
Abstract
Glycosphingolipids are amphiphilic plasma membrane components formed by a glycan linked to a specific lipid moiety. In this chapter we report on these compounds, on their role played in our cells to maintain the correct cell biology.
In detail, we report on their structure, on their metabolic processes, on their interaction with proteins and from this, their property to modulate positively in health and negatively in disease, the cell signaling and cell biology.
Ganglioside nomenclature is in accordance with IUPAC-IUB recommendations (Chester 1998).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Glycosphingolipids
The history of glycosphingolipids (GSL) dates back to the late nineteenth century by J.L.W. Thudichum (1884), who reported, for the first time, on the isolation of unknown compounds from human brain. These compounds were found to be cerebrosides, that is, monoglycosylceramides (Fig. 3.1), but the knowledge of their structure remained enigmatic for a long time, before being resolved. It soon became clear that glycosphingolipids are a very large family of amphiphilic compounds, which present complex structure of glycans and lipids, involved in the positive and negative regulation of the biology of our cells (Thudichum 1884; Sonnino et al. 2006).

Structure of glucosylceramide Glc-β-(1-1)-Cer (GlcCer) (a) and galactosylceramide Gal-β-(1-1)-Cer (GalCer) (b)
2 General Structure of Natural Glycosphingolipids
The term “glycosphingolipid ” refers to lipids containing at least one monosaccharide residue and a sphingoid (Fig. 3.2) or a ceramide (Fig. 3.3). Ceramides (Cer) are N-acylated sphingoids. The carbohydrate residue is attached to O-1 of the sphingoid by a glycosidic linkage. The trivial name of lyso-glycosylceramides is used for the glycosylsphingoid. The vast majority of glycosphingolipids are glycosylceramides but some glycosylsphingoids have been found in nature, which appear to be more abundant in patients with lysosomal storage diseases (Hikita et al. 2002).

Structure of sphingosine (2S,3R,4E)-2-aminooctadec-4-ene-1,3-diol,(Sph)

Structure of ceramide (Cer)
Glycosphingolipids are classified as neutral or acidic. Among the latter, the main ones contain sialic acid (Fig. 3.4) (sialosylglycosphingolipids or gangliosides) or sulfuric acid (sulfoglycosphingolipids or sulfatides), but some contain uronic acid.

Structure of: α -Neu5Ac (a) and α-Neu5Ac-(2-8)-α-Neu5A (b)
Sphingoids (Sph) are long-chain aliphatic amino alcohols, and the most common is the compound (2S,3R,4E)-2-aminooctadec-4-ene-1,3-diol. The abbreviation is d18:1 or C18-sphingosine. The corresponding saturated sphingoid (2S,3R)-2-aminoocta-1,3-diol originally called “dihydrosphingosine” is commonly coded as sphinganine, C18-sphinganine or d18:0 (Charter et al. 1947). Small amount of sphingoids composed by different number of carbon atoms, with multiple double bonds, or hydroxyl, oxo, methyl substituents have been found in humans.
The main fatty acids of naturally occurring ceramides vary in chain length from C16 to C26 and may contain one or more double bonds and/or hydroxyl substituents at C-2 (Grösch et al. 2012; Wertz 2018). The neutral chain of glycosphingolipids on the basis of its structure defines the series of the glycosphingolipid to which additional sugars (e.g., sialic acid) or sulfate are connected. Table 3.1 shows the glycan series.
In all the glycans, sugars are defined with Roman numbering from I to V starting from the saccharide linked to ceramide , and the linkage position of an additional glycose residue to the sugar of the glycan series is coded with an apical Arabic number as reported in Table 3.2.
Sulfoglycosphingolipids are glycosphingolipids carrying a sulfate ester group, formerly called “sulfatides.” The nomenclature of sulfatides is shown in Table 3.3. More detailed information on the nomenclature of glycosphingolipids established by the IUPAC-IUB are available in (Chester 1998).
3 Glycosphingolipid Metabolic Processes
Glycosphingolipids are widely distributed in all the cellular compartments, being membrane components of the subcellular fractions but are particularly abundant in the outer layer of the cell plasma membrane (PM). A small amount is also present in the cell cytosol in form of complexes with proteins (Sonnino et al. 1979; Merrill 2011).
De novo biosynthesis of glycosphingolipids first occurs in the endoplasmic reticulum (ER) where the highly hydrophobic ceramide is synthesized from serine and palmitoyl-CoA (in some tissues even stearoyl-CoA is the acceptor of serine) and following in the Golgi compartment, where the hydrophilic oligosaccharide core is built up (Merrill 2011). The oligosaccharide chains are extremely variable displaying differences related to the number, structure and sequence of sugars, as well as the position of the sugar linkage and its configuration (Figs. 3.5 and 3.6). Figure 3.7 shows the general scheme for the biosynthesis of glycosphingolipids.

Structure of: gangliotetraosylceramide, β-Gal-(1-3)-β-GalNAc-(1-4)-β-Gal-(1-4)-β-Glc-(1-1)-Cer, Gg4Cer (a) and globotetraosylceramide, β-GalNAc-(1-3)-α-Gal-(1-4)-β-Gal-(1-4)-β-Glc-(1-1)-Cer, Gb4Cer (b)

Structure of GD3 α-Neu5Ac-(2-8)-α-Neu5Ac-(2-3)-β-Gal-(1-4)-β-Glc-(1-1)-Cer (a) and 9-O-Acetyl-GD3, α-Neu5,9Ac2-(2-8)-α-Neu5Ac-(2-3)-β-Gal-(1-4)-β-Glc-(1-1)-Cer (b)

Biosynthesis of glycosphingolipids: a general scheme
The biosynthesis of ceramide requires pyridoxal phosphate (vitamin B6), NADPH (vitamin B3), and oxygen (Stoffel 1970). The ceramide moves to the cytosolic layer of Golgi membrane transported by specific proteins (CERT) and by vesicle transport (Perry and Ridgway 2005). Here, the ceramide is first glycosylated into the cerebroside glucosylceramide (GlcCer) by GlcCer synthase or into the galactosylceramide (GalCer) by galactosylceramide synthase and the corresponding highly reactive UDP-sugar.
A portion of the GlcCer is directly transported to the PM inner layer by specific GlcCer cytosolic transferring proteins. At the PM, a specific flippase places GlcCer into the outer layer. Vesicular transport, flip flop, and/or four-phosphate adaptor protein 2 (FAPP2)-mediated transport allow most of GlcCer and GalCer to reach the lumen of the late-Golgi compartments where the synthesis of more complex glycosphingolipids takes place by sequential activity of glycosyltransferases specific for the acceptor and the nucleotide activated sugar (D’Angelo et al. 2007). Neosynthesized sphingolipids move via vesicular transport to the PMs, becoming components of the external leaflet (Capasso et al. 2017). Catabolism of sphingolipids occurs in lysosomes, from which simpler products, obtained in the degradation pathway, can escape.
Lysosomes are acidic membrane-provided intracellular organelles residing mainly in the perinuclear region. They contain about 60 different acid hydrolases, whose variety reflects the capability of the lysosomes to degrade multiple kinds of macromolecules, including nucleic acids, lipids, proteins, and glycosaminoglycans (Saftig and Klumperman 2009). Most of these enzymes are soluble, except for those involved in the lipid catabolism that are principally associated with the lysosomal membrane.
Lysosomal hydrolases are synthesized in the ER and then transported to the Golgi apparatus, where they are glycosylated and tagged with mannose-6-phosphate residues in the terminal position of the oligosaccharide chains (Fig. 3.8) (Braulke and Bonifacino 2009). Mannosylated enzymes are recognized by specific mannose-6-phosphate receptors (M6PRs) in the trans-Golgi network (Ghosh et al. 2003), and the M6PR–enzyme complexes are transferred to lysosomes through clathrin-coated vesicles. In the pre-lysosomal compartment, the increased acidity induces the release of enzymes from the M6PRs, which are recycled again in the Golgi apparatus. Another transport mechanism, mediated by Lysosomal Integral Membrane Protein 2 (LIMP-2), has been identified for the lysosomal enzyme β-glucocerebrosidase, responsible for the hydrolysis of the simplest glycosphingolipid glucosylceramide into glucose and ceramide (Reczek et al. 2007).

Lysosome biogenesis and trafficking of hydrolytic enzymes. Lysosome biogenesis originates from the combination of biosynthetic and endocytic pathways as fusion between endolysosome and late endosome. Most of hydrolytic enzymes are synthesized in the endoplasmic reticulum and are glycosylated. In the Golgi apparatus they are tagged with mannose-6-phosphate (M6P) residues and, by M6P receptor (M6PR) mediated transport, can reach the endolysosomes in clathrin-coated vesicles. The acidity induces the release of enzymes from the M6PRs that is recycled to the Golgi apparatus. β-glucocerebrosidase is transported in a M6PR-independent way, by the interaction with the lysosomal integral membrane protein 2 (LIMP-2)
Lysosomes degrade intra- and extracellular macromolecules (Settembre et al. 2013) that reach these organelles by two main processes: autophagy and endocytosis. The main autophagic pathway is characterized by the formation of a double-membrane vesicle, called autophagosome, around the damaged organelle to be destroyed. Then, the fusion of the autophagosome with a lysosome allows the degradation of its content. On the other hand, endocytosis is the process involved in the internalization of extracellular material (Doherty and McMahon 2009). Endosomes undergo a maturation process characterized by multiple changes including exchange of membrane components, perinuclear localization, and decrease in luminal pH. After that, late endosomes fuse with lysosomes and lysosomal enzymes that degrade the extracellular material (Fig. 3.8).
Another important physiological role of endocytosis is its involvement in the plasma membrane turnover with particular regards for the transport of glycosphingolipids into the lysosomes for their degradation (Kolter and Sandhoff 2005). Within the lysosomes, glycosidases sequentially remove the glycosidic residues from the nonreducing end of the oligosaccharide chains.
For the proper glycosphingolipids catabolism, hydrolytic enzymes require the presence of activator proteins, a group of four small nonenzymatic glycoproteins called sphingolipid activator proteins (SAPs) all deriving from the same precursor gene, plus the GM2 activator protein (Fig. 3.9) (Kishimoto et al. 1992). At the end of the degradative pathway of the oligosaccharide chain of glycosphingolipids, ceramide is hydrolyzed by acid ceramidase and saposin D to sphingosine and fatty acid (Ferlinz et al. 2001). The final products of glycosphingolipid degradation (sphingosine, fatty acids, and sugars) leave the lysosomes through specific membrane proteins and, in part, are recycled for the biosynthesis of new sphingolipids (Sonderfeld et al. 1985; Riboni et al. 1996; Tettamanti 2004; Kitatani et al. 2008; Maceyka et al. 2012).

Lysosomal glycosphingolipids’ catabolism. The diagram reports the lysosomal catabolism of ganglioside GM1. Glycosidases sequentially remove the glycosidic residues from the non-reducing end of the oligosaccharide chains, thanks to the presence of activator proteins (Saposins – SAPs or GM2 activator protein) needed for the hydrolysis, whereas ceramide is hydrolyzed to sphingosine and fatty acid. In cytosol, sphingosine is phosphorylated and becomes substrate of a lyase, which converts sphingosine-1-phosphate in phosphoethanolamine and hexadecanal
The catabolism of glycosphingolipids depends on the recycling of the PM. Nevertheless, it is necessary to recall that only a minor portion of the endocytic vesicles becomes lysosomes, while the larger part rapidly reassociates with the PM. The glycosphingolipids’ half-life is short in neurons, in fact for some gangliosides it is about 1 h, whereas in the case of fibroblasts it is around 2–3 days. In addition, it has been determined that up to 7–8% of the total cell sphingolipids are shed daily from cultured cells (Chigorno et al. 1997, 2006; Prinetti et al. 2000). Some of these are partially taken up by cells, becoming components of the membranes and contributing to modify their composition.
Accordingly, the regulation of PM sphingolipid content and pattern involves neosynthesis, catabolism, complex intracellular trafficking, and exchanges with the extracellular environment. Any change in these pathways contributes to the composition and organization of PM glycosphingolipids. In addition, both catabolic (hydrolases) and biosynthetic (transferases) glycosphingolipid enzymes have been found to be associated with the PMs, where they have been shown to exhibit activity on the membrane components.
A possible regulation of the glycosphingolipid biosynthesis at the transcriptional level through the control of the expression of glycosyltransferases or transporter proteins must be considered. This seems to be demonstrated by the parallelism that exists in the changes in the expression of glycosyltransferases and the corresponding glycosphingolipid patterns that occur during neuronal development, oncogenic transformation, or the acquisition of drug resistance in cancer cells.
