
Abstract
Telomeres form protective caps at the ends of linear chromosomes to prevent nucleolytic degradation, end-to-end fusion, irregular recombination, and chromosomal instability. Telomeres are composed of repetitive DNA sequences (TTAGGG)n in humans, that are bound by specialized telomere binding proteins. Telomeres lose capping function in response to telomere shortening, which occurs during each division of cells that lack telomerase activity—the enzyme that can synthesize telomeres de novo. Telomeres have a dual role in cancer: telomere shortening can lead to induction of chromosomal instability and to the initiation of tumors, however, initiated tumors need to reactivate telomerase in order to stabilize chromosomes and to gain immortal growth capacity. In this review, we summarize current knowledge on the role of telomeres in the maintenance of chromosomal stability and carcinogenesis.
Cagatay Günes and Karl Lenhard Rudolph equally contributed to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Telomeres and Telomerase
1.1 Telomeres Are Protective Caps at the Ends of Linear Chromosomes
Cells with linear chromosomes have to meet several challenges: (i) cellular repair mechanism have to discriminate between broken ends as a result of DNA damage and the ends of the chromosomes, (ii) the ends of chromosomes must be protected against degradation by nucleases, and (iii) as the conventional DNA polymerase cannot replicate the most extreme ends of chromosomes (end-replication-problem), loss of genetic material must be compensated by some end maintenance mechanism. Work by Hermann Muller and Barbara McClintock provided the first evidence that the ends of linear chromosomes must be capped by a specialized structure to prevent chromosome fusions (McClintock 1939, 1941; Muller 1938). Müller introduced the term ‘telomere’ to emphasize this specific function of chromosome ends. A telomere is functionally defined as a region of DNA at the molecular end of a linear chromosome that is required for replication and stability of the chromosome (Blackburn and Szostak 1984). It has become clear now that the telomeres are composed of short repetitive DNA-sequences and specific proteins, the shelterin, that bind this sequence to protect the ends of linear chromosomes (de Lange 2010).
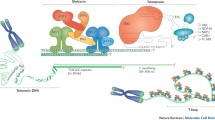
Telomeric DNA consists of a tandem array of GT-rich repeats (e.g. TTGGGG in Tetrahymena and TTAGGG in humans and other vertebrates). The number of the repeats and consequently the length of telomeric DNA varies among species (Fig. 1) ranging from 36 nucleotides present at the ends of macronuclear chromosomes of ciliated protozoans (Klobutcher et al. 1981), ~300 bp in Saccharomyces cerevisiae (Zakian 1989) to ~150,000 bp in mice (Kipling and Cooke 1990). In human somatic cells telomeres consist of 7000–10,000 bp telomeric DNA and of about 20,000 bp in germ cells (Allshire et al. 1989; Moyzis et al. 1988).

Telomere-DNA is an evolutionarily conserved GT-rich repetitive sequence. Basically all eukaryotes use GT-rich repetitive sequences at the ends of their chromosomes (telomeres). Despite considerable variation in telomere length and telomere-sequence, telomerase activity is the major telomere maintenance mechanism among the eukaryotes, with only few exceptions (Drosophila melanogaster). Modified from Meyne et al. (1989)
One important feature of telomeres, which is conserved in all eukaryotes, is that they posses a protruding 3′ single-stranded overhang due to the mechanism of the lagging strand DNA replication (Makarov et al. 1997; McElligott and Wellinger 1997). In mammalian cells, this single stranded overhang may fold back and invade into the preceding double stranded telomeric DNA to form a unique D-loop and T-loop structure (Griffith et al. 1999). Telomere looping may be a bona fide end protecting mechanism since it has been observed in mammals, plants and several lower eukaryotes (Cesare et al. 2003; Griffith et al. 1999; Munoz-Jordan et al. 2001; Murti and Prescott 1999). This special structure functions to seal the ends of chromosomes thus protecting them from hazardous cellular actions. In its absence, the 3′-overhang is simply occupied with specific telomere-binding proteins protecting the chromosome ends from DNA-damage.
To date a series of proteins have been described to be associated with telomeres (Fig. 2). In human cells, six proteins, TRF1, TRF2, TIN2, TPP1, RAP1 and POT1, form the shelterin complex and interact with several other proteins for telomere length regulation (de Lange 2005). Among the latter, proteins involved in DNA double-strand break repair (Ku-proteins) and non-homologous-end-joining (RAD50-NBS1-Mre11 complex) are found. It is not yet clear whether these proteins are present at the telomeres at all times or in a cell-cycle dependent manner. Among these proteins TRF1 and TRF2 form a platform for the binding and function of other telomere specific factors (Fig. 2).

