer vaccines. These principles were applied to the next generation of vaccines.
Cytokine Adjuvants
Specific cytokines activate the adaptive immune response and establish T cell memory. Among multiple signaling layers of antigen presentation and coreceptor ligation, cytokines mobilize immune cells. Granulocyte-macrophage colony-stimulating factor (GM-CSF)—an immune stimulatory cytokine—enhances proliferation of T cells and IFN-γ and IL-2 secretion in multiple myeloma patients immunized with autologous myeloma immunogen [142]. GM-CSF enhanced antibody response to CEA antigen and protected against metastatic CRC. All nine patients subcutaneously immunized and treated with GM-CSF demonstrated stronger dose-dependent IgG antibody responses, T cell proliferation, and IFN-γ secretion when compared to those not given GM-CSF [143]. Another study inserted epithelial cellular adhesion molecule (Ep-CAM)—commonly expressed on CRC cells—into replicative-deficient recombinant avipoxvirus to ensure the antigen is presented through MHC class I and CTL pathway. All six patients received GM-CSF, and five generated Ep-CAM-specific IFN-γ+ T cells and no IL-4 secreting T cells [144].
Dendritic Cell Vaccines
The previous studies suggest that successful vaccination against tumor antigens may be dependent on GM-CSF-mobilized immune cells. Introduced earlier in this review, DCs reside in dermal tissues and lymph nodes, present antigens, induce protective T cell responses, and reprogram immunosuppression in the tumor environment and elsewhere in the body. Early attempts to direct a memory response against antigen-presenting tumor cells using DC-based vaccines have been promising [133]. DCs require harvesting, propagation, and specific ex vivo manipulation to be coaxed into effective primers of type-1 T cells [145].
A series of clinical trials successfully activated CTL responses by vaccinating CRC patients with antigen-loaded DCs. Patients with metastatic tumors receiving CEA-pulsed GM-CSF/IL-4-derived DCs (GM/IL4-DCs) demonstrated no signs of autoimmune disease or acute toxicities [146]. Other studies used alternative means to generate DCs with similar results: Flt3 ligand [147] and IL-13 [148]. Either way, the majority of recent DC vaccines are generated using GM-CSF, IL-4, and CEA peptide [149–152].
Theoretically, antigen loading via messenger RNA (mRNA) introduces a broader range of antigenic epitopes than peptide loading, thereby generating a more potent T cell repertoire. In contrast, however, mRNA-transfected GM/IL4-DCs did not produce CEA-reactive T cells in CRC patients [153]. Regardless of the loading method, GM/IL4-DCs with CEA class I HLA-A2-restricted peptide, CAP-1, or with mRNA encoding CEA triggered similar antigen-specific T cell activities in vitro [154].
The method of antigen loading may not primarily dictate effective DC vaccination. Inactivated DCs remain ineffective at promoting protective T cells and can induce tolerance to target antigens [155]. Immunopotentiators optimize DC-based vaccines and enhance production of the type-1 T cell mediator IL-12. These include Toll-like receptor agonists poly I:C, LPS, Pam3Cys, and R848 [156–158]. The final study to be discussed in this review designed a vaccine with hyperstimulatory potential by loading DCs with a poxvector containing genes for CEA and other costimulatory molecules, including CD80. Amazingly, this vaccine induced CEA-specific immune T cells in 10 out of 12 patients [159]. The clinical studies mentioned in this review are summarized in Table 3.
Combined, these studies fortify the assumptions that (1) naturally processed peptides via class I MHC to CTLs are sufficient for T cell priming, (2) DCs can be generated with effective stimulating capacity through multiple means, and (3) properly activated DCs induce protective immunity and avoid inducing tolerance to tumor antigens. DC vaccines are efficient at generating tumor-specific CD8+ T cells in vivo; however, the question still remains to whether these are effective at countering the multiple layers of immunosuppression inherent in the tumor environment.
Conclusion
The population of colorectal tumors is heterogeneous in regard to immune cell infiltrates. This explains why directly manipulating only one immune mechanism limits success. Most studies described in this review administered experimental treatments to groups of mCRCs only to witness diseases relapse. Confounding treatment, particular mutations within cancer genes distinctly construe different tumor subtypes with unique histology and gene expression. Antibody therapy can only unlock one layer of suppression at a time, CAR T cell therapy targets only one antigen, and DC-based vaccines activate CTLs that eventually become muted once migrated to the tumor. This Sisyphean cycle of trail and failure mainly proves that tumor environments eventually return to an immunosuppressive state. Improved perception of the multiple immunosuppressive forces interwoven throughout each tumor environment, as seen in mismatch repair defective tumors [107••], will facilitate progress of experimental immunotherapies past the trial stage. Effective immunotherapy needs to counter all layers of immunosuppression; therefore, patients may benefit from combinational therapy, barring severe toxicities (Fig. 2).

