
Abstract
Mitogen-activated protein kinases (MAPKs) are key regulators that have been linked to cell survival and death. Among the main classes of MAPKs, c-jun N-terminal kinase (JNK) has been shown to mediate cell stress responses associated with apoptosis.
In Vitro, hypoxia induced a significant increase in 661W cell death that paralleled increased activity of JNK and c-jun. 661W cells cultured in presence of the inhibitor of JNK (D-JNKi) were less sensitive to hypoxia-induced cell death.
In vivo, elevation in intraocular pressure (IOP) in the rat promoted cell death that correlated with modulation of JNK activation. In vivo inhibition of JNK activation with D-JNKi resulted in a significant and sustained decrease in apoptosis in the ganglion cell layer, the inner nuclear layer and the photoreceptor layer. These results highlight the protective effect of D-JNKi in ischemia/reperfusion induced cell death of the retina.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Neuronal cell death following excitotoxicity is a common feature of neurodegenerative and ischemic diseases of the central nervous system and of a variety of ocular diseases, such as glaucoma. Glaucoma is characterized by a slowly progressive loss of retinal ganglion cells (RGC) and their axons and is often associated with elevated intraocular pressure (IOP). Retinal ischemia/reperfusion (I/R) induced by experimental elevation of IOP leads to damage and cell death in the different layers of the retina.
Among the signaling events downstream of the excitotoxic cascade, the three main classes of mitogen-activated protein kinases (MAPKs) , extracellular signal-regulated kinase (ERK), p38 and the c-Jun N-terminal kinase (JNK) were reported to be increased after cerebral ischemia (Sugino et al. 2000; Wu et al. 2000) as well as in the retina (Peterson et al. 2000; Roth et al. 2003) . The observation that JNK was activated in ischemic neurons highlighted its potential involvement in the apoptotic process following cerebral ischemia (Borsello et al. 2003) .MAPKs activation has also been investigated in retinas after ischemia. Zhang et al. first made the observation that both JNK and p38 activation could be attenuated by ischemic preconditioning, suggesting that these two MAPKs were implicated in the deleterious effects induced by ischemia in the retina (Zhang et al. 2002) .The use of D-JNKi provided a significant protection against neuronal loss after optic nerve crush in mice and in a model of retinopathy of prematurity (Tezel et al. 2004; Guma et al. 2009) .
Here, we investigated the functional consequence of JNK activation in Vitro and In vivo and showed that JNK activity is a critical contributor to ischemic-induced retinal damages and that its inhibition resulted in the reduction of cell death.
2 Materials and Methods
2.1 Animal Handling and Surgery
All animal experiments were approved by the Veterinary Office of the State of Valais. The procedure to induce transient ischemia followed by reperfusion has previously been described (Produit-Zengaffinen et al. 2009) . Animals were divided into a control and an I/R group. In the control group, rats were sham-operated by inserting a needle into in the anterior chamber of the left eye without elevation of the IOP. In the I/R group, the needle was introduced in the left eye and the pressure was increased. Animals were divided into a control and an I/R group. In the control group, rats were sham-operated by inserting a needle into in the anterior chamber of the l after reperfusion, rats were euthanized as described previously (Produit-Zengaffinen et al. 2009).
2.2 Cell Culture
In order to evaluate the consequences of hypoxia on 661W survival, cells were cultured in DMEM, 1 % FBS, 1 mM glucose and incubated for 48 h in normoxic (21 % O2) or hypoxic (3 % O2) incubators (Hypoxic Workstation Whitley H35).
2.3 ATPlite, LDH Assay, Western Blot Analysis
Cell survival, cell death and western blot analyses were performed as previously described. Anti-phospho JNK, anti-phospho cjun and anti-cjun were obtained from Cell Signaling Technology, anti-JNK was purchased from Santa Cruz Biotechnology.
2.4 Immunohistology
Eyes were fixed as previously described. Detection of apoptosis was performed using an in situ cell death detection kit (Roche Diagnostics). TUNEL staining was performed according to the manufacturer’s instructions and images were viewed under a fluorescence microscope equipped with a digital camera using appropriate filters.
2.5 Statistic
Results are presented as mean ± standard error of the mean (SEM) of the indicated number of independent experiments. Statistical analysis was performed using Student’s t-test. Differences were considered significant at p values of 0.05 or less.
3 Results
3.1 Hypoxia Decreased 661W Viability in vitro
We first examined the effect of hypoxia on cell viability. 661W cells cultured in hypoxia for 48 h were compared to cells cultured in normoxic conditions for the same period of time. After 48 h, 661W cells cultured in hypoxia showed a 50 % decrease in cell viability as demonstrated by ATPlite measurements: 1 vs. 0.5 ± 0.04, p < 0.001. This reduced viability could be attributed to an elevation in cell death induced by hypoxia as shown by LDH release measurements: 1 vs. 4.94 ± 1.3, p<0.05.