Telomeres, Protein-DNA complexes at the ends of linear chromosomes, may form a lariat structure, the telomere-loop (T-loop). A schematic representation of the D-loop, T-loop structure at the chromosome ends. Lagging strand blue, leading strand red. The 3′-protruding end of the lagging strand may invade into the double stranded telomeric DNA and result in the displacement of the double strand (displacement-loop: D-loop). A large lariat structure can be observed at the telomeres (telomere-loop: T-loop). Several proteins have been localized to the telomeres. The number, temporal and spatial localization of these proteins at the telomeres is not completely understood. There is evidence that T-loop formation is facilitated by TRF2 (Doksani et al. 2013; Griffith et al. 1999) whereas RTEL1 helicase activity is required for the faithful T-loop resolution during replication (Vannier et al. 2012)
1.2 Telomerase
Telomerase is a ribonucleo-protein complex with reverse transcriptase activity with conserved sequence homology to non-LTR and LTR reverse transcriptases (Shippen-Lentz and Blackburn 1990). The activity of telomerase is necessary to overcome the ‘end replication problem’. The human telomerase enzyme is composed of two essential components, the RNA component (TERC: Telomerase RNA) which acts as a template for reverse transcription (Blasco et al. 1995); and the catalytic subunit Telomerase reverse transcriptase (TERT) with the reverse transcriptase activity (Meyerson et al. 1997; Nakamura et al. 1997). In recent years, a number of additional factors, including dyskerin, TCAB1, NOP10 and TPP1 have been identified to be constantly or transiently associated with the telomerase complex and have important functions in telomerase recruitment to telomeres or subcellular localization of the telomerase complex (Cohen et al. 2007; Collins and Mitchell 2002; Nandakumar and Cech 2013; Venteicher et al. 2009; Zhong et al. 2011; Gonzalez et al. 2014).
Telomerase is active in a variety of tumor cell lines and transformed cells in culture but not in normal fibroblasts (Morin 1989) or embryonic kidney cells (Counter et al. 1992) and most somatic human tissues do not exhibit telomerase activity (Djojosubroto et al. 2003; Kim et al. 1994; Meyerson et al. 1997; Shay and Wright 1996; Weise and Gunes 2006). In human, telomerase activity is down-regulated during embryogenesis and cellular differentiation through repression of its catalytic subunit (Gunes et al. 2000; Wright et al. 1996; Sirma et al. 2011). Due to the lack of telomerase, telomeres shorten during aging in human tissues in vivo and telomere length sets a limit to the proliferative capacity of human fibroblasts (HFs) in vitro involving the p53 and Rb pathways (Chang and Harley 1995; Harley et al. 1990; Shay et al. 1991). In this line, cells devoid of these two major pathways exhibit extended life-span but telomeres continue to shorten until a ‘crisis’ checkpoint. Cells that survive the crisis checkpoint possess telomerase activity or activate an alternative mechanism of telomere maintenance (ALT) (Counter et al. 1992). Based on these observations, Allsopp et al. (1992) proposed a model for of telomere hypothesis of ‘cell ageing and immortalization’ (Fig. 3).

Telomere hypothesis of senescence and cancer. Proliferation-dependent telomere shortening leads to telomere dysfunction, manifested by non-reciprocal-translocations and end-to-end fusions, resulting in the activation of DNA-damage checkpoints, and induction of senescence in telomerase negative, check-point proficient human cells. Checkpoint-deficient cells continue to proliferate experiencing further telomere shortening and eventually end up in crisis, characterized by apoptotic cell death, in the absence of a telomere maintenance mechanism. Activation of telomerase (or the ALT mechanism) is one of the key events to overcome crisis during tumourigenesis to stabilize telomere length and for the continuous proliferation of malignant cells
These observations together with the findings that telomerase activity can be detected in early human development but is absent in most normal somatic cells have led to the hypothesis that the down-regulation of telomerase activity in somatic cells may be a tumor-protective mechanism. In line with this hypothesis it was shown that telomerase is required for tumorigenic conversion of primary human cells (Hahn et al. 1999a). In adult human tissues some cell types maintain weak but detectable telomerase activity or telomerase activity may be induced upon stimulation. These include bone marrow stem cells, germline cells in testes, activated peripheral blood lymphocytes, skin epidermis and intestinal crypt cells (Chiu et al. 1996; Hiyama et al. 1995, 1996; Morrison et al. 1996; Ramirez et al. 1997; Ravindranath et al. 1997; Ritz et al. 2005; Weise and Gunes 2009).
Although telomerase activity could be detected in the vast majority of human cancers, it is worth mentioning that about 10–15 % of human tumors do not express detectable levels of telomerase activity. Tumors that lack telomerase activity, maintain their telomere length via a recombination-based mechanism (ALT for Alternative Lengthening of Telomeres) (Bryan et al. 1997). Experimental data indicate that telomere maintenance is required for continuous tumor cell proliferation and tumor progression (Greenberg et al. 1999; Hahn et al. 1999b; Rudolph et al. 2001). The prominent occurrence of telomerase in human cancers and data from mouse models on its requirement for tumor progression motivated the development of telomerase inhibitors to suppress tumor growth in pre-clinical studies (Damm et al. 2001; Dikmen et al. 2005; Djojosubroto et al. 2005; Herbert et al. 2002; Kumar et al. 2013; Norton et al. 1996; Zahler et al. 1991). One of these inhibitors, a lipid-conjugated 13-mer oligonucleotide that is complementary to the RNA template of telomerase, thereby directly inhibiting telomerase activity is a promising candidate and has evaluated safety, tolerability and pharmacokinetics in Phase I clinical trials. This inhibitor, Imetelstat, was developed by Geron Inc. and is now being tested to treat Hematologic Myeloid Malignancies in Phase II clinical trials. As a potential drawback, experimental studies on mouse models showed that deletion of telomerase in tumors provokes the activation of ALT as an adaptive response in cancer cells (Hu et al. 2012). It is therefore essential to explore and understand the factors that control the ALT pathway.