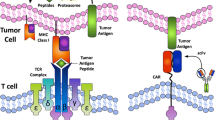
Immunotherapies used to treat metastatic CRC. Immunotherapies designed to direct cytotoxic T cells to eradicate colorectal tumor cells. Antibody therapy against CRC has two popular targets. Anti-CTLA-4 (purple) liberates CD28 to ligate to CD80/CD86 for CTL (red) activation. Anti-PD-1 (purple) prevents tumor cells (gray) and DCs (violet) from inducing tolerance, unresponsiveness, and exhaustion of T cell activities. Adoptive cell transfer requires autologous peripheral or sentinel lymph node T cells (T H 1-cells, blue) to be isolated and propagated in vitro via IL-2 and antigen (red curved bar) stimulation, then delivered back into the patient. Allo-SCT requires introducing HSCs (mustard) into cancer patients. HSCs mature into lymphoid progenitors (PL, green) and then into functional T cells (and other lymphocytes) to induce a GvT effect. This effect requires donor CTLs to proliferate in vivo and target malignancies. DC vaccines program the immune system to mount a durable memory response against tumor antigens. DCs are derived ex vivo from peripheral immune cells precursor, then propagated and challenged with tumor antigens. Mature and activated DCs are delivered back into the patient and migrate to the lymph nodes to prime and educate naive T cells against tumor antigens. In the lymph node, primed TH1 cells release IFN-γ (dark green) to activate CD8+ T cells. Once mature, CTLs migrate to tumor cells and release cytotoxic mediators (dark red) upon recognition of antigen-MHC complex (light green) on the tumor cell surface
Currently, chemotherapy may provide a better option for altering the tumor environment. Major findings are limited to mouse studies; however, MDSCs are inhibited by 5-fluorouracil, which relinquishes its suppressive effects on T cells [160]. In contrast, irinotecan—a topoisomerase inhibitor [161] used in a cocktail with 5-fluorouracil and leucovorin called FOLFIRI—reverses the inhibitory effects of 5-fluorouracil on MDSCs [162•]. Therefore, the specific effects of combinational chemotherapy on the tumor environment should be fully understood to provide immunotherapies the potency to abrogate all suppressive barriers. Effective immunotherapy must reprogram the tumor environment by disrupting either immune cells that induce tolerance or stromal and tumor cells that anchor immunosuppression.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015.
Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, et al. Colorectal cancer. Lancet. 2010;375(9719):1030–47.
Steele Jr G, Ravikumar TS. Resection of hepatic metastases from colorectal cancer. Biologic perspective. Ann Surg. 1989;210(2):127–38.
Fong Y, Cohen AM, Fortner JG, Enker WE, Turnbull AD, Coit DG, et al. Liver resection for colorectal metastases. J Clin Oncol. 1997;15(3):938–46.
Van Cutsem E, Nordlinger B, Adam R, Kohne CH, Pozzo C, Poston G, et al. Towards a pan-European consensus on the treatment of patients with colorectal liver metastases. Eur J Cancer. 2006;42(14):2212–21.
Kahn KL, Adams JL, Weeks JC, Chrischilles EA, Schrag D, Ayanian JZ, et al. Adjuvant chemotherapy use and adverse events among older patients with stage III colon cancer. JAMA. 2010;303(11):1037–45.
Ogi C, Aruga A. Immunological monitoring of anticancer vaccines in clinical trials. Oncoimmunology. 2013;2(8):e26012.
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9.
Jass JR. Lymphocytic infiltration and survival in rectal cancer. J Clin Pathol. 1986;39(6):585–9.
Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Chiba T, Ohtani H, Mizoi T, Naito Y, Sato E, Nagura H, et al. Intraepithelial CD8+ T-cell-count becomes a prognostic factor after a longer follow-up period in human colorectal carcinoma: possible association with suppression of micrometastasis. Br J Cancer. 2004;91(9):1711–7.
Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–66.