4 Glycosphingolipid–Receptor Interactions
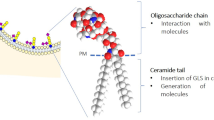
As described above, glycosphingolipids are amphiphilic molecules mainly present in the PM with the hydrophobic ceramide inserted into the lipid bilayer and with the oligosaccharide portion protruding in the extracellular environment. Indeed, they not only are membrane structural components, but also play a fundamental role in signaling and regulatory pathways. Their chemical structure makes them ideal players in the role of mediators of information across the PM. The hydrophilic portion provides recognition sites for the interaction with other molecules into the lipid bilayer, while the hydrophobic portion, in its peculiar position, allows them to interact with the other components of the plasma membrane.
The unique lipid composition of a specific cell membrane could affect the function of a protein in two independent and distinct ways, either by modifying the properties of the membrane to achieve the physical parameters required for proper protein function or by specific protein–lipid interactions (Coskun and Simons 2011). These interactions may lead to different outcomes: (1) allosteric regulation of the protein conformation; (2) regulation of protein multimerization; (3) protein segregation to membrane domains; and (4) clustering of signaling molecules in proximity to their effectors (Russo et al. 2016).
For many years, it has been hypothesized that glycosphingolipids can modulate cell signaling pathways by interacting with hormones, membrane enzymes, intracellular transducers as well as transmembrane receptors, thus modulating their properties. With the advancement of methodological technologies, some of these interactions and the participation of glycosphingolipids to the cell signaling have been studied in detail and explained. Below are some of the best known and well characterized interactions between glycosphingolipids and receptor proteins associated with the cell PM. Particularly, the role of sialic acid containing glycosphingolipids, that is, gangliosides, in regulating the trophic factor-stimulated dimerization, autophosphorylation, and subsequent signal transduction of several tyrosine-kinase receptors is discussed.
4.1 Ganglioside GM1 and Trk Receptor
Neurotrophins and their receptors have been shown to be ganglioside GM1 interactors, contributing to explain the neurotrophic and protective effects of GM1, thanks to a specific protein–oligosaccharide interaction in the extracellular space (Ferrari et al. 1995; Mutoh et al. 1995; Farooqui et al. 1997; Bachis et al. 2002; Chiricozzi et al. 2017). Specifically, GM1 ganglioside (II3Neu5Ac-Gg4Cer, β-Gal-(1-3)-β-GalNAc-(1-4)-[α-Neu5Ac-(2-3)]-β-Gal-(1-4)-β-Glc-Cer) was shown to be necessary for the function of the tyrosine kinase receptor TrkA, specific for the nerve growth factor (NGF) neurotrophin. Earlier studies demonstrated that GM1 was able to activate TrkA receptor in different cell lines (Rabin and Mocchetti 1995; Mutoh et al. 1995) and primary neurons (Da Silva et al. 2005), suggesting that GM1 may mimic NGF activity in inducing neuronal differentiation, neuritogenesis, and neuroprotection (Sokolova et al. 2014). Importantly, using PC12 cells, with a very low GM1 content, it has been shown that the exogenous administration of GM1 strongly enhances NGF-mediated TrkA activation (Mutoh et al. 1995; Farooqui et al. 1997). Moreover, in cells lacking endogenous GM1, it has been demonstrated that NGF did not induce the autophosphorylation of TrkA, but the rescue of GM1 content recovered the responsiveness of Trk to its ligand (Mutoh et al. 2002). This evidence strongly suggests that GM1 is necessary for the normal functioning of Trk protein.
Colocalization of GM1 and Trk receptors in lipid rafts has been proposed to be necessary for TrkA phosphorylation in cultured cells (Mutoh et al. 1995), brain tissues (Duchemin et al. 2008), and in vivo (Duchemin et al. 2002; Mo et al. 2005). However, it has been recently shown that in neuroblastoma cells Neuro2a (N2a), TrkA does not belong to lipid rafts together with GM1, but its interaction with GM1 involves only the GM1 oligosaccharide chain and the extracellular portion of TrkA, which may flop down on the PM approaching the GM1 oligosaccharide chain (Chiricozzi et al. 2019a). Accordingly, 30 years ago, Schengrund observed that the GM1 oligosaccharide chain alone was able to induce neuritogenesis process in a neuroblastoma cell line (Schengrund and Prouty 1988). Following, Fantini and Yahi pointed out the presence of a GM1-binding domain in the extracellular domain of Trk, suggesting that GM1 could act as an endogenous activator of Trk receptors (Fantini and Yahi 2015). Specifically, Chiricozzi and coworkers recently reported that the specific sugar code of GM1-oligosaccharide (II3Neu5Ac-Gg4, β-Gal-(1-3)-β-GalNAc-(1-4)-[α-Neu5Ac-(2-3)]-β-Gal-(1-4)-β-Glc, OligoGM1) acts as a bridge between the NGF and the TrkA receptor, directly participating in and stabilizing the interaction that leads to TrkA phosphorylation and to the activation of MAPK signaling (Fig. 3.10). This event leads to the activation of complex biochemical pathways that finally bring to neuronal differentiation and protection both in neuroblastoma cells and primary neurons (Chiricozzi et al. 2019a, b; Di Biase et al. 2020a; Fazzari et al. 2020).

Diagram of the GM1-TrkA mediated functions. GM1 ganglioside through its oligosaccharide chain stabilizes the TrkA-NGF complex on the cell surface triggering the phosphorylation of TrkA on tyrosine 490 (Tyr490) promoting MAPK signaling. This induces the activation of multiple intracellular pathways that finally lead to neuronal differentiation, protection and restoration. (Modified from Chiricozzi et al. 2017, 2019b). Glycosphingolipid sugar code is according to Varki et al. (2015)
Furthermore, GM1 also associates with TrkB, the tyrosine kinase receptor for brain-derived neurotrophic factor, BDNF (Pitto et al. 1998; Bachis et al. 2002) and with the GDNF receptor complex comprised of Ret, the tyrosine kinase component, and GFRα, a GPI-anchored coreceptor. The important role of GM1 in interaction with neurotrophic receptors is highlighted once again since the Ret association with GFRα was severely impaired in neurons totally or even partially devoid of GM1 (Hadaczek et al. 2015).
4.2 GM3 Ganglioside and EGF Receptor (EGFR)
Specific carbohydrate–carbohydrate and carbohydrate–protein interactions are known to be involved in the molecular mechanism mediating the inhibitory effect of GM3 ganglioside on the epidermal growth factor receptor (EGFR). Ganglioside GM3 ([α-Neu5Ac-(2-3)]-β-Gal-(1-4)-β-Glc) has been reported to inhibit EGF-dependent cell proliferation in a variety of cell lines. Both in vitro and in vivo, this glycosphingolipid blocks the kinase activity of the EGF receptor (Miljan and Bremer 2002; Miljan et al. 2002). The ganglioside interaction site seems to be distinct from the receptor EGF-binding site, so this effect is not due to inhibition of binding of EGF to its receptor. It has been shown that GM3 interferes with the EGFR dimerization through a direct interaction (Yednak and Bremer 1994). It was reported that EGFR forms two different specific lateral interactions with GM3 at the extracellular plasma membrane level. One is a protein–carbohydrate interaction involving the terminal N-acetylneuraminic acid of the GM3 and the lysine 642 of EGFR localized in proximity to its transmembrane domain. Through this interaction, GM3 maintains the EGFR in its inactive state preventing receptor dimerization and activation without affecting ligand binding (Coskun et al. 2011). Second, GM3 interacts with the EGFR also by a carbohydrate–carbohydrate interaction involving the sialylated galactose of GM3 and the multi terminal N-acetylglucosamine residues on EGFR N-glycans. This interaction, although weaker, was demonstrated to inhibit EGFR autophosphorylation and activation (Fig. 3.11) (Kawashima et al. 2009; Russo et al. 2016).

Diagram of EGFR inhibition mechanism by GM3 ganglioside. On the left: EGF receptor association with sphingolipid cholesterol domains prevents the aberrant activation of EGFR signaling. EGF ligand binding promotes EGFR dimerization leading to the formation of an active dimer. On the right: When GM3 is present in the bilayer, the direct association of GM3 with the EGFR domain leads to the inactivation of the EGFR kinase activity. (Modified from Coskun and Simons 2011). Glycosphingolipid sugar code is according to Varki et al. (2015)
4.3 Ganglioside GM3 and Insulin Receptor (IR)
Of clinical relevance is also the protein–carbohydrate interaction of ganglioside GM3 with the insulin receptor (IR) in adipocytes in a state of inflammation-induced insulin resistance (Nojiri et al. 1991; Tagami et al. 2002). An inhibitory effect of GM3 on insulin signaling has been reported both in cell systems (Tagami et al. 2002) and in mice lacking GM3 synthase that exhibit enhanced insulin signaling and are unaffected by insulin resistance induced by a high-fat diet (Yamashita et al. 2003).
Specifically, IRs are highly concentrated in caveolae microdomains where their binding to Cav1 is necessary for successful insulin signaling in adipocytes (Fig. 3.12) (Cohen et al. 2003). In adipocytes, in a state of TNFα-induced insulin resistance, there is an accumulation of ganglioside GM3 that is responsible for the elimination of IR from the caveolae microdomains and the inhibition of insulin metabolic signaling (Tagami et al. 2002). The dissociation of the IR–Cav1 complex is caused by the interaction between the lysine 944 residue of IR, located just above the transmembrane domain, and the increased GM3 clustered at the cell surface (Kabayama et al. 2007).

Diagram of proposed mechanism behind the shift of Insulin receptor (IR) from the caveolae to the GM3 lipid rafts in adipocytes during a state of insulin resistance. Top: IR may be constitutively resident in caveolae via its binding to the scaffolding domain of Cav1 through the caveolin binding domain in its cytoplasmic region. Binding of IR and Cav1 is necessary for successful insulin metabolic signaling. Bottom: in adipocytes the localization of IR in the caveolae is interrupted by elevated levels of the endogenous ganglioside GM3 during a state of induced insulin resistance. (Modified from Kabayama et al. 2007). Glycosphingolipid sugar code is according to Varki et al. (2015)
4.4 Lactosylceramide, Lyn, and CD11b/CD18 Integrins
Neutral glycosphingolipids expressed on PM have been considered by many studies as the cell antenna for the recognition of pathogenic microorganisms (i.e., bacteria, fungi, and viruses). This interaction occurring at the cell surface represents the starting point for the cell signal response. Neutral glycosphingolipids can modulate the reactivity to stimuli both by acting as a membrane receptor for an extracellular ligand and by interacting with the other components of the cell PM.
Lactosylceramide [LacCer; β-Gal-(1-4)-β-Glc-(1-1)-Cer] is the most abundant neutral glycosphingolipid and is highly expressed on PM of human mature neutrophils (Brackman et al. 1995; Kniep and Skubitz 1998; Spychalska et al. 2003). It has been reported to act as a pattern recognition receptor (PRR) able to recognize pathogen-associated molecular patterns (PAMPs), leading to the activation of phagocytes’ functions such as chemotaxis, phagocytosis, and superoxide generation (Iwabuchi et al. 2008; Yoshizaki et al. 2008; Nakayama et al. 2013). In particular, LacCer containing very long fatty acid chain of ceramide , C24:0 and C24:1, is the major molecular species of LacCer in human neutrophils (Iwabuchi et al. 2008). Specifically, C:24-LacCer has been demonstrated to form plasma membrane microdomains where specifically and directly interacts with Lyn protein, which is associated with the cytoplasmic layer via myristic/palmitic chains, whose activation finally leads to phagocytosis, superoxide formations, and migration mechanisms as response to pathogen infection (Fig. 3.13) (Iwabuchi and Nagaoka 2002; Iwabuchi et al. 2008; Chiricozzi et al. 2015). The long fatty acid of LacCer has been proposed to interdigitate with the acyl chains of the cytosolic layer, reducing membrane thickness. The starting process required for LacCer-immunological function is the binding of pathogen ligand with the carbohydrate moiety that triggers transmembrane signaling. Consequently, a change in the thermodynamic properties of the distal carbons of the long fatty acid acyl chain allows direct interaction with the Src kinase Lyn on the cytosolic membrane surface. This is the first evidence of a direct interaction between a glycosphingolipid resident in the outer layer of the PM with a cytosolic-associated protein.

Diagram of the LacCer-lipid raft mediated function in inflammatory response. Lactosylceramide (LacCer) with long fatty acid chain (C:24) presents into the lipid rafts of plasma membranes of neutrophil cells directly associates with the cytoplasmic protein Lyn via myristic/palmitic chains promoting its phosphorylation and activation of other proteins (Gi family protein). This specific C:24-LacCer/Lyn interaction finally leads to phagocytosis, superoxide generation and cell migration following CD11b/CD18 integrin translocation in C:24-LacCer microdomain in response to bacterial infection . (Modified from Chiricozzi et al. 2018)
Importantly C:24-LacCer enriched PM microdomains are also fundamental for the CD11b/CD18 integrin mediated phagocytosis of nonopsonized microorganisms by human neutrophils (Iwabuchi and Nagaoka 2002; Nakayama et al. 2008). Upon binding of PAMPs expressed on pathogens, CD11b/CD18 is activated and undergoes a conformational change, resulting in the rearrangement of cytoskeletal proteins. The CD11b/CD18 subsequently translocates into LacCer-enriched membrane microdomains, allowing CD11b/CD18 to transmit stimulatory signals to Lyn through the interaction of LacCer with residues 514–553 in the C-terminal portion of the conserved domain of CD18 (Lu et al. 2001; Nakayama et al. 2008). These signaling cascades lead to the formation of actin-enriched phagocytic cups, resulting in phagosome formation (Nakayama et al. 2013).
4.5 Gb3 Globotriaosylceramide and Fas (CD95) Receptor
Another important neutral glycosphingolipid involved in the regulation of cell signaling response across the PM is globotriaosylceramide [Gb3, α-Gal(1-4)-β-Gal(1-4)-β-Glc-Cer] through its interaction with Fas receptor. Fas (CD95/APO-1/TNFRSF6) is a TNF receptor superfamily member, known as a key apoptosis activator, upon binding with its specific ligand (FasL). An efficient Fas activation of the apoptotic mechanism is subsequent to its internalization. Specifically, this process requires the interaction between Fas and a specific glycosphingolipid-rich membrane Gb3 raft. In particular, it has been demonstrated that Fas receptor contains a conserved extracellular glycosphingolipid-binding motif that interacts specifically with Gb3 regulating its internalization route and consequently the signals transmitted upon ligand binding (Lee et al. 2006; Chakrabandhu et al. 2008). When interacting with Gb3, Fas bounded to its ligand is internalized by clathrin-dependent endocytosis, which allows the transduction of cell death signal deriving from caspase-8 cascade activation (Kischkel et al. 1995; Mayor and Pagano 2007). In the absence of the lipid–receptor interaction, ligand-bound Fas is internalized by ezrin-mediated endocytosis, activating a progrowth signaling through MAPK cascade pathway (Chakrabandhu et al. 2008).
5 Aging: Glycosphingolipids in Span Life
Aging is defined as the progressive decline of the body’s physiological functions, not necessarily linked to the chronological age (Gladyshev 2016). This is accompanied by an ever-decreasing ability of the organism to respond adequately to stimuli and thus the increasing of mortality rate (Flatt 2012). Aging is the first risk factor for a series of pathologies such as neurodegenerative disorders, cardiovascular and immune system diseases, and cancer (Ferrucci et al. 2020). Many factors are thought to contribute to mammalian aging including changes in gene expression, mitochondrial dysfunction, oxidative stress, shortening of telomeres, and accumulation of advanced glycation end-products. These processes are accompanied by substantial molecular and cellular changes that also affect glycosphingolipids. Changes in the content and composition of glycosphingolipids during aging have been mainly studied in few tissues, both in humans and in experimental in vivo and in vitro models. Here, we will make an overview on the variations that glycosphingolipids undergo during aging in different tissues.
5.1 Glycosphingolipids in Brain
Sialic acid-containing glycosphingolipids are particularly enriched in the nervous system playing both a structural and an essential functional role for the adequate development and maintenance of the nervous system (Schengrund 2015). The ganglioside content in the brain increases as development proceeds and simultaneously the expression pattern of gangliosides shifts from simple gangliosides, such as GM3 and GD3, which are mainly expressed in embryos and along the early post-natal life, to complex gangliosides, with GM1, GD1a, GD1b, and GT1b becoming the most represented ganglioside species in the adults (Ngamukote et al. 2007). In humans the content of gangliosides remains barely constant from 20 to 70 years, after when undergoes a progressive decline (Svennerholm 1994). Ganglioside changes in the brain are region-specific (Kracun et al. 1991; Svennerholm and Gottfries 1994). In fact, the frontal and temporal cortices and respective white matters show a reduction mostly of GD1a and GM1, and an increase of GM3 and GD1b within the cortices and of GD3 in the white matters. Additionally, a moderate GD1a and GM1 decrease in aged human hippocampus was detected (Kracun et al. 1991). Noteworthy, GD1a and GM1 deficiency has been reported to predispose to Parkinsonism (Wu et al. 2012; Ledeen and Wu 2018b). On the other hand, the cerebellar cortex showed a decrease in b-series gangliosides (GT1b and GD1b) during human aging (Kracun et al. 1991).
Moreover, imaging mass spectrometry studies highlighted the increase in the C:20 to C:18 ratio of fatty acid chain of ganglioside ceramide during brain aging (Sugiura et al. 2008), in accordance with previous founding in aged rat cerebellar granule neurons (Valsecchi et al. 1996). This, together with a progressive increase in the expression of the 3-keto-sphinganine synthase that uses stearoyl-CoA as substrate, leads to an increase in the C:20 to C:18 ratio of sphingosine. All this makes the brain gangliosides more hydrophobic and, consequently, the plasma membranes more rigid during aging. In rodents, the trend of brain ganglioside levels is similar but not identical to that of humans (Ohsawa 1989). In general, from 6 to 24 months of life, GD1a, GD1b, and GT1b remain constant and then drastically collapse up to 30 months. GM1, on the other hand, tends to increase up to 24 months and then only slightly decrease. Interestingly, GM1 tends to increase in the murine aged hippocampus and hippocampal synaptosomes (Yamamoto et al. 2008) such an increment supposedly to be associated with a seeding activity to the aggregation of misfolded proteins.
Considering ganglioside localization at the level of the presynaptic neuronal terminals modulating survival and neuronal transmission processes, the alteration of their content and composition associates with the reduction of neuronal function mainly through two mechanisms: trophic support insufficiency, since neurotrophic receptor activity is known to be modulated by gangliosides, and reduction of neuronal plasticity deriving from a limited vesicles capability to fuse with the membrane resulting in the lowering of synaptic vesicle release upon stimuli (Ledesma et al. 2012) This reflects in the cognitive decline that occurs with aging. Besides gangliosides, age-related increase in LacCer and GlcCer has been found in rodent brain together with a reduced acidic glucosylcerebrosidase activity (GCase, EC number 3.2.1.45) (Hallett et al. 2018). GCase is encoded by GBA gene and GBA haploinsufficiency is the highest genetic risk factor to develop Parkinson’s disease (Sidransky and Lopez 2012), and interestingly, a significant reduction in GCase activity was found within the substantia nigra and putamen of elder people that could increase the vulnerability of dopamine neurons and lower the threshold for developing Parkinson’s disease (Rocha et al. 2015).
5.2 Glycosphingolipids in Bone
Gangliosides were also described to be expressed in bone marrow mesenchymal stem cells (MSCs) and osteoblasts (Bergante et al. 2014, 2018). It has been documented that a-series ganglioside GD1a contributes to the regulation of osteoblast differentiation in MSCs (Kim et al. 2008). Series b gangliosides (GD3, GD2, GD1b, andGT1b) were found in osteoclast precursors and primary murine osteoclasts, while they are not expressed in osteoblasts (Yo et al. 2019). Moreover, a downregulation of GD3 synthase and b-series gangliosides is observed following induction of osteoclastogenesis mediated by receptor activator of nuclear factor kappa-B ligand (RANKL). Interestingly, the inhibition of GD3 synthase with the following ablation of b-series gangliosides appeared to prevent the age-related bone resorption in mice (Yo et al. 2019).
5.3 Glycosphingolipids in Kidney
Hernández-Corbacho and coworkers (Hernández-Corbacho et al. 2011) first described alterations in kidney sphingolipids in aged mice. Their data revealed that long-chain hexosilceramide (HexCer) and LacCer significantly increase during renal aging as well as in mouse aging brain and liver and in human fibroblasts obtained from elderly individuals. Even though the underlying mechanism associated with the age-related deterioration of kidney function and any other tissues is not known, some studies report HexCer and LacCer acting as mediators of inflammatory and apoptotic processes (Zhang and Kiechle 2004; Wennekes et al. 2009). Still scanty is the mechanism by which the alteration of glycosphingolipid metabolism occurs. Diet could play an in important role, as the caloric restriction was found to limit the accumulation of both renal HexCer and LacCer in aged mice (Hernández-Corbacho et al. 2011).
5.4 Glycosphingolipids in Endothelial Cells
Changes in glycosphingolipid composition of endothelial cells (EC) have been first reported by Sasaki and coworkers (Sasaki et al. 2015). Immunocytochemical and cytofluorimetric analysis revealed a GM1 increase in human cultured senescent EC. In addition, B4GalNT1 gene (GM2-syntase, EC number 2.4.1.92) was upregulated. Furthermore, a decrease in the abundance of GD3 was observed in senescent ECs, suggesting that the GM2 and GM1 synthetic pathway is predominant in senescence. This is accompanied by impaired insulin signaling in senescent EC, with a slight reduction in the insulin receptor. Treatment of senescent EC with AMP-DNM, a glycosphingolipids’ synthesis inhibitor, restored insulin signaling suggesting a possible negative impact of GM1 level alteration induced by aging. Even though this evidence remains to investigate more in detail the content of GM3 in the EC deriving from elderly subjects since GM3 is known to directly modulate the insulin receptor signaling (Kabayama et al. 2007).
5.5 Glycosphingolipids in Skin
The outer human epidermis layer, namely stratum corneum (SC), is enriched in lipids and is composed of 50% ceramides, 25% cholesterol, and 15% free fatty acids (Rogers et al. 1996). All three components are required for skin integrity, especially the ceramides, which play a crucial role in bilayer system formation (Feingold and Elias 2014). The SC ceramide derives from the metabolism mostly of GlcCer and sphingomyelin (SM) present in the innermost layers of the epidermis. These substrates are then metabolized by the action of glucosylceramidase and sphingomyelinase, respectively (Holleran et al. 2006). During aging, the skin becomes thinner and dry and decreases the ability to recover from lesions. An overall reduction in SC lipid content has been found in elder subjects as well as in aged mice, without any alteration in composition of each lipid species (Ghadially et al. 1995). A decrease activity of the enzymes involved in the synthesis and catabolism of GlcCer have been found in aged skin and pathological conditions suggesting a lower substrate availability for ceramide production (Holleran et al. 2006). Interestingly, the oral administration of cereal lipid extract enriched in GlcCer appeared to increase ceramide content in the SC of aged mice and to ameliorate recovery ability after injury (Bizot et al. 2017).
6 Glycosphingolipids in Disease
In the previous paragraphs, we reported the role played by glycosphingolipids in regulating signal transduction. Hence, here comes the importance of the correct glycosphingolipidic content and pattern of living cells and the correct glycosphingolipid topology. These three points depend on the balanced expression of a multitude of enzymes necessary for their biosynthesis, catabolism, and intracellular transport. Any change in the metabolic pathway could affect glycosphingolipids’ content and composition resulting in their reduced availability on the cell surface and/or overexpression of some species. All these events predispose to impairment of cell functioning leading to disease onset.
6.1 Glycosphingolipids and Neurodegeneration
Considering the increase of population age, in the last decades, a huge number of studies have focused on neurodegeneration, a phenomenon consisting in the progressive loss of structure and function of neurons leading to the onset of neurodegenerative disorders.
Since sphingolipids are key cell signaling molecules enriched in neuronal membranes, their biology and alterations have been extensively studied in the context of neurodegeneration (Piccinini et al. 2010; Breiden and Sandhoff 2018; Magistretti et al. 2019). Indeed, different neurodegenerative disorders are characterized by central and peripheral alterations of the composition and metabolism of sphingolipids, particularly of glycosphingolipids such as glycosylceramide, sulfatides, and gangliosides (Wang and Bieberich 2018).
Altered levels of sphingolipids have been found both in acute and in chronic neurodegeneration.
In response to acute injuries (i.e., stroke, spinal cord injury, and traumatic brain injury), the activation of sphingomyelinases and glycosidases accompanied by increased levels of ceramide and glycolipids has been reported (Novgorodov and Gudz 2009; Horres and Hannun 2012; Gu et al. 2013; Brunkhorst et al. 2015; Ong et al. 2015; Jones and Ren 2016; Roux et al. 2016; Barbacci et al. 2017; Abe et al. 2018; Sajja et al. 2018). On the other hand, loss-of-function alterations of enzymes involved in the hydrolytic degradation of glycosphingolipids can lead finally to chronic neurodegeneration due to sphingolipid aberrant accumulation within lysosomes and related cellular compartments (Ariga et al. 2008; Mielke and Lyketsos 2010; Haughey 2010; Mencarelli and Martinez-Martinez 2013; Farfel-Becker et al. 2014; Schnaar 2016; Spassieva and Bieberich 2016; Sandhoff 2016; Arenz 2017; Di Pardo and Maglione 2018; Grassi et al. 2019) (see section “Glycosphingolipidosis”).
Besides its association in sphingolipidosis-derived neurodegeneration, several evidence highlighted the central role of gangliosides both as therapeutic agents and as putative initiators of neurodegeneration via subnormal levels (Ledeen and Wu 2018a, b; Magistretti et al. 2019).
The involvement of a-series gangliosides, and in particular of GM1 and its catabolic precursor GD1a, represents a common denominator for at least three neurodegenerative disorders: Parkinson’s disease (PD), Alzheimer’s disease (AD ), and Huntington’s disease (HD) (Ledeen and Wu 2018a, b; Magistretti et al. 2019).
6.1.1 Parkinson’s Disease
PD is a disorder characterized by accumulation of α-synuclein (α-syn) fibrillary aggregates and progressive degeneration of nigrostriatal dopaminergic neurons that finally lead to motor and cognitive dysfunctions (Del Tredici et al. 2002; Ledeen and Wu 2018a, b; Zesiewicz 2019).
About 5 to 10% of PD cases have a known genetic origin, while the vast majority, defined as sporadic PD, has only age as the major risk factor (Stern et al. 2012; Ledeen and Wu 2018b). Up to now, the most common genetic rick factor of PD is represented by mutations in the GBA gene, coding for the lysosomal β-glucocerebrosidase enzyme (GCase), for which the 3–5% of PD patients carry heterozygous mutations.
Until 20 years ago, the GBA gene was considered responsible for only the Gaucher’s disease (GD), one of the most common lysosomal storage disorders. GCase catalyzes the hydrolysis of GlcCer to ceramide and glucose mostly within lysosomes and partially at PM level. Homozygous or, more commonly, combined heterozygous mutations in GBA gene, generally, cause the enzyme to be defective leading to the accumulation of the substrate, responsible for the multiorgan clinical manifestations of the disease.
The initial discovery of Parkinsonism in a subset of adult onset type I GD patients suggested a possible pathogenic link between the two disorders (Neudorfer et al. 1996; Tayebi et al. 2003; Sidransky 2005). Neuropathological analysis of these patients revealed the presence of α-syn positive Lewy bodies (Wong et al. 2004), suggesting the involvement of αsyn aggregation. Importantly, several additional genetic studies in large patient cohorts demonstrated that patients with Parkinsonism have an increased incidence of GBA mutations (Lill et al. 2009).
Despite the efforts of researchers, to date the mechanism underlying the relation between GBA mutations and the development of PD is far to be completely understood and different hypotheses have been explored. Since GCase resides and acts within endolysosomal compartment, its incorrect folding could induce an impairment and engulfment of ER and lysosomes which in turn would trigger a stress response in the dopaminergic neurons leading to their damage and death (McNeill et al. 2014). In addition in GBA-PD and non-GBA-PD, GCase shows a significant reduction of its catalytic activity resulting in a loss of function that seems to be associated with a gain of a toxic–cytotoxic function resulting in an aberrant positive feedback loop involving GCase, alpha-synuclein (α-syn) and the nonmetabolized GlcCer (Mazzulli et al. 2011). The intracellular GlcCer levels control the formation of soluble toxic α-syn assemblies in cultured neurons and mouse and human brain, leading to neurodegeneration. The elevation and formation of α-syn assemblies further contributes to a pathogenic cycle by inhibiting the lysosomal maturation and activity of normal GCase, resulting in additional GlcCer accumulation and augmented α-syn oligomer formation. The central role of not correctly metabolized GlcCer accumulation in the α-syn pathology continues to emerge from recent reports according to which the accumulation of GlcCer even in the absence of a direct GBA mutation is sufficient to induce a conformational change of the physiological aggregates of α-syn in pathological oligomers, with a tendency to form fibrils. Even more important for the therapeutic perspectives, the reduction of GlcCer induces a reversion of the pathological conformers of α-syn into the physiological ones (Zunke et al. 2018). It follows that strategies to restore/maintain GCase activity and reduce GlcCer levels are currently under exploration to treat the misfolding and pathogenic aggregation of α-syn in PD (Riboldi and Di Fonzo 2019).
Although sporadic PD etiopathogenesis is complex and to date poorly understood, and both genetic and environmental factors have been reported to play a synergistic role, a new theory confers a crucial role to GM1 ganglioside. This ganglioside highly enriched in neurons exhibits a physiological progressive decline with aging and/or epigenetic influences (Svennerholm and Gottfries 1994). However, its decrease below critical thresholds may induce the deregulation of key molecular mechanisms leading to neuropathological dysfunction and finally to PD onset (Ledeen and Wu 2018a, b; Chiricozzi et al. 2020). Accordingly, PD patients reported a decreased expression of genes involved in GM1 synthesis, such as B4galnt1, B3galnt4, and St3gal2, accompanied by a reduction of GM1 in central and peripheral nervous tissues (Ledeen and Wu 2018b; Schneider 2018). Besides the clinical data, the consequence of GM1 insufficiency is illustrated in a newly presented mouse model of sporadic PD that was obtained from the heterozygous disruption of the B4galnt1 gene causing a partial depletion of GM1 comparable to those found in PD patients (Wu et al. 2012; Hadaczek et al. 2015; Ledeen and Wu 2018a, b). This condition is sufficient to accurately recapitulate behavioral and biochemical PD phenotype that was found to be recovered by administration of LIGA-20, a membrane-permeable analogue of GM1 (Wu et al. 2012; Ledeen and Wu 2018b).
The neurorestorative and neuroprotective potential of GM1 ganglioside was highlighted by different studies using widely accepted in vivo PD models, including mice and nonhuman primates exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a neurotoxin impairing mitochondrial function (Hadjiconstantinou et al. 1989; De Girolamo et al. 2001; Nicotra and Parvez 2002; Meredith and Rademacher 2011), and rats overexpressing human A53T mutant α-syn via adeno-associated viral vector (Schneider et al. 2019). At molecular level, several neuronal functions can become gradually compromised as GM1 levels recede (Ledeen and Wu 2018a, b; Chiricozzi et al. 2020). In PD context, a particular attention was given to the signaling mediated by neurotrophic receptors and the α-syn interaction. Indeed, it has been hypothesized that the reduced level of plasma membrane GM1 in PD neurons may trigger the neurodegenerative process due to a failure in trophic signaling together with a reduction of α-syn clearance (Bartels et al. 2014; Hadaczek et al. 2015). Accordingly, GDNF signaling, found to be impaired both in the substantia nigra from PD patients and B4galnt1 PD mouse model, was rescued through the administration of the GM1 analogue LIGA20 (Hadaczek et al. 2015). In addition, studies have highlighted the specific inhibition of α-syn fibril formation mediated by GM1 binding and the key role of N-acetylation of human α-syn in enhancing GM1 binding and specificity (Martinez et al. 2007; Bartels et al. 2014).
However, although a direct correlation between GM1 and the loss of neurotrophic signal is quite evident both in vitro and in vivo (Schengrund and Prouty 1988; Schneider and DiStefano 1994; Wu et al. 2004, 2005, 2012; Schneider et al. 2015a), to date the molecular mechanisms underlying these GM1 effects remain to be elucidated. In this context, difference in vitro and in vivo evidence pointed out that the functional moiety responsible for activating the GM1 neurofunctions resides in its oligosaccharide chain (Schengrund and Prouty 1988; Schneider and DiStefano 1994; Wu et al. 2004, 2005, 2012; Hadaczek et al. 2015; Schneider et al. 2015b; Di Biase et al. 2020b; Fazzari et al. 2020). This evidence led to hypothesize that sporadic PD pathogenesis may be due to PM GM1 deficiency that causes an alteration of the molecular interaction between the GM1 oligosaccharide and specific PM proteins. Since GM1 is involved in the regulation of numerous neuronal pathways, the loss of specific GM1 oligosaccharide–protein interactions may gradually compromise several functions.
As reported above, recently a new theory emerged regarding a possible role for ganglioside GM1 and its precursor (i.e., ganglioside GD1a) in the etiopathogenesis of sporadic form of PD, due to a decrease of their expression under a specific threshold level during aging (Ledeen and Wu 2018a, b).
Accordingly, this reflects the partial success of the GM1 replacement therapy in clinic. Starting from the 90s, Schneider and coworkers carried out a trial with the aim to check the short- (24 weeks) and long-term (120 weeks) effects of GM1 treatment on PD: improve symptoms, delay disease progression, and partially restore damaged brain cells (ClinicalTrials.gov NCT00037830; (Schneider et al. 2010). Up to 100 patients were daily treated intravenously or subcutaneously with 100–200 mg of GM1 for up to 5 years (Schneider et al. 2010). The GM1 treatment brought positive effects, like a partial restoration of dopamine transporter functional level in the striatum, improvement of motor symptoms, and lowering the disease symptom progression (Schneider et al. 2010, 2013, 2015a). The study results provided indications that GM1 may improve motor symptoms and may also have potentially disease modifying effects on PD patients. However, since the small cohort of patients enrolled in these trials, the data obtained were not clear enough to authorize GM1 as an official drug for the treatment of PD.
Additionally, an important and fundamental problem in the peripheral administration of GM1 ganglioside is that for its chemophysical properties (i.e., amphiphilic characteristic) it hardly crosses the blood–brain barrier (BBB) and thus a too small quantity of GM1 may reach the damaged neurons. It is plausible to think that the positive results obtained in GM1 trials on PD patients are mainly due to the altered-inflamed BBB areas, typically observed in patients with advanced pathology (Saulino and Schengrund 1994; Desai et al. 2007; Erdö et al. 2017).
In conclusion, although different studies and clinical data reported the therapeutic action of systemically administered GM1 (Schneider et al. 1992, 2015a; Herrero et al. 1993; Rothblat and Schneider 1998), it remains to be verified whether GM1 can reach brain regions by crossing intact BBB (Ghidoni et al. 1986; Svennerholm et al. 1990; Saulino and Schengrund 1994; Polo et al. 1994).
To facilitate the brain internalization different strategies can be developed, including the optimization of drug delivery and the design of modified-GM1 able to more efficiently cross BBB.
In 2012, Wu et al. demonstrated that the addition of a dichloroacetyl group linked to the sphingosine amino group instead of the acyl chain generates a membrane-permeable analogue of GM1, LIGA-20, able to maintain different GM1 neuroproperties (Wu et al. 2012). In addition, while the toxicity associated with long-term treatments hampered its use, the effectiveness of LIGA-20 suggested that the ceramide moiety is not critical for preserving the GM1 functions (Schneider and DiStefano 1994; Wu et al. 2004, 2005; Hadaczek et al. 2015). In well accordance, recent studies have highlighted that the oligosaccharide portion of ganglioside GM1 is able to replicate the neurotrophic and neuroprotective properties of the entire GM1 both in vitro and in vivo (Chiricozzi et al. 2017, 2019a, b, c; Di Biase et al. 2020a, b; Fazzari et al. 2020). In this scenario, the GM1 oligosaccharide maintains all the beneficial effects of the ganglioside from which it derives but loses its amphiphilicity acquiring the ability to effectively reach the central nervous system, thus demonstrating a strong therapeutic potential.
6.1.2 Alzheimer’s Disease
AD is the most common neurodegenerative disorder characterized by impairment in memory and cognition due to the presence of abundant extracellular amyloid β plaques and neurofibrillary tangles triggering neuronal degeneration (Serrano-Pozo et al. 2011).
In this context, the analysis of samples from patients affected by dementia of Alzheimer type compared to controls led to identification of a significant reduction of all gangliosides in hippocampal, entorhinal, posterior cingulate, visual and prefrontal cortex, with a preferential decrease of b-series gangliosides (GD1b, GT1b) (Crino et al. 1989). On the other hand, the ganglioside composition in AD frontal cortex was found to be similar to controls, with minor exception for GQ1b and GT1b that resulted decreased (Brooksbank and McGovern 1989). By employing a peculiar method for ganglioside quantification (TLC–Blot and MALDI-TOF mass spectrometry), the depletion in b-series gangliosides was identified in AD hippocampal gray matter, while the level of the a-series gangliosides (GM1 and GD1a) was not affected (Valdes-Gonzalez et al. 2011).
The decrease of all gangliosides in the temporal and frontal cortex and nucleus basalis of Meynert (Kracun et al. 1991) or a mild decrease of a-series gangliosides in the temporal lobe of late-onset patients (Svennerholm and Gottfries 1994) were also reported. In addition, another study identified an increase of simpler gangliosides (i.e. GM2 and GM3) in AD cortex indicating, possibly, the acceleration of gangliosides lysosomal degradation and/or the presence of reactive astrogliosis typical of neuronal death process (Kracun et al. 1992). Data regarding the GM1 levels and function are controversial: it has been both negatively and positively linked to AD pathogenesis (Ariga et al. 2008). Different studies have reported the GM1 capability to bind amyloid β-protein and seed its fibrillogenesis to the aggregated form, the neuropathological hallmark of AD tissues (Hayashi et al. 2004; Ngamukote et al. 2007; Ariga et al. 2008). In addition, a cytotoxic effect was reported in mouse embryonic neural stem cell coexposed with GM1 and amyloid β (1–40) peptide (Yanagisawa et al. 2010). On the other hand, independent in vitro studies reported the protective action of GM1 ganglioside in the presence of amyloid β fibrillae (Ariga et al. 2008; Ledeen and Wu 2018b), indicating the GM1 capability to increase cell viability (Sokolova et al. 2007), to modulate inflammation by inhibiting amyloid β-induced release of proinflammatory cytokines (Ariga and Yu 1999), and to prevent toxicity triggered by fibrillar amyloid β via modulating the GSK3β-induced apoptotic pathway (Kreutz et al. 2011).
In addition, the crossing of homozygous GD3 synthase knockout mice, lacking the b- and c-series gangliosides, with a double-transgenic (APP/PSEN1) mouse model of AD has led to the elimination of amyloid β plaques and to a behavioral performance overlapped to wild-type mice (Bernardo et al. 2009). These results suggested not only that GD3 synthase could be a valuable therapeutic target to counteract AD but also that these improvements might be attributed to increase of GM1 and GD1a seen in the triple-transgenic mice (Bernardo et al. 2009).
Accordingly, GM1 administration to Wistar rat injected with amyloid β1-40 improved spatial learning and memory deficits and inhibited the oxidative stress and lipid peroxidation (Yang et al. 2013). Further in vivo studies suggested that the beneficial effects of GM1 against amyloid β-derived toxicity could reside in the enhanced of amyloid β clearance via autophagy (Dai et al. 2017) or the elimination by brain phagocyte microglia of amyloid β trapped in glycosphingolipid-rich exosomes (Yuyama et al. 2014).
Finally, the intraventricular injection of GM1 in five early-onset AD patients determined a significant amelioration of motor performance and neuropsychological assessments (Yuyama et al. 2014). The experimental and clinical evidence on the GM1 positive effect suggest a possible role not only for plasma membrane GM1 deficiency but also for AD pathogenesis that, in our opinion, should be investigated more in detail.
6.1.3 Huntington’s Disease
Altered levels of glycosphingolipids content have been correlated to a third neurodegenerative disorder: HD (Desplats et al. 2007; Maglione et al. 2010; Ledeen and Wu 2018a; Magistretti et al. 2019). This condition, characterized primarily by involuntary movements and impaired motor coordination, is caused by an autosomal dominant mutation in the huntingtin (HTT) gene determining the expansion of poliQ stretch at N-terminus of the protein that results in HTT misfolding and aggregation (Magistretti et al. 2019).
By analyzing postmortem caudate HD samples, Desplats and coworkers identified a decreased expression of enzymes involved in ganglioside synthesis, including St3gal5, St8sia3, St3gal2, and B4galnt1 (Desplats et al. 2007). Consequently, an overall remodeling of ganglioside expression and a significant elevation of GD3 ganglioside accompanied by decreased level of GM1 were found (Desplats et al. 2007). The analyses of different HD transgenic mouse models confirmed the alteration of ganglioside pattern with a GM1 reduction in the striatum and cortex, two regions involved in HD (Desplats et al. 2007; Maglione et al. 2010), and a significant decrease of cerebrosides and sulfatides in the forebrain suggesting abnormalities in myelin content (Desplats et al. 2007).
In vitro administration of GM1 protected HD cells from apoptosis via PI3K/AKT pathway activation (Maglione et al. 2010) and, accordingly, the intraventricular infusion of GM1 was able to improve motor and anxiety behaviors and cognitive functions in HD mice models (Maglione et al. 2010; Alpaugh et al. 2017). In line with the phenotypic amelioration, different molecular events were modulated by GM1 exposure. Depending on the HD model used, GM1 administration led to reduction of ferritin levels (Alpaugh et al. 2017) that correlate to striatal and cortical atrophy in HD patients (Magistretti et al. 2019), to restoration of expression and phosphorylation of DARPP-32 protein involved in modulation of dopamine signaling. Additionally, GM1 treatment of HD model shows to modulate different neurotransmitters levels (Alpaugh et al. 2017), to attenuate HTT toxicity by inducing the phosphorylation at Ser13 and Ser16 that allow the decrease of HTT aggregates (Di Pardo et al. 2012) and by reducing both soluble and insoluble mutant HTT (Alpaugh et al. 2017).
6.1.4 Peripheral Neuroprotection
From 1976, gangliosides were used as commercial drug for the therapy of peripheral neuropathies, but at the beginning of the 1990s, gangliosides were claimed by the medical society as responsible for inducing the Guillain-Barré syndrome (Sonnino et al. 2017) and the drug was withdrawn in many countries. Today, the idea that the administration of gangliosides is not associated with the onset of Guillain-Barré syndrome seems to be sounder (Magistretti et al. 2019), and in recent years, clinical trials at different phases still involve GM1 ganglioside. One of these, related to the association of GM1 with oxaliplatin chemotherapy seems to give very promising results.
Oxaliplatin is a very powerful drug against gastrointestinal tumors; however, it is characterized by severe peripheral neurotoxicity. Sixty patients with gastrointestinal tumors were injected daily with 100 mg of GM1 for the 3 days following the chemotherapy treatment. The treatment showed some protection against the oxaliplatin neurotoxicity (Zhu et al. 2013).
6.2 Glycosphingolipidoses
For a long time, lysosomes have been considered the final destination of degradative pathways, but it is now clear that they are also crucial regulators of cell homeostasis and their impairment causes the onset of severe pathology called lysosomal storage disorders (LSDs) (Perera and Zoncu 2016).
LSDs are caused by a reduced or absent enzymatic activity of catabolic lysosomal enzymes, by defect in the nonenzymatic lysosomal activator proteins or in nonlysosomal proteins involved in glycohydrolases maturation. These defects result in the intralysosomal accumulation of undegraded metabolites that leads to the onset of cell damage (Platt et al. 2012).
Classically, LSDs are classified based on the nature of the accumulated substrate: mucopolysaccharidoses (accumulation of mucopolysaccharides), sphingolipidoses (sphingolipids), and oligosaccharidoses also known as glycoproteinoses (oligosaccharides) (Filocamo and Morrone 2011). More recently LSDs have also been classified by the molecular defect, including more pathologies recognized now as LSDs: (1) nonenzymatic lysosomal defects, (2) transmembrane protein defects (transporters and structural proteins), (3) lysosomal enzyme protection defects, (4) post-translational processing defects of lysosomal enzymes, (5) trafficking defects in lysosomal enzymes and (6) polypeptide degradation defects (Table 3.4).
LSDs clinical manifestations involve multiple organs and systems (Wang et al. 2011). The principal pathological phenotypes are represented by hepatosplenomegaly, corneal or lenticular opacities, retinal dystrophy, optic nerve atrophy, glaucoma, blindness, bone dysplasia, abnormalities of bone density, and osteonecrosis (Parenti et al. 2015). About two-thirds of patients affected by LSDs also show an important neurological deficiency, which is extremely variable and heterogeneous ranging from progressive neurodegeneration and severe cognitive deficit to psychiatric and behavioral disorders (Parenti et al. 2015). This is particularly true for lysosomal storage disorders caused by defects in SL metabolism (sphingolipidoses); reviewed in (Futerman et al. 2004; Futerman and Van Meer 2004; Kolter and Sandhoff 2006). The onset of symptoms can occur before the birth, for the most severe phenotypes, or during the adulthood for the late-onset mild forms. Severity and age of onset in LSDs depend by several factors including: residual enzyme activity, distribution of tissue-specific and cell-specific substrates, cell turnover rate, defective protein expression, and other mechanisms that influence the lifespan of affected cells (Jakóbkiewicz-Banecka et al. 2014).
The complexity of LSDs, at the phenotypic and molecular level, particularly considering the aspects related to neuronal dysfunctions, clearly indicates that, even if it is undoubtedly clear that the intralysosomal accumulation of unmetabolized substrates is the primary cause of the disease, other, still obscure, molecular mechanisms might lead from this event to the pathology.
Since SL metabolism and traffic is a complex network of interdependent events, and the recycle of catabolic fragments originated in the lysosome for biosynthetic purposes is quantitatively relevant, it can been expected that the blockade of proper sphingolipids catabolism at the lysosomal level leads to the jamming of the overall flow of metabolites, with consequences on the sphingolipids composition in all cellular districts, including the plasma membrane. Interestingly, in confirmation of this, more than 70% of LSDs is characterized by the secondary accumulation of glycosphingolipids that is not confined to the lysosome but also occurs at the plasma membrane level.
The cell machinery responsible for cell sphingolipids pattern is complex and still not completely understood in its details. The Golgi de novo biosynthesis, that is necessary to replace the molecules catabolized in the lysosomes as the consequence of membrane turnover, is considered the heart of the metabolic process, and is regarded as the main regulatory site. Nevertheless, in recent years many other processes capable to modulate the final PM composition have been included to the complex sphingolipids biosynthetic pathway: (1) partially catabolized compounds can leave the lysosomes and can be transported to the Golgi for reassembling or to any other membrane (Kitatani et al. 2008); (2) both sphingolipids synthases and hydrolases are associated with the PM allowing changes of the content of ceramide , SM, and gangliosides directly in situ; (3) PM sphingolipids can be shed in the extracellular milieu ad can be partially re-associated with the same membrane or associated with the membranes of neighboring cells; (4) intracellular vesicles (endosomes, lysosomes, exosomes, and other vesicles) can fuse with PM; and (5) lipids, including sphingolipids, can undergo active trafficking between different intracellular membranes mediated by membrane contact sites (MCSs).
Interestingly, using an artificial model of lysosomal impairment represented by human fibroblasts loaded with sucrose, it has been described that the impaired lysosomes are not able to catabolize the sphingolipids leading to their lysosomal accumulation (Samarani et al. 2018). Nevertheless, as already demonstrated in a cellular model of Niemann Pick type A disease (Gabandé-Rodríguez et al. 2014), it has been found that the SL accumulation not only is confined to lysosomes but also affects the PM. The molecular mechanism responsible for the migration of noncatabolized sphingolipids from lysosomes to the PM is not understood so far, even if several line of evidence support the activation of two mechanisms: (1) the intralysosomal vesicles accumulating SL could exchange monomers with the internal leaflet of the lysosomal membrane that, by the fusion with the cell surface, becomes component of the PM; (2) the lysosomal exocytosis may induce the release in the extracellular milieu of the intralysosomal vesicles accumulating sphingolipids that, by uptake, could be inserted into the outer leaflet of the cell membrane. This study points out also on the importance of the PM sphingolipids alteration in determining the onset of cell damage in lysosomal storage disorders that is independent by the primary accumulated substrate due to the genetic defect.
Indeed, in LSDs the activation of the lysosomal exocytosis not only is responsible for an increased content of glycosphingolipids but drives also lysosomal hydrolases at the external leaflet of the PM. The concomitant aberrant presence of glycosphingolipids and its hydrolases induces conformational changes in the membrane architecture that are responsible for the onset of cells damage (Samarani et al. 2018) (Fig. 3.14).

Diagram showing the lysosome-PM axis suggested to explain the mechanism linking lysosomal storage and the onset of cell damage resulting in cell growth arrest
The bioactive role of PM sphingolipids is mainly related to the lipids-rafts concept. The lateral segregation of sphingolipids, and particularly of gangliosides, is considered a major driving force for the formation of lipid domains (Simons and Ikonen 1997; Sonnino et al. 2006; Sonnino and Prinetti 2013), where they modulate the activity of a number of cell surface receptors and enzymes (Simons and Toomre 2000; Prinetti et al. 2009). The resulting sphingolipids-enriched membrane domains with non-physiological composition might be responsible for altered signaling events involved in the onset of the cellular damage and of tissue pathology.
This hypothesis has been recently confirmed by several observations: (1) in a cell model of GD, impaired lysosomal catabolism of GlcCer led to the accumulation of GlcCer at the plasma membrane level in lipid rafts, possibly explaining the altered lipid and protein sorting observed in this pathological condition (Hein et al. 2008); moreover, it has been reported that GD is associated with insulin resistance (Langeveld et al. 2008). Since insulin receptor function is regulated by its interaction in lipid rafts with GSL (Kabayama et al. 2007) and in particular, GM3 ganglioside, this suggests that the altered lipid rafts’ organization in Gaucher cells might be responsible for altered responsiveness to insulin; (2) psychosine (galactosylsphingosine) is one of the galactoslylsphingolipids that accumulates in the brain of Krabbe disease (human globoid cell leukodystrophy) patients due to the deficient activity of β-galactosylceramidase. Psychosine accumulates in lipid rafts from brain and sciatic nerve from twitcher mice (the animal model for the infantile variant of the disease) and from human Krabbe patients, leading to an altered distribution of lipid raft proteins and to inhibition of protein kinase C (White et al. 2009); (3) in brains from ASMKO mice, an animal model for Niemann–Pick disease type A (due to deficient activity of the lysosomal acid sphingomyelinase) (Schuchman 2007), in addition to the expected SM accumulation, we observed an unexpected remodeling of the fatty acid composition of the accumulated SM and a significant increase in ganglioside content, mainly due to the accumulation of monosialogangliosides GM3 and GM2, leading to a non-conventional lipid raft organization (Scandroglio et al. 2008; Buccinnà et al. 2009). Taken together, these observations open a new scenario on the existence of a lysosome-PM axis fundamental to maintain the cell homeostasis that in case of alterations is responsible for the onset of cell damage.
Starting from the ‘90, the enzyme replacement therapy has been introduced for treatment of glycogen storage disease type II, the Pompe disease caused by a deficiency of the lysosomal acid α-glucosidase, of a few several mucopolysaccharidoses and a few glycosphingolipidoses. Nevertheless, the treatment could not generally be applied to the LSDs with serious damage of the central nervous system due the obstacle posed by the BBB (Li 2018; Concolino et al. 2018).
Administration of glucocerebrosidase to type 1 GD, that causes damages to liver, spleen, kidney, lung and bone, increases the activity of this enzyme intracellularly, and ameliorates symptoms. Several recombinant approved forms of glucocerebrosidase are available reporting modification of the glycosylation sites to have mannose at the end of the chain. This is necessary, to be recognized by mannose receptors present on the surface of macrophages and be then taken up. Side effects are mild, but present, such as, diarrhea, back pain, and many others (Revel-Vilk et al. 2018).
A similar approach has been used in building an α-galactosidase necessary to treat the Fabry disease, in which the enzyme deficiency in the lysosomal leads to the accumulation of globotriaosylceramide, with progressive damage of kidney and other organs, and to peripheral neuropathy.
Long-term treatment slows down disease progression, but many complications develop in most patients (Ortiz et al. 2018). Therefore, several new treatment approaches are under development and study: chaperone therapy aimed to reduce the catabolism of incorrect enzyme and increase its availability into the lysosomes; administration of the enzyme mRNA to help stimulate production of the enzyme; substrate reduction therapies by administration of inhibitors of glucosylceramide synthase.
6.3 Cancer
The origin and progression of tumors are characterized by several steps and features, including limitless replicative potential (the result of the combination of different factors, such as self-sufficiency in growth signals on one hand, and insensitivity to growth-inhibitory signals associated with escape from programmed cell death on the other hand), sustained angiogenesis, interplay with immune system cells, and eventually, in some cases, tissue invasion and metastasis. The initial events that trigger tumorigenic transformation are digging deeply into the genome (e.g. genetic mutations of oncogenes and tumor suppressor genes) of the cells undergoing transformation. On the other hand, in all the steps leading to cancer disease progression tumor-host interactions are of crucial importance. In the interactions between tumor cells and the surrounding microenvironment, the relevant interface is represented by the cellular surfaces. The surface of the tumor cell, the site where interactions between the cell and the extracellular environment are organized and transduced into signals able to modify the cell properties influencing the tumor phenotype. But also the surface of the incredible variety of cells present in the tumor stromal compartment, including endothelial cells, fibroblasts, and resident and recruited inflammatory and immune system cells. Thus not surprisingly, the glycocalyx of tumor cells is characterized by multiple and complex alterations, and the altered expression of carbohydrate epitopes at the tumor cell surface (“aberrant glycosylation ”) is involved in many, if not all, of the steps of tumor progression. The term “aberrant glycosylation ” indicates an altered expression of oligosaccharide epitopes, associated with both glycolipids and glycoproteins. Independent reports from several authors, but in particular the extensive work in this area of Dr. Sen-itiroh Hakomori and his scholars allowed to define aberrant glycosylation as a general feature of human cancer (Hakomori 1985), and to elucidate the main metabolic alterations responsible for aberrant glycosylation in cancer . At least two different metabolic mechanisms contribute to the generation of tumor-associated carbohydrate epitopes: (1) the impairment of specific glycosylation steps (“incomplete synthesis”); (2) the induction of genes encoding for glycosyltransferases or, less frequently, for carbohydrate transporters (“neo-synthesis”) (Hakomori 1996). As the result, tumors are characterized by the appearance and/or accumulation of carbohydrate epitopes that are often present in tumor-associated antigens. Indeed, glycan epitopes associated with aberrant glycosylation in tumors were in most cases originally defined by their ability to raise the production of specific antibodies, and only later their molecular structures have been characterized. The discovery of oligosaccharide tumor-associated antigens and the development of different antibodies reacting against them provided useful diagnostic and research tools, and opened the field of tumor glycobiology, that developed tremendously in the following decades.
As already mentioned, aberrant glycosylation in tumors do affect carbohydrate epitopes associated with both glycolipids and glycoproteins. Sphingolipids, and in particular glycosphingolipids, mediate several aspects of the interactions between a cell and the extracellular environment or other cells under physiological conditions. They are known as cell surface antigens, as mediators of cell-cell recognition and cell adhesion and as modulators of several aspects of signal transduction processes involved in cell survival/proliferation and differentiation. Aberrant glycosylation in GSL seems to play very relevant roles under pathological conditions, and several lines of evidence point on the importance of aberrantly expressed GSL in the formation and progression of tumors. Indeed, the notion that GSL metabolism and expression is dramatically changed during neoplastic transformation and tumor progression was probably the origin of the concept of aberrant glycosylation associated with tumors, and it is rooted into multifaceted experimental evidence. In several tumors, total ganglioside-bound sialic acid levels was found to be higher than in the corresponding normal tissue (e.g., in breast tumor tissues v. normal mammary tissues (Marquina et al. 1996). The chemical structure of sialic acid associated with tumor tissue gangliosides was different in several tumors, in particular gangliosides containing N-glycolylneuraminic acid, usually expressed at low levels in humans, were relatively abundant (Hakomori 1989) (this finding led to the hypothesis that consumption of foods rich in N-glycolylneuraminic acid could favor the onset of certain tumors). Moreover, the accumulation of high levels of specific gangliosides and/or neutral GSLs in specific types of cancer was reported: this is the case for GD3, GD2 and GM3 gangliosides in human and mouse melanoma, GD2 in neuroblastoma, Gg3 in human and mouse lymphoma, fucosyl-GM1 in small cell lung carcinoma, globo-H in breast and ovarian carcinoma, disialosylgalactosylgloboside in renal cell carcinoma. Several pieces of evidence pointed out that aberrant glycosphingolipid expression is not simply an epiphenomenon accompanying neoplastic transformation, but rather suggested that aberrant GSL expression could actively contribute to the tumor phenotype. In vitro, it has been shown that tumor cell lines with higher tumorigenic potential are characterized by higher ganglioside levels (Ladisch et al. 1987; Deng et al. 2000), and that the artificially induced increase in cellular ganglioside levels enhanced the ability to form experimental tumors. On the other hand, in human cancer patients correlations between the expression levels of some GSL structures and tumor malignancy and/or patient survival rates have been observed (e.g. disialosylgalactosylgloboside in renal cell carcinoma (Satoh et al. 1996), galactosylgloboside in seminoma (Ohyama et al. 1996) and GM3 ganglioside in bladder cancer – the latter example will be further discussed in this section) (Hakomori 1996, 2002). Another intriguing aspect of aberrant GSL behavior in tumors is represented by the ability of tumor cells to shed significant amounts of selected ganglioside species mostly in the form of vesicles (Ladisch et al. 1994, 1997; Chang et al. 1997). GSL shedding from cells is known for long time and is also present in non-tumor cells, however tumor cells do shed much higher amounts of GSL, and, as mentioned, there is apparently a selection of the molecular species that are preferentially shed. Many Authors argued that GSL shedding might reflect a non-physiological membrane turnover typical of cultured cells, however GSL and in particular gangliosides are found in human serum, and, remarkably, serum ganglioside levels in cancer patients were found higher than in healthy individuals (Ladisch et al. 1994, 1997; Chang et al. 1997). When GSL shedding was discovered, researchers still had a quite naïve view about the interplay between the tumor and the immune system, that is indeed very complicated and crucial for the progression of the tumor itself. At that time, it has been hypothesized that tumor-derived gangliosides released in the host environment might play a role in eluding the aggression by the host immune system. Now it is becoming evident that release of selected ganglioside species from tissues characterized by chronic low-grade inflammation (a feature considered a tumor hallmark) do interact with specific immune system cells subpopulations, and in particular do affect the function of innate immune response, likely contributing to the progression of TLR4-mediated diseases, possibly including some forms of cancer (Kanoh et al. 2020).
The physiological roles of GSL in modulating cell properties are multiple and at least in part highly specific for a given GSL molecular species. Thus, not surprisingly, the consequences of aberrant GSL expression on tumor cell phenotype are as well multifaceted. The modification of glycosphingolipid expression deeply affects several properties of tumor cells, that are directly relevant to the growth and progression of the tumor , and to metastasis formation: cell proliferation/survival, cell adhesion (to other cells in the original tumor mass for solid tumors, to the extracellular matrix or to the endothelium of blood vessels), motility, recognition and invasion of host tissues. This heterogeneity at the cellular level is reflected by the corresponding heterogeneity in the underlying interactions at the molecular levels.
At the molecular level, the phenotypic alteration in tumor cells explained most straightforwardly by aberrant GSL expression is probably the proliferation sustained by the interaction of GSL, in particular gangliosides, with classical tyrosine kinase growth factor receptors. The ability of GSL, and gangliosides in particular, to modulate the activity of receptor tyrosine kinases, has been widely documented (Hakomori and Igarashi 1995; Yates and Rampersaud 1998). Still to be elucidated in most cases remain the molecular aspects of GSL-protein interactions underlying the modulatory effect of GSL. However, at least for some growth factor receptors, the interaction with gangliosides is facilitated by the co-clustering of the receptor itself and of the interacting lipids within lipid rafts. The sustained proliferation of tumor cells is supported by a network of different soluble growth factors produced from different cells present or recruited in the tumor stromal compartment. Among those, the EGFR is particularly relevant, and prognosis of patients affected by several solid tumors (for example, small cell lung carcinoma) is determined by mutations in the EGFR. EGFRis effectively inhibited by GM3 (and, at lesser extent, by other gangliosides) (Bremer et al. 1986). GM3 inhibited receptor autophosphorylation but not receptor dimerization (Zhou et al. 1994), neither its binding with the ligand, EGF (Yednak and Bremer 1994). The sialyllactose oligosaccharide is essential for ganglioside-receptor interaction (Miljan et al. 2002), being involved in side-by-side carbohydrate-carbohydrate interactions with N-linked glycan bearing multiple GlcNAc terminal residues on the receptor (Yoon et al. 2006a, b). GM3/EGFR interaction is facilitated by the enrichment of EGFR in GM3-enriched lipid rafts (Ringerike et al. 2002; Roepstorff et al. 2002). However, it is becoming clear that different subpopulations of EGFR associated with distinct membrane domains exist. Interestingly, high levels of GM3 seem to be able to negatively regulate EGF-dependent proliferation of tumor cells with other mechanism, in addition to the direct inhibition of the receptor as described above. In some cell types, GM3 overexpression favors the interaction of caveolin-1, and important modulator of tumor phenotype, with EGFR, in turn causing inhibition of EGFR tyrosine phosphorylation and dimerization (Wang et al. 2002). Thus, GM3 influences EGFR signaling by a second molecular mechanism, clearly distinct from the direct inhibition, by modulating EGFR/caveolin-1 association.
The modulation of cell adhesion, motility and recognition seems to be modulated through a wide variety of GSL-binding proteins and/or GSL-GSL interactions (Hakomori 1996, 2002; Hakomori et al. 1998). In particular, GSL might contribute to the modulation of integrin-dependent interactions of tumor cells (determining their adhesion, motility and invasiveness, but also affecting their proliferation potential) with the extracellular matrix as well as with host cells present in the stromal compartment of the tumor . GSL might also be involved in selectin- or galectin-dependent adhesion of tumor cells to endothelial cells, a crucial step in the extravasation of circulating tumor cells and in the initiation of metastasis. At least in some cases, trans GSL-GSL interactions are also important in determining the motility and metastatic potential of tumor cells, while in others GSL at the tumor cell surface have anti-adhesive properties. For example, GM3-GM3 interaction-mediated repulsion of tumor cells was implicated in the release of cells from the tumor mass, playing a role in initiation of metastasis. On the other hand, adhesion of circulating melanoma cells to endothelial cells, a critical step in melanoma cell metastasis, was mediated by the heterophilic trans interaction between GM3 at the surface of melanoma cells and lactosylceramide and/or Gg3 at the surface of endothelial cells. Melanoma cells usually do express high levels of GM3, while in turn endothelial cells express LacCer and Gg3. In vitro, GM3-rich B16 melanoma cells were able to adhere to LacCer- or Gg3 coated dishes. GM3-dependent adhesion is probably the best documented example of trans head-to-head carbohydrate-carbohydrate interaction. Remarkably, GM3-dependent adhesion (in a similar way to integrin-dependent adhesion) is not merely a physical link. B16 adhesion mediated by GM3-LacCer or GM3-Gg3 interactions and led to initiation of signal transduction, in particular FAK activation and enhanced GTP loading on Rho and Ras, with consequent phenotypic changes in terms of motility and morphology, suggesting that B16 GM3-dependent adhesion is associated with enhanced B16 cell motility and thereby initiates metastasis (Kojima et al. 1992; Iwabuchi et al. 1998). In these cells, GM3 is closely associated with signaling proteins such as c-Src, Rho and Ras within sphingolipid-enriched membrane domains, and binding with Gg3 or anti-GM3 antibody stimulates focal adhesion kinase phosphorylation and c-Src activity. Activation of c-Src and FAK with enhanced motility and invasiveness was induced in MCF-7 breast carcinoma cells by anti monosialyl-Gb5 monoclonal antibody, but not by antibodies to other glycosphingolipids (anti-Gb3, anti-Gb5, anti-GM2) (Steelant et al. 2002).
Recently, it has been proposed that many aspects of tumor cell social life are mediated by cell surface signaling complexes regulated by GSL. Again, lipid rafts seem to play a relevant role in the organization of these signaling complexes, favoring the clustering of different interactors, made possible only when certain GSL are expressed above a given quantitative threshold. Apparently, highly hydrophobic adapter membrane proteins such as tetraspanins or caveolins are essential for the organization of these complexes. On the other hand, their function seems strictly dependent on their GSL composition, providing clues about the molecular mechanisms underlying the phenotypic effects associated with aberrant glycosylation .
In the case of GM3 ganglioside, a number of studies have shown that its expression levels do affect the tumor phenotype in multiple ways, in particular by controlling of tumor cell motility, invasiveness and survival. As mentioned above, in the case of GM3-dependent adhesion of melanoma cells, it has been shown that GM3 is closely associated with c-Src, Rho and Ras within Triton X-100 insoluble ganglioside-rich lipid rafts and binding to Gg3 triggers the phosphorylation of the focal adhesion kinase and stimulates c-Src activity (Iwabuchi et al. 1998). A similar functional association between a sialoglycolipid and c-Src and other related signaling molecules was observed for GM3 also in neuroblastoma cells (Prinetti et al. 1999), for disialylgalactosylgloboside in renal carcinoma cells (Satoh et al. 2000), and for monosialyl-Gb5 in breast carcinoma cells (Steelant et al. 2002).
In bladder cancer , GM3 is highly expressed in non-invasive, superficial tumors compared with invasive tumors, as result of the upregulation of relevant glycosyltransferases (Satoh et al. 1996; Satoh et al. 2001; Kawamura et al. 2001). In a non-invasive cell line (KK47) originated from superficial human bladder cancer , GM3 levels were higher than in the invasive YTS1 human bladder cancer cell line. In this kind of tumor , the regulation of adhesion/motility by GM3 requires a multimolecular signaling complex organized by members of the tetraspan membrane protein superfamily (“tetraspanins”). Tetraspanins are highly hydrophobic integral membrane proteins strongly interacting with GSL (indeed, they were originally defined as “proteolipids”) (Kawakami et al. 2002), and the best characterized member, CD9, has been frequently described as interacting with integrin receptors (Hemler 1998). Tetraspanin CD9 and integrin α3 or α5 are associated within the same Brij 98-insoluble glycolipid-enriched domain. However, the physical and functional interaction between the tetraspanin and integrins, and the resisting downstream signaling controlling cell motility, are strongly affected by the cellular levels of GM3. In fact, knock down of CD9 or GM3 depletion in non-invasive KK47 cells induced their phenotypic conversion into invasive variants. On the other hand, exogenous GM3 addition induces the phenotypic reversion of the highly invasive and metastatic cell lines YTS1 to low motility variants. The changes in cell motility observed upon manipulation of GM3 levels were strictly correlated with the association of CD9 with α3 integrin. The level of interaction was positively modulated by GM3 and was thus higher in non-invasive than in highly invasive cells. In turn, CD9/α3 integrin association was reduced by GM3 depletion in KK47 and conversely enhanced by exogenous GM3 addition in YTS1 cells.
GM3 levels in these cells control not only CD9/α3 integrin association, but also the downstream signaling events by controlling the activation state of c-Src. The activity of the non-receptor tyrosine kinase c-Src is usually very high in malignant, invasive tumors, where c-Src is frequently constitutively overexpressed. The extendt of c-Src association to the CD9/α3 complex is negatively determined by the cellular GM3 levels. Moreover, with a still unclear mechanism, GM3 levels determine the extent of translocation and recruitment of the kinase Csk to the CD9/α3/c-Src complex. C-Src phosphorylation by Csk represents the main inhibitory mechanism for c-Src activity. c-Src is present in higher amount in the glycosynapse fraction in YTS1 cells, and it is activated in cells with low GM3 levels and high invasive potential (YTS1 or GM3-depleted KK47). On the other hand, exogenous addition of GM3 to YST1 cells caused Csk translocation to the lipid raft fraction and consequent inactivation of c-Src, influencing cell motility (Mitsuzuka et al. 2005).
Thus, GM3 complexed with CD9 and integrin receptors seems to play a crucial role in the control of tumor cell motility and invasiveness. This role of CD9/GM3 complexes in the regulation of integrin-mediated cell adhesion and signal transduction in oncogenic transformation, was confirmed by experiments showing the effect of the manipulation of GM3 levels in transformed cells. v-Jun-transformed mouse and chicken embryo fibroblasts were characterized by lower GM3 levels and down–regulated GM3 synthase mRNA levels respect to the non-transformed counterparts (Miura et al. 2004) and reversion of v-Jun oncogenic phenotype could be achieved by enhanced GM3 synthase gene transfection. During phenotypic reversion of v-Jun-transformed cells induced by GM3 synthase transfection, the association of CD9 and integrin receptors complex was increased.
Most notably, artificially increasing cellular GM3 levels (by loading cells with exogenous GM3 (Kawamura et al. 2001) or by pharmacological treatments (Satoh et al. 2001; Nojiri et al. 2002) led to a strong reduction of the tumorigenic activity and/or of the invasive potential of different human tumor cells lines. Consistently with this observation, the stable overexpression of GM3 synthase (SAT-I) in a mouse bladder carcinoma cell line reduced cell proliferation, motility and invasion with concomitant increase in the number of apoptotic cells (Watanabe et al. 2002). High expression levels of GM3 with concomitant expression of the tetraspanin CD9 inhibited cell motility in colorectal (Ono et al. 1999, 2001) cancer cells, and in CHO mutants the coexpression of CD9 and GM3 is essential for the downregulation of cell motility.
All these data support the notion that altered ganglioside levels can affect the tumor phenotype in terms of adhesion/motility by regulation the organization and function of the integrin signaling machinery. Recently it has been shown that integrin signaling can be controlled by other kinds of GSL/tetraspanin complexes. GM2 ganglioside complexed with a different tetraspanin, CD82, inhibits the activation of Met tyrosine kinase induced by hepatocyte growth factor (Todeschini et al. 2007). This suggests that ganglioside-controlled integrin signaling complexes might be a general paradigm controlling cell motility via tyrosine kinase signaling.
Compliance with Ethical Standards
Funding
This work was supported by funds of the Department of Medical Biotechnology and Translational Medicine of the University of Milano deriving by an analytical service directed by SS.
Disclosure of Interests
All authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with animal/human participants performed by any of the authors.
References
Abe T, Niizuma K, Kanoke A et al (2018) Metabolomic analysis of mouse brain after a transient middle cerebral artery occlusion by mass spectrometry imaging. Neurol Med Chir (Tokyo) 58:384–392. https://doi.org/10.2176/nmc.oa.2018-0054
Alpaugh M, Galleguillos D, Forero J et al (2017) Disease-modifying effects of ganglioside GM1 in Huntington’s disease models. EMBO Mol Med 9:1537–1557. https://doi.org/10.15252/emmm.201707763
Arenz C (2017) Recent advances and novel treatments for sphingolipidoses. Future Med Chem 9:1685–1698
Ariga T, Yu RK (1999) GM1 inhibits amyloid β-protein-induced cytokine release. Neurochem Res 24:219–226. https://doi.org/10.1023/A:1022557920150
Ariga T, McDonald MP, Yu RK (2008) Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease – a review. J Lipid Res 49:1157–1175
Bachis A, Rabin SJ, Fiacco MD, Mocchetti I (2002) Gangliosides prevent excitotoxicity through activation of TrkB receptor. Neurotox Res 4:225–234. https://doi.org/10.1080/10298420290015836
Barbacci DC, Roux A, Muller L et al (2017) Mass spectrometric imaging of ceramide biomarkers tracks therapeutic response in traumatic brain injury. ACS Chem Neurosci 8:2266–2274. https://doi.org/10.1021/acschemneuro.7b00189
Bartels T, Kim NC, Luth ES, Selkoe DJ (2014) N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS One 9. https://doi.org/10.1371/journal.pone.0103727
Bergante S, Torretta E, Creo P et al (2014) Gangliosides as a potential new class of stem cell markers: the case of GD1a in human bone marrow mesenchymal stem cells. J Lipid Res 55:549–560. https://doi.org/10.1194/jlr.M046672
Bergante S, Creo P, Piccoli M et al (2018) GM1 ganglioside promotes osteogenic differentiation of human tendon stem cells. Stem Cells Int 2018. https://doi.org/10.1155/2018/4706943
Bernardo A, Harrison FE, McCord M et al (2009) Elimination of GD3 synthase improves memory and reduces amyloid-β plaque load in transgenic mice. Neurobiol Aging 30:1777–1791. https://doi.org/10.1016/j.neurobiolaging.2007.12.022
Bizot V, Cestone E, Michelotti A, Nobile V (2017) Improving skin hydration and age-related symptoms by oral administration of wheat glucosylceramides and digalactosyl diglycerides: a human clinical study. Cosmetics 4:37. https://doi.org/10.3390/cosmetics4040037
Brackman D, Lund-Johansen F, Aarskog D (1995) Expression of leukocyte differentiation antigens during the differentiation of HL-60 cells induced by 1,25-dihydroxyvitamin D3: comparison with the maturation of normal monocytic and granulocytic bone marrow cells. J Leukoc Biol 58:547–555. https://doi.org/10.1002/jlb.58.5.547
Braulke T, Bonifacino JS (2009) Sorting of lysosomal proteins. Biochim Biophys Acta, Mol Cell Res 1793:605–614
Breiden B, Sandhoff K (2018) Ganglioside metabolism and its inherited diseases. In: Methods in molecular biology. Humana Press Inc., Clifton, pp 97–141
Bremer EG, Schlessinger J, Hakomori S (1986) Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J Biol Chem 261:2434–2440
Brooksbank BWL, McGovern J (1989) Gangliosides in the brain in adult Down’s syndrome and Alzheimer’s disease. Mol Chem Neuropathol 11:143–156. https://doi.org/10.1007/BF03160048
Brunkhorst R, Friedlaender F, Ferreirós N et al (2015) Alterations of the ceramide metabolism in the peri-infarct cortex are independent of the sphingomyelinase pathway and not influenced by the acid sphingomyelinase inhibitor fluoxetine. Neural Plast 2015:503079
Buccinnà B, Piccinini M, Prinetti A et al (2009) Alterations of myelin-specific proteins and sphingolipids characterize the brains of acid sphingomyelinase-deficient mice, an animal model of Niemann-Pick disease type A. J Neurochem 109:105–115. https://doi.org/10.1111/j.1471-4159.2009.05947.x
Capasso S, Sticco L, Rizzo R et al (2017) Sphingolipid metabolic flow controls phosphoinositide turnover at the trans -Golgi network. EMBO J 36:1736–1754. https://doi.org/10.15252/embj.201696048
Chakrabandhu K, Huault S, Garmy N et al (2008) The extracellular glycosphingolipid-binding motif of Fas defines its internalization route, mode and outcome of signals upon activation by ligand. Cell Death Differ 15:1824–1837. https://doi.org/10.1038/cdd.2008.115
Chang F, Li R, Ladisch S (1997) Shedding of gangliosides by human medulloblastoma cells. Exp Cell Res 234:341–346. https://doi.org/10.1006/excr.1997.3619
Charter HE, Glick FJ, Norris WP, Philips GE (1947) Biochemistry of the sphingolipides: III. Structure of sphingosine. J Biol Chem 170:285–294
Chester MA (1998) IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature of glycolipids – recommendations 1997. Eur J Biochem 257:293–298. https://doi.org/10.1046/j.1432-1327.1998.2570293.x
Chigorno V, Riva C, Valsecchi M et al (1997) Metabolic processing of gangliosides by human fibroblasts in culture – formation and recycling of separate pools of sphingosine. Eur J Biochem 250:661–669. https://doi.org/10.1111/j.1432-1033.1997.00661.x
Chigorno V, Sciannamblo M, Mikulak J et al (2006) Efflux of sphingolipids metabolically labeled with [1-3H] sphingosine, L-[3-3H]serine and [9,10-3H]palmitic acid from normal cells in culture. Glycoconj J 23:159–165. https://doi.org/10.1007/s10719-006-7921-7
Chiricozzi E, Ciampa MG, Brasile G et al (2015) Direct interaction, instrumental for signaling processes, between LacCer and Lyn in the lipid rafts of neutrophil-like cells. J Lipid Res 56:129–141. https://doi.org/10.1194/jlr.M055319
Chiricozzi E, Pomè DY, Maggioni M et al (2017) Role of the GM1 ganglioside oligosaccharide portion in the TrkA-dependent neurite sprouting in neuroblastoma cells. J Neurochem 143:645–659. https://doi.org/10.1111/jnc.14146
Chiricozzi E, Loberto N, Schiumarini D et al (2018) Sphingolipids role in the regulation of inflammatory response: from leukocyte biology to bacterial infection. J Leukoc Biol 103:445–456
Chiricozzi E, Biase E Di, Maggioni M, et al (2019a) GM1 promotes TrkA-mediated neuroblastoma cell differentiation by occupying a plasma membrane domain different from TrkA. J Neurochem 149:231–241. https://doi.org/10.1111/jnc.14685
Chiricozzi E, Maggioni M, di Biase E, et al (2019b) The neuroprotective role of the GM1 oligosaccharide, II3Neu5Ac-Gg4, in neuroblastoma cells. Mol Neurobiol 56:6673–6702. https://doi.org/10.1007/s12035-019-1556-8
Chiricozzi E, Mauri L, Lunghi G et al (2019c) Parkinson’s disease recovery by GM1 oligosaccharide treatment in the B4galnt1 +/− mouse model. Sci Rep 9. https://doi.org/10.1038/s41598-019-55885-2
Chiricozzi E, Lunghi G, Di Biase E et al (2020) GM1 ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int J Mol Sci 21
Cohen AW, Razani B, Wang XB et al (2003) Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol 285. https://doi.org/10.1152/ajpcell.00006.2003
Concolino D, Deodato F, Parini R (2018) Enzyme replacement therapy: efficacy and limitations. Ital J Pediatr 44. https://doi.org/10.1186/s13052-018-0562-1
Coskun Ü, Simons K (2011) Cell membranes: the lipid perspective. Structure 19:1543–1548
Coskun Ü, Grzybek M, Drechsel D, Simons K (2011) Regulation of human EGF receptor by lipids. Proc Natl Acad Sci U S A 108:9044–9048. https://doi.org/10.1073/pnas.1105666108
Crino PB, Ullman MD, Vogt BA et al (1989) Brain gangliosides in dementia of the Alzheimer type. Arch Neurol 46:398–401. https://doi.org/10.1001/archneur.1989.00520400054019
D’Angelo G, Polishchuk E, Tullio G Di, et al (2007) Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 449:62–67. https://doi.org/10.1038/nature06097
Da Silva JS, Hasegawa T, Miyagi T et al (2005) Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat Neurosci 8:606–615. https://doi.org/10.1038/nn1442
Dai R, Zhang S, Duan W et al (2017) Enhanced autophagy contributes to protective effects of GM1 ganglioside against Aβ1-42-induced neurotoxicity and cognitive deficits. Neurochem Res 42:2417–2426. https://doi.org/10.1007/s11064-017-2266-0
De Girolamo LA, Hargreaves AJ, Ellen Billett E (2001) Protection from MPTP-induced neurotoxicity in differentiating mouse N2a neuroblastoma cells. J Neurochem 76:650–660. https://doi.org/10.1046/j.1471-4159.2001.00066.x
Del Tredici R, Vos D et al (2002) Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 61. https://doi.org/10.1093/JNEN/61.5.413
Deng W, Li R, Ladisch S (2000) Influence of cellular ganglioside depletion on tumor formation. J Natl Cancer Inst 92:912–917
Desai BS, Monahan AJ, Carvey PM, Hendey B (2007) Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: implications for drug therapy. In: Cell transplantation. Cognizant Communication Corporation, Amsterdam, pp 285–299
Desplats PA, Denny CA, Kass KE et al (2007) Glycolipid and ganglioside metabolism imbalances in Huntington’s disease. Neurobiol Dis 27:265–277. https://doi.org/10.1016/j.nbd.2007.05.003
Di Biase E, Lunghi G, Fazzari M et al (2020a) Gangliosides in the differentiation process of primary neurons: the specific role of GM1-oligosaccharide. Glycoconj J:329–343. https://doi.org/10.1007/s10719-020-09919-x
Di Biase E, Lunghi G, Maggioni M et al (2020b) GM1 oligosaccharide crosses the human blood-brain barrier in vitro by a paracellular route. Int J Mol Sci 21. https://doi.org/10.3390/ijms21082858
Di Pardo A, Maglione V (2018) Sphingolipid metabolism: a new therapeutic opportunity for brain degenerative disorders. Front Neurosci 12
Di Pardo A, Maglione V, Alpaugh M et al (2012) Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc Natl Acad Sci U S A 109:3528–3533. https://doi.org/10.1073/pnas.1114502109
Doherty GJ, McMahon HT (2009) Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. https://doi.org/10.1146/annurev.biochem.78.081307.110540
Duchemin AM, Ren Q, Mo L et al (2002) GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J Neurochem 81:696–707. https://doi.org/10.1046/j.1471-4159.2002.00831.x
Duchemin AM, Ren Q, Neff NH, Hadjiconstantinou M (2008) GM1-induced activation of phosphatidylinositol 3-kinase: involvement of Trk receptors. J Neurochem 104:1466–1477. https://doi.org/10.1111/j.1471-4159.2007.05088.x
Erdö F, Denes L, De Lange E (2017) Age-associated physiological and pathological changes at the blood-brain barrier: a review. J Cereb Blood Flow Metab 37:4–24
Fantini J, Yahi N (2015) Lipid regulation of receptor function. In: Brain lipids in synaptic function and neurological disease, first edit. Elsevier, San Diego, pp 163–181
Farfel-Becker T, Vitner EB, Kelly SL et al (2014) Neuronal accumulation of glucosylceramide in a mouse model of Neuronopathic Gaucher disease leads to neurodegeneration. Hum Mol Genet 23:843–854
Farooqui T, Franklin T, Pearl DK, Yates AJ (1997) Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J Neurochem 68:2348–2355. https://doi.org/10.1046/j.1471-4159.1997.68062348.x
Fazzari M, Audano M, Lunghi G et al (2020) The oligosaccharide portion of ganglioside GM1 regulates mitochondrial function in neuroblastoma cells. Glycoconj J 37:293–306. https://doi.org/10.1007/s10719-020-09920-4
Feingold KR, Elias PM (2014) Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim Biophys Acta Mol Cell Biol Lipids 1841:280–294
Ferlinz K, Kopal G, Bernardo K et al (2001) Human acid ceramidase: processing, glycosylation, and lysosomal targeting. J Biol Chem 276:35352–35360. https://doi.org/10.1074/jbc.M103066200
Ferrari G, Anderson BL, Stephens RM et al (1995) Prevention of apoptotic neuronal death by G(M1) ganglioside. Involvement of Trk neurotrophin receptors. J Biol Chem 270:3074–3080. https://doi.org/10.1074/jbc.270.7.3074
Ferrucci L, Gonzalez-Freire M, Fabbri E et al (2020) Measuring biological aging in humans: a quest. Aging Cell 19
Filocamo M, Morrone A (2011) Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics 5:156–169
Flatt T (2012) A new definition of aging? Front Genet 3:1–2
Futerman AH, Van Meer G (2004) The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 5:554–565
Futerman AH, Sussman JL, Horowitz M et al (2004) New directions in the treatment of Gaucher disease. Trends Pharmacol Sci 25:147–151. https://doi.org/10.1016/j.tips.2004.01.004
Gabandé-Rodríguez E, Boya P, Labrador V et al (2014) High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ 21:864–875. https://doi.org/10.1038/cdd.2014.4
Ghadially R, Brown BE, Sequeira-Martin SM et al (1995) The aged epidermal permeability barrier. Structural, functional, and lipid biochemical abnormalities in humans and a senescent murine model. J Clin Invest 95:2281–2290. https://doi.org/10.1172/JCI117919
Ghidoni R, Trinchera M, Venerando B et al (1986) Incorporation and metabolism of exogenous G(M1) ganglioside in rat liver. Biochem J 237:147–155. https://doi.org/10.1042/bj2370147
Ghosh P, Dahms NM, Kornfeld S (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4:202–212
Gladyshev VN (2016) Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15:594–602
Grassi S, Chiricozzi E, Mauri L et al (2019) Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem 148:600–611. https://doi.org/10.1111/jnc.14540
Grösch S, Schiffmann S, Geisslinger G (2012) Chain length-specific properties of ceramides. Prog Lipid Res 51:50–62
Gu LZ, Huang BS, Shen W et al (2013) Early activation of nSMase2/ceramide pathway in astrocytes is involved in ischemia-associated neuronal damage via inflammation in rat hippocampi. J Neuroinflam 10. https://doi.org/10.1186/1742-2094-10-109
Hadaczek P, Wu G, Sharma N et al (2015) GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp Neurol 263:177–189. https://doi.org/10.1016/j.expneurol.2014.10.010
Hadjiconstantinou M, Mariani AP, Neff NH (1989) GM1 ganglioside-induced recovery of nigrostriatal dopaminergic neurons after MPTP: an immunohistochemical study. Brain Res 484:297–303. https://doi.org/10.1016/0006-8993(89)90373-9
Hakomori S (1985) Aberrant glycosylation in cancer cell membranes as focused on glycolipids: overview and perspectives. Cancer Res 45:2405–2414
Hakomori S-I (1989) Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv Cancer Res 52:257–331. https://doi.org/10.1016/S0065-230X(08)60215-8
Hakomori SI (1996) Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res 56:5309–5318
Hakomori S (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A 99:10231–10233
Hakomori SI, Igarashi Y (1995) Functional role of glycosphingolipids in cell recognition and signaling. J Biochem 118:1091–1103. https://doi.org/10.1093/oxfordjournals.jbchem.a124992
Hakomori SI, Handa K, Iwabuchi K et al (1998) New insights in glycosphingolipid function: “Glycosignaling domain,” a cell surface assembly of glycosphingolipids with signal transducer molecules, involved in cell adhesion coupled with signaling [1]. Glycobiology 8
Hallett PJ, Huebecker M, Brekk OR et al (2018) Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol Aging 67:189–200. https://doi.org/10.1016/j.neurobiolaging.2018.02.028
Haughey NJ (2010) Sphingolipids in neurodegeneration. NeuroMolecular Med 12:301–305
Hayashi H, Kimura N, Yamaguchi H et al (2004) A seed for Alzheimer amyloid in the brain. J Neurosci 24:4894–4902. https://doi.org/10.1523/JNEUROSCI.0861-04.2004
Hein LK, Duