
Abstract
Current knowledge on gonadal development and sex determination is the product of many decades of research involving a variety of scientific methods from different biological disciplines such as histology, genetics, biochemistry, and molecular biology. The earliest embryological investigations, followed by the invention of microscopy and staining methods, were based on histological examinations. The most robust development of histological staining techniques occurred in the second half of the nineteenth century and resulted in structural descriptions of gonadogenesis. These first studies on gonadal development were conducted on domesticated animals; however, currently the mouse is the most extensively studied species. The next key point in the study of gonadogenesis was the advancement of methods allowing for the in vitro culture of fetal gonads. For instance, this led to the description of the origin of cell lines forming the gonads. Protein detection using antibodies and immunolabeling methods and the use of reporter genes were also invaluable for developmental studies, enabling the visualization of the formation of gonadal structure. Recently, genetic and molecular biology techniques, especially gene expression analysis, have revolutionized studies on gonadogenesis and have provided insight into the molecular mechanisms that govern this process. The successive invention of new methods is reflected in the progress of research on gonadal development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Gonad
- Histology
- Reporter genes
- In situ hybridization
- Immunolabeling
- In vitro culture
- Gene expression analysis
- PCR
- Transgenic animals
14.1 An Outline of the History of Gonadal Development Studies
For centuries investigators have tried to elucidate the processes responsible for the creation of organ structure. Although Hippocrates (460–370 BC) and Aristotle (384–322 BC) made the first descriptions of fetal development, the question of how the differences between males and females arise had remained unanswered (Needham 1959). Later in the modern period, it became evident that the gonads are the first structures in the body to undergo sexual differentiation and they are primarily responsible for orchestrating the development of sex-specific features. It was interesting to study the mechanisms leading to the appearance of the earliest sex-specific traits, i.e., the differences between the testes and ovaries—organs that arise from common sexually undifferentiated anlage. Figure 14.1 summarizes main events and discoveries in the field of studies on the gonadal development.

A timeline depicting invention of new research methods that appeared crucial for studies on the gonadal development, and the main discoveries in the field of gonadogenesis studies
The earliest observations of gonadal development were made during the first histological studies of mammalian fetuses. These early studies were possible after the invention of microscopy and staining techniques. The first staining and microscopic observations were made in the seventeenth century by Leeuwenhoek who used dyes isolated from plants (madder, saffron, indigo). In 1856, Perkin invented the first synthetic dye (mauveine later known as toluidine blue). In 1858, von Gerlach stained cerebellum with carmine isolated from cochineal insects. In 1876, Waldeyer used hematoxylin as a histological stain. In 1876, Wissowsky used hematoxylin and eosin as the first complex histological stain. This shows that in the second half of the nineteenth century, there was significant development of histological methods, which resulted in acceleration of embryological discoveries. Many early descriptions of gonadogenesis and meiosis come from this period. The earliest stages of gonadogenesis and genital ridge formation were described in rabbit (Coert 1898; Egli 1876; Janošik 1885, 1887a, b, 1890a, b, 1894), swine (Janošik 1885, 1887a, b, 1890a, b, 1894), sheep (Janošik 1885, 1887a, b, 1890a, b, 1894), cat (Coert 1898), and humans (Nagel 1889a, b). Sexual differentiation was also described early on in rabbit (Coert 1898; Janošik 1885), swine (Allen 1904), sheep (Janošik 1885, 1887a, b, 1890a, b, 1894), and humans (Nagel 1889a, b). Felix (1911) and Fuss (1911) were the first to describe the extragonadal origin of PGCs in human embryos. Diagnostic features useful for sex recognition of a developing gonad had been defined. The fetal testis had been described as an organ covered by a thick connective tissue capsule under superficial epithelium and filled with cords inside. The fetal ovary had been identified by structural division into the cortex and medulla, the division which according to current knowledge seems oversimplified and misleading. Today, the majority of studies on gonadal development focus on the laboratory mouse (Mus musculus). Despite the differences between human and mouse, the latter species became a powerful research tool owing to easy genetic modification, a sequenced genome, small size, quick reproduction, and easy breeding. The mouse is also a convenient model in the field of developmental biology. The availability of many species-specific probes, antibodies, and markers makes research on mice fast, very efficient, and cost-effective. In 1902, Cuénot conducted the first genetic study on mice. In 1909, Castle and Little developed the first “lab mouse”—inbred strain DBA (Castle and Little 1910). After World War I, Strong bred C3H and CBA mice in 1909, and Little in 1921 developed the C57BL strain (Crow 2002). The C57BL/6 (B6) mouse is the first mammal whose complete genome sequence was published (Chinwalla et al. 2002; Waterston et al. 2002). Most studies concerning murine gonadogenesis were conducted on the C57BL/6 strain, a few on the CD-1 strain (Bendel-Stenzel et al. 2000; Best et al. 2008; Bradford et al. 2009; Brennan et al. 2002; Combes et al. 2009a, b; Cool et al. 2008; DeFalco et al. 2011, 2014, 2015; Di Carlo et al. 2000; Karl and Capel 1998; Kim et al. 2006; Koubova et al. 2006; Martineau et al. 1997; Merchant-Larios and Moreno-Mendoza 1998; Munger et al. 2013; Schmahl and Capel 2003; Schmahl et al. 2000), and only exceptionally on FVB (Anderson et al. 1999; Bendel-Stenzel et al. 2000; Bishop et al. 2000; Bullejos and Koopman 2005), CBA (Buehr et al. 1993; Willerton et al. 2004), MF1 (Best et al. 2008), 129S1/SvImJ (Fleming et al. 2012), AKR/J (Bouma et al. 2007; Bullejos and Koopman 2005; Bouma et al. 2005), DBA/2J (Bouma et al. 2007), and Balb/C (Salas-Cortés et al. 1999) strains. The C57BL/6 mouse strain deserves special attention due to its exceptional sensitivity to disturbances in early gonadogenesis and is thus an efficacious tool for identifying genes involved in sex determination (Bouma et al. 2007). Valuable results are obtained when Y chromosomes, derived from other mouse strains, are placed on a different genetic background, e.g., the Y chromosome from Mus domesticus poschiavinus (YPOS) carried on a C57BL/6 background causes ovarian development in XY but not on DBA/2J. This sensitivity is even stronger when the Y chromosome from AKR/J mice (YAKR) is on a C57BL/6 background (Bouma et al. 2007; Bullejos and Koopman 2005).
In the twentieth century, the rapid development of new methods, especially in the field of cytology, genetics, and molecular biology, propelled embryological studies and led to at least two crucial discoveries in research on sex determination. First, the Y chromosome was discovered in 1905 independently by American geneticist Nettie Maria Stevens and American zoologist and geneticist Edmund B. Wilson (Wilson 1905; Morgan 1912). Using cytogenetic methods and microscopy, they noticed that males of mealworms produce two kinds of sperm differing in the presence of a large or small variant of one of the chromosomes. It was deduced that the sex depends on the presence of the small (Y) chromosome. A second significant breakthrough in this discipline was the discovery of the SRY (the sex-determining region on the Y chromosome) gene in humans (Sinclair et al. 1990). This discovery was possible due to genetic analysis of four male patients with an XX karyotype; SRY was found in a translocated fragment of the Y chromosome. At that time, the Sry gene was described in mice (Gubbay et al. 1990). The role of Sry in sex determination, as a primary switch, was proved by XX male mice with an introduced Sry gene (Koopman et al. 1991).
14.2 Sex Markers
The Sry gene automatically became a suitable sex marker; however, before its discovery other markers were used to define the sex of an individual. Molecular sex markers were always eligible for studying the genes and processes involved in sex determination and development of the gonads due to difficulties or even incapability in distinguishing between male and female fetuses using structural features. Molecular sex markers can identify or predict the sex of an individual even before the gonads appear. However, before the widespread availability and use of sex-linked genes, the sex of fetuses was identified by the presence of Barr bodies (X chromatin bodies). Sex chromatin was discovered by Canadian physicians Murray L. Barr and Ewart G. Bertram in 1949 (Barr and Bertram 1949). By applying histological staining to neurons, they noticed an intranuclear intrusion present in about half of the studied animals. In gonadal development studies requiring the identification of the sex of an individual, the amnion membranes were dissociated and cell suspension was air dried on slides, stained with orcein or Giemsa dye, and Barr bodies were detected under a light microscope (Capel and Batchvarov 2008; Karl and Capel 1998; Palmer and Burgoyne 1991; Yao et al. 2004). Another sexing method involved the detection of activity of the X-linked enzyme glucose-6-phosphate dehydrogenase (G6PDH), used to identify the genetic sex in mice (Williams 1986).
Some studies on gonadogenesis rely on easy sexing based on the presence/absence of testis cords. These elongated structures begin forming in the murine testes at 11.8 dpc (stage of 21 tail somites); however, 12.25 dpc is the earliest moment in which the sex can be recognized by the presence/absence of the testis cords under a dissecting microscope (Nel-Themaat et al. 2009; Wilhelm et al. 2005). A lack of cords indicates ovarian differentiation.
The aforementioned methods have been replaced by more accurate molecular techniques. Nowadays, the most extensively used method to define the sex of an individual is genotyping relying on sex-specific gene identification by PCR (polymerase chain reaction). This procedure involves tissue collection (e.g., a piece of earlobe or tail ideally from 10- to 21-day-old mice), DNA isolation, and simplex (one pair of primers for one template) or multiplex (two or more primer pairs) PCR. A series of genes are used as sex markers. First, Y-linked sequences, such as Sry (Chaboissier et al. 2004; Greenlee et al. 1998; Kunieda et al. 1992; Lambert et al. 2000; Lavrovsky et al. 1998; McClive and Sinclair 2001) or Zfy (Greenlee et al. 1998; Kunieda et al. 1992; Nagamine et al. 1990; Nef et al. 2005), can be amplified and detected indicating the male genetic sex. Since Sry is present only on the Y chromosome and has no equivalent on the X, and the Zfy gene has a homolog on the X chromosome (Zfx) with equal size, amplification of X-linked or autosomal genes is needed as a control (McFarlane et al. 2013; Palmer et al. 1990). The X-linked gene Dxnds3 is an example of such an internal control in multiplex PCR (Greenlee et al. 1998; Kunieda et al. 1992). There are currently only three simplex PCR methods used for sex genotyping in mice. Sex identification is possible through the amplification of homologous genes Uba1 and Ube1y1 linked to the X and Y chromosomes, respectively, using a set of primers designed to amplify a deleted region in the Ube1y1 gene, which results in two products of amplification in males and one large product in females with a small size difference (19 bp) between two amplicons (Best et al. 2008; Chuma and Nakatsuji 2001; McFarlane et al. 2013). Another method is based on amplification of X-linked Kdm5c (also known as Smcx, Jarid1c) and its Y-linked equivalent Kdm5d (Smcy, Jarid1d) with a 29 bp size difference between amplicons (Barrionuevo et al. 2006; Bullejos and Koopman 2005; Clapcote and Roder 2005; Jameson et al. 2012; Mroz et al. 1999; Munger et al. 2009). A third example of simplex PCR sex genotyping involves a size difference between the Y-linked Sly gene and its X-linked equivalent Xlr that results from a 405-bp deletion in Sly (McFarlane et al. 2013). The primers were designed upstream and downstream of the deleted region giving two different size products in each sex. The last method is the most accurate due to size differences large enough to be easily detected during electrophoresis.
Previously, an alternative method of identification of Sry fragments via Southern blot was used to define the sex (Fechner et al. 1993; Gubbay et al. 1990; Tanoue et al. 1992). Identification of H-Y antigen encoded by Kdm5d gene was also used as a male-specific marker (Wolf 1998).
14.3 Staging and Timing of Early Gonadogenesis in Mouse and Other Vertebrates
Precise fetal age determination is essential for embryological studies especially if accurate spatiotemporal pattern of gene expression or description of structural changes in developing organs is required. Precise staging is crucial for studying the embryology of animals with a rapid rate of development such as rodents, in which some genes are expressed in a narrow time window.
In mice, it is necessary to use timed matings with E0.5 (or 0.5 dpc, dies post coitum) representing noon on the day a mating plug was detected. Usually mouse pairs are set up at ~5:00 pm; the presence of vaginal plugs is checked the next morning and noon is counted as 0.5 dpc. More accurate staging relies on counting the tail somites (ts). The first tail somite is counted as the first one posterior to the junction of the hindlimbs. In mice, early genital ridges begin to be visible around 10.5 dpc which corresponds to the presence of 8 ts (±2 ts); and onward 11.5 dpc corresponds to 18 ts (±2 ts), and 12.5 dpc corresponds to 30 ts (±3 ts) (Hacker et al. 1995). To obtain the key stages for the study of sexual differentiation of murine gonads, e.g., stages 15–17 ts, mice are sacrificed at 7:00 am on the 11th dpc, 18–20 ts at noon on the 11th day, and 28–30 ts at noon on the 12th day. Additionally, tables of Theiler stages (TS; from TS01 to TS28) are useful in determining the embryonic age, especially in fetuses older than 13.5 dpc staged by forelimb and hindlimb morphology (Theiler 1989). Mouse gonads start developing at TS16 (i.e., 9.5–10.75 dpc, corresponding to 30–34 total somite number), sex determination takes place at TS17–TS18 (i.e., 35–44 total somite number), and testis cords start forming at TS19 (i.e., 45–47 total somite number) and become discernible at TS20 (i.e., 48–51 total somite number).
Various standardized systems of staging are used for different vertebrates. For example, Carnegie stages are used to determine the human fetal age. These stages were applied in studies on human gonadogenesis (Hanley et al. 2000) but can be used for other vertebrates. Carnegie stages range from C01 to C23 covering the first 56 days of human gestation and later stages are described using the term of gestation. Sexual differentiation of gonads in humans occurs at C18–23. For chicken embryonic development, Hamburger and Hamilton stages (1951) are used and range from HH01 (laying) to HH46 (hatching). Undifferentiated gonads of chickens appear at HH25 (embryonic day E4.5), while sexual differentiation of gonads occurs at HH29 (E6) (Morais da Silva et al. 1996). Staging according to Greenbaum (2002) was used in a study of gonadal development in the red-eared slider (Trachemys scripta) (Smith et al. 2008b). In this reptile, the temperature-sensitive period begins at Greenbaum stage 14, and sexual differentiation of gonads begins at stages 18/19. In most anuran amphibians, embryonic and larval development is defined according to the Gosner system (from stage 1 to 46 at completion of metamorphosis) (1960). Undifferentiated gonads appear at about Gosner stage 26; however, the time of sexual differentiation varies among anuran species (Piprek 2013). The tadpoles of the model anuran species the African clawed frog (Xenopus laevis) and the Western clawed frog (Silurana tropicalis) differ in morphology from most anuran tadpoles and therefore another staging system is used to describe their development, i.e., Nieuwkoop and Faber stages (1967). This system ranges from NF1 (egg) to NF66 (completed metamorphosis). Undifferentiated gonads appear at NF49 and sexual differentiation occurs at about NF52/53 (Piprek 2013). In the model zebrafish (Danio rerio), development is staged according to somite number or time after fertilization. In this species, primordial germ cells accumulate at the sites of gonad formation at 2 weeks pf (post-fertilization), at 4 weeks pf the first ovarian development features are present, and at 7 weeks pf the first signs of testicular differentiation are visible (Kimmel et al. 1995; Maack and Segner 2003).