Pages F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol. 2009;27(35):5944–51.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4.
Koelzer VH, Lugli A, Dawson H, Hadrich M, Berger MD, Borner M, et al. CD8/CD45RO T-cell infiltration in endoscopic biopsies of colorectal cancer predicts nodal metastasis and survival. J Transl Med. 2014;12:81,5876-12-81.
Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29(6):610–8.
Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–48.
Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76(2):287–99.
Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101.
Street NE, Mosmann TR. Functional diversity of T lymphocytes due to secretion of different cytokine patterns. FASEB J. 1991;5(2):171–7.
Rousalova I, Krepela E. Granzyme B-induced apoptosis in cancer cells and its regulation (review). Int J Oncol. 2010;37(6):1361–78.
Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–58.
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–50.
Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138(4):1429–40.
Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187(1):129–34.
Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187(6):875–83.
Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187(12):2009–21.
Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2):207–15.
Musha H, Ohtani H, Mizoi T, Kinouchi M, Nakayama T, Shiiba K, et al. Selective infiltration of CCR5(+)CXCR3(+) T lymphocytes in human colorectal carcinoma. Int J Cancer. 2005;116(6):949–56.
Zimmermann T, Moehler M, Gockel I, Sgourakis GG, Biesterfeld S, Muller M, et al. Low expression of chemokine receptor CCR5 in human colorectal cancer correlates with lymphatic dissemination and reduced CD8+ T-cell infiltration. Int J Color Dis. 2010;25(4):417–24.
Schimanski CC, Moehler M, Gockel I, Zimmermann T, Lang H, Galle PR, et al. Expression of chemokine receptor CCR5 correlates with the presence of hepatic molecular metastases in K-ras positive human colorectal cancer. J Cancer Res Clin Oncol. 2011;137(7):1139–45.
Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996;35(11):3362–7.
Catalfamo M, Karpova T, McNally J, Costes SV, Lockett SJ, Bos E, et al. Human CD8+ T cells store RANTES in a unique secretory compartment and release it rapidly after TcR stimulation. Immunity. 2004;20(2):219–30.
Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347(6294):669–71.
Zumwalt TJ, Arnold M, Goel A, Boland CR. Active secretion of CXCL10 and CCL5 from colorectal cancer microenvironments associates with GranzymeB+ CD8+ T-cell infiltration. Oncotarget. 2015;6(5):2981–91.
Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Sato H, et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Valpha14 NKT cells. Proc Natl Acad Sci U S A. 1998;95(10):5690–3.
Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, et al. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol. 1996;14:619–48.
Sconocchia G, Arriga R, Tornillo L, Terracciano L, Ferrone S, Spagnoli GC. Melanoma cells inhibit NK cell functions. Cancer Res. 2012;72(20):5428,9; author reply 5430.
Benevolo M, Mottolese M, Tremante E, Rollo F, Diodoro MG, Ercolani C, et al. High expression of HLA-E in colorectal carcinoma is associated with a favorable prognosis. J Transl Med. 2011;9:184,5876-9-184.
Pugh SA, Harrison RJ, Primrose JN, Khakoo SI. T cells but not NK cells are associated with a favourable outcome for resected colorectal liver metastases. BMC Cancer. 2014;14:180,2407-14-180.
Papanikolaou IS, Lazaris AC, Apostolopoulos P, Kavantzas N, Papas MG, Mavrogiannis C, et al. Tissue detection of natural killer cells in colorectal adenocarcinoma. BMC Gastroenterol. 2004;4:20.
Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. 2011;17(4):678–89.
Sconocchia G, Zlobec I, Lugli A, Calabrese D, Iezzi G, Karamitopoulou E, et al. Tumor infiltration by FcgammaRIII (CD16) + myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer. 2011;128(11):2663–72.
Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222(4):350–66.
Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59(7):926–33.
Guidoboni M, Gafa R, Viel A, Doglioni C, Russo A, Santini A, et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathol. 2001;159(1):297–304.
Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology. 2009;137(4):1270–9.
Ropponen KM, Eskelinen MJ, Lipponen PK, Alhava E, Kosma VM. Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer. J Pathol. 1997;182(3):318–24.
Huh JW, Lee JH, Kim HR. Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch Surg. 2012;147(4):366–72.
Chang EY, Dorsey PB, Frankhouse J, Lee RG, Walts D, Johnson W, et al. Combination of microsatellite instability and lymphocytic infiltrate as a prognostic indicator in colon cancer. Arch Surg. 2009;144(6):511–5.