2 Telomere Shortening Impairs Proliferation of Transformed Cells but Dysfunctional Telomeres Can Initiate Cancer Formation
The role of telomeres in human biology was unclear until the discovery of telomerase and subsequent demonstration that telomeres shorten during aging due to the end-replication problem (Greider and Blackburn 1985; Harley et al. 1990; Hastie et al. 1990). As discussed above, telomere shortening limits the proliferation capacity of human cells, referred to as ‘Hayflick Limit’. At this stage, cells exhibit a ‘cellular senescence’ phenotype characterized by morphological changes and by the accumulation of aneuploidy, polyploidy and chromosomal fusions (Benn 1976; Saksela and Moorhead 1963; Thompson and Holliday 1975). Telomerase negative human cells that can overcome the senescence checkpoint by the expression of viral oncoproteins continue to accumulate chromosomal instability during the extended proliferation period (Counter et al. 1992). These observations indicated a pivotal role of functional telomeres in genome stability and telomerase activity thwarted telomere shortening and genomic instability (Harley 1991).
Dysfunctional telomeres can result either from alterations in the telomere-associated proteins required for end-capping function, or from alterations that promote the gradual or sudden loss of sufficient repeat sequence necessary to maintain proper telomere structure. The identification of mammalian telomerase components in the mid 90s enabled to experimentally address the functional role of telomere shortening in aging and cancer formation in vivo (Blasco et al. 1997; Rudolph et al. 1999).
Telomerase knockout mice exhibit progressive shortening of telomeres resulting in loss of telomere capping function (also referred to as telomere dysfunction) in 3rd–6th generation of knockout mice. In vivo studies supported the observations from HFs that dysfunctional telomeres are recognized by the DNA-damage-response (DDR) machinery leading to activation of p53 and Rb dependent checkpoints inhibiting tumorigenesis in cancer mouse models (Chin et al. 1999; Greenberg et al. 1999). A formal experimental prove of telomere-dysfunction induced tumor suppression in vivo was provided by studies where overexpression of c-Myc oncogene in mice with short telomeres induced genomic instability as determined by increased end-to-end fusions, non-reciprocal translocations and anaphase bridges. These genomic instability induced senescence in the presence of wild-type p53 (Feldser and Greider 2007). In fact, the tumor suppressor function was dependent on the senescence-activation function of p53 (Cosme-Blanco et al. 2007). During aging or in the absence of functional checkpoints, however, (i.e., loss of p53) or by the co-expression of oncogenic mutations, telomere dysfunction promotes genomic instability and initiates tumorigenesis (Artandi et al. 2000; Chin et al. 1999; Rudolph et al. 1999, 2001). The studies with telomerase deficient mice also underpinned the need for telomere stability—either by activating telomerase or by the ALT mechanism—for continuous tumor cell proliferation in vivo (Begus-Nahrmann et al. 2012; Ding et al. 2012; Greenberg et al. 1999; Jaskelioff et al. 2009; Rudolph et al. 2001).
In human, telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities and therefore is a cancer predisposition factor (Gisselsson et al. 2001; Wu et al. 2003). The cellular basis of telomere dysfunction induced genomic instability is explained by chromosomal breakage-fusion-bridge (BFB) cycles (McClintock 1939, 1941). Persistent or transient telomere dysfunction in telomerase knockout mice can result in increased mutation rates and induce BFB-cycles resulting in gains and losses of chromosomes (Blasco et al. 1997; Hackett et al. 2001; Lee et al. 1998; Rudolph et al. 2001). Although BFB-cycles seem to be the major physiological outcome of dysfunctional telomeres, persistent telomere dysfunction can induce genomic instability via cytokinesis failure and tetraploidy (Davoli et al. 2010; Pampalona et al. 2012).
Progressive telomere shortening may also result from mutations in shelterin proteins and telomerase have been shown to be associated with human pathologies. Mutations in telomerase components (TERT, TERC, DKC1) telomerase associated factors (NOP10, NHP2, WRAP53) or the shelterin components (TRF1, TRF2, POT1) forms a bigger portion of several human diseases, like dyskeratosis congenita, aplastic anemia, pulmonary fibrosis, malignant melanoma and late stage liver cirrhosis (Hartmann et al. 2011; Savage et al. 2008; Shi et al. 2014; Vulliamy et al. 2001, 2004, 2005; Walne et al. 2007, 2008; Yamaguchi et al. 2005, 2010; Zhong et al. 2011). Mutations in telomerase components result in reduced telomerase activity and accelerated telomere shortening and thus accelerated stem cell exhaustion with age, accompanied by an increased frequency of chromosomal breaks and chromosomal aberrations and increased risk for cancer formation (Calado et al. 2012).
Together, both, mice and human studies indicate that telomere dysfunction induced genetic instability occurs through persistent bridge-breakage events, leading to a continuous reorganization of the tumor genome. These findings also show that senescence and apoptosis induced by telomere dysfunction and p53 activation contribute to tumor suppression.