14.4 Histology: Light and Electron Microscopy
As previously mentioned, the earliest studies of gonadal development were based on structural analysis using histological techniques. Today, the histological studies have been replaced by molecular studies. Nevertheless, histological techniques are indispensable for instance if the effects of a gene mutation or knockout need to be assessed. Despite a wide range of staining methods, hematoxylin and eosin staining (H&E, HE) on paraffin sections (usually 3–8 μm thick) is mostly used. Sample preparation for histological analysis begins with tissue dissection and fixation. Fetal gonads are usually fixed in 4 % paraformaldehyde (PFA in PBS at 4 °C overnight) (Bagheri-Fam et al. 2008; Chang et al. 2008; Hummitzsch et al. 2013; Moniot et al. 2009; Nef et al. 2003; Nel-Themaat et al. 2009) or in Bouin’s solution (room temp. or 4 °C for 1–2 days) (Britt et al. 2002; Chassot et al. 2008; El Jamil et al. 2008; Gassei et al. 2008; Hiramatsu et al. 2009; Koubova et al. 2006; Maatouk et al. 2008; MacLean et al. 2009; McLaren and Southee 1997; Qing et al. 2008; Tang et al. 2008). Other fixation methods are rarely used, e.g., Serra fixation (Barrionuevo et al. 2006, 2009). Bouin-fixed samples are dehydrated and embedded in paraffin; however, PFA-fixed samples can be both frozen or dehydrated and embedded in paraffin. Bouin’s solution provides strong fixation of gonadal tissue ensuring maintenance of structure; poorer resolution is usually obtained in PFA fixation. PFA-fixed frozen sections are the most suitable for immunolocalization. Histological staining on cryosections and dyes other than H&E are rarely used. Trichrome staining (e.g., Masson’s trichrome; Britt et al. 2002) provides more structural detail showing three different colors in the tissue (differently stained nuclei, cytoplasm, and extracellular matrix). Histological images analyzed via specialist software can also be used for 3D reconstruction of internal organ structure. This method was applied in the reconstruction of the structure of testis cords within developing mouse testes (Nel-Themaat et al. 2009) and ovarian follicles in Xenopus (Bilinski et al. 2010).
Ultrastructure of gonads can be studied using transmission electron microscopy (TEM) techniques (Best et al. 2008; Britt et al. 2002; El Jamil et al. 2008; Hummitzsch et al. 2013; Merchant-Larios and Moreno-Mendoza 1998; Ottolenghi et al. 2005; Tanimura and Iwasawa 1991). In the majority of these studies, tissues were fixed in Karnovsky’s fixative (PFA, glutaraldehyde, cacodylate buffer), then postfixed in osmium tetroxide, dehydrated, and embedded in resin (Ito and Karnowsky 1968). Such samples can be cut for semithin (0.5–1 μm) or ultrathin sections (50–70 nm). Semithin sections are placed on microscope slides, stained with methylene blue and azur II and viewed under a light microscope. Ultrathin sections are placed on copper (sometimes nickel for immunolocalization) grids, contrasted with uranyl acetate and lead citrate, and viewed with TEM. Other histological staining techniques and even immunolocalization can also be used for both semithin (on slides) and ultrathin (on grids) sections; however, chemical removal of resin is necessary.
Scanning electron microscopy (SEM) has been used to study gonadal development and especially the external structure of gonads (Piprek et al. 2014; Wylie et al. 1976). In this procedure, samples are usually fixed in Karnovsky’s fixative, postfixed in osmium tetroxide, dehydrated, dried using liquid carbon dioxide, sputter-coated with gold, and viewed using SEM (Ito and Karnowsky 1968).
14.5 Molecule Identification: Cytochemistry, Immunolocalization
Cyto- and histochemistry is the science of localization of chemical compounds within cells or tissues using chemical reactions or specific antibodies. Among cytochemical techniques, PAS staining, alkaline phosphatase detection, and especially immunohistochemistry are widely used in gonadal development studies. The PAS (periodic acid-Schiff reaction) method detects polysaccharides such as glycogen, glycoproteins, glycolipids, and mucins in tissues. Primordial germ cells (PGCs) have abundant cytoplasmic glycogen and thus the PAS method can be used to identify these cells (Chassot et al. 2008; De Felici et al. 2004; Gassei et al. 2008; Hiramatsu et al. 2009).
Alkaline phosphatase (AP, ALP) is an enzyme removing phosphate groups from many types of molecules and acts most efficiently in an alkaline environment. This enzyme is especially widespread on the surfaces of stem cells including PGCs and thus has been used to identify migrating PGCs prior to gonad development (Chiquoine 1954; De Felici and Dolci 1989; Di Carlo et al. 2000; Hanley et al. 2000; Maitland and Ullmann 1993).
Specific recognition and detection of proteins via antibodies became an extensively used method in embryological research and revolutionized this discipline. In direct immunolabeling, an antibody is covalently labeled with a marker enabling detection; in indirect immunolabeling, a primary antibody binds an antigen and a secondary antibody conjugated with a marker binds the primary antibody. Secondary antibodies can be labeled with (1) an enzyme that produces a colored compound (usually alkaline phosphatase with BCIP/NBT substrate, or horseradish peroxidase—HRP with DAB as a substrate) detected in bright-field light microscopy, (2) fluorochrome (usually derivatives of fluorescein or rhodamine) detectable in fluorescence microscopy, and (3) colloid gold for transmission electron microscopy (TEM), scanning electron microscopy (SEM), or light microscopy (LM). Several compounds such as biotin, avidin, and streptavidin are used to increase the number of bound secondary antibodies or enzymes and thus to intensify signal. Double immunolabeling is possible due to the application of two primary antibodies (differing in species origin) against two different proteins. This technique enables the detection of potential co-localization of two proteins. Interestingly, the first use of antibodies for detection was reported in 1934 (Marrack 1934), the first fluorescent detection using antibodies took place in 1941 (Coons et al. 1941), and the first enzymatic labeling was described in 1966 (Avrameas and Uriel 1966).
Immunolabeling can be classified as (1) immunocytochemistry (ICC)—immunolabeling of cells usually after in vitro culture, (2) immunohistochemistry (IHC)—detection in tissues on paraffin or cryosections or in whole organs (whole mount, WM-IHC), (3) immunofluorescence (IF)—localization via fluorochrome using fluorescence (also confocal) microscopy, and (4) immunogold staining (IGS)—detection of colloid gold under an electron or light microscope.
There are two types of immunolabeling in respect to sequence of labeling and embedding: pre-embedding and post-embedding. In the pre-embedding method, immunolabeling is done prior to embedding and sectioning (i.e., fixation, immunolabeling, embedding in paraffin, resin, or freezing, cutting, imaging). In the post-embedding method, the tissue after fixation is first embedded in paraffin, resin, or frozen, then cut, and immunolabeling is performed on sections (i.e., fixation, embedding, cutting, immunolabeling, imaging).
There is a plethora of gonadal development studies in which immunolabeling was used, usually IHC or IF (Abramyan et al. 2009; Bagheri-Fam et al. 2008; Barrionuevo et al. 2006, 2009; Bendel-Stenzel et al. 2000; Best et al. 2008; Beverdam and Koopman 2006; Bogani et al. 2009; Bradford et al. 2009; Britt et al. 2002; Chang et al. 2008; Chassot et al. 2008; Chen et al. 2012; Childs et al. 2011; DeFalco et al. 2015; DiNapoli and Capel 2007; Dumond et al. 2011; Dupont et al. 2000; Fleming et al. 2012; Fröjdman et al. 1992; Hanley et al. 2000; Hiramatsu et al. 2009; Hummitzsch et al. 2013; Jameson et al. 2012; Karl and Capel 1998; Nef et al. 2005; Li et al. 2012; Li and Kim 2004; Liu et al. 2009, 2015; Maatouk et al. 2008; MacLean et al. 2009; Matzuk et al. 1995; Molyneaux et al. 2003; Moniot et al. 2009; Morais da Silva et al. 1996; Mork et al. 2014; Nicol and Yao 2015; Ohe et al. 2002; Oshima et al. 2005; Ross et al. 2009; Salas-Cortés et al. 1999; Schmahl et al. 2004; Smith et al. 2008a, b; Soyal et al. 2000; Tevosian et al. 2002; Wilhelm et al. 2005; Willerton et al. 2004; Yao et al. 2004). For IHC, in most cases tissues are fixed with 4 % PFA or Bouin’s solution, dehydrated, and embedded in paraffin. Such strong fixation entails later antigen retrieval which is usually done in citrate buffer at ~95 °C (HIER, heat-induced epitope retrieval). Samples for IF are usually fixed in 4 % PFA, frozen, and later cryosectioned; in the case of cryosections, antigen retrieval is unnecessary. Cryosections and deparaffinized sections after antigen retrieval, both on slides, are usually blocked with 6 % BSA in PBS (instead of BSA, Casein or serum from species matching the secondary antibody can be used), which is followed by incubation with primary antibodies (usually overnight at 4 °C), and then after rinsing (PBS with detergent), incubation with secondary antibodies is done (usually for 1–2 h at room temp.); after rinsing, color development (for IHC) and counterstain (for IHC and IF) is carried out.
In the pre-embedding version of IHC, also known as whole mount immunohistochemistry (WM-IHC or WIHC), the whole fetal gonads or urogenital ridges (gonads attached to mesonephroi) are fixed usually in 4 % PFA (or in a methanol:DMSO mixture), permeabilized in a solution of detergent to allow large molecules to penetrate, and then immersed in blocking buffer followed by incubation with primary and then secondary antibodies (Albrecht and Eicher 2001; Barske and Capel 2010; Bendel-Stenzel et al. 2000; Bogani et al. 2009; Bouma et al. 2005, 2007; Brennan et al. 2002; Bullejos and Koopman 2005; Combes et al. 2009a, b, 2011; Coveney et al. 2008; Cool et al. 2008; DeFalco et al. 2014, 2015; Karl and Capel 1998; Maatouk et al. 2008; Martineau et al. 1997; Nicol and Yao 2015; Schmahl et al. 2000; Schmahl and Capel 2003; Tang et al. 2008; Tevosian et al. 2002; Warr et al. 2009; Wilhelm et al. 2007). After immunolabeling, the gonads can be embedded and sectioned.
Immunogold staining (IGS) permits the detection of a protein using electron microscopy (TEM or SEM). Colloid gold or nanogold conjugated to secondary antibody are used in this technique. Both the pre-embedding and post-embedding versions of the IGS method were presented in detail by Bilinski et al. (2010). In most commonly used post-embedding IGS methods, the tissue is gently fixed in PFA with or without glutaraldehyde (strong fixation and osmium tetroxide postfixation should be omitted in order not to disrupt immunoreactivity), dehydrated, embedded in hydrophilic resin, and cut on formvar-carbon-coated nickel grids; the grids with ultrathin sections are applied onto drops of the following solutions: blocking solution (BSA), primary antibodies, rinsing, secondary antibodies, rinsing, and contrast staining with uranyl acetate and lead citrate. The subcellular localization of a protein of interest can be investigated with this method (Best et al. 2008).
The most important processes involved in development include cell proliferation and apoptosis. Both can be studied using appropriate antibodies. Cell divisions can be detected via immunolocalization of proliferation markers such as nuclear protein PCNA (Bogani et al. 2009), phosphohistone-H3 (pHH3) (Bogani et al. 2009; Manuylov et al. 2007), and nuclear antigen Ki-67 (Dupont et al. 2000). However, the most popular technique used to identify proliferating cells is immunolocalization of bromodeoxyuridine (BrdU) after injection of this compound. In studies on cell proliferation in developing gonads of the mouse, BrdU is injected intraperitoneally (IP) to a pregnant female (in amount 10–50 mg/kg) and is incorporated into DNA during replication (phase S) in female and fetal tissues. Females are sacrificed 2–5 h after injection. Fetal gonads are fixed and immunolocalization with anti-BrdU is performed on paraffin or frozen sections (DeFalco et al. 2015; Dupont et al. 2000; Nef et al. 2003; Schmahl et al. 2000). BrdU pulse chase is a special version of this assay, which can determine when precursors for specific cell types were dividing (Schmahl et al. 2000). There is also a series of descriptions of proliferation studies in developing gonads in in vitro culture (DeFalco et al. 2011; Hiramatsu et al. 2009; Martineau et al. 1997; Schmahl et al. 2004; Schmahl and Capel 2003). In such assays, BrdU is dissolved in culture medium, and after 1–3 h, organs are rinsed and cultured in medium without BrdU for 9–12 h and then fixed and immunolabeled.
Administration of proliferation inhibitors enables to test if proliferation plays a role in a specific developmental process. Proliferation inhibitors such as 5-fluorouracil or methotrexate (MTX) were administered intraperitoneally to pregnant mice to study the role of proliferation in gonad development (Schmahl and Capel 2003). Alternatively, Affi-gel beads soaked in aphidicolin, a substance that inhibits the cell cycle at the S phase, demonstrated the role of proliferation in testis development (Schmahl and Capel 2003).
The contribution of apoptosis in development is typically studied using the TUNEL assay (Li and Kim 2004; Manuylov et al. 2007; Matzuk et al. 1995). In the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) method, the fragmentation of DNA, common for cell death, is detected. Terminal deoxynucleotidyl transferase TdT binds 3′ nicks in DNA and catalyzes the addition of dUTPs labeled with a marker. This visualizes apoptotic cells on frozen or paraffin sections using chromogen or fluorochrome. Additionally, immunolocalization of an apoptosis marker—caspase3—is useful in cell death studies (Tang et al. 2008).
14.6 Identification of mRNA: In Situ Hybridization
Protein identification is especially useful for cell-specific marker identification and structure viewing; however, it is less accurate if gene expression is studied because a protein is present even after gene expression has ceased and moreover a protein can be detected in places other than its site of expression (e.g., in secreted proteins). Therefore, identification of specific mRNA is a more useful and accurate method in gene expression analyses.
The most commonly used method to detect gene expression in situ (in tissue) is RNA in situ hybridization (ISH). The first report of detection of DNA:RNA hybridization was given by Gall and Pardue (1969). This method is frequently used in gonadal development studies in both the post-embedding version (ISH) (Abramyan et al. 2009; Best et al. 2008; Bradford et al. 2009; Chassot et al. 2008; El Jamil et al. 2008; Hanley et al. 2000; Kim et al. 2006; Maatouk et al. 2008; Martineau et al. 1997; Matzouk et al. 1995; Moniot et al. 2009; Morais da Silva et al. 1996; Nef et al. 2003; Oulad-Abdelghani et al. 1996; Parma et al. 2006; Schmahl et al. 2004; Sekido et al. 2004; Wilhelm et al. 2005) and pre-embedding or whole mount-ISH (WM-ISH or WISH) (Barrionuevo et al. 2006; Bishop et al. 2000; Bogani et al. 2009; Combes et al. 2009a,2011; Coveney et al. 2008; Jameson et al. 2012; Koubova et al. 2006; Manuylov et al. 2007; Matsuyama et al. 2005; Nef et al. 2005; Rolland et al. 2011; Smith et al. 2008a, b; Stebler et al. 2004). The core idea in the ISH technique is the hybridization of a labeled complementary probe to mRNA allowing the identification of a specific gene transcript under a microscope. A probe consists of complementary DNA (cDNA) or more commonly complementary RNA (cRNA, riboprobe). This analysis can be done on paraffin sections, frozen sections on slides for bright-field light or fluorescence microscopy, or even on grids for electron microscopy (TEM) (Kloc et al. 2001). This method involves probe design (250–1500 nt), probe in vitro synthesis, and probe labeling usually with digoxigenin (DIG) or biotin. DIG is a steroid with high antigenicity recognized by antibodies that can be conjugated with fluorochrome or enzyme (AP or HRP). Alternatively, probes can be biotinylated, i.e., conjugated to biotin which is bound by avidin or streptavidin conjugated to an enzyme. This method is termed CIHS (chromogenic in situ hybridization) in the case of enzymatic labeling and the appearance of color product indicating the presence of the mRNA of interest. In the case of fluorescence labeling, ISH is termed FISH (fluorescence in situ hybridization). In ISH, tissues are usually fixed in 4 % PFA, rinsed, frozen, or dehydrated and embedded in paraffin. Sections on slides undergo hybridization with probes overnight at 65 °C and samples are viewed under a microscope after labeling with antibodies. In WM-ISH, the whole organ is fixed in 4 % PFA, rinsed, hybridized in buffers, and after labeling the sample is viewed in toto under a dissection microscope or dehydrated, embedded, and sectioned and viewed on slides under light or fluorescence microscopes. In situ hybridization can also be used to localize a DNA fragment on chromosomes, e.g., in sex determination studies (Bishop et al. 2000; Parma et al. 2006; Yoshimoto et al. 2008).