Dahlin AM, Henriksson ML, Van Guelpen B, Stenling R, Oberg A, Rutegard J, et al. Colorectal cancer prognosis depends on T-cell infiltration and molecular characteristics of the tumor. Mod Pathol. 2011;24(5):671–82.
Morris M, Platell C, Iacopetta B. Tumor-infiltrating lymphocytes and perforation in colon cancer predict positive response to 5-fluorouracil chemotherapy. Clin Cancer Res. 2008;14(5):1413–7.
Zlobec I, Karamitopoulou E, Terracciano L, Piscuoglio S, Iezzi G, Muraro MG, et al. TIA-1 cytotoxic granule-associated RNA binding protein improves the prognostic performance of CD8 in mismatch repair-proficient colorectal cancer. PLoS ONE. 2010;5(12):e14282.
Anitei MG, Zeitoun G, Mlecnik B, Marliot F, Haicheur N, Todosi AM, et al. Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin Cancer Res. 2014 Apr 1;20(7):1891–9. The authors demonstrate that evaluating densities of CD3 + and CD8 + lymphocytes more accurately predicts recurrence and survival than TNM staging for rectal cancer patients.
Menon AG, Janssen-van Rhijn CM, Morreau H, Putter H, Tollenaar RA, van de Velde CJ, et al. Immune system and prognosis in colorectal cancer: a detailed immunohistochemical analysis. Lab Invest. 2004;84(4):493–501.
Suzuki H, Chikazawa N, Tasaka T, Wada J, Yamasaki A, Kitaura Y, et al. Intratumoral CD8(+) T/FOXP3 (+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol Immunother. 2010;59(5):653–61.
Correale P, Rotundo MS, Botta C, Del Vecchio MT, Tassone P, Tagliaferri P. Tumor infiltration by chemokine receptor 7 (CCR7)(+) T-lymphocytes is a favorable prognostic factor in metastatic colorectal cancer. Oncoimmunology. 2012;1(4):531–2.
Mlecnik B, Bindea G, Pages F, Galon J. Tumor immunosurveillance in human cancers. Cancer Metastasis Rev. 2011;30(1):5–12.
Lugli A, Karamitopoulou E, Panayiotides I, Karakitsos P, Rallis G, Peros G, et al. CD8+ lymphocytes/ tumour-budding index: an independent prognostic factor representing a ‘pro-/anti-tumour’ approach to tumour host interaction in colorectal cancer. Br J Cancer. 2009;101(8):1382–92.
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–4.
Yoon HH, Orrock JM, Foster NR, Sargent DJ, Smyrk TC, Sinicrope FA. Prognostic impact of FoxP3+ regulatory T cells in relation to CD8+ T lymphocyte density in human colon carcinomas. PLoS ONE. 2012;7(8):e42274.
Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27(2):186–92.
Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer. 2010;126(11):2635–43.
Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother. 2010;33(4):435–41.
Kim M, Grimmig T, Grimm M, Lazariotou M, Meier E, Rosenwald A, et al. Expression of Foxp3 in colorectal cancer but not in Treg cells correlates with disease progression in patients with colorectal cancer. PLoS ONE. 2013;8(1):e53630.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182:207–14.
Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389(6652):737–42.
Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–10.
Murakami M, Sakamoto A, Bender J, Kappler J, Marrack P. CD25+CD4+ T cells contribute to the control of memory CD8+ T cells. Proc Natl Acad Sci U S A. 2002;99(13):8832–7.
Clarke SL, Betts GJ, Plant A, Wright KL, El-Shanawany TM, Harrop R, et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE. 2006;1:e129.
Golgher D, Jones E, Powrie F, Elliott T, Gallimore A. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol. 2002;32(11):3267–75.
de Leeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18(11):3022–9.
Loddenkemper C, Schernus M, Noutsias M, Stein H, Thiel E, Nagorsen D. In situ analysis of FOXP3+ regulatory T cells in human colorectal cancer. J Transl Med. 2006;4:52.
Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F. Human FOXP3 and cancer. Oncogene. 2010;29(29):4121–9.
Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother. 2011;60(7):909–18.
Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med. 2011;9:216,5876-9-216.
Ong SM, Tan YC, Beretta O, Jiang D, Yeap WH, Tai JJ, et al. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur J Immunol. 2012;42(1):89–100.
Bauer K, Michel S, Reuschenbach M, Nelius N, von Knebel Doeberitz M, Kloor M. Dendritic cell and macrophage infiltration in microsatellite-unstable and microsatellite-stable colorectal cancer. Fam Cancer. 2011;10(3):557–65.
Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res. 2002;62(23):7042–9.
Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11(10):702–11.
Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS ONE. 2013;8(2):e57114.
Sun HL, Zhou X, Xue YF, Wang K, Shen YF, Mao JJ, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012;18(25):3303–9.
Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–21.
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–99.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–90.
Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13(5):1472–9.
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003.
Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137(5):1142–62.
Palucka AK, Ueno H, Fay JW, Banchereau J. Taming cancer by inducing immunity via dendritic cells. Immunol Rev. 2007;220:129–50.
Li D, Romain G, Flamar AL, Duluc D, Dullaers M, Li XH, et al. Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10-producing suppressive CD4+ T cells. J Exp Med. 2012;209(1):109–21.
Taylor CR, Levenson RM. Quantification of immunohistochemistry—issues concerning methods, utility and semiquantitative assessment II. Histopathology. 2006;49(4):411–24.
Schwaab T, Weiss JE, Schned AR, Barth Jr RJ. Dendritic cell infiltration in colon cancer. J Immunother. 2001;24(2):130–7.
Dadabayev AR, Sandel MH, Menon AG, Morreau H, Melief CJ, Offringa R, et al. Dendritic cells in colorectal cancer correlate with other tumor-infiltrating immune cells. Cancer Immunol Immunother. 2004;53(11):978–86.
Yuan A, Steigen SE, Goll R, Vonen B, Husbekk A, Cui G, et al. Dendritic cell infiltration pattern along the colorectal adenoma-carcinoma sequence. APMIS. 2008;116(6):445–56.
Wu X, Zhang H, Xing Q, Cui J, Li J, Li Y, et al. PD-1(+) CD8(+) T cells are exhausted in tumours and functional in draining lymph nodes of colorectal cancer patients. Br J Cancer. 2014;111(7):1391–9.
Scurr M, Ladell K, Besneux M, Christian A, Hockey T, Smart K, et al. Highly prevalent colorectal cancer-infiltrating LAP(+) Foxp3(−) T cells exhibit more potent immunosuppressive activity than Foxp3(+) regulatory T cells. Mucosal Immunol. 2014;7(2):428–39. By screening for intra-tumoral regulatory T cells, the authors identified these cells to express CTLA-4 and LAG-3 and secrete TFG-β and IL-10.
Frumento G, Rotondo R, Tonetti M, Ferrara GB. T cell proliferation is blocked by indoleamine 2,3-dioxygenase. Transplant Proc. 2001;33(1–2):428–30.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
O’Mahony D, Morris JC, Quinn C, Gao W, Wilson WH, Gause B, et al. A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin Cancer Res. 2007;13(3):958–64.
Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010;28(21):3485–90.
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–75.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Llosa NJ, Cruise M, Tam A, Wick EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2014. By screening for immune checkpoints in CRCs, the authors suggest that active T H 1/CTL activities are countered by immunosuppressive barriers.
Chen J, Chen Z. The effect of immune microenvironment on the progression and prognosis of colorectal cancer. Med Oncol. 2014;31(8):82,014-0082-9. Epub 2014 Jul 18.
Prigent P, El Mir S, Dreano M, Triebel F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur J Immunol. 1999;29(12):3867–76.
Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. 2006;12(4):1144–51.
Satoh K, Kan N, Okino T, Mise K, Yamasaki S, Harada T, et al. The therapeutic effect of OK-432-combined adoptive immunotherapy against liver metastases from gastric or colorectal cancers. Biotherapy. 1993;6(1):41–9.
Gardini A, Ercolani G, Riccobon A, Ravaioli M, Ridolfi L, Flamini E, et al. Adjuvant, adoptive immunotherapy with tumor infiltrating lymphocytes plus interleukin-2 after radical hepatic resection for colorectal liver metastases: 5-year analysis. J Surg Oncol. 2004;87(1):46–52.
Kono K, Ichihara F, Iizuka H, Sekikawa T, Matsumoto Y. Expression of signal transducing T-cell receptor zeta molecules after adoptive immunotherapy in patients with gastric and colon cancer. Int J Cancer. 1998;78(3):301–5.