3.2 661 W Cultured in Hypoxia Showed Increased JNK Activation
The effect of hypoxia on JNK activation was measured at the protein level by western blot. 661W cells cultured in hypoxic conditions for 48 h underwent a 2.5 fold increase in JNK activity (1 ± 0.2 vs. 2.59 ± 0.32, p < 0.005). The efficiency of JNK activity could be further visualized on c-jun phosphorylation, where hypoxia induced a similar increase in c-jun activity (1 vs. 1.8 ± 0.33, p < 0.05).
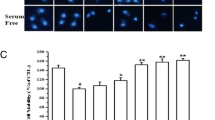
3.3 D-JNKi Prevented Hypoxia Induced Cell Death
We assessed the physiological relevance of JNK activation in the initiation of damages induced by the hypoxic stress. As ATPlite assay was not sensitive enough to measure D-JNKi effect on cell viability, we quantified hypoxia induced cell death in presence or absence of D-JNKi in living cells nuclei stained with propidium iodie (PI) and Hoechst. Cell death was increased about 8 folds after 48 h in hypoxia (1 vs. 7.7 ± 1.7, p < 0.05). A significant protective effect against cell death was obtained when cells were incubated in the presence of D-JNKi, (p < 0.05) (2.67 ± 0.73 vs. 7.7 ± 1.7 cell death in non-treated cells).
3.4 Retinal Ischemia Enhanced Apoptosis 24 h after Reperfusion
To evaluate whether this in vitro action was also effective In vivo, we analyzed the effect of I/R on retinal cell survival. In order to exclude any variation induced by the experimental method, we compared each measure to values obtained from anesthetized sham-operated rats. Twenty-four hours after reperfusion, TUNEL staining revealed a robust increase in the number of apoptotic cells in the innermost retinal layers, mainly in GCL and INL, and to a lower level, in the outer nuclear layer (ONL) (Fig. 90.1). Cells from the INL were the most sensitive to I/R (14.7 % in apoptosis ± 1.3), whereas 3.8 % ± 0.4 of GCL and 3.7 % ± 1.3 of the cells within the ONL were in apoptosis. No TUNEL positive cells could be observed in the sham-operated retina. Increased apoptosis was paralleled with increased JNK phosphorylation (2.39 ± 0.18 vs. 1 ± 0.16, p < 0.05). This was confirmed by an increase activity of cjun visible by immunohistochemistry on retinal sections.

Cell death after retinal ischemia. Retinal ischemia enhanced apoptosis 24 h after I/R. a TUNEL staining from sham-operated retina on the left and from I/R retina on the right, showed a robust increase in apoptosis in the GCL and INL from ischemic retina. b Quantification of TUNEL positive cells in the different layers of the retina. Scale bar 100 μM
3.5 D-JNKi Reduced JNK Activation in Vivo
As increased apoptotic cells correlated with elevated pJNK, we further examined the significance of JNK activity on cell death induced by I/R. We injected serial concentrations of D-JNKi in the vitreous cavity of the eye, immediately after the 1-h ischemic stress. D-JNKi was able to reduce JNK phosphorylation In vivo in a dose-dependent ability (1 ± 0.07 vs. 0.75 ± 0.18; 0.51 ± 0.15 and 0.52 ± 0.04 in non treated vs. D-JNKi 20 μdose-dependent ability (1 ± 0.07 vs. 0.75 ± 0.18; 0.51 ± 0.15 andificant at 500 μ dose-dependent 20 μM, 100 μM and 500 μM, respectively).
3.6 D-JNKi Prevented Retinal Ischemia-Induced Apoptosis 24 h after Reperfusion by Reducing the Activity of JNK
As shown in Fig. 90.2, the number of apoptotic cells within the INL was reduced by 33 % (p < 0.05) independently of the concentration of D-JNKi used (14.7 ± 1.3 vs. 7.9 ± 2.1; 10.1 ± 1.0 and 9.0 ± 1.1 in non treated eyes vs. D-JNKi 20 μ4.7 ± 1.3 vs. 7.9 ± 2.1; 10.1). In GCL, the number of apoptotic cells was also reduced by almost 30 %, probably in a dose-dependent manner, but was only statistically significant (p <0.05) at the highest concentration of D-JNKi used (3.8 ± 0.4 vs. 2.3 ± 0.5; 2.1 ± 0.7 and 1.3 ± 0.7 in non treated vs. D-JNKi 20 μM, 100 μM and 500 μM treated eyes, respectively).