14.7 Reporter Genes
One of the most advanced methods currently applied for visualization and specific cell type identification is the utilization of reporter genes. This method involves the use of transgenic animals. A reporter gene gives an easily identifiable product. The most commonly used reporter genes are LacZ and Gfp. These genes enable identifying the expression of a gene of interest and can also be used to trace specific cell lines. A reporter gene is introduced into a genome and is expressed under the control of a given promoter of interest.
LacZ encodes β-galactosidase (β-gal), an enzyme converting X-gal substrate to a blue product. LacZ with a promoter of the gene of interest is introduced into a genome. After LacZ expression, the β-gal protein is present in cells in which the gene of interest is normally expressed. Organs of the transgenic animal are dissected and fixed in PFA, and X-gal staining can be conducted on slides or in the whole organ (whole mount technique). This method is widely used in studies of gonadogenesis to visualize cells of a special type or to investigate the spatiotemporal pattern of gene expression (Anderson et al. 1999; Brennan et al. 2002; Capel et al. 1999; Liu et al. 2015; Maatouk et al. 2013; Manuylov et al. 2007; Martineau et al. 1997; McLaren and Southee 1997; Merchant-Larios and Moreno-Mendoza 1998; Miyamoto et al. 2008; Tang et al. 2008). This method was also applied in an assessment of the action of β-catenin in sex determination (Chassot et al. 2008).
Fluorescent proteins such as GFP (green fluorescent protein) act in a different manner. A signal is detectable after excitation (e.g., ultraviolet for GFP). The Gfp gene was isolated from the jellyfish Aequorea victoria in 1960 (Tsuji 2010). Several mutations were introduced in order to modify and improve GFP fluorescence and thus GFP derivatives were obtained, e.g., EGFP (enhanced GFP) and YFP (yellow fluorescent protein). Red fluorescent proteins include mCherry or tdTomato. In gonadogenesis studies, fluorescent reporter genes are used to identify and trace specific cells, to visualize gonadal structure, and to isolate cells of interest (Albrecht and Eicher 2001; Anderson et al. 1999; Best et al. 2008; Beverdam and Koopman 2006; Cool et al. 2008; DeFalco et al. 2011, 2014, 2015; Jameson et al. 2012; Maatouk et al. 2013; Matzuk et al. 1995; Molyneaux et al. 2001; Nef et al. 2005; Nel-Themaat et al. 2009; Qing et al. 2008; Rolland et al. 2011). For example GFP, YFP, mCherry, and tdTomato were used to detect cells expressing Sf1, Sry, Sox9, Oct4, or αSma, markers of germ cells, or gonadal somatic cells (Albrecht and Eicher 2001; Beverdam and Koopman 2006; Cool et al. 2008; DeFalco et al. 2014; Jameson et al. 2012; Matzuk et al. 1995; Molyneaux et al. 2001; Nef et al. 2005; Nel-Themaat et al. 2009; Qing et al. 2008; Rolland et al. 2011).
Luciferase may be used as a reporter gene because of light emission via the process of bioluminescence after oxidation of substrate (luciferin) by this enzyme. A luciferase reporter gene assay was used to trace the expression of Sry and Amh (Miyamoto et al. 2008; Wilhelm et al. 2007).
14.8 Cell Markers
The aforementioned methods such as IHC, ISH, and reporter genes can be used for the identification of a cell of interest due to the presence of cell markers. Each cell type has its own unique transcriptome and proteome, and thus, cell-specific markers, unique for a given cell type, can be identified. Such cell markers can be used for cell visualization in an organ, for tracing migrating cells, cell sorting, and isolation. It must be emphasized that cell markers revolutionized gonadal development studies and have been used to describe the origins and fates of cell lines during gonadogenesis.
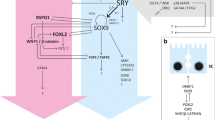
A series of markers useful for identification of specific cell lines (e.g., Sertoli cells, follicular cells, fetal Leydig cells, endothelial cells, germ line cells, meiotic cells) in developing gonads are summarized in Table 14.1. The most crucial cell markers include GATA4, LHX9, SF1, and WT1 for coelomic epithelium-derived gonadal precursor cells; SRY, SOX9, and AMH for pre-Sertoli cells; 3βHSD for fetal Leydig cells; OCT4 for germ cells; PECAM1 for germ and endothelial cells; and SYN/COR for meiotic cells.
14.9 Structure Visualization Using Cell Markers and Tracers
Labeled cells can be visualized continuously under in vitro conditions. This valuable method traces migrating cells within developing gonads in a time-lapse course (Coveney et al. 2008; DeFalco et al. 2011; Molyneaux et al. 2001; Nel-Themaat et al. 2009, 2010). Coveney et al. (2008) studied the dynamics of testis morphogenesis using an in vitro culture of fetal gonads of transgenic mice with expression of GFP in endothelial cells. In this study, Cre recombinase was expressed under a control of Tie2 or Flk1 promoters (markers of endothelium) and cleaved a stop codon upstream of GFP gene, which led to GFP presence in the endothelial cells.
An assembly of cell labeling images by specialist software enables the three-dimensional modeling of the internal structure of the developing gonads. This stereological analysis technique was especially useful in describing the process of testis cord formation. Barrionuevo et al. (2009) conducted 3D visualization of developing testes via immunolabeling for P450scc (a marker of Leydig cells); DeFalco et al. (2015) used immunolocalization of several cell markers to obtain a 3D model of the developing testis (Sertoli cells—TUBB3, PMCs—ACTA2, macrophages—MHCII, spermatogonia—ZBTB16, blood vessels and basement membranes—collagen IV). Nel-Themaat et al. (2009) used transgenic mice with expression of EGFP under control of the Sox9 gene to visualize Sertoli cells and to present a 3D model of testiculogenesis.
14.10 In Vitro Culture Methods
In vitro organ culture is one of the most commonly used techniques to study the mechanisms of gonadogenesis in mice. In this method, developing gonads are isolated from fetus, placed in medium, and cultured for up to several days. This enables (1) observation of development in a time-lapse course, (2) studying cell migration between the gonad reassembled with the mesonephros (migration assay in coculture), and (3) analysis of the molecular mechanisms governing gonadogenesis via culturing gonads in medium supplemented with growth factors, their inhibitors, or antibodies blocking given proteins.
In vitro culture techniques were originally developed for embryological trials. In vitro culture of chicken embryo organs by Roux in 1885 and frog embryonic cells by Harrison in 1907 were maintained ex vivo at first. Byskov and Saxen (1976) conducted early in vitro cultures of developing gonads. Today, several techniques of in vitro culture of developing gonads are used, among which culture on agar blocks is the most popular. Dissected fetal mouse gonads are always cultured under conditions standard for mammal cells, i.e., at 37 °C and 5 % CO2/95 % air. In most studies, developing gonads are cultured in DMEM medium.
In an early trial, developing gonads were maintained in vitro “glued” to a nucleopore filter using 1 % agar in Eagle’s balanced salt solution (EBSS) supplemented with 10 % fetal calf serum (FCS) (Byskov and Saxen 1976). In subsequent studies, gonads were cultured at an air-medium interface on agar blocks (Evans et al. 1982; McLaren and Buehr 1990). The first recombinant cultures (a gonad reassembled with the mesonephros) were used by Buehr et al. (1993) and showed migration of mesonephric cell into developing testes (Combes et al. 2009b; Cool et al. 2008; Martineau et al. 1997). Buehr et al. (1993) also provided the original description of fetal gonad culture on agar blocks, currently the most common method for maintaining gonads in vitro (Barrios et al. 2010; Best et al. 2008; Bogani et al. 2009; Capel and Batchvarov 2008; Combes et al. 2009b; Coveney et al. 2008; DeFalco et al. 2011; Di Carlo et al. 2000; Hiramatsu et al. 2009; Karl and Capel 1998; Kim et al. 2006; Koubova et al. 2006; Martineau et al. 1997; McLaren and Southee 1997; Nishino et al. 2001; Wilhelm et al. 2005; Tang et al. 2008). Alternatively, a droplet method is used to maintain whole fetal gonads under in vitro conditions (Cupp et al. 2003; DeFalco et al. 2014; Maatouk et al. 2008). An organ is cultured in a droplet on the lid of Petri dish while the dish is floating on water in a larger Petri dish. Fetal gonads were also cultured on Millicell filters (Cupp et al. 2003; MacLean et al. 2009; Nel-Themaat et al. 2009). A slice method of in vitro culture was used to study the regulation of PGC migration from the hindgut to the genital ridges (Bendel-Stenzel et al. 2000; Molyneaux et al. 2001, 2003).
The fetal gonads are usually cultured for 48 h on 1.5 % agar blocks in DMEM medium supplemented with 10 % FCS or FBS and ampicillin. Exceptionally, F12/DME medium supplemented with 10 % FBS is used (Liu et al. 2015; Nishino et al. 2001). This method of fetal gonad culture on agar blocks was also used for studying fetal T. scripta gonads (Mork et al. 2014). In this case, the L15 medium was supplemented with 10 % FBS. However, in vitro culture of fetal chicken gonads was done on an isopore filter in DMEM medium supplemented with FCS and chicken serum (Smith et al. 2008a).
14.11 In Vitro Studies of Cell Signaling
In vitro gonad culture was also effective in determining the function of signaling molecules in the regulation of gonad development. The method is based on culturing dissected gonads in medium supplemented with exogenous growth factors, their inhibitors, or antibodies binding and thus blocking the action of particular proteins. This technique was used to study the role of FGF9 in testis development. Gonads were cultured in medium supplemented with FGF9 (25, 50, or 100 ng/ml) (Barrios et al. 2010; Hiramatsu et al. 2009; Kim et al. 2006; Wilhelm et al. 2005) or in the vicinity of agarose beads soaked with FGF9 growth factor for up to 2 days (Bogani et al. 2009; DeFalco et al. 2011). The function of neurotrophin 3 in gonadogenesis was studied by implementing a blocker of its receptor (AG879) added to medium (Cupp et al. 2003). In a study on neurotrophic tyrosine receptor kinases, the medium was supplemented with neurotrophin 3, antibodies binding and blocking neurotrophin 3, antibodies binding receptor NTRK3, inhibitors of tyrosine kinase, NGF, or antibodies binding NGF (Gassei et al. 2008). γ-secretase inhibitor (DAPT) added to medium inhibited Notch signaling and elucidated the function of this pathway in progenitor Leydig cells (Tang et al. 2008). The role of hedgehog (HH) signaling was examined via addition of cyclopamine (inhibitor of HH pathway) to medium (Liu et al. 2015). Sex determination was also investigated by the addition of prostaglandin D2 to medium (Wilhelm et al. 2005). Interestingly, the role of blood vessels in gonad morphogenesis was tested via blocking of vasculature formation with an inhibitor of VEGF receptor tyrosine kinase (VEGFR TKI II) (DeFalco et al. 2014). Moreover, the influence of blood vessels on gonadogenesis was studied by addition of anti-VE-cadherin antibodies to medium which disrupted vasculature formation (Combes et al. 2009b). WNT signaling was investigated by adding LiCl to medium, which increased the function of β-catenin and WNT pathways (Maatouk et al. 2008). The method of signaling modulation in vitro was also useful for studying the induction of meiosis by retinoic acid. Medium was supplemented with all-trans retinoic acid (RA), agonists of retinoic acid receptors (RAR), and ketoconazole that inhibits the Cyp26 enzyme from degrading RA (Bowles et al. 2006; Koubova et al. 2006; Li and Kim 2004; Piprek et al. 2013).
14.12 Cell Tracing and Migration Assay
Cell tracing is a method revealing cell migration within developing organs. This technique also provides evidence for the origin of different cells or cell types. In gonadal development studies, cell tracing involves cell labeling with a marker (usually fluorescent dye applied onto the surface of the gonad) (DeFalco et al. 2011; Karl and Capel 1998; Li and Kim 2004; Yao et al. 2004). The fluorescent dye penetrates the superficial cells of the developing gonad and then labeled cells carry the marker while migrating inward into the developing organ. Identification of labeled cells after in vitro culture of the whole organ shows the ingression of superficial cells. This method demonstrated the origin of Sertoli and granulosa cells from coelomic epithelium (Karl and Capel 1998). Karl and Capel (1998) incorporated fluorescent CM-DiI dye to superficial gonadal cells using ionophoresis. A single superficial cell was touched by an electrode with the applied dye; the current was pulsed until a single cell was positive for CM-DiI. In the same study, an alternative method of surface cell labeling was used; the dye was pipetted onto the surface of the developing gonad, and after washing, the gonads were cultured in vitro for 30–48 h. The labeled cells were localized under a microscope. DeFalco et al. (2011) labeled superficial cells of developing mouse gonads via pipetting MitoTracker Orange dye onto the gonads dissected from mouse fetuses; after 45 min of incubation with dye, gonads were washed and cultured in vitro for 24–48 h and then viewed. Two studies used a mixture of two fluorescent dyes (MitoTracker Red and rhodamine derivative, 5-carboxytetramethylrhodamine, succinimidyl ester) applied onto the surface of the developing gonad of T. scripta and X. laevis (Piprek 2013; Yao et al. 2004). After incubation, gonads were washed and cultured in vitro for 48 h, then fixed, and processed for immunohistochemistry and imaging.
Different cell tracing methods were used to study cell migration from the fetal mesonephros to developing gonads (migration assay). In this technique, in vitro coculture of the fetal gonad assembled with the mesonephros (usually derived from another individual) is conducted, with expression of reporter genes enabling cell identification or with labeled cells. The first experiment showing cell migration from the mesonephros to the fetal gonads was carried out by Buehr et al. (1993). They detached developing mouse gonads from the adjacent mesonephroi and cultured isolated gonads and mesonephroi in vitro separately, or the organs were reassembled and grafted into the fragments of limb or heart, or cultured while separated by a membrane. The migration of cells from mesonephroi into developing testes was observed in reassembly of wild-type embryonic testes with transgenic mesonephroi from murine embryos carrying a nuclear marker (~1000 copies of the β-globin gene). Also other techniques, such as in situ hybridization and immunolocalization, permit identification of mesonephros-derived cells in the testes. Li and Kim (2004) tested mesonephric cell migration to developing rat gonads via in vitro culture of reassembled embryonic gonads with mesonephros labeled with fluorescent cell-permeable dye. The mesonephros, isolated from a fetus, was cultured in medium with dye (5(6)-carboxyfluorescein diacetate, succinimidyl ester, CFDA-SE) for 20 min; after washing, the stained mesonephros was reassembled with a fetal gonad and cultured in vitro for 3 days, and then the organs were fixed and viewed using fluorescent microscopy. Capel et al. (1999) and Martineau et al. (1997) studied cell migration from the mesonephros to the developing testes using in vitro culture of mesonephros from ROSA26 transgenic mice showing constitutive expression of β-gal reassembled with fetal wild-type mouse gonads. After 42–48 h of in vitro culture, the organs were fixed, stained for β-gal, embedded, and viewed for detection of (mesonephros-derived) blue cells in the testes. Brennan et al. (2002) used coculture of wild-type gonads with mesonephroi from mice with endothelial cells marked with β-gal expressed under the control of endothelial markers (Flt1 or Tie2).
Most recent studies on cell migration to developing gonads reassemble fetal wild-type mouse gonads with mesonephros derived from transgenic mice with constitutional expression of fluorescent protein GFP (Bogani et al. 2009; Brennan et al. 2002; Combes et al. 2009a; Cool et al. 2008; Cui et al. 2004; DeFalco et al. 2011; Hiramatsu et al. 2009; Nishino et al. 2001; Yao et al. 2004). After 24–64 h of in vitro culture, the organs are fixed, processed for immunolabeling, and viewed to detect green signal in the gonads. Yao et al. (2004) studied gonads isolated from the slider T. scripta reassembled with mouse mesonephros or mouse gonads and mesonephros in various sandwich combinations.