Karlsson M, Marits P, Dahl K, Dagoo T, Enerback S, Thorn M, et al. Pilot study of sentinel-node-based adoptive immunotherapy in advanced colorectal cancer. Ann Surg Oncol. 2010;17(7):1747–57.
Marits P, Karlsson M, Dahl K, Larsson P, Wanders A, Thorn M, et al. Sentinel node lymphocytes: tumour reactive lymphocytes identified intraoperatively for the use in immunotherapy of colon cancer. Br J Cancer. 2006;94(10):1478–84.
Sheen AJ, Sherlock DJ, Irlam J, Hawkins RE, Gilham DE. T lymphocytes isolated from patients with advanced colorectal cancer are suitable for gene immunotherapy approaches. Br J Cancer. 2003;88(7):1119–27.
Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res. 2001;83:117–58.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–51.
Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–6.
Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–9.
Ridolfi L, Petrini M, Granato AM, Gentilcore G, Simeone E, Ascierto PA, et al. Low-dose temozolomide before dendritic-cell vaccination reduces (specifically) CD4+CD25++Foxp3+ regulatory T-cells in advanced melanoma patients. J Transl Med. 2013;11:135,5876-11-135.
Scurr M, Bloom A, Pembroke T, Srinivasan R, Brown C, Smart K, et al. Escalating regulation of 5T4-specific IFN-gamma CD4 T cells distinguishes colorectal cancer patients from healthy controls and provides a target for therapy. Cancer Immunol Res. 2013;1(6):10.1158/2326,6066.CIR-13-0035.
Kojima R, Kami M, Hori A, Murashige N, Ohnishi M, Kim SW, et al. Reduced-intensity allogeneic hematopoietic stem-cell transplantation as an immunotherapy for metastatic colorectal cancer. Transplantation. 2004;78(12):1740–6.
Hashino S, Kobayashi S, Takahata M, Onozawa M, Nakagawa M, Kawamura T, et al. Graft-versus-tumor effect after reduced-intensity allogeneic hematopoietic stem cell transplantation in a patient with advanced colon cancer. Int J Clin Oncol. 2008;13(2):176–80.
Nishida T, Hudecek M, Kostic A, Bleakley M, Warren EH, Maloney D, et al. Development of tumor-reactive T cells after nonmyeloablative allogeneic hematopoietic stem cell transplant for chronic lymphocytic leukemia. Clin Cancer Res. 2009;15(14):4759–68.
Barkholt L, Danielsson R, Calissendorff B, Svensson L, Malihi R, Remberger M, et al. Indium-111-labelled donor-lymphocyte infusion by way of hepatic artery and radio-frequency ablation against liver metastases of renal and colon carcinoma after allogeneic hematopoietic stem-cell transplantation. Transplantation. 2004;78(5):697–703.
Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8(6):575–81.
Zetterquist H, Hentschke P, Thorne A, Wernerson A, Mattsson J, Uzunel M, et al. A graft-versus-colonic cancer effect of allogeneic stem cell transplantation. Bone Marrow Transplant. 2001;28(12):1161–6.
Carnevale-Schianca F, Cignetti A, Capaldi A, Vitaggio K, Vallario A, Ricchiardi A, et al. Allogeneic nonmyeloablative hematopoietic cell transplantation in metastatic colon cancer: tumor-specific T cells directed to a tumor-associated antigen are generated in vivo during GVHD. Blood. 2006;107(9):3795–803.
Hentschke P, Barkholt L, Uzunel M, Mattsson J, Wersall P, Pisa P, et al. Low-intensity conditioning and hematopoietic stem cell transplantation in patients with renal and colon carcinoma. Bone Marrow Transplant. 2003;31(4):253–61.
Aglietta M, Barkholt L, Schianca FC, Caravelli D, Omazic B, Minotto C, et al. Reduced-intensity allogeneic hematopoietic stem cell transplantation in metastatic colorectal cancer as a novel adoptive cell therapy approach. The European group for blood and marrow transplantation experience. Biol Blood Marrow Transplant. 2009;15(3):326–35.
Park G, Choi YJ, Lee SE, Lim JY, Lee C, Choi EY, et al. A paradoxical pattern of indoleamine 2,3-dioxygenase expression in the colon tissues of patients with acute graft-versus-host disease. Exp Hematol. 2014;42(9):734–40.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–77.