3 Activation of Checkpoints as a Consequence of Telomere Dysfunction
Due to their structure and shielding by shelterin components telomeres are protected from irregular repair activities. Studies on shelterin components have identified at least six different DNA damage repair pathways that protect telomeres from irregular recombination events (Martinez et al. 2012; Sfeir and de Lange 2012). The choice of the repair pathway is dependent on the type of DNA-damage and the cell type and dictates the cellular consequences in response to telomere dysfunction. Mammalian DSBs are repaired primarily by homologous recombination (HR) or non homologous end joining (NHEJ). Gene knockout studies have revealed that loss of the shelterin components TRF1 and TRF2 activates ATM/ATR signaling for NHEJ whereas dysfunctional telomeres due to loss of POT1 trigger ATR-signaling or the activation of homologous DNA repair. Activation of the classical (c-NHEJ) or alternative (alt-NHEJ) non homologous end-joining repair pathways involving MRN complex (MRE11, NBS and Rad50), DNA-PK and Lig4 (c-NHEJ) or Lig3 or CtIP (alt-NHEJ) (Rai et al. 2010) initiate end-to-end fusions but repair activities at dysfunctional telomeres leads to chromosomal fusions, which are not stable during the cell cycle and can be a source of genetic instability (d’Adda di Fagagna et al. 2004; Takai et al. 2003). Upstream protein kinases such as ataxia telangiectasia mutated (ATM) and ATR as well as the downstream protein kinases CHK1 and CHK2 are also involved in the 5′-end-resection at dysfunctional telomeres causing a G1 cell cycle arrest or the senescence response by activating the tumour suppressor p53 pathway. In the absence of p53BP, a target of the ATM kinase that accumulates at the sides of DNA damage and suppresses end-resection, the classical NHEJ pathway is inhibited and may direct the repair mechanism towards the homologous repair, resulting in increased recombination at dysfunctional telomeres, a phenotype observed in telomerase negative, ALT-positive tumor cells (Dimitrova et al. 2008; Martinez et al. 2012).
Whether the same pathways are activated as a consequence of physiological telomere shortening remains to be shown but some data exist indicating that the alt-NHEJ is the major pathway to repair DNA damage at naturally occurring dysfunctional telomeres (Rai et al. 2010). The p16/INK4a-Rb pathway has been implemented to contribute to the detection of telomere-induced DNA damage, activating the senescence pathway and recent data show that p16/INK4a protects cells against dysfunctional telomere–induced ATR-dependent DDR in Pot1b deficient mice but the contribution of p16 remains still elusive yet (Shay et al. 1991; Wang et al. 2013). Elucidating the DDR pathways in response to physiological telomere dysfunction would be crucial to better understand the role of genomic instability to tumorigenesis during aging.
4 Telomere-Dysfunction and Induction of Senescence as a Tumor Suppressor Mechanism
As discussed above, telomere shortening is regarded as the main cause of telomere dysfunction leading to induction of replicative senescence in aging cells. There is now emerging evidence that the accumulation of telomeric DNA damage in response to DNA replication stress can also contribute to induction of senescence. The induction of this checkpoint involves abrupt induction of replication stress at telomeres, which appears to be independent of classical telomere shortening (Fig. 4).

Telomerase activity alleviates telomere replication stress and facilitates to overcome oncogene-induced senescence. Oncogene activation leads to abrupt accumulation of DNA damage at telomeres resulting in senescence and tumour suppression. Telomerase-positive stem cells could be resistant to oncogene-induced senescence and may be selected as the cell type of origin of tumour development
Dysfunctional telomeres can be detected by the accumulation of telomere dysfunction-induced foci (TIF) at the telomeres (d’Adda di Fagagna et al. 2003; Takai et al. 2003). These foci include 53BP1 and phosphorylated H2AX (gamma-H2AX) at the dysfunctional telomeres. Interestingly, recent observations show the accumulation of persistent TIFs upon oncogene-induced senescence (OIS) or stress-induced senescence (Fumagalli et al. 2012; Hewitt et al. 2012; Suram et al. 2012). We recently showed that aneuploidy-induced senescence (AIS) involves replication stress and TIF formation at telomeres indicating that telomeres seem to mediate (AIS) (Meena et al. 2015). These new findings may provide a unifying mechanism for senescence as a general tumor suppressor mechanism whereby telomeres may converge different kinds of cellular stress in one pathway (Reviewed in Gunes and Rudolph 2012, 2013).
The biological basis for this function of telomeres as a sensor of replication defects may be due to their specific sequence composition and structure. Telomeres can form G-quadruplex structures (G4) by intra-molecular Hoogsteen G-G base pairs. G4 structures increase in a cell cycle dependent manner in human cells (Biffi et al. 2013) and preferentially form at the 3′-end of chromosomes (Tang et al. 2008), are highly stable. G4 structures are thought difficult to resolve during replication and may provoke replication fork stalling and chromosome fragility (Tarsounas and Tijsterman 2013). Fragile sites are particularly prone to chromosomal breakage and recombination events as a result of replication stress (O’Keefe and Richards 2006). Replication stress can be induced by inappropriate proliferation signaling such as oncogene activation or loss of cell cycle inhibitors that deregulate transcription and generate DNA damage (Bermejo et al. 2012; Di Micco et al. 2006). Telomeres are difficult to replicate and may lead to fork stalling during replication upon inflated proliferation signals (Suram et al. 2012). Consistently, replication stress at telomeres and thus inefficient replication of telomeric DNA could attract DDR and induce the senescence checkpoints as a tumor suppressor mechanism. In cells defective in functional repair mechanisms or faithful telomere replication, however, dysfunctional telomeres can initiate genome instability.
5 TRF1 and Telomerase in the Context of Telomere Replication Stress
There is emerging experimental evidence that replication through difficult replicating sites requires coordinated action of telomerase activity, telomere binding proteins and specific helicases that are recruited to the telomeres for faithful replication. TRF1 plays a key role in this context. Loss of mammalian TRF1 or its fission yeast counterpart Taz1 leads to stalled replication forks and fragile telomere phenotype (Martinez et al. 2009; Miller et al. 2006; Sfeir et al. 2009). Importantly, MEFs from TRF1 deficient mice exhibited a premature senescence phenotype compared to their wild type counterparts; in the absence of cellular checkpoints, i.e., in cells expressing SV40-LT, the senescence phenotype was rescued but led to increased chromosomal instability (Martinez et al. 2009). At organismal level, mice lacking TRF1 in the stratified epithelia (TRF1flox/flox × K5-Cre transgenic bitransgenic mice) showed dysfunctional telomeres associated with skin hyperpigmentation and epithelial dysplasia but died perinatally. When these mice were crossed with p53 null mice, they could survive but exhibited an increase in squamous cell carcinoma. Together, these studies indicate that telomere replication is facilitated by the shelterin factor TRF1 to prevent replication fork stalling and that telomeric replication stress generates fragile telomeres that can instigate genomic instability and cancer.