14.13 Separation of Cell Lines
The gonad consists of cells of many lines, which differ in origin, structure, function, and gene expression pattern. The first analyses of gene expression were based on RNA isolated from whole developing gonads and thus genes expressed in many different cell lines were studied. Nowadays, the use of cell markers facilitates the isolation of a single cell line from the heterogeneous mass of tissues forming the gonad. Moreover, even a single cell can be removed from the whole organ or from a section on a microscopic slide (LMD, laser microdissection) and its transcriptome can be studied. For instance, genes expressed in epithelial cells covering developing gonads were studied after laser dissection of frozen sections (Kusaka et al. 2010).
Isolation of a specific cell line from an organ was propelled by the application of transgenic animals with the expression of a reporter gene in a cell type of interest. This method requires isolation of the gonads, their enzymatic digestion, and cell sorting (e.g., fluorescence-activated cell sorting—FACS or cell isolation with magnetic beads). Tissue digestion is usually conducted by treatment with one or more enzymes at 37 °C followed by mechanical dissociation (pipetting, filtering) and cell resuspension in medium or PBS. Trypsin/EDTA, collagenase, DNase, and dispase are enzymes used for dissociation of developing gonads (Beverdam and Koopman 2006; Childs et al. 2011; DeFalco et al. 2014, 2015; Jameson et al. 2012; McLaren and Southee 1997; Molyneaux et al. 2003; Munger et al. 2013; Nef et al. 2005; Nishino et al. 2001; Qing et al. 2008; Rolland et al. 2011; Smith et al. 2008a; Wilhelm et al. 2005).
FACS is a specialized type of flow cytometry that can separate a heterogeneous mixture of cells of various lines into two or more components based upon specific light emission of the cells. For example, in this method a fetal gonad of a transgenic mouse with expression of GFP under control of the Sox9 gene (pre-Sertoli cell marker) can be dissected and digested; the mixture of cells can be sorted with FACS and two pools of cells (GFP+ pre-Sertoli cells and the rest—GFP–) are obtained. Such isolated cells (of the same type) can be used for protein or RNA isolation and thus for gene expression analysis. FACS was used to separate cell lines from developing gonads in several studies (Anderson et al. 1999; Bendel-Stenzel et al. 2000; Beverdam and Koopman 2006; DeFalco et al. 2014, 2015; Jameson et al. 2012; Nef et al. 2005; Nishino et al. 2001; Rolland et al. 2011). In an excellent study, Jameson et al. (2012) presented a method to separate the main cell lines from developing mouse gonads. They used several transgenic mouse strains to specifically label cell lines; EGFP under control of the Sry promoter (Sry:EGFP) resulted in a green signal in XY but also XX supporting cells and enabled the separation of these cells (Albrecht and Eicher 2001); Sox9:EGFP can be used to isolate XY supporting cells, Mafb:EGFP and α-Sma:EYFP to isolate stromal/interstitial cells, Flk1:mCherry to isolate endothelial cells, and Oct4:EGFP to isolate germ cells. As an alternative method, cell purification via magnetic beads was used to isolate primordial germ cells (PGCs) (Pellegrini et al. 2008; Pesce and De Felici 1995; Qing et al. 2008). In this technique, anti-SSEA1 (a marker of PGCs and other stem cells) primary antibodies are added to a mix of cells after organ digestion, and then magnetic beads coated with secondary antibodies are introduced; the beads facilitate isolation of PGCs. Pellegrini et al. (2008) used CD117 magnetic microbeads to isolate Kit-positive spermatogonia.
Previously, several methods allowing gonadal cell isolation and separation were developed. Separation of somatic and germ cells isolated from fetal or newborn gonads is based on differences in their adhesion to a culture dish (O and Baker 1978). Somatic cells have a tendency to adhere to a culture dish, whereas the germ cells do not; thus, during rinsing the germ cells can be removed from the culture. A method for Sertoli cell isolation via a sequence of gradual enzymatic treatment and centrifugation of tissue has been described (Chang et al. 2008; Chapin et al. 1987; Gassei et al. 2008; Hadley et al. 1985; Mackay et al. 1999; Willerton et al. 2004). Enzymes used to digest gonadal tissue include collagenase, DNase, trypsin, and hyaluronidase. Tissue dissociation via enzyme and EDTA treatment was used for germ cell isolation from gonads (Barrios et al. 2010; De Felici and McLaren 1982; Di Carlo et al. 2000; Pellegrini et al. 2008).
14.14 Gene Expression Analysis
Contemporary analyses of gene expression take advantage of a series of techniques that provide a wealth of knowledge about an organ or cell of interest. From early development, the gonad is composed of many cells with different gene expression patterns. The large number of genes expressed in a given cell complicates gene expression analysis. For instance, more than 15,000 genes are expressed in human testis (Ramsköld et al. 2009). Some gene expression techniques allow an analysis of only one or a few genes (low-plex analyses such as the use of reporter genes, Northern blot, ISH, RT-PCR, qPCR, RNase protection assay); however, other techniques enable simultaneous analyses of the expression of many or all genes in a given organ, tissue, or even a single cell (high-plex analyses such as SAGE, DNA microarray, RNA-Seq). Most of these methods have been crucial for studying the mechanisms driving gonadal development.
In this chapter, several techniques of gene expression analysis were already mentioned, such as in situ hybridization (ISH) and the use of reporter genes. Immunolocalization of proteins also provides data on spatiotemporal gene expression. These three methods are based on gene expression product analysis in situ, in a single cell, tissue, or whole organ. Another series of techniques of gene expression analysis entails RNA isolation from the whole organ, tissue, or cell and is conducted ex situ, “in a test tube.” In studies on gonadal development, the gonads are dissected and immediately immersed in a solution (usually RNAlater) permeating the tissue and thereby stabilizing and protecting RNA due to the inactivation of ubiquitous RNases. Later the gonads are homogenized and RNA is isolated using column-based techniques or rarely using oligo(dT)-conjugated magnetic beads (Anderson et al. 1999). The most commonly used solution for RNA isolation is TRIzol (Best et al. 2008; Cupp et al. 2003; Dumond et al. 2011; Hiramatsu et al. 2009; Hummitzsch et al. 2013; Koubova et al. 2006; Maatouk et al. 2008; Molyneaux et al. 2003; Munger et al. 2013; Nef et al. 2003; Schmahl et al. 2004; Smith et al. 2008a; Tang et al. 2008). The RNA isolation method via TRIzol is based on guanidinium thiocyanate–phenol–chloroform extraction. The amount of mRNA in fetal gonads is very low and sometimes pooling of samples from several individuals is necessary. The dissection of a gonad and its separation from adjoining tissues (mesonephros) is especially difficult at early stages. In this case, RNA isolation is facilitated by removing the gonads along with adjoining tissues and initially immersing them in RNA later solution in which the gonads harden and become easy to separate from the mesonephros. In the meantime, the individuals can be genotyped and then pooled according to sex and stages. After tissue homogenization followed by column-based RNA isolation, RNA suspended in RNase-free water can be stored frozen. Total RNA is obtained after isolation (including all types of RNA of which only 1–5 % constitutes mRNA representing the expression of protein-coding genes). In order to analyze protein-coding genes, mRNA can be isolated from total RNA by hybridizing the mRNA poly(A) tail to oligo(d)T connected to a carrier (e.g., magnetic beads) (Anderson et al. 1999; Parma et al. 2006).
14.14.1 Northern Blot
Among ex situ, low-plex methods (i.e., analysis of one or a few transcripts after RNA isolation from cells), the Northern blot assay (introduced in 1977) enables identification of individual RNA in a heterogeneous RNA sample separated by electrophoresis. In this method, RNA samples are separated by agarose gel electrophoresis and then transferred onto a nylon membrane through a vacuum or capillary blotting system. RNA is immobilized by linkage to the membrane using UV light or heat and a labeled probe is applied onto the membrane leading to the visualization of a transcript. A probe consists of cDNA or cRNA complementary to the sequence of interest and can be labeled with radioactive (32P) or a chemiluminescent signal. The Northern blot assay was useful in studying the expression of genes in developing gonads (Gallardo et al. 2007; Higgy et al. 1995; Pask et al. 2000; Seligman and Page 1998).
14.14.2 RNase Protection Assay
The RNase protection assay (RPA) is another method used for the identification of individual RNA molecules accompanied by the degradation of other RNAs with enzymes. In RPA, isolated RNA is mixed with a labeled complementary probe (cRNA or cDNA) and hybridized to form double-stranded structures (cRNA:mRNA or cDNA:mRNA). Then RNases (e.g., RNase A and/or RNase T1) are added to the mixture. The enzymes digest only single-stranded molecules leaving double-stranded molecules intact. The RNA molecules from double-stranded hybrids are analyzed using gel electrophoresis. This method helped to describe the exact timing of Sry expression in the developing gonads of mice (Hacker et al. 1995) and also the expression of other genes (Hacker et al. 1995; Morais da Silva et al. 1996; Smith et al. 2009; Soyal et al. 2000).
PCR (polymerase chain reaction) (known since 1971) methods are also commonly used to analyze gene expression. Standard PCR and its implementation in genotyping were previously described in this chapter. However, gene expression analysis uses RT-PCR and qRT-PCR. The basis of all types of PCR is amplification of a given sequence using specific primers, a polymerase enzyme, a template (DNA or RNA), and reaction substrates. Amplification proceeds by repeated cycling of three steps: denaturation (at ~95 °C the DNA helix is split into single threads), annealing (at 45–65 °C primers bind to complementary sequences), and extension (at ~72 °C polymerase extends primers by adding nucleotides in a sequential manner). The products of amplification are detected at a subsequent stage.
14.14.3 PCR-Based Methods of Gene Expression Analysis
In RT-PCR (reverse transcription PCR), RNA as a template is converted into complementary cDNA using reverse transcriptase (RT) and resulting cDNA is amplified as in typical PCR. Gene expression data are collected by analysis of bands after agarose gel electrophoresis. RT-PCR is commonly used to study gene expression in gonadogenesis (Abramyan et al. 2009; Albrecht and Eicher 2001; Anderson et al. 1999; Barrionuevo et al. 2006; Barrios et al. 2010; Bendel-Stenzel et al. 2000; Best et al. 2008; Beverdam and Koopman 2006; Bradford et al. 2009; Chang et al. 2008; Chassot et al. 2008; Childs et al. 2011; Coveney et al. 2008; DeFalco et al. 2014; Dumond et al. 2011; Dupont et al. 2000; Hummitzsch et al. 2013; Jameson et al. 2012; Kocer et al. 2008; Koubova et al. 2006; Li et al. 2012; Molyneaux et al. 2003; Moniot et al. 2009; Nef et al. 2003, 2005; Nishino et al. 2001; Oshima et al. 2005; Oulad-Abdelghani et al. 1996; Parma et al. 2006; Rolland et al. 2011; Schmahl et al. 2004; Smith et al. 2008a, b; Tang et al. 2008; Wilhelm et al. 2005; Yoshimoto et al. 2008, 2010).
In qRT-PCR (quantitative reverse transcriptase PCR or real-time PCR, qPCR), the first stage consists of converting RNA into cDNA via PCR with reverse transcriptase; however, during the following steps the fluorescently labeled primers or probes are used, enabling monitoring of the amount of product during amplification. qRT-PCR is commonly used to study gene expression during gonadogenesis (Abramyan et al. 2009; Barrionuevo et al. 2006; Barske and Capel 2010; Beverdam and Koopman 2006; Bogani et al. 2009; Bouma et al. 2005, 2007; Bradford et al. 2009; Britt et al. 2002; Chang et al. 2008; Chassot et al. 2008; Chen et al. 2012; Childs et al. 2011; Combes et al. 2011; Cupp et al. 2003; DeFalco et al. 2014; Hiramatsu et al. 2009; Houmard et al. 2009; Koubova et al. 2006; Li et al. 2012; Liu et al. 2015; Maatouk et al. 2008; Manuylov et al. 2007; Molyneaux et al. 2003; Moniot et al. 2009; Munger et al. 2013; Nef et al. 2003, 2005; Nicol and Yao 2015; Parma et al. 2006; Rolland et al. 2011; Ross et al. 2009; Smith et al. 2008a, b, 2009; Tang et al. 2008; Tevosian et al. 2002; Wilhelm et al. 2005). In the majority of studies of gonad development, primers labeled with SYBR Green fluorochrome are used although TaqMan probes have also been applied (Bogani et al. 2009; Liu et al. 2015; Smith et al. 2008a, b). Multigene qRT-PCR is used to analyze numerous genes simultaneously (Bouma et al. 2004, 2005, 2007). The accuracy of results obtained by qRT-PCR strongly depends on accurate normalization using stably expressed genes (Guenin et al. 2009). In studies on gonad development, normalization is usually carried out with Gapdh (Chang et al. 2008; DeFalco et al. 2014; Dumond et al. 2011; Hiramatsu et al. 2009; Liu et al. 2015; Manuylov et al. 2007; Nef et al. 2003, 2005; Moniot et al. 2009; Munger et al. 2013), Hprt1 (Albrecht and Eicher 2001; Bogani et al. 2009; Chassot et al. 2008; Coveney et al. 2008; Nef et al. 2003; Smith et al. 2008a,2009; Tang et al. 2008), 18S rRNA (Liu et al. 2015; Wilhelm et al. 2005), Tbp (Barrionuevo et al. 2006), or Actb (Houmard et al. 2009).
14.14.4 SAGE
SAGE (serial analysis of gene expression) developed in 1995 is a high-plex ex situ method, enabling analysis of the transcriptome and the expression of even thousands of genes in a single assay (Yamamoto et al. 2001). In the SAGE method, RNA is reverse transcribed using biotinylated primers, and then the biotin-labeled cDNAs are bound by streptavidin beads and cleaved by restriction enzymes (anchoring enzyme, AE). In result, partially digested cDNAs of different length remain on the beads; this material is divided into two portions (A and B) and exposed to one of two adapter oligonucleotides (A or B). After ligation, cDNAs with adapters are removed from the beads using tagging enzyme (TE), and in effect short tags of original cDNAs are detached from beads. Cleaved cDNA tags are repaired with DNA polymerase to produce blunt ends; tags from A and B samples are mixed and ditags form due to blunt ends. Ditags are then cleaved with AE and linked with other ditags to form a cDNA concatemer; concatemers are transformed into bacteria and thus amplified. Isolated concatemers are sequenced using high-throughput DNA sequencers; bioinformatics methods can quantify the recurrence of individual tags (Lee et al. 2010), yielding quantitative data. SAGE methods were used to study the transcriptome in cell lines in developing gonads (Chan et al. 2006; Lee et al. 2006; Wu et al. 2004).
14.14.5 Microarray
Another high-plex method is the microarray technique (developed in 1983) that is based on applying a sample onto a solid surface (usually a glass slide) with an array of probes (Trevino et al. 2007). A series of microarray techniques can be used for analysis of gene expression (DNA microarray), protein identification (protein microarray), tissue imaging (tissue array), cell biology (cellular microarray), etc. All subtypes of microarrays miniaturize and multiplex the analysis ensuring the study of multiple samples simultaneously. Gene expression analysis via this method has the capacity to cover thousands of transcripts at once on a single microarray (or gene chip) wherein each spot contains multiple identical strands of DNA, representing a single gene. In gonadogenesis studies, DNA microarrays have provided a large amount of data and have significantly contributed to knowledge on gene expression in developing gonads. DNA microarray techniques require RNA isolation and its conversion into cDNA through reverse transcription; cDNA is labeled with fluorochrome, then applied onto chips, and hybridized to probes (cDNA or oligonucleotides) previously attached to the chip; after rinsing, only labeled sequences that bound complementary probes remain on the chip and signals emitted by them are detected. In gonadogenesis studies, microarrays were used for gene expression analysis of RNA isolated from whole developing gonads and thus from many different cell lines (Combes et al. 2011; Coveney et al. 2008; Gallardo et al. 2007; Garcia-Ortiz et al. 2009; Grimmond et al. 2000; Houmard et al. 2009; Liu et al. 2015; Munger et al. 2013; Nicol and Yao 2015; Ottolenghi et al. 2007; Rolland et al. 2011; Small et al. 2005), whereas other microarray studies were conducted on particular gonadal cell lines, e.g., on germ cells (Jameson et al. 2012; Molyneaux et al. 2003; Pan et al. 2005), pre-Sertoli and pre-granulosa cells (Beverdam and Koopman 2006; Bouma et al. 2007; Jameson et al. 2012; Nef et al. 2005), epithelial cells of gonadal primordium (Hummitzsch et al. 2013; Kusaka et al. 2010), and interstitial and endothelial cells (Jameson et al. 2012). The microarray technique was also used to identify noncoding RNAs in developing gonads (Chen et al. 2012).