Conry RM, Curiel DT, Strong TV, Moore SE, Allen KO, Barlow DL, et al. Safety and immunogenicity of a DNA vaccine encoding carcinoembryonic antigen and hepatitis B surface antigen in colorectal carcinoma patients. Clin Cancer Res. 2002;8(9):2782–7.
Moulton HM, Yoshihara PH, Mason DH, Iversen PL, Triozzi PL. Active specific immunotherapy with a beta-human chorionic gonadotropin peptide vaccine in patients with metastatic colorectal cancer: antibody response is associated with improved survival. Clin Cancer Res. 2002;8(7):2044–51.
Rocha-Lima CM, de Queiroz Marques Junior E, Bayraktar S, Broome P, Weissman C, Nowacki M, et al. A multicenter phase II study of G17DT immunogen plus irinotecan in pretreated metastatic colorectal cancer progressing on irinotecan. Cancer Chemother Pharmacol. 2014;74(3):479–86.
Morse MA, Chaudhry A, Gabitzsch ES, Hobeika AC, Osada T, Clay TM, et al. Novel adenoviral vector induces T-cell responses despite anti-adenoviral neutralizing antibodies in colorectal cancer patients. Cancer Immunol Immunother. 2013;62(8):1293–301.
Hoover Jr HC, Brandhorst JS, Peters LC, Surdyke MG, Takeshita Y, Madariaga J, et al. Adjuvant active specific immunotherapy for human colorectal cancer: 6.5-year median follow-up of a phase III prospectively randomized trial. J Clin Oncol. 1993;11(3):390–9.
Vermorken JB, Claessen AM, van Tinteren H, Gall HE, Ezinga R, Meijer S, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. Lancet. 1999;353(9150):345–50.
Schlag P, Manasterski M, Gerneth T, Hohenberger P, Dueck M, Herfarth C, et al. Active specific immunotherapy with Newcastle-disease-virus-modified autologous tumor cells following resection of liver metastases in colorectal cancer. First evaluation of clinical response of a phase II-trial. Cancer Immunol Immunother. 1992;35(5):325–30.
Schulze T, Kemmner W, Weitz J, Wernecke KD, Schirrmacher V, Schlag PM. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: results of a prospective randomized trial. Cancer Immunol Immunother. 2009;58(1):61–9.
Osterborg A, Yi Q, Henriksson L, Fagerberg J, Bergenbrant S, Jeddi-Tehrani M, et al. Idiotype immunization combined with granulocyte-macrophage colony-stimulating factor in myeloma patients induced type I, major histocompatibility complex-restricted, CD8- and CD4-specific T-cell responses. Blood. 1998;91(7):2459–66.
Samanci A, Yi Q, Fagerberg J, Strigard K, Smith G, Ruden U, et al. Pharmacological administration of granulocyte/macrophage-colony-stimulating factor is of significant importance for the induction of a strong humoral and cellular response in patients immunized with recombinant carcinoembryonic antigen. Cancer Immunol Immunother. 1998;47(3):131–42.
Ullenhag GJ, Frodin JE, Mosolits S, Kiaii S, Hassan M, Bonnet MC, et al. Immunization of colorectal carcinoma patients with a recombinant canarypox virus expressing the tumor antigen ep-CAM/KSA (ALVAC-KSA) and granulocyte macrophage colony-stimulating factor induced a tumor-specific cellular immune response. Clin Cancer Res. 2003;9(7):2447–56.
von Mehren M. Colorectal cancer vaccines: what we know and what we don’t yet know. Semin Oncol. 2005;32(1):76–84.
Morse MA, Deng Y, Coleman D, Hull S, Kitrell-Fisher E, Nair S, et al. A phase I study of active immunotherapy with carcinoembryonic antigen peptide (CAP-1)-pulsed, autologous human cultured dendritic cells in patients with metastatic malignancies expressing carcinoembryonic antigen. Clin Cancer Res. 1999;5(6):1331–8.
Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A. 2001;98(15):8809–14.
Kavanagh B, Ko A, Venook A, Margolin K, Zeh H, Lotze M, et al. Vaccination of metastatic colorectal cancer patients with matured dendritic cells loaded with multiple major histocompatibility complex class I peptides. J Immunother. 2007;30(7):762–72.
Babatz J, Rollig C, Lobel B, Folprecht G, Haack M, Gunther H, et al. Induction of cellular immune responses against carcinoembryonic antigen in patients with metastatic tumors after vaccination with altered peptide ligand-loaded dendritic cells. Cancer Immunol Immunother. 2006;55(3):268–76.