D-JNKi effect on ischemia-induced cells death. The functional effect of D-JNKi was assessed by TUNEL staining on retinal section. Quantification of TUNEL positive cells vs. DAPI was performed in each retinal cell layer separately. Statistically significant neuroprotective effect of the inhibition of JNK activation was obtained in the INL (p < 0.05) at each D-JNKi concentration tested and in the GCL (p < 0.05) at 500 μM
4 Discussion
In the present study, D-JNKi, a specific inhibitor of JNK activation, was evaluated for its ability to reduce hypoxic cell death and neuronal degeneration induced by I/R in the retina. In these two models, induction of cell death was mediated through the activation of JNK. Our results indicated that treatment with D-JNKi significantly protected hypoxic 661W cells from apoptosis. This protection was also observed In vivo in rat retina when D-JNKi was injected intravitreoulsy at the end of a 1-h I/R stress.
We and others previously showed that cell death programs are induced after I/R (Buchi 1992; Zhang et al. 2002; Produit-Zengaffinen et al. 2009) . Recent studies have shown that cell death induced by I/R in the retina occurs through apoptosis (Rosenbaum et al. 1998; Zheng et al. 2007) , necrosis (Buchi 1992; Dvoriantchikova et al. 2010) and, more recently, through necroptosis, a caspase-independent form of apoptosis (Rosenbaum et al. 2010) .
Our results showed that 661W cells cultured in hypoxia have a significantly reduced cell viability, that was, at least in part, the result of JNK activation. Inhibition of JNK activation with D-JNKi significantly improved cell viability in response to hypoxia in vitro. We also established that D-JNKi was effective in a model of retinal ischemia In vivo, decreasing apoptosis within GCL, INL. We also demonstrated that the mechanisms induced in vitro by hypoxia were similar to that observed In vivo, which bestow new perspectives to study the molecular mechanisms induced by retinal ischemia.
References
Borsello T, Clarke PG, Hirt L et al (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nature Med 9:1180–1186
Buchi ER (1992) Cell death in the rat retina after a pressure-induced ischaemia-reperfusion insult: an electron microscopic study. I. Ganglion cell layer and inner nuclear layer. Exp Eye Res 55:605–613
Dvoriantchikova G, Barakat DJ, Hernandez E et al (2010) Liposome-delivered ATP effectively protects the retina against ischemia-reperfusion injury. Mol Vis 16:2882–2890
Guma M, Rius J, Duong-Polk KX et al (2009) Genetic and pharmacological inhibition of JNK ameliorates hypoxia-induced retinopathy through interference with VEGF expression. Proc Natl Acad Sci U S A 106:8760–8765
Peterson WM, Wang Q, Tzekova R et al (2000) Ciliary neurotrophic factor and stress stimuli activate the Jak-STAT pathway in retinal neurons and glia. J Neurosci 20:4081–4090
Produit-Zengaffinen N, Pournaras CJ, Schorderet DF (2009) Retinal ischemia-induced apoptosis is associated with alteration in Bax and Bcl-x(L) expression rather than modifications in Bak and Bcl-2. Mol Vis 15:2101–2110
Rosenbaum DM, Rosenbaum PS, Gupta H et al (1998) The role of the p53 protein in the selective vulnerability of the inner retina to transient ischemia. Investig Ophthalmol Vis Sci 39:2132–2139
Rosenbaum DM, Degterev A, David J et al (2010) Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 88:1569–1576
Roth S, Shaikh AR, Hennelly MM et al (2003) Mitogen-activated protein kinases and retinal ischemia. Investig Ophthalmol Vis Sci 44:5383–5395
Sugino T, Nozaki K, Takagi Y et al (2000) Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci 20:4506–4514
Tezel G, Yang X, Yang J et al (2004) Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res 996:202–212
Wu DC, Ye W, Che XM et al (2000) Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cere Blood Flow Metab 20:1320–1330
Zhang C, Rosenbaum DM, Shaikh AR et al (2002) Ischemic preconditioning attenuates apoptotic cell death in the rat retina. Investig Ophthalmol Vis Sci 43:3059–3066
Zheng L, Gong B, Hatala DA et al (2007) Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Investig Ophthalmol Vis Sci 48:361–367
Acknowledgments
The authors wish to thank Nicole Gilodi for expert technical help and Dr Al-Ubaidi, Univ. of Oklahoma, for providing the 661W cells. This work was supported by a grant from the ProVisu Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this paper
Cite this paper
Produit-Zengaffinen, N., Favez, T., Pournaras, C., Schorderet, D. (2016). JNK Inhibition Reduced Retinal Ganglion Cell Death after Ischemia/Reperfusion In Vivo and after Hypoxia In Vitro . In: Bowes Rickman, C., LaVail, M., Anderson, R., Grimm, C., Hollyfield, J., Ash, J. (eds) Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology, vol 854. Springer, Cham. https://doi.org/10.1007/978-3-319-17121-0_90
Download citation
DOI: https://doi.org/10.1007/978-3-319-17121-0_90
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-17120-3
Online ISBN: 978-3-319-17121-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)