14.14.6 RNA Sequencing
RNA sequencing (RNA-Seq) is one of the latest high-throughput methods used for gene expression analysis (from 2008). RNA-Seq involves next-generation sequencing (NGS) to reveal a snapshot of RNA presence and quantity. In this method, mRNA is isolated and converted to cDNA; sequencing adapters are added and then a cDNA library is prepared; sequencing of cDNA is performed using an NGS platform. RNA-Seq has been used in studies on sex determination and gonad development (Ayers et al. 2015; Gong et al. 2013).
14.15 Protein Analysis Methods
Proteins can be identified in situ, i.e., in a given organ, tissue, or cell, and examples of such methods (immunohistochemistry, IHC, or immunocytochemistry, ICC) were previously described in this chapter. However, there is also a wide range of ex situ methods based on protein analysis in vitro after their isolation from organs, tissues, or cells. In many trials, it is crucial to isolate proteins separately from the nucleus or the cytoplasm (Miyamoto et al. 2008; Niksic et al. 2004; Oulad-Abdelghani et al. 1996; Tremblay and Viger 2003).
14.15.1 Western Blot
There are thousands of different proteins in a single cell type; thus before analysis proteins are usually separated using the Western blot technique (used since 1979). In this method, proteins are separated via vertical electrophoresis in a polyacrylamide gel. An anionic detergent, SDS (sodium dodecyl sulfate), is added to a sample to linearize proteins and to impart an even distribution of charge per unit mass which results in proteins being fractioned in the electric field only by their size. Electrophoresis in a polyacrylamide gel in the presence of SDS is called SDS-PAGE. Analogically to nucleic acid electrophoresis, in Western blot smaller proteins migrate from the cathode (–) toward the anode (+) faster than larger proteins. Proteins separated via electrophoresis are transferred onto a membrane (usually nitrocellulose) using an electric current. The proteins of interest are identified using specific primary and secondary antibodies; the latter are usually linked to horseradish peroxidase (HRP) or alkaline phosphatase (AP). The most commonly used HRP enzyme cleaves a chemiluminescent substrate (e.g., luminol); the intensity of the luminescent signal is proportional to the amount of protein. Western blot analysis was useful in many studies on gonadal development (Bradford et al. 2009; Barrios et al. 2010; Chang et al. 2008; Dumond et al. 2011; Dupont et al. 2000; Kocer et al. 2008; Miyamoto et al. 2008; Oulad-Abdelghani et al. 1996; Paranko and Pelliniemi 1992; Parma et al. 2006; Salas-Cortés et al. 1999; Tremblay and Viger 2003).
14.15.2 Phosphorylation Analysis
Proteins are modified by, e.g., phosphorylation, glycosylation, SUMOylation, etc. Such posttranslational modifications change the function or determine the subcellular location of a protein. In gonadogenesis studies, phosphorylation of proteins was tested using antibodies specifically binding phosphorylated forms of a given protein and has revealed the presence and subcellular location of unphosphorylated vs. phosphorylated proteins. Phosphorylation of proteins can also be studied using recombinant proteins under in vivo and in vitro conditions (Tremblay and Viger 2003).
14.16 Methods for Studying Interactions Between Molecules
14.16.1 EMSA
EMSA (electrophoretic mobility shift assay) is a method used to study protein–DNA or protein–RNA interactions and to test if a protein is capable of binding a given sequence of DNA or RNA. In this technique, a mixture of protein–DNA or protein–RNA is electrophoretically separated on a polyacrylamide or agarose gel for a short period. Protein–DNA interactions (binding) result in an increase of molecular weight and lower velocity of migration in an electric current. Thus, a shift in bands indicates an interaction between a protein and given sequence of DNA (or RNA). Protein interactions with DNA were studied via EMSA to elucidate mechanisms of gonadogenesis (Bernard et al. 2008; Miyamoto et al. 2008; Wilhelm et al. 2007; Yoshimoto et al. 2010).
14.16.2 Immunoprecipitation
Immunoprecipitation (pull-down techniques) encompasses a group of similar techniques enabling the precipitation of a protein out of a solution using antibodies specifically binding to a particular protein.
Individual protein immunoprecipitation (IP) involves using specific antibody that binds a particular protein in a mixture of many different proteins (Kaboord and Perr 2008). Proteins are usually precipitated using agarose beads that are coated with protein A or G which show affinity to immunoglobins. In studies on gonad development, IP is typically used to isolate recombinant proteins (Barrios et al. 2010; Tremblay and Viger 2003).
Protein complex immunoprecipitation (Co-IP) is useful in identifying protein–protein interactions. In this technique, an antibody specifically binding to a particular protein that is thought to form a complex with other proteins is added to a mixture of different proteins (e.g., all proteins isolated from an organ). After antibody binding, the whole protein complex is pulled out of the mixture. The complex is disintegrated and proteins of the complex are separated via SDS-PAGE. Then the separated proteins are isolated from bands and identified using liquid chromatography-mass spectrometry (LC-MS). Co-immunoprecipitation has been valuable in studies on direct interactions between proteins involved in sex determination and other aspects of gonad development, such as interactions between factors including SOX9, GATA4, WT1, FOXL2, vinexin, and E-cadherin (Di Carlo et al. 2000; Matsuyama et al. 2005; Miyamoto et al. 2008; Uhlenhaut et al. 2009).
Chromatin immunoprecipitation (ChIP) can determine DNA binding sites in the genome for a protein of interest. Thus, this technique reveals DNA–protein interactions and is especially useful in studying transcription factors and histones that bind DNA. In this method, formaldehyde is used to cross-link proteins to DNA and then cells are lysed and DNA is fragmented via sonication. Specific antibodies are used to precipitate a particular protein in a complex with specific DNA sequences. Subsequently, the isolated DNA–protein complexes are heated to release cross-linking and purified DNA is then amplified and sequenced. Considering the great number of binding sites in a genome, DNA microarray techniques (ChIP-on-chip) can be useful in detecting all binding sites for a given protein. In studies on gonad development, ChIP has been useful in studying factors binding to promoters, e.g., SOX9 binding to the Pdgs promoter, GATA4 to the Sry promoter, and SRY and SOX9 binding to the Cerbelin4 promoter (Bradford et al. 2009; Miyamoto et al. 2008; Wilhelm et al. 2007).
The RNA immunoprecipitation (RIP) method examines protein–RNA interactions. Cells are lysed and the protein of interest along with bound RNA is precipitated using specific antibodies. RNA is separated from protein, extracted, reverse transcribed, and analyzed using cDNA sequencing or RT-PCR. RNA–protein interactions were intensively studied with this method in sex determination of Drosophila (Vied et al. 2003).
14.17 Genetic Engineering of Cells Cultured In Vitro
14.17.1 Tagged Proteins (Fusion Proteins) and In Vitro Cell Culture
A pull-down assay can be facilitated by using tagged proteins. A protein tag is a sequence genetically grafted to the N- or C-terminus. The recombinant protein, recognized by a specific antibody due to the presence of a tag, is easy to detect or purify. Several tags can be used for different purposes. One of the most commonly applied protein tags is glutathione-S-transferase (GST), which was used to study such proteins as SRY or GATA4 (Bernard et al. 2008; Best et al. 2008; Salas-Cortés et al. 1999; Tremblay and Viger 2003). A protein of interest along with bound GST constitutes a fusion protein, purified via the pull-down assay owing to GST affinity to GSH (glutathione); usually GSH-coated beads are used. After pull-down, a tag can be removed by a proteolytic enzyme. Other protein tags used in studies on gonad development include Myc-tag (Bradford et al. 2009; Kim et al. 2006; Sekido et al. 2004), FLAG-tag (Bernard et al. 2008; Matsuyama et al. 2005), HA-tag (Bernard et al. 2008), His-tag (Miyamoto et al. 2008), and TRX-tag (Dumond et al. 2011). Such fusion proteins can be purified using antibodies specifically binding the tags. Fluorescent protein tags, such as GFP or YFP, can also be used to visualize a protein and to study its subcellular localization. As previously mentioned, GFP and YFP are used to study a protein in the organ or to visualize or isolate a given cell line; however, these tags can also be used to identify a recombinant protein in in vitro cell culture, e.g., Cerbelin4 and Sdmg1 in a study on mouse sex determination (Best et al. 2008; Bradford et al. 2009). Recombinant proteins may also be used to produce specific antibodies (Best et al. 2008; Matsuyama et al. 2005; Salas-Cortés et al. 1999).
Fusion proteins are synthesized in cells under in vitro conditions via cell transfection. The most commonly used cell lines in studies on sex determination and mechanisms of gonad development are HEK293 (Barrios et al. 2010; Bernard et al. 2008; Munger et al. 2013; Wilhelm et al. 2007), HeLa (Moniot et al. 2009; Ohe et al. 2002; Salas-Cortés et al. 1999; Wilhelm et al. 2005), COS7 (Li and Kim 2004; Ohe et al. 2002; Wilhelm et al. 2005), NT2/D1 (Bernard et al. 2008; Moniot et al. 2009; Ohe et al. 2002), NIH3T3 (Best et al. 2008), C3H10T1/2 (Matsuyama et al. 2005), MCF7 (Bradford et al. 2009), and Sertoli cells SK11 (Best et al. 2008). These cell lines are usually maintained in DMEM medium supplemented with 10 % FBS at 37 °C and 5 % CO2. Lipofection is typically applied to transfect these cells with an exogenous genetic construct; here liposomes containing genetic material merge with the cell membrane. In some cases, fusion proteins were synthesized in E. coli (Best et al. 2008; Dumond et al. 2011; Wilhelm et al. 2007).
Cells cultured in vitro are sometimes maintained on a layer of cells that produce nutrients supporting cell growth. Such feeder cells include embryonic primary fibroblasts (EMFI) used for culture of mesonephros-derived migrating cells (Nishino et al. 2001) or the STO fibroblast cell line used for culture of PGCs (Di Carlo et al. 2000). Cells can also be cultured on a three-dimensional matrix scaffold, e.g., Matrigel. A 3D cell culture was used to culture primary Sertoli cells to reconstitute their 3D aggregations (Gassei et al. 2008).
14.17.2 RNA Interference
Cells cultured in vitro are useful in studying gene function via silencing of expression, i.e., genetic knockdown (KD). For example, RNA interference (RNAi) is used to silence gene expression. In studies on the role of genes in sex determination, shRNA (small hairpin RNA) was introduced via viral vectors or lipotransfection to cells cultured in vitro to silence the expression of a gene of interest. shRNA binds the RISC complex that recognizes mRNA due to the antisense sequence of shRNA and degrades mRNA leading to a decrease in gene expression. For instance, Munger et al. (2013) used lentiviruses to introduce shRNA to gonadal primary cells cultured in vitro. shRNA was designed to silence the expression of candidate genes presumably involved in sex determination in mice. Best et al. (2008) used shRNA introduced into Sertoli SK11 cells via lipotransfection in order to silence the expression of the Sdmg1 gene. Smith et al. (2009) used an avian retroviral vector to introduce shRNA to chicken egg to silence Dmrt1 gene expression, which resulted in feminization of ZZ individuals and proved the role of Dmrt1 in avian sex determination (Smith et al. 2009).
14.17.3 Labeling of Cells Cultured In Vitro (Immunocytochemistry)
Proteins can be detected in cells cultured in vitro using antibodies. In immunocytochemical labeling (ICC), cells attached to microscope slides are usually fixed with 2–4 % PFA, permeabilized with detergent, blocked with BSA, incubated with primary and then secondary antibodies, and viewed (Barrios et al. 2010; Best et al. 2008; Gassei et al. 2008; Qing et al. 2008; Willerton et al. 2004). Alternatively, incubation of non-permeabilized cells with antibodies may precede fixation (Best et al. 2008).
14.18 Genetically Modified Mice
Genetic modification is a powerful tool for studies in the field of developmental biology. The first transgenic mouse was obtained in 1974 by Rudolf Jaenish via viral DNA insertion into an early-stage mouse embryo (Jaenisch and Mintz 1974). Since then, genetically modified mice have revolutionized studies on gonad development. The aforementioned mice with expression of GFP or other reporter genes are useful for studying gene expression and to trace the organogenesis process. A reporter gene such as GFP or LacZ can be expressed in all cells of an individual (Capel et al. 1999; Combes et al. 2009b; Martineau et al. 1997; Merchant-Larios and Moreno-Mendoza 1998; Nishino et al. 2001; Qing et al. 2008). Alternatively, a reporter gene can be expressed only in a cell line of interest; e.g., expression of EGFP under control of the Oct4 promoter is useful to trace and even isolate primordial germ cells (Anderson et al. 1999; Maatouk et al. 2013; Molyneaux et al. 2001; Rolland et al. 2011). A cell line giving rise to Sertoli cells can be visualized and isolated employing strains with EGFP under control of Sf1 (Beverdam and Koopman 2006; Nef et al. 2005), Sry (Albrecht and Eicher 2001; Kim et al. 2006), or Sox9 (Nel-Themaat et al. 2009). Lists of numerous transgenic strains with labeled cell lines facilitating studies on gonad development have been published (Brennan et al. 2002; DeFalco et al. 2011, 2014, 2015; Jameson et al. 2012).
Transgenic mice also facilitate the functional annotation of a gene of interest. The best method to study gene function is to delete this gene from a genome or introduce a nonfunctional sequence of a gene (knockout, KO). There are two types of KO: constitutive and conditional. Ablation of gene expression in all cells of an individual determines a constitutive knockout and has been used to study the function of Wnt4, Foxl2, and Fgf9 in gonadogenesis (Coveney et al. 2008; Kim et al. 2006; Matzuk et al. 1995; Ottolenghi et al. 2005). However, a disadvantage of this method is the lethality caused by deletion of some genes, precluding analysis due to the early death of mutant embryos. A conditional knockout can sidestep lethality after gene deletion. In the conditional knockout, gene expression is depleted only in a particular cell type. There is a series of methods of such tissue-specific knockout, among which the Cre-loxP system is commonly applied in embryological studies. In this method, Cre recombinase, expressed under the control of a particular promoter, cleaves and thus deletes a given gene that is flanked with loxP sequences. This method has been used to study mouse gonadal development (Bagheri-Fam et al. 2008; Barrionuevo et al. 2006, 2009; Bouma et al. 2005; Buscara et al. 2009; Chang et al. 2008; Chassot et al. 2008; Coveney et al. 2008; Liu et al. 2009, 2015; Maatouk et al. 2008, 2013; Moniot et al. 2009; Tang et al. 2008). A gene knockout in the precursors of Sertoli and granulosa cells is possible due to Cre expression under the control of promoters of steroidogenic factor 1—Sf1 (Chassot et al. 2008; Liu et al. 2009; Maatouk et al. 2008; Tang et al. 2008) and cytokeratin 19—Ck19 (Bagheri-Fam et al. 2008; Barrionuevo et al. 2006; Moniot et al. 2009); however, a knockout in differentiating Sertoli cells can be achieved with the aid of Amh-Cre (Barrionuevo et al. 2009; Chang et al. 2008; Moniot et al. 2009). The Oct4-Cre strain permits gene deletion in the germ cell line (Sabour et al. 2014). In these studies, genes such as Sox9, Fgf9, Fgfr2, Wt1, and Ctnnb were successfully deleted in a tissue-specific manner. A tissue-specific knockout is usually induced by tamoxifen injected into an individual. Cre recombinase is fused with estrogen receptor (ER), and after intraperitoneal injection of tamoxifen, this compound binds ER and activates Cre recombinase which deletes a specific gene. Tamoxifen-inducible recombination has improved investigations owing to the possibility of controlling the timing of gene deletion.