Liu KJ, Wang CC, Chen LT, Cheng AL, Lin DT, Wu YC, et al. Generation of carcinoembryonic antigen (CEA)-specific T-cell responses in HLA-A*0201 and HLA-A*2402 late-stage colorectal cancer patients after vaccination with dendritic cells loaded with CEA peptides. Clin Cancer Res. 2004;10(8):2645–51.
Itoh T, Ueda Y, Kawashima I, Nukaya I, Fujiwara H, Fuji N, et al. Immunotherapy of solid cancer using dendritic cells pulsed with the HLA-A24-restricted peptide of carcinoembryonic antigen. Cancer Immunol Immunother. 2002;51(2):99–106.
Lesterhuis WJ, de Vries IJ, Aarntzen EA, de Boer A, Scharenborg NM, van de Rakt M, et al. A pilot study on the immunogenicity of dendritic cell vaccination during adjuvant oxaliplatin/capecitabine chemotherapy in colon cancer patients. Br J Cancer. 2010;103(9):1415–21.
Lesterhuis WJ, De Vries IJ, Schreibelt G, Schuurhuis DH, Aarntzen EH, De Boer A, et al. Immunogenicity of dendritic cells pulsed with CEA peptide or transfected with CEA mRNA for vaccination of colorectal cancer patients. Anticancer Res. 2010;30(12):5091–7.
Nair SK, Hull S, Coleman D, Gilboa E, Lyerly HK, Morse MA. Induction of carcinoembryonic antigen (CEA)-specific cytotoxic T-lymphocyte responses in vitro using autologous dendritic cells loaded with CEA peptide or CEA RNA in patients with metastatic malignancies expressing CEA. Int J Cancer. 1999;82(1):121–4.
Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449(7161):419–26.
Lim SN, Kuhn S, Hyde E, Ronchese F. Combined TLR stimulation with Pam3Cys and poly I: C enhances Flt3-ligand dendritic cell activation for tumor immunotherapy. J Immunother. 2012;35(9):670–9.
Nourizadeh M, Masoumi F, Memarian A, Alimoghaddam K, Moazzeni SM, Yaghmaie M, et al. In vitro induction of potent tumor-specific cytotoxic T lymphocytes using TLR agonist-activated AML-DC. Target Oncol. 2014;9(3):225–37.
Brosbol-Ravnborg A, Bundgaard B, Hollsberg P. Synergy between vitamin D(3) and toll-like receptor agonists regulates human dendritic cell response during maturation. Clin Dev Immunol. 2013;2013:807971.
Morse MA, Clay TM, Hobeika AC, Osada T, Khan S, Chui S, et al. Phase I study of immunization with dendritic cells modified with fowlpox encoding carcinoembryonic antigen and costimulatory molecules. Clin Cancer Res. 2005;11(8):3017–24.
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–61.
Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17(5):421–33.
Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, et al. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014;74(21):6022–35. The authors demonstrate that CP11 encourages detrimental immune regulation that counters the benefits of 5FU.
Grant Funding
This work was supported by grants R01 CA72851, CA181572 and U01 CA187956 from the National Cancer Institute, National Institutes of Health, pilot grant from Charles A Sammons Cancer Center, and funds from the Baylor Research Institute to AG.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Timothy J. Zumwalt and Ajay Goel declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Therapeutic Approaches to Metastatic Colorectal Cancers
Rights and permissions
About this article

Cite this article
Zumwalt, T.J., Goel, A. Immunotherapy of Metastatic Colorectal Cancer: Prevailing Challenges and New Perspectives. Curr Colorectal Cancer Rep 11, 125–140 (2015). https://doi.org/10.1007/s11888-015-0269-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-015-0269-2
Keywords
- Metastatic colorectal cancer
- T cell
- Cytotoxic T lymphocyte
- Helper T cell
- Regulatory T cell
- Immunotherapy
- Dendritic cell
- Macrophage
- Myeloid-derived suppressor cell
- Vaccine
- Antibody therapy
- Adoptive T cell therapy
- Allogeneic stem cell transplant
- Ipilimumab
- CTLA-4
- PD-1/L1
- Immunosuppression
- Immunosurveillance
- Biomarker
- Anti-tumor immunity
- Adoptive immunotherapy