Interestingly, BLM helicase, which is also able to bind and resolve G4 structures, interacts with TRF1 and is recruited to telomeres during replication in late S/G2 and cells lacking BLM accumulate dysfunctional telomeres and telomere-dependent chromosome fusions (Barefield and Karlseder 2012). RTEL1 is another helicase that facilitates faithful telomere replication, potentially by resolving the G-quadruplex structures at the T-loop (Vannier et al. 2012, 2013). Other helicases with G4 resolving activity include the recQ helicases WRN, RECQL4 and DNA2. DNA2 deficiency results in defective telomere replication, leading to elevated fragile telomeres, telomeres loss, and telomere DNA damage response (Lin et al. 2013). In the same line, it has recently been demonstrated that the activity of the Pif1 helicase, that can associate with telomerase, is required to open telomeric G4 structures and that the enzymatic activity of telomerase is crucial for this function indicating that the damage present at telomeres is repaired by telomerase (Chang et al. 2009; Mateyak and Zakian 2006; Paeschke et al. 2011). It remains speculative whether Pif1 activity precedes and facilitates telomere replication or it is required to resolve structures generated during replication. Studies in the ciliate Stylonychia lemnae indicate that telomerase recruitment by the telomere binding protein-ß, the homologue of the mammalian shelterin protein TPP1, facilitates unfolding G4-structures. However, the exact mechanisms how these helicases act to resolve telomeric G4 and their differential functions remain elusive.
Recent studies indicate that BRCA2 and RAD51 act in concert to heal fragile telomeres in mouse cells, probably by enabling the restart of replication at stalled replication forks that are processed by HR during the S-phase (Badie et al. 2010). BRCA2 recruits RAD51 to the telomeres during replication in S-phase and both factors are required for maintenance of telomere length in mouse embryonic fibroblasts (MEFs). Consistently, MEFs lacking BRCA2 or RAD51 exhibited an increased fragility, telomere shortening and telomere dysfunction induced DNA damage foci (TIF) indicative of loss of telomere protection. Interestingly, telomerase positive cells showed higher fragility in the context of BRCA2 mice when compared to telomerase negative cells with shorter telomeres from late generation telomerase knockout cells. This result indicates that longer telomeres have a greater chance to accumulate fragile telomeres in the absence of repair mechanisms and in the presence of telomerase. In conclusion, the adult stem cells, the main cell type that retains telomerase activity in adult human tissues may represent the cell type of origin of cancer formation (Fig. 4).
Together, telomeres have a dual role in cancer formation. Telomere shortening and telomere replication stress in malignant cell clones serve as a tumor suppressor mechanism by activating senescence and or crisis checkpoints. In contrast, telomere shortening in aging tissues can also lead to an induction of chromosomal instability by promoting chromosomal fusion and fusion-bridge-breakage cycles. In addition, the inhibition of cell proliferation in aging tissues can also increase the selective pressure for clonal outgrowth of (pre-) malignant cell clones by changing the tissue environment and by impairing proliferative competition of non-transformed cells (Bilousova et al. 2005; Braig et al. 2014; Ju and Rudolph 2006). The influence of telomeres on tumor protection/tumor promotion may depend on the lifetime. Early in life when telomeres are long cancer protective effects of telomere shortening/replication stress in malignant cell clones may be dominant. In contrast, tumor-promoting effects of telomere shortening may become dominant in aged tissue and tissues experiencing telomere shortening in response to chronic diseases such as liver cirrhosis in response to hepatitis or progressive stages of ulcerative colitis (Rabinovitch et al. 1999; Rudolph et al. 2009). It remains to be investigated whether targeting of senescence checkpoints in response to telomere shortening or telomere replication stress could lead to development of novel anti-cancer therapies and how these approaches affect tissue aging. Studies in mouse models indicate that it is possible to improve tissue maintenance without increasing cancer risk by inhibiting downstream checkpoint responses (Cdkn1a/p21) that limit proliferation of cells in response to telomere shortening (Choudhury et al. 2007). In addition, it was shown that p21 deletion can have anti-tumor effects in mouse models of leukemia or irradiated human tumor cells (Lazzarini et al. 2008; Viale et al. 2009; Waldman et al. 1996). It is possible that the tumor inhibiting effects of p21 deletion involve the increase in telomere replication stress in genomically instable tumor cells. Together, these studies suggest that it should be possible to define molecular targets that can improve both tissue maintenance and cancer protection in aging tissues.
Important areas of future research include the delineation of (i) distinct cellular stress factors that cause telomere replication stress, (ii) molecular mechanisms that are involved in the induction of replication stress, (iii) activation of checkpoints in response to replication stress at telomeres, and (iv) mechanism how telomerase contributes to the suppression of telomere replication stress.
References
Allshire RC, Dempster M, Hastie ND (1989) Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res 17:4611–4627
Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, Harley CB (1992) Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 89:10114–10118
Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA (2000) Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406:641–645
Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, Suram A, Jaco I, Benitez J, Herbig U et al (2010) BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol 17:1461–1469
Barefield C, Karlseder J (2012) The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res 40:7358–7367
Begus-Nahrmann Y, Hartmann D, Kraus J, Eshraghi P, Scheffold A, Grieb M, Rasche V, Schirmacher P, Lee HW, Kestler HA et al (2012) Transient telomere dysfunction induces chromosomal instability and promotes carcinogenesis. J Clin Investig 122:2283–2288
Benn PA (1976) Specific chromosome aberrations in senescent fibroblast cell lines derived from human embryos. Am J Hum Genet 28:465–473
Bermejo R, Kumar A, Foiani M (2012) Preserving the genome by regulating chromatin association with the nuclear envelope. Trends Cell Biol 22:465–473
Biffi G, Tannahill D, McCafferty J, Balasubramanian S (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5:182–186
Bilousova G, Marusyk A, Porter CC, Cardiff RD, DeGregori J (2005) Impaired DNA replication within progenitor cell pools promotes leukemogenesis. PLoS Biol 3:e401
Blackburn EHS, Szostak JW (1984) The molecular structure of centromeres and telomeres. Ann Rev Biochem 53:163–194
Blasco MA, Funk W, Villeponteau B, Greider CW (1995) Functional characterization and developmental regulation of mouse telomerase RNA. Science 269:1267–1270
Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91:25–34
Braig M, Pallmann N, Preukschas M, Steinemann D, Hofmann W, Gompf A, Streichert T, Braunschweig T, Copland M, Rudolph KL et al (2014) A ‘telomere-associated secretory phenotype’ cooperates with BCR-ABL to drive malignant proliferation of leukemic cells. Leukemia
Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3:1271–1274
Calado RT, Cooper JN, Padilla-Nash HM, Sloand EM, Wu CO, Scheinberg P, Ried T, Young NS (2012) Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia 26:700–707
Cesare AJ, Quinney N, Willcox S, Subramanian D, Griffith JD (2003) Telomere looping in P. sativum (common garden pea). Plant J 36:271–279 (for cell and molecular biology)
Chang E, Harley CB (1995) Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci USA 92:11190–11194
Chang M, Luke B, Kraft C, Li Z, Peter M, Lingner J, Rothstein R (2009) Telomerase is essential to alleviate pif1-induced replication stress at telomeres. Genetics 183:779–791
Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97:527–538
Chiu CP, Dragowska W, Kim NW, Vaziri H, Yui J, Thomas TE, Harley CB, Lansdorp PM (1996) Differential expression of telomerase activity in hematopoietic progenitors from adult human bone marrow. Stem Cells 14:239–248
Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J et al (2007) Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet 39:99–105
Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR (2007) Protein composition of catalytically active human telomerase from immortal cells. Science 315:1850–1853
Collins K, Mitchell JR (2002) Telomerase in the human organism. Oncogene 21:564–579
Cosme-Blanco W, Shen MF, Lazar AJ, Pathak S, Lozano G, Multani AS, Chang S (2007) Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep 8:497–503
Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 11:1921–1929
d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426:194–198
d’Adda di Fagagna F, Teo SH, Jackson SP (2004) Functional links between telomeres and proteins of the DNA-damage response. Genes Dev 18:1781–1799
Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H, Niestroj C, Daiber C, Enenkel B, Guilliard B et al (2001) A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J 20:6958–6968
Davoli T, Denchi EL, de Lange T (2010) Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 141:81–93
de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19:2100–2110
de Lange T (2010) How shelterin solves the telomere end-protection problem. Cold Spring Harb Symp Quant Biol 75:167–177
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A et al (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642
Dikmen ZG, Gellert GC, Jackson S, Gryaznov S, Tressler R, Dogan P, Wright WE, Shay JW (2005) In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res 65:7866–7873
Dimitrova N, Chen YC, Spector DL, de Lange T (2008) 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456:524–528
Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu GC, Wang G, Lu X, Labrot ES et al (2012) Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell 148:896–907
Djojosubroto MW, Choi YS, Lee HW, Rudolph KL (2003) Telomeres and telomerase in aging, regeneration and cancer. Mol Cells 15:164–175
Djojosubroto MW, Chin AC, Go N, Schaetzlein S, Manns MP, Gryaznov S, Harley CB, Rudolph KL (2005) Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 42:1127–1136
Doksani Y, Wu JY, de Lange T, Zhuang X (2013) Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155:345–356
Feldser DM, Greider CW (2007) Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 11:461–469
Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM et al (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14:355–365
Gisselsson D, Jonson T, Petersen A, Strombeck B, Dal Cin P, Hoglund M, Mitelman F, Mertens F, Mandahl N (2001) Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci USA 98:12683–12688
Gonzalez OG, Assfalg R, Koch S, Schelling A, Meena JK, Kraus J, Lechel A, Katz SF, Benes V, Scharffetter-Kochanek K, Kestler HA, Gunes C, Iben S (2014) Telomerase stimulates ribosomal DNA transcription in hyperproliferative conditions. Nat Commun 5:4599
Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA (1999) Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 97:515–525
Greider CW, Blackburn EH (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43:405–413
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999) Mammalian telomeres end in a large duplex loop. Cell 97:503–514
Gunes C, Rudolph KL (2012) Telomere dysfunction puts the brakes on oncogene-induced cancers. EMBO J 31:2833–2834
Gunes C, Rudolph KL (2013) The role of telomeres in stem cells and cancer. Cell 152:390–393
Gunes C, Lichtsteiner S, Vasserot AP, Englert C (2000) Expression of the hTERT gene is regulated at the level of transcriptional initiation and repressed by Mad1. Cancer Res 60:2116–2121
Hackett JA, Feldser DM, Greider CW (2001) Telomere dysfunction increases mutation rate and genomic instability. Cell 106:275–286
Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999a) Creation of human tumour cells with defined genetic elements. Nature 400:464–468
Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA (1999b) Inhibition of telomerase limits the growth of human cancer cells. Nat Med 5:1164–1170
Harley CB (1991) Telomere loss: mitotic clock or genetic time bomb? Mutat Res 256:271–282
Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345:458–460
Hartmann D, Srivastava U, Thaler M, Kleinhans KN, N’Kontchou G, Scheffold A, Bauer K, Kratzer RF, Kloos N, Katz SF et al (2011) Telomerase gene mutations are associated with cirrhosis formation. Hepatology 53:1608–1617
Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC (1990) Telomere reduction in human colorectal carcinoma and with ageing. Nature 346:866–868
Herbert BS, Pongracz K, Shay JW, Gryaznov SM (2002) Oligonucleotide N3′ → P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene 21:638–642
Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3:708
Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyama E, Piatyszek MA, Shay JW, Ishioka S, Yamakido M (1995) Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol 155:3711–3715
Hiyama E, Tatsumoto N, Kodama T, Hiyama K, Shay J, Yokoyama T (1996) Telomerase activity in human intestine. Int J Oncol 9:453–458
Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, Ding Z, Ying H, Boutin AT, Zhang H et al (2012) Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148:651–663
Jaskelioff M, Song W, Xia J, Liu C, Kramer J, Koido S, Gendler SJ, Calderwood SK, Gong J (2009) Telomerase deficiency and telomere dysfunction inhibit mammary tumors induced by polyomavirus middle T oncogene. Oncogene 28:4225–4236
Ju Z, Rudolph KL (2006) Telomeres and telomerase in stem cells during aging and disease. Genome Dyn 1:84–103
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266:2011–2015
Kipling D, Cooke HJ (1990) Hypervariable ultra-long telomeres in mice. Nature 347:400–402
Klobutcher LA, Swanton MT, Donini P, Prescott DM (1981) All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and an unusual 3′ terminus. Proc Natl Acad Sci USA 78:3015–3019
Kumar M, Witt B, Knippschild U, Koch S, Meena JK, Heinlein C, Weise JM, Krepulat F, Kuchenbauer F, Iben S et al (2013) CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis. Int J Cancer 132:2032–2043
Lazzarini R, Moretti S, Orecchia S, Betta PG, Procopio A, Catalano A (2008) Enhanced antitumor therapy by inhibition of p21waf1 in human malignant mesothelioma. Clin Cancer Res 14:5099–5107 (an official journal of the American Association for Cancer Research)
Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, DePinho RA (1998) Essential role of mouse telomerase in highly proliferative organs. Nature 392:569–574
Lin W, Sampathi S, Dai H, Liu C, Zhou M, Hu J, Huang Q, Campbell J, Shin-Ya K, Zheng L et al (2013) Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J 32:1425–1439
Makarov VL, Hirose Y, Langmore JP (1997) Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 88:657–666
Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA (2009) Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 23:2060–2075
Martinez P, Flores JM, Blasco MA (2012) 53BP1 deficiency combined with telomere dysfunction activates ATR-dependent DNA damage response. J Cell Biol 197:283–300
Mateyak MK, Zakian VA (2006) Human PIF helicase is cell cycle regulated and associates with telomerase. Cell Cycle 5:2796–2804
McClintock B (1939) The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc Natl Acad Sci USA 25:405–416
McClintock B (1941) The stability of broken ends of chromosomes in zea mays. Genetics 26:234–282
McElligott R, Wellinger RJ (1997) The terminal DNA structure of mammalian chromosomes. EMBO J 16:3705–3714
Meena JK, Cerutti A, Beichler C, Morita Y, Bruhn C, Kumar M, Kraus JM, Speicher MR, Wang ZQ, Kestler HA, d'Adda di Fagagna F, Günes C, Rudolph KL (2015) Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J 10:1371–1384
Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q et al (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90:785–795
Meyne J, Ratliff RL, Moyzis RK (1989) Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci USA 86:7049–7053
Miller KM, Rog O, Cooper JP (2006) Semi-conservative DNA replication through telomeres requires Taz1. Nature 440:824–828
Morin GB (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59:521–529
Morrison SJ, Prowse KR, Ho P, Weissman IL (1996) Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity 5:207–216
Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA 85:6622–6626
Muller HJ (1938) The remaking of chromosomes. Collect Net 13:181–195
Munoz-Jordan JL, Cross GA, de Lange T, Griffith JD (2001) t-loops at trypanosome telomeres. EMBO J 20:579–588
Murti KG, Prescott DM (1999) Telomeres of polytene chromosomes in a ciliated protozoan terminate in duplex DNA loops. Proc Natl Acad Sci USA 96:14436–14439
Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science 277:955–959
Nandakumar J, Cech TR (2013) Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol 14:69–82
Norton JC, Piatyszek MA, Wright WE, Shay JW, Corey DR (1996) Inhibition of human telomerase activity by peptide nucleic acids. Nat Biotechnol 14:615–619
O’Keefe LV, Richards RI (2006) Common chromosomal fragile sites and cancer: focus on FRA16D. Cancer Lett 232:37–47
Paeschke K, Capra JA, Zakian VA (2011) DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145:678–691
Pampalona J, Frias C, Genesca A, Tusell L (2012) Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet 8:e1002679
Rabinovitch PS, Dziadon S, Brentnall TA, Emond MJ, Crispin DA, Haggitt RC, Bronner MP (1999) Pancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res 59:5148–5153
Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S (2010) The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J 29:2598–2610
Ramirez RD, Wright WE, Shay JW, Taylor RS (1997) Telomerase activity concentrates in the mitotically active segments of human hair follicles. J Invest Dermatol 108:113–117
Ravindranath N, Dalal R, Solomon B, Djakiew D, Dym M (1997) Loss of telomerase activity during male germ cell differentiation. Endocrinology 138:4026–4029
Ritz JM, Kuhle O, Riethdorf S, Sipos B, Deppert W, Englert C, Gunes C (2005) A novel transgenic mouse model reveals humanlike regulation of an 8-kbp human TERT gene promoter fragment in normal and tumor tissues. Cancer Res 65:1187–1196
Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA (1999) Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96:701–712
Rudolph KL, Millard M, Bosenberg MW, DePinho RA (2001) Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet 28:155–159
Rudolph KL, Hartmann D, Opitz OG (2009) Telomere dysfunction and DNA damage checkpoints in diseases and cancer of the gastrointestinal tract. Gastroenterology 137:754–762
Saksela E, Moorhead PS (1963) Aneuploidy in the degenerative phase of serial cultivation of human cell strains. Proc Natl Acad Sci USA 50:390–395
Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP (2008) TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82:501–509
Sfeir A, de Lange T (2012) Removal of shelterin reveals the telomere end-protection problem. Science 336:593–597
Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90–103
Shay JW, Wright WE (1996) The reactivation of telomerase activity in cancer progression. Trends Genet TIG 12:129–131
Shay JW, Pereira-Smith OM, Wright WE (1991) A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res 196:33–39
Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac-de Paillerets B, Nagore E, Avril MF et al (2014) Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 46:482–486
Shippen-Lentz D, Blackburn EH (1990) Functional evidence for an RNA template in telomerase. Science 247:546–552
Sirma H, Kumar M, Meena JK, Witt B, Weise JM, Lechel A, Ande S, Sakk V, Guguen-Guillouzo C, Zender L et al (2011) The promoter of human telomerase reverse transcriptase is activated during liver regeneration and hepatocyte proliferation. Gastroenterology 141:326–337, 337, e321–323
Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL et al (2012) Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J 31:2839–2851
Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol CB 13:1549–1556
Tang J, Kan ZY, Yao Y, Wang Q, Hao YH, Tan Z (2008) G-quadruplex preferentially forms at the very 3′ end of vertebrate telomeric DNA. Nucleic Acids Res 36:1200–1208
Tarsounas M, Tijsterman M (2013) Genomes and G-quadruplexes: for better or for worse. J Mol Biol 425:4782–4789
Thompson KV, Holliday R (1975) Chromosome changes during the in vitro ageing of MRC-5 human fibroblasts. Exp Cell Res 96:1–6
Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ (2012) RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149:795–806
Vannier JB, Sandhu S, Petalcorin MI, Wu X, Nabi Z, Ding H, Boulton SJ (2013) RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342:239–242
Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE (2009) A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323:644–648
Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, Ronchini C, Ronzoni S, Muradore I, Monestiroli S et al (2009) Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 457:51–56
Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I (2001) The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413:432–435
Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I (2004) Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 36:447–449
Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I (2005) Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis 34:257–263
Waldman T, Lengauer C, Kinzler KW, Vogelstein B (1996) Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature 381:713–716
Walne AJ, Vulliamy T, Marrone A, Beswick R, Kirwan M, Masunari Y, Al-Qurashi FH, Aljurf M, Dokal I (2007) Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet 16:1619–1629
Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I (2008) TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112:3594–3600
Wang Y, Sharpless N, Chang S (2013) p16(INK4a) protects against dysfunctional telomere-induced ATR-dependent DNA damage responses. J Clin Investig 123:4489–4501
Weise JM, Gunes C (2006) Telomeres and telomerase. A survey about methods and recent advances in cancer diagnostic and therapy. Histol Histopathol 21:1249–1261
Weise JM, Gunes C (2009) Differential regulation of human and mouse telomerase reverse transcriptase (TERT) promoter activity during testis development. Mol Reprod Dev 76:309–317
Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW (1996) Telomerase activity in human germline and embryonic tissues and cells. Dev Genet 18:173–179
Wu X, Amos CI, Zhu Y, Zhao H, Grossman BH, Shay JW, Luo S, Hong WK, Spitz MR (2003) Telomere dysfunction: a potential cancer predisposition factor. J Natl Cancer Inst 95:1211–1218
Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS (2005) Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 352:1413–1424
Yamaguchi H, Inokuchi K, Takeuchi J, Tamai H, Mitamura Y, Kosaka F, Ly H, Dan K (2010) Identification of TINF2 gene mutations in adult Japanese patients with acquired bone marrow failure syndromes. Br J Haematol 150:725–727
Zahler AM, Williamson JR, Cech TR, Prescott DM (1991) Inhibition of telomerase by G-quartet DNA structures. Nature 350:718–720
Zakian VA (1989) Structure and function of telomeres. Annu Rev Genet 23:579–604
Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, Artandi SE (2011) Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 25:11–16
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Meena, J., Rudolph, K.L., Günes, C. (2015). Telomere Dysfunction, Chromosomal Instability and Cancer. In: Ghadimi, B., Ried, T. (eds) Chromosomal Instability in Cancer Cells. Recent Results in Cancer Research, vol 200. Springer, Cham. https://doi.org/10.1007/978-3-319-20291-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-20291-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20290-7
Online ISBN: 978-3-319-20291-4
eBook Packages: MedicineMedicine (R0)