Alternatively, knowledge on gene function can be obtained by the introduction of additional copies of a gene into a genome leading to the overexpression of a gene (knockin, KI). For instance, the effects of overexpression of goat Rspo1 introduced into mice deprived of Rspo1 gene expression (Rspo1 –/–, Rspo1-KO) have been studied (Buscara et al. 2009). The KI method has also revealed the function of Gata4 in gonad development after the insertion of a sequence encoding GATA4 with disrupted potentiality to bind the FOG2 cofactor (Tevosian et al. 2002). Insertion of reporter gene sequences into the genome is also an example of gene knockin.
There are two main methods to obtain genetically modified organisms. First, transgenic mice can be generated via injection of genetic material to a male pronucleus in the zygote (Albrecht and Eicher 2001; Anderson et al. 1999; Buscara et al. 2009). Second, a transgene can be introduced via injection or electroporation to ES cells (embryonic stem cells) that are then introduced into a blastocyst. The second method was used to generate Cre-loxP knockout mice in studies on gonad development (Chassot et al. 2008; Dupont et al. 2000; Maclean et al. 2009; Nel-Themaat et al. 2009; Ottolenghi et al. 2005).
14.19 Methods for Studies of Steroidogenesis
One of the most crucial processes during gonad development is differentiation of steroidogenic cells and the onset of sex steroid synthesis orchestrating the establishment of secondary sex features. The first method to study steroidogenesis is identification of steroidogenic enzymes via immunolabeling or their expression via RT-PCR, qRT-PCR, or in situ hybridization. The most important steroidogenic enzymes for studies on gonad development include cytochrome P450scc (CYP11a1), 3β-hydroxysteroid dehydrogenase (3β-HSD), 17β-hydroxysteroid dehydrogenase (17β-HSD), 3α-hydroxysteroid dehydrogenase (3α-HSD), and cytochrome P450 17A1 (CYP17a1), which mark steroidogenic cells such as Leydig and theca cells and aromatase (CYP19a1) expressed mainly in granulosa cells (Chassot et al. 2008; Payne et al. 1992; Tang et al. 2008). Immunolabeling or gene expression analysis, however, only suggests steroid hormone production; other methods are needed to prove sex steroid synthesis. The assessment of hormone synthesis can be done by investigation of enzymatic activity via histochemical methods or by assaying hormone levels in the blood.
The histochemical method for studying steroidogenic enzyme activity is usually carried out on frozen sections containing tissue gently fixed in 4 % PFA. A staining solution is applied onto the sample to conduct a chromogenic reaction (Baillie et al. 1965, 1966). The staining solution contains a substrate (e.g., dehydroepiandrosterone for 3β-HSD; testosterone for 17β-HSD; androsterone for 3α-HSD), nicotinamide adenine dinucleotide (NAD+), and NBT/BCIP. Enzymatic activity gives rise to NADH and H+, which leads to reduction of yellow NBT to blue insoluble diformazan. A blue signal indicates enzyme activity.
Steroid hormone concentration can be assessed via the RIA (radioimmunoassay) method (Habert and Picon 1982; Migrenne et al. 2012). Rosalyn Yalow, the inventor of this method, was awarded the Nobel Prize in 1977. RIA is a very sensitive technique measuring the concentration of hormones, e.g., in blood plasma. In this method, a known quantity of antigen (hormone) is labeled with the radioactive isotope of iodine attached to tyrosine. Radiolabeled antigen is mixed with a known quantity of antibody specifically binding the antigen. Then a tested sample with an unknown quantity of antigen is added to the mixture. The unlabeled antigens from the tested sample compete with the radiolabeled antigen for binding to antibodies. As the concentration of unlabeled antigen increases, it displaces radiolabeled antigen. This leads to a decrease of the ratio between antibody-bound radiolabeled antigen to free radiolabeled antigen. The radioactivity of unbound radiolabeled antigen in the supernatant is measured, indicating the quantity of hormone in the tested sample.
Another method allowing for the detection of steroid hormones or any other antigen is ELISA (enzyme-linked immunosorbent assay). In this method, a multi-well plate is used; each well is coated with capture antigen (indirect ELISA) or antibody that specifically binds a given antigen (sandwich ELISA) (Voller et al. 1978). A plate with antibody-coated wells is used to study the presence and concentration of antigen such as steroid hormone in blood plasma. Samples are applied to each well, and after incubation and rinsing, the enzyme-conjugated secondary antibody is added to trigger a signal, which permits the detection of the antigen. A plate is then analyzed in an imager, enabling quantitative measurement of the steroid. This method was used by Liu et al. (2015) as a steroid hormone multiplex immunoassay to study the concentrations of DHEA, estradiol, progesterone, and testosterone during the ovarian development.
14.20 Germ Cell Depletion
There is a series of studies based on gonadogenesis analysis after the removal of germ cells. Such studies elucidate the role of germ cells in gonadal development. Several methods are used to deplete germ cells in developing gonads. Usually chemical ablation of germ cells or mutated mice deprived of germ cells are used.
Chemical depletion of germ cells is typically acquired via busulfan treatment. Busulfan (butane-1,4-diyl dimethanesulfonate) is an alkylating compound that forms cross-links between the DNA bases A-G and G-G (Iwamoto et al. 2004). DNA cross-linking prevents DNA replication. Moreover, due to the presence of intrastrand cross-links, DNA cannot be repaired in the treated cells, leading to cell apoptosis. Originally it was shown that the intraperitoneal administration of busulfan in rat caused a lack of germ cells (Bollag 1953). Germ cell depletion and infertility is a serious drawback for busulfan use for human chemotherapy (López-Ibor and Schwartz 1986). Nevertheless, busulfan has been useful in studying the effects of germ cell depletion in many vertebrate species. To obtain sterile mouse embryos, pregnant females are intraperitoneally (IP) injected with a solution of busulfan (usually 20–50 mg/kg in 50 % DMSO) at 9.5, 10.5, or 11.5 dpc (Koubova et al. 2006; Liu et al. 2015; Maatouk et al. 2013; Nef et al. 2005). In chicken, busulfan treatment and germ cell depletion is done by administration of the solution onto the surface of the egg (DiNapoli and Capel 2007). In anuran amphibians, germ cell depletion can be achieved by the addition of busulfan dissolved in DMSO to water with living tadpoles to a final concentration of 0.12 mM (Piprek et al. 2012).
Another method of obtaining sterile gonads involves using mice with mutation. Here a viable allelic form W v in the dominant white spotting (W) is used in a homozygous configuration (W v/W v) resulting in almost complete sterility (Russell 1979). Sterile individuals (W v/W v) are obtained by mating heterozygotes W v/+. The factor originally termed W is the c-Kit proto-oncogene and is a type of transmembrane receptor tyrosine-protein kinase. Signaling from the c-Kit receptor is essential for primordial germ cell (PGC) survival (De Miguel et al. 2002). W v/W v mutants have been used to study sterile gonads (Chen et al. 2012; Garcia-Ortiz et al. 2009; Koubova et al. 2006; Rolland et al. 2011).
Uhlenhaut et al. (2009) used transgenic mice to delete oocytes in postnatal ovaries. A strain with Cre recombinase expression under the Gdf9 promoter was mated with the R26DTA strain. Gdf9 is expressed in oocytes and thus Cre recombinase in these cells removes the stop codon triggering the expression of diphtheria toxin receptor (DTR) in oocytes. Eight-week-old females were injected with diphtheria toxin (DT) in PBS (50 μg/kg). DT via DTR receptor disrupts protein synthesis and thus causes cell death. Here diphtheria toxin receptor-mediated targeted cell ablation led to the total absence of oocytes in postnatal ovaries.
14.21 Conclusion
In this chapter, I showed how various techniques have been used to investigate the mechanisms controlling gonad development and how scientific methods have evolved through the decades. Figure 14.1 shows how new discoveries in this field correlate with inventions of new scientific methods and tools. Initially, exclusively histological methods were used to study gonad development and thus only structural changes in gonad development were described. Advances in cell and molecular biology drastically accelerated the rate of embryological studies. Some of the most powerful tools revealing the molecular mechanisms controlling development involve techniques of gene expression analysis, such as reverse transcription PCR, quantitative PCR, in situ hybridization, and DNA microarray. Additionally, transgenic animals have significantly contributed to our understanding of the genetic control of gonadogenesis and sex determination. Knockout mice have been especially informative concerning the functions of some genes in these processes. In vitro culture of gonads was valuable for studying the development of these organs. This method helped trace cell migration shedding light on their origins and contributions to gonad structures. Immunolabeling and reporter genes have been applied in investigations of changes taking place in gonad structure during development. These methods enabled not only visualization of gonad structure, but also facilitated the isolation of particular cell lines and thus also advanced the study of gene expression in a given cell type. The issues raised in this chapter reveal that although we accumulated a lot of information about the mechanisms orchestrating gonad development and sex determination, there are still many developmental mechanisms awaiting discovery. It is known that molecular mechanisms are complex and involve a large number of interactions between the genes driving development. Further studies of gonad development will undoubtedly lead to the discovery of new mechanisms controlling the development of an individual into a male or female.
References
Abramyan J, Feng CW, Koopman P (2009) Cloning and expression of candidate sexual development genes in the cane toad (Bufo marinus). Dev Dyn 238:2430–2441
Albrecht KH, Eicher EM (2001) Evidence that Sry is expressed in pre-Sertoli cells and Sertoli and granulosa cells have a common precursor. Dev Biol 240:92–107
Allen BM (1904) The embryonic development of the ovary and testis of the mammalia. Am J Anat 3:89–146
Anderson R, Fassler R, Georges-Labouesse E et al (1999) Mouse primordial germ cells lacking β1 integrins enter the germline but fail to migrate normally to the gonads. Development 126:1655–1664
Avrameas S, Uriel J (1966) Method of antigen and antibody labelling with enzymes and its immunodiffusion application. C R Acad Sci Hebd Seances Acad Sci D 262:2543–2545
Ayers KL, Lambeth LS, Davidson NM et al (2015) Identification of candidate gonadal sex differentiation genes in the chicken embryo using RNA-seq. BMC Genomics 16:704
Bagheri-Fam S, Sim H, Bernard P et al (2008) Loss of Fgfr2 leads to partial XY sex reversal. Dev Biol 314:71–83
Baillie AH, Calman KC, Ferguson MM et al (1965) Histochemical demonstration od 20β-hydroxysteroid dehydrogenase. J Endocrinol 32:337–339
Baillie AH, Calman KC, Ferguson MM et al (1966) Histochemical utilization of 3α-, 6β-, 11α-, 12α-, 16α-, 17α-, 21- and 24-hydroxysteroids. J Endocrinol 34:1–12
Barr ML, Bertram EG (1949) A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 163:676–677
Barrionuevo F, Bagheri-Fam S, Klattig J et al (2006) Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biol Reprod 74:195–201
Barrionuevo F, Georg I, Scherthan H et al (2009) Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev Biol 15:301–312
Barrios F, Filipponi D, Pellegrini M et al (2010) Opposing effects of retinoic acid and Fgf9 on Nanos2 expression and meiotic entry of mouse germ cells. J Cell Sci 123:871–880
Barske LA, Capel B (2010) Estrogen represses SOX9 during sex determination in the red-eared slider turtle Trachemys scripta. Dev Biol 341:305–314
Bendel-Stenzel MR, Gomperts M, Anderson R et al (2000) The role of cadherins during primordial germ cell migration and early gonad formation in the mouse. Mech Dev 91:143–152
Bernard P, Sim H, Knower K et al (2008) Human SRY inhibits beta-catenin-mediated transcription. Int J Biochem Cell Biol 40:2889–2900
Best D, Sahlender DA, Walther N et al (2008) Sdmg1 is a conserved transmembrane protein associated with germ cell sex determination and germline-soma interactions in mice. Development 135:1415–1425
Beverdam A, Koopman P (2006) Expression profiling of purified mouse gonadal somatic cells during the critical time window of sex determination reveals novel candidate genes for human sexual dysgenesis syndromes. Hum Mol Genet 15:417–431
Bilinski SM, Jaglarz MK, Dougherty MT, Kloc M (2010) Electron microscopy, immunostaining, cytoskeleton visualization, in situ hybridization, and three-dimensional reconstruction of Xenopus oocytes. Methods 51:11–19
Birk OS, Casiano DE, Wassif CA et al (2000) The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 403:909–913
Bishop CE, Whitworth DJ, Qin Y et al (2000) A transgenic insertion upstream of sox9 is associated with dominant XX sex reversal in the mouse. Nat Genet 26:490–494
Bogani D, Siggers P, Brixey R (2009) Loss of mitogen-activated protein kinase kinase kinase 4 (MAP3K4) reveals a requirement for MAPK signalling in mouse sex determination. PLoS Biol 7(9):e1000196
Bollag W (1953) The effect of myleran on rat gonads. Experientia 9:268
Bouma GJ, Hart GT, Washburn LL et al (2004) Using real time RT-PCR analysis to determine multiple gene expression patterns during XX and XY mouse fetal gonad development. Gene Expr Patterns 5:141–149
Bouma GJ, Albrecht KH, Washburn LL et al (2005) Gonadal sex reversal in mutant Dax1 XY mice: a failure to upregulate Sox9 in pre-Sertoli cells. Development 132:3045–3054
Bouma GJ, Washburn LL, Albrecht KH et al (2007) Correct dosage of Fog2 and Gata4 transcription factors is critical for fetal testis development in mice. Proc Natl Acad Sci U S A 104:14994–14999
Bowles J, Knight D, Smith C et al (2006) Retinoid signaling determines germ cell fate in mice. Science 312:596–600
Bradford ST, Hiramatsu R, Maddugoda MP et al (2009) The cerebellin 4 precursor gene is a direct target of SRY and SOX9 in mice. Biol Reprod 80:1178–1188
Brennan J, Karl J, Capel B (2002) Divergent vascular mechanisms downstream of Sry establish the arterial system in the XY gonad. Dev Biol 244:418–428
Brennan J, Tilmann C, Capel B (2003) Pdgfr-alpha mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev 17:800–810
Britt KL, Kerr J, O’Donnell L et al (2002) Estrogen regulates development of the somatic cell phenotype in the eutherian ovary. FASEB J 16:1389–1397
Buehr M, Gu S, McLaren A (1993) Mesonephric contribution to testis differentiation in the fetal mouse. Development 117:273–281
Bullejos M, Koopman P (2005) Delayed Sry and Sox9 expression in developing mouse gonads underlies B6-Y(DOM) sex reversal. Dev Biol 278:473–481
Buscara L, Montazer-Torbati F, Chadi S et al (2009) Goat RSPO1 over-expression rescues sex-reversal in Rspo1-knockout XX mice but does not perturb testis differentiation in XY or sex-reversed XX mice. Transgenic Res 18:649–654
Byskov AG, Saxen L (1976) Induction of meiosis in fetal mouse testis in vitro. Dev Biol 52:193–200
Byskov AG, Høyer PE, Yding Andersen C et al (2011) No evidence for the presence of oogonia in the human ovary after their final clearance during the first two years of life. Hum Reprod 26:2129–2139
Capel B, Batchvarov J (2008) Sex chromatin staining in amnion cells. CSH Protoc 3:11
Capel B, Albrecht KH, Washburn LL et al (1999) Migration of mesonephric cells into the mammalian gonad depends on Sry. Mech Dev 84:127–131
Castle WE, Little CC (1910) On a modified Mendelian ration among yellow mice. Science 32:868–870
Chaboissier MC, Kobayashi A, Vidal VI et al (2004) Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 131:1891–1901
Chan WY, Lee TL, Wu SM et al (2006) Transcriptome analyses of male germ cells with serial analysis of gene expression (SAGE). Mol Cell Endocrinol 250:8–19
Chang H, Gao F, Guillou F et al (2008) Wt1 negatively regulates β-catenin signaling during testis development. Development 135:1875–1885
Chapin RE, Phelps JL, Miller BE et al (1987) Alkaline phosphatase histochemistry discriminates peritubular cells in primary rat testicular cell culture. J Androl 8:155–161
Chassot AA, Ranc F, Gregoire EP et al (2008) Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum Mol Genet 17:1264–1277
Chen H, Palmer JS, Thiagarajan RD et al (2012) Identification of novel markers of mouse fetal ovary development. PLoS One 7(7):e41683
Childs AJ, Cowan G, Kinnell HL et al (2011) Retinoic acid signalling and the control of meiotic entry in the human fetal gonad. PLoS One 6:e20249
Chinwalla AT, Cook LL, Delehaunty KD et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562
Chiquoine AD (1954) The identification, origin, and migration of the primordial germ cells in the mouse embryo. Anat Rec 118:135–146
Chuma S, Nakatsuji N (2001) Autonomous transition into meiosis of mouse fetal germ cells in vitro and its inhibition by gp130-mediated signaling. Dev Biol 229:468–479
Clapcote SJ, Roder JC (2005) Simplex PCR assay for sex determination in mice. Biotechniques 38:702–706
Coert HJ (1898) Over de Ontwikkeling en den Bouw van de Geschlachtsklier bij de Zoogdieren meer in het bijzonder van den Eierstok. Dissertation, Leiden University
Combes AN, Lesieur E, Harley VR et al (2009a) Three-dimensional visualization of testis cord morphogenesis, a novel tubulogenic mechanism in development. Dev Dyn 238:1033–1041
Combes AN, Wilhelm D, Davidson TL et al (2009b) Endothelial cell migration directs testis cord formation. Dev Biol 326:112–120
Combes AN, Bowles J, Feng CW et al (2011) Expression and functional analysis of Dkk1 during early gonadal development. Sex Dev 5:124–130
Cool J, Carmona FD, Szucsik JC, Capel B (2008) Peritubular myoid cells are not the migrating population required for testis cord formation in the XY gonad. Sex Dev 2:128–133
Coons AH, Creech HJ, Jones RN (1941) Immunological properties of an antibody containing a fluorescent group. Proc Soc Exp Biol Med 47:200–202
Coveney D, Cool J, Oliver T, Capel B (2008) Four-dimensional analysis of vascularization during primary development of an organ, the gonad. Proc Natl Acad Sci U S A 105:7212–7217
Crow JF (2002) C.C. Little, cancer and inbred mice. Genetics 161:1357–1361
Cuénot L (1902) La loi de Mendel et l’hérédité de la pigmentation chez les souris. Arch Zool Exp Gen 3:27–30
Cui S, Ross A, Stallings N et al (2004) Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development 131:4095–4105
Cupp AS, Uzumcu M, Skinner MK (2003) Chemotactic role of neurotropin 3 in the embryonic testis that facilitates male sex determination. Biol Reprod 68:2033–2037
DeFalco T, Takahashi S, Capel B (2011) Two distinct origins for Leydig cell progenitors in the fetal testis. Dev Biol 352:14–26
DeFalco T, Bhattacharya I, Williams AV et al (2014) Yolk-sac-derived macrophages regulate fetal testis vascularization and morphogenesis. Proc Natl Acad Sci U S A 111:E2384–E2393
DeFalco T, Potter S, Williams AV et al (2015) Macrophages contribute to the spermatogonial niche in the adult testis. Cell Rep 12:1107–1119
De Felici M, Dolci S (1989) In vitro adhesion of mouse fetal germ cells to extracellular matrix components. Cell Differ Dev 26:87–96
De Felici M, McLaren A (1982) Isolation of mouse primordial germ cells. Expl Cell Res 142:476–482
De Felici M, Scaldaferri ML, Lobascio M (2004) In vitro approaches to the study of primordial germ cell lineage and proliferation. Hum Reprod Update 10:197–206
De Miguel M, Cheng L, Holland EC et al (2002) Dissection of the c-Kit signaling pathway in mouse primordial germ cells by retroviral-mediated gene transfer. Proc Natl Acad Sci U S A 99:10458–10463
Di Carlo AD, Travia G, De Felici M (2000) The meiotic specific synaptonemal complex protein SCP3 is expressed by female and male primordial germ cells of the mouse embryo. Int J Dev Biol 44:241–244
DiNapoli L, Capel B (2007) Germ cell depletion does not alter the morphogenesis of the fetal testis or ovary in the red-eared slider turtle (Trachemys scripta). J Exp Zool B Mol Dev Evol 308:236–241
Dumond H, Al-Asaad I, Chesnel A et al (2011) Temporal and spatial SOX9 expression patterns in the course of gonad development of the caudate amphibian Pleurodeles waltl. J Exp Zool B Mol Dev Evol 316B:199–211
Dupont S, Krust A, Gansmuller A et al (2000) Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 127:4277–4291
Egli T (1876) Beitrage zur Anatomie der Entwickelungsgeschichte der Geschlechtsorgane. I. Zur Entwickelung des Urogenitalsystems beim Kaninchen. Dissertation, University of Zurich
El Jamil A, Kanhoush R, Magre S et al (2008) Sex-specific expression of SOX9 during gonadogenesis in the amphibian Xenopus tropicalis. Dev Dyn 237:2996–3005
Evans CW, Robb DI, Tuckett F, Challoner S (1982) Regulation of meiosis in the foetal mouse gonad. J Embryol Exp Morphol 68:59–67
Fechner PY, Marcantonio SM, Jaswaney V et al (1993) The role of the sex-determining region Y gene in the etiology of 46, XX maleness. J Clin Endocrinol Metab 76:690–695
Felix W (1911) Die Entwicklung der Harn- und Geschlechtsorgane. In: Keibel F, Mall FP (eds) Handbuch der Entwicklungageschichte des Menshen, vol 2. Hirzel, Leipzig, pp 732–795
Fleming A, Ghahramani N, Zhu MX et al (2012) Membrane β-catenin and adherens junctions in early gonadal patterning. Dev Dyn 241:1782–1798
Fröjdman K, Paranko J, Virtanen I, Pelliniemi LJ (1992) Intermediate filaments and epithelial differentiation of male rat embryonic gonad. Differentiation 50:113–123
Fuss A (1911) Uber extraregionare Geschlechtszellen bei einem menschlichen Embryo von 4 Wochen. Anat Am 39:407–409
Gall JG, Pardue ML (1969) Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A 63:378–383
Gallardo T, Shirley L, John GB, Castrillon DH (2007) Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis 45:413–417
Garcia-Ortiz JA, Pelosi E, Omari S et al (2009) Foxl2 functions in sex determination and histogenesis throughout mouse ovary development. BMC Dev Biol 9:36
Gassei K, Ehmcke J, Wood MA et al (2008) Immature rat seminiferous tubules reconstructed in vitro express markers of Sertoli cell maturation after xenografting into nude mouse hosts. Mol Hum Reprod 16:97–110
Gong W, Pan L, Lin Q et al (2013) Transcriptome profiling of the developing postnatal mouse testis using next-generation sequencing. Sci China Life Sci 56:1–12
Gosner KL (1960) A simplified table for staging anuran embryos and larvae with notes on identification. Herpetologica 16:183–190
Greenbaum E (2002) A standardized series of embryonic stages for the emydid turtle Trachemys scripta. Can J Zool 80:1350–1370
Greenlee AR, Krisher RL, Plotka ED (1998) Rapid sexing of murine preimplantation embryos using a nested, multiplex polymerase chain reaction (PCR). Mol Reprod Dev 49:261–267
Grimmond S, Van Hateren N, Siggers P et al (2000) Sexually dimorphic expression of protease nexin-1 and vanin-1 in the developing mouse gonad prior to overt differentiation suggests a role in mammalian sexual development. Hum Mol Genet 9:1553–1560
Gubbay J, Collignon J, Koopman P et al (1990) A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346:245–250
Guenin S, Mauriat M, Pelloux J et al (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot 60:487–493
Habert R, Picon R (1982) Control of testicular steroidogenesis in foetal rat: effect of decapitation on testosterone and plasma luteinizing hormone-like activity. Acta Endocrinol 99:466–473
Hacker A, Capel B, Goodfellow P, Lovell-Badge R (1995) Expression of Sry, the mouse sex determining gene. Development 121:1603–1614
Hadley MA, Byers SW, Suarez-Quian CA et al (1985) Extracellular matrix regulates Sertoli cell differentiation, testicular cord formation, and germ cell development in vitro. J Cell Biol 101:1511–1522
Hamburger V, Hamilton HL (1951) A series of normal stages in the development of the chick embryo. J Morphol 88:49–92
Hanley NA, Hagan DM, Clement-Jones M et al (2000) SRY, SOX9, and DAX1 expression patterns during human sex determination and gonadal development. Mech Dev 91:403–407
Hatzirodos N, Hummitzsch K, Irving-Rodgers HF, Rodgers RJ (2015) Transcriptome comparisons identify new cell markers for theca interna and granulosa cells from small and large antral ovarian follicles. PLoS ONE 10(3):e0119800
Higgy NA, Zackson SL, van der Hoorn FA (1995) Cell interactions in testis development: overexpression of c-mos in spermatocytes leads to increased germ cell proliferation. Dev Genet 16:190–200
Hiramatsu R, Matoba S, Kanai-Azuma M et al (2009) A critical time window of Sry action in gonadal sex determination in mice. Development 136:129–138
Houmard B, Small C, Yang L et al (2009) Global gene expression in the human fetal testis and ovary. Biol Reprod 81:438–443
Hu YC, Okumura LM, Page DC (2013) Gata4 is required for formation of the genital ridge in mice. PLoS Genet 9(7), e1003629
Hummitzsch K, Irving-Rodgers HF, Hatzirodos N et al (2013) A new model of development of the mammalian ovary and follicles. PLoS One 8(2):e55578
Ito S, Karnowsky MJ (1968) Formaldehyde glutaraldehyde fixatives containing trinitro compounds. J Cell Biol 36:168
Iwamoto T, Hiraku Y, Oikawa S et al (2004) DNA intrastrand cross-link at the 5’-GA-3’ sequence formed by busulfan and its role in the cytotoxic effect. Cancer Sci 95:454–458
Jaenisch R, Mintz B (1974) Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proc Natl Acad Sci U S A 71:1250–1254
Jameson SA, Natarajan A, Cool J et al (2012) Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad. PLoS Genet 8(3):e1002575
Janošik J (1885) Histologish embryologische Untersuchungen über das Urogenitalsystem. Sitz-Ber Akad Wiss Wien Math Nat Kl 91:32–38
Janošik J (1887a) Zur Histologie des Ovariums. Sitz-Ber Akad Wiss Wien Math Nat Kl 96:45–48
Janošik J (1887b) Zwei junge menschliche Embryonen. Arch Mikr Anat 30:87–90
Janošik J (1890a) Berichtigung zu Nagels’Arbeit: Ueber die Entwickelung des Urogenitalsystems des Menschen. Ebenda 35:3–9
Janošik J (1890b) Bemerkungen über die Entwickelung des Genitalsystems. Sitz-Ber Akad Wiss Wien Math Nat Kl 99:46–54
Janošik J (1894) Ueber die Entwickelung der Vorniere und des Vornierenganges bei Säugern. Arch Mikr Anat 64:214–234
Kaboord B, Perr M (2008) Isolation of proteins and protein complexes by immunoprecipitation. Methods Mol Biol 424:349–364
Karl J, Capel B (1998) Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev Biol 203:323–333
Kim Y, Kobayashi A, Sekido R (2006) Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol 4:e187
Kimmel CB, Ballard WW, Kimmel SR et al (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203:253–310
Kitamura K, Yanazawa M, Sugiyama N et al (2002) Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 32:359–369
Kloc M, Bilinski S, Chan AP, Etkin LD (2001) Mitochondrial ribosomal RNA in the germinal granules in Xenopus embryos revisited. Differentiation 67:80–83
Kocer A, Pinheiro I, Pannetier M et al (2008) R-spondin1 and FOXL2 act into two distinct cellular types during goat ovarian differentiation. BMC Dev Biol 8:36
Koopman P, Gubbay J, Vivian N et al (1991) Male development of chromosomally female mice transgenic for Sry. Nature 351:117–121
Koubova J, Menke DB, Zhou Q et al (2006) Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc Natl Acad Sci U S A 103:2474–2479
Kunieda T, Xian M, Kobayashi E et al (1992) Sexing of mouse preimplantation embryos by detection of Y chromosome-specific sequences using polymerase chain reaction. Biol Reprod 46:692–697
Kusaka M, Katoh-Fukui Y, Ogawa H et al (2010) Abnormal epithelial cell polarity and ectopic epidermal growth factor receptor (EGFR) expression induced in Emx2 KO embryonic gonads. Endocrinology 151:5893–5904
Lambert JF, Benoit BO, Colvin GA et al (2000) Quick sex determination of mouse fetuses. J Neurosci Methods 95:127–132
Lavrovsky Y, Song CS, Chatterjee B, Roy AK (1998) A rapid and reliable PCR-based assay for gene transmission and sex determination in newborn transgenic mice. Transgenic Res 7:319–320
Lee TL, Alba D, Baxendale V et al (2006) Application of transcriptional and biological network analyses in mouse germ-cell transcriptomes. Genomics 88:18–33
Lee TL, Li Y, Alba D et al (2009) Developmental staging of male murine embryonic gonad by SAGE analysis. J Genet Genomics 36:215–227
Lee TL, Li Y, Cheung HH et al (2010) GonadSAGE: a comprehensive SAGE database for transcript discovery on male embryonic gonad development. Bioinformatics 26:585–586
Li H, Kim KH (2004) Retinoic acid inhibits rat XY gonad development by blocking mesonephric cell migration and decreasing the number of gonocytes. Biol Reprod 70:687–693
Li Y, Taketo T, Lau YF (2012) Isolation of fetal gonads from embryos of timed-pregnant mice for morphological and molecular studies. Methods Mol Biol 825:3–16
Liu CF, Bingham N, Parker K, Yao HH (2009) Sex-specific roles of beta-catenin in mouse gonadal development. Hum Mol Genet 18:405–417
Liu C, Peng J, Matzuk MM, Yao HH (2015) Lineage specification of ovarian theca cells requires multicellular interactions via oocyte and granulosa cells. Nat Commun 6:6934
López-Ibor B, Schwartz AD (1986) Gonadal failure following busulfan therapy in an adolescence girl. Am J Pediatr Hematol Oncol 8:85–87
Luo X, Ikeda Y, Parker KL (1994) A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 77:481–490
Maack G, Segner H (2003) Morphological development of the gonads in zebrafish. J Fish Biol 62:895–906
Maatouk DM, DiNapoli L, Alvers A (2008) Stabilization of beta-catenin in XY gonads causes male-to-female sex-reversal. Hum Mol Genet 17:2949–2955
Maatouk DM, Mork L, Chassot AA et al (2013) Disruption of mitotic arrest precedes precocious differentiation and transdifferentiation of pregranulosa cells in the perinatal Wnt4 mutant ovary. Dev Biol 383:295–306
Mackay S, Booth SH, MacGowan A, Smith RA (1999) Ultrastructural studies demonstrate that epithelial polarity is established in cultured mouse pre-Sertoli cells by extracellular matrix components. J Electron Microsc 48:159–165
Maclean G, Dolle P, Petkovich M (2009) Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev Dyn 238:732–745
Maitland P, Ullmann SL (1993) Gonadal development in the opossum, Monodelphis domestica: the rete ovarii does not contribute to the steroidogenic tissues. J Anat 183:43–56
Manuylov NL, Fujiwara Y, Adameyko II et al (2007) The regulation of Sox9 gene expression by the GATA4/FOG2 transcriptional complex in dominant XX sex reversal mouse models. Dev Biol 307:356–367
Marrack J (1934) Nature of antibodies. Nature 133:292
Martineau J, Nordqvist K, Tilmann C et al (1997) Male specific cell migration into the developing gonad. Curr Biol 7:958–968
Matsuyama M, Mizusaki H, Shimono A et al (2005) A novel isoform of Vinexin, Vinexin gamma, regulates Sox9 gene expression through activation of MAPK cascade in mouse fetal gonad. Genes Cells 10:421–434
Matzuk MM, Finegold MJ, Mishina Y et al (1995) Synergistic effects of inhibins and müllerian-inhibiting substance on testicular tumorigenesis. Mol Endocrinol 9:1337–1345
McClive PJ, Sinclair AH (2001) Rapid DNA extraction and PCR-sexing of mouse embryos. Mol Reprod Dev 60:225–226
McFarlane L, Truong V, Palmer JS, Wilhelm D (2013) Novel PCR assay for determining the genetic sex of mice. Sex Dev 7:207–211
McLaren A, Buehr M (1990) Development of mouse germ cells in cultures of fetal gonads. Cell Diff Dev 31:185–195
McLaren A, Southee D (1997) Entry of embryonic germ cells into meiosis. Dev Biol 187:107–113
Merchant-Larios H, Moreno-Mendoza N (1998) Mesonephric stromal cells differentiate into Leydig cells in the mouse fetal testis. Exp Cell Res 244:230–238
Migrenne S, Moreau E, Pakarinen P et al (2012) Mouse testis development and function are differently regulated by follicle-stimulating hormone receptors signaling during fetal and prepubertal life. PLoS One 7(12):e53257
Miyamoto Y, Taniguchi H, Hamel F et al (2008) A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol Biol 9:44
Miyamoto N, Yoshida M, Kuratani S et al (1997) Defects of urogenital development in mice lacking Emx2. Development 124:1653–1664
Molyneaux KA, Stallock J, Schaible K, Wylie C (2001) Time-lapse analysis of living mouse germ cell migration. Dev Biol 240:488–498
Molyneaux K, Zinszner H, Kunwar P et al (2003) The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development 130:4279–4286
Moniot B, Declosmenil F, Barrionuevo F et al (2009) The PGD2 pathway, independently of FGF9, amplifies SOX9 activity in Sertoli cells during male sexual differentiation. Development 136:1813–1821
Morais da Silva S, Hacker A, Harley V et al (1996) Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet 14:62–68
Morgan TH (1912) The scientific work of Miss N. M. Stevens. Science 36:468–470
Mork L, Czerwinski M, Capel B (2014) Predetermination of sexual fate in a turtle with temperature-dependent sex determination. Dev Biol 386:264–271
Mroz K, Carrel L, Hunt PA (1999) Germ cell development in the XXY mouse: evidence that X chromosome reactivation is independent of sexual differentiation. Dev Biol 207:229–238
Munger SC, Aylor DL, Syed HA et al (2009) Elucidation of the transcription network governing mammalian sex determination by exploiting strain-specific susceptibility to sex reversal. Genes Dev 23:2521–2523
Munger SC, Natarajan A, Looger LL et al (2013) Fine time course expression analysis identifies cascades of activation and repression and maps a putative regulator of mammalian sex determination. PLoS Genet 9(7):e1003630
Nagamine CM, Chan K, Hake LE, Lau YF (1990) The two candidate testis-determining Y genes (Zfy-1 and Zfy-2) are differentially expressed in fetal and adult mouse tissues. Genes Dev 4:63–74
Nagel W (1889a) Ueber das Vorkommen von Primordialeiern auberhalb der Keimdrusenanlage beim Menschen. Anat Anz 4:12–19
Nagel W (1889b) Ueber die Entwickelung des Urogenitalsystmes des Menschen. Arch Mikr Anat 34:269–384
Needham J (1959) A history of embryology. Abelard-Schuman, New York
Nef S, Verma-Kurvari S, Merenmies J et al (2003) Testis determination requires insulin receptor family function in mice. Nature 426:291–295
Nef S, Schaab O, Stallings NR et al (2005) Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev Biol 287:361–377
Nel-Themaat L, Vadakkan TJ, Wang Y et al (2009) Morphometric analysis of testis cord formation in Sox9-EGFP mice. Dev Dyn 238:1100–1110
Nel-Themaat L, Gonzalez G, Akiyama H, Behringer RR (2010) Illuminating testis morphogenesis in the mouse. J Androl 31:5–10
Nicol B, Yao HH (2015) Gonadal Identity in the Absence of Pro-Testis Factor SOX9 and Pro-Ovary Factor Beta-Catenin in Mice. Biol Reprod 93:35
Nieuwkoop PD, Faber J (1967) Normal table of Xenopus laevis (Daudin): a systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis, 2nd edn. North Holland, Amsterdam
Niksic M, Slight J, Sanford JR et al (2004) The Wilms’ tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum Mol Genet 13:463–471
Nishino K, Yamanouchi K, Naito K, Tojo H (2001) Characterization of mesonephric cells that migrate into the XY gonad during testis differentiation. Exp Cell Res 267:225–232
O WS, Baker TG (1978) Germinal and somatic cell interrelationships in gonadal sex differentiation. Ann Biol Anim Biochim Biophys 18:351–357
Ohe K, Lalli E, Sassone-Corsi P (2002) A direct role of SRY and SOX proteins in pre-mRNA splicing. Proc Natl Acad Sci U S A 99:1146–1151
Oshima Y, Hayashi T, Tokunaga S, Nakamura M (2005) Wnt4 expression in the differentiating gonad of the frog Rana rugosa. Zoolog Sci 22:689–693
Ottolenghi C, Omari S, Garcia-Ortiz JE et al (2005) Foxl2 is required for commitment to ovary differentiation. Hum Mol Genet 14:2053–2062
Ottolenghi C, Pelosi E, Tran J et al (2007) Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum Mol Genet 16:2795–2804
Oulad-Abdelghani M, Bouillet P, Décimo D et al (1996) Characterization of a premeiotic germ cell-specific cytoplasmic protein encoded by Stra8, a novel retinoic acid-responsive gene. J Cell Biol 135:469–477
Palmer SJ, Burgoyne PS (1991) The Mus musculus domesticus Tdy allele acts later than the Mus musculus musculus Tdy allele: a basis for XY sex-reversal in C57BL/6-YPOS mice. Development 113:709–714
Palmer MS, Berta P, Sinclair AH et al (1990) Comparison of human ZFY and ZFX transcripts. Proc Natl Acad Sci U S A 87:1681–1685
Pan H, O’Brien MJ, Wigglesworth K et al (2005) Transcript profiling during mouse oocyte development and the effect of gonadotropin priming and development in vitro. Dev Biol 286:493–506
Paranko J, Pelliniemi LJ (1992) Differentiation of smooth muscle cells in fetal rat testis and ovary: localization of alkaline phosphatase, smooth muscle myosin, F-actin, and desmin. Cell Tissue Res 268:521–530
Parma P, Radi O, Vidal V et al (2006) R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 38:1304–1309
Pask AJ, Harry JL, Renfree MB, Marshall Graves JA (2000) Absence of SOX3 in the developing marsupial gonad is not consistent with a conserved role in mammalian sex determination. Genesis 27:145–152
Payne DW, Packman JN, Adashi EY (1992) Follicle-stimulating hormone inhibits granulosa activity. J Biol Chem 267:13348–13355
Pellegrini M, Filipponi D, Gori M et al (2008) ATRA and KL promote differentiation toward the meiotic program of male germ cells. Cell Cycle 7:3878–3888
Pesce M, De Felici M (1995) Purification of mouse primordial germ cells by MiniMACS magnetic separation system. Dev Biol 170:722–725
Piprek RP (2013) Gonadogenesis in anuran amphibians: cellular and molecular mechanisms of sexual differentiation of gonads. Dissertation, Jagiellonian University, Krakow
Piprek RP, Pecio A, Kubiak JZ, Szymura JM (2012) Differential effects of busulfan on gonadal development in five divergent anuran species. Reprod Toxicol 34:393–401
Piprek RP, Pecio A, Laskowska-Kaszub K et al (2013) Retinoic acid homeostasis regulates meiotic entry in developing anuran gonads and in Bidder’s organ through Raldh2 and Cyp26b1 proteins. Mech Dev 130:613–627
Piprek RP, Pecio A, Kloc M et al (2014) Evolutionary trend for metamery reduction and gonad shortening in Anurans revealed by comparison of gonad development. Int J Dev Biol 58:929–934
Qing T, Liu H, Wei W et al (2008) Mature oocytes derived from purified mouse fetal germ cells. Hum Reprod 23:54–61
Ramsköld D, Wang ET, Burge CB, Sandberg R (2009) An Abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5(12):e1000590
Rastetter RH, Bernard P, Palmer JS et al (2014) Marker genes identify three somatic cell types in the fetal mouse ovary. Dev Biol 394:242–252
Rolland AD, Lehmann KP, Johnson KJ et al (2011) Uncovering gene regulatory networks during mouse fetal germ cell development. Biol Reprod 84:790–800
Ross DG, Bowles J, Hope M et al (2009) Profiles of gonadal gene expression in the developing bovine embryo. Sex Dev 3:273–283
Russell ES (1979) Hereditary anemias of the mouse: a review for geneticists. Adv Genet 20:357–459
Sabour D, Xu X, Chung ACK et al (2014) Germ cell nuclear factor regulates gametogenesis in developing gonads. PLoS One 9(8):e103985
Salas-Cortés L, Jaubert F, Barbaux S et al (1999) The human SRY protein is present in fetal and adult Sertoli cells and germ cells. Int J Dev Biol 43:135–140
Schmahl J, Capel B (2003) Cell proliferation is necessary for the determination of male fate in the gonad. Dev Biol 258:264–276
Schmahl J, Eicher EM, Washburn LL, Capel B (2000) Sry induces cell proliferation in the mouse gonad. Development 127:65–73
Schmahl J, Kim Y, Colvin JS et al (2004) Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 131:3627–3636
Sekido R, Bar I, Narvaez V et al (2004) SOX9 is up-regulated by the transient expression of SRY specifically in Sertoli cell precursors. Dev Biol 274:271–279
Seligman J, Page DC (1998) The Dazh gene is expressed in male and female embryonic gonads before germ cell sex differentiation. Biochem Biophys Res Commun 245:878–882
Sharpe RM, McKinnell C, Kivlin C, Fisher JS (2003) Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction 125:769–784
Sinclair AH, Berta P, Palmer MS et al (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346:240–244
Small CL, Shima JE, Uzumcu M et al (2005) Profiling gene expression during the differentiation and development of the murine embryonic gonad. Biol Reprod 72:492–501
Smith CA, McClive PJ, Hudson Q, Sinclair AH (2005) Male-specific cell migration into the developing gonad is a conserved process involving PDGF signalling. Dev Biol 284:337–350
Smith CA, Roeszler KN, Bowles J et al (2008a) Onset of meiosis in the chicken embryo; evidence of a role for retinoic acid. BMC Dev Biol 8:85
Smith CA, Shoemaker CM, Roeszler KN et al (2008b) Cloning and expression of R-Spondin1 in different vertebrates suggests a conserved role in ovarian development. BMC Dev Biol 8:72
Smith CA, Roeszler KN, Ohnesorg T et al (2009) The avian Z-linked gene DMRT1 is required for male sex determination in the chicken. Nature 461:267–271
Soyal SM, Amleh A, Dean J (2000) FIGalpha, a germ cell-specific transcription factor required for ovarian follicle formation. Development 127:4645–4653
Stebler J, Spieler D, Slanchev K et al (2004) Primordial germ cell migration in the chick and mouse embryo: the role of the chemokine SDF-1/CXCL12. Dev Biol 272:351–361
Tang H, Brennan J, Karl J et al (2008) Notch signaling maintains Leydig progenitor cells in the mouse testis. Development 135:3745–3753
Tanimura A, Iwasawa H (1991) Proliferative activity of somatic cells during gonadal development in the Japanese pond frog, Rana nigromaculata. J Exp Zool 259:365–370
Tanoue A, Nakamura T, Endo F et al (1992) Sex-determining region Y (SRY) in a patient with 46, XX true hermaphroditism. Jpn J Hum Genet 37:311–320
Teerds KJ, de Boer-Brouwer M, Dorrington JH et al (1999) Identification of markers for precursor and leydig cell differentiation in the adult rat testis following ethane dimethyl sulphonate administration. Biol Reprod 60:1437–1445
Tevosian SG, Albrecht KH, Crispino JD et al (2002) Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2. Development 129:4627–4634
Theiler K (1989) The house mouse. Development and normal stages from fertilization to 4 weeks of age, 2nd edn. Springer, Berlin
Tremblay JJ, Viger RS (2003) Transcription factor GATA-4 is activated by phosphorylation of serine 261 via the cAMP/PKA pathway in gonadal cells. J Biol Chem 278:22128–22135
Trevino V, Falciani F, Barrera-Saldana HA (2007) DNA microarrays: a powerful genomic tool for biomedical and clinical research. Mol Med 13:527–541
Tsuji FI (2010) Early history, discovery, and expression of Aequorea green fluorescent protein, with a note on an unfinished experiment. Microsc Res Tech 73:785–796
Uhlenhaut NH, Jakob S, Anlag K et al (2009) Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 139:1130–1142
Vied C, Halachmi N, Salzberg A, Horabin JI (2003) Antizyme is a target of sex-lethal in the Drosophila germline and appears to act downstream of hedgehog to regulate sex-lethal and cyclin B. Dev Biol 253:214–229
Voller A, Bartlett A, Bidwell DE (1978) Enzyme immunoassays with special reference to ELISA techniques. J Clin Pathol 31:507–520
Warr N, Siggers P, Bogani D et al (2009) Sfrp1 and Sfrp2 are required for normal male sexual development in mice. Dev Biol 326:273–284
Waterston RH, Lindblad-Toh K, Birney E et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562
Wilhelm D, Martinson F, Bradford S et al (2005) Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev Biol 287:111–124
Wilhelm D, Hiramatsu R, Mizusaki H et al (2007) SOX9 regulates prostaglandin D synthase gene transcription in vivo to ensure testis development. J Biol Chem 282:10553–10560
Wilhelm D, Washburn LL, Truong V et al (2009) Antagonism of the testis- and ovary-determining pathways during ovotestis development in mice. Mech Dev 126:324–336
Willerton L, Smith RA, Russell D, Mackay S (2004) Effects of FGF9 on embryonic Sertoli cell proliferation and testicular cord formation in the mouse. Int J Dev Biol 48:637–643
Williams TJ (1986) A technique for sexing mouse embryos by a visual colorimetric assay of the X-linked enzyme, glucose 6-phosphate dehydrogenase. Theriogenology 25:733–739
Wilson EB (1905) Studies on chromosomes. I. The behavior of the idiochromosomes in hemiptera. J Exp Zool 2:371–405
Wolf U (1998) The serologically detected H-Y antigen revisited. Cytogenet Cell Genet 80:232–235
Wu SM, Baxendale V, Chen Y et al (2004) Analysis of mouse germ-cell transcriptome at different stages of spermatogenesis by SAGE: biological significance. Genomics 84:971–981
Wylie CC, Bancroft M, Heasman J (1976) The formation of the gonadal ridge in Xenopus laevis. II. A scanning electron microscope study. J Embryol Exp Morphol 35:139–148
Yamamoto M, Wakatsuki T, Hada A, Ryo A (2001) Use of serial analysis of gene expression (SAGE) technology. J Immunol Methods 250:45–66
Yao HH, DiNapoli L, Capel B (2004) Cellular mechanisms of sex determination in the red-eared slider turtle, Trachemys scripta. Mech Dev 121:1393–13401
Yao HH, Whoriskey W, Capel B (2002) Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev 16:1433–1440
Yoshimoto S, Okada E, Umemoto H et al (2008) A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc Natl Acad Sci U S A 105:2469–2474
Yoshimoto S, Ikeda N, Izutsu Y et al (2010) Opposite roles of DMRT1 and its W-linked paralogue, DM-W, in sexual dimorphism of Xenopus laevis: implications of a ZZ/ZW-type sex-determining system. Development 137:2519–2526
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Piprek, R.P. (2016). Methods for the Study of Gonadal Development. In: Piprek, R. (eds) Molecular Mechanisms of Cell Differentiation in Gonad Development. Results and Problems in Cell Differentiation, vol 58. Springer, Cham. https://doi.org/10.1007/978-3-319-31973-5_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-31973-5_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31971-1
Online ISBN: 978-3-319-31973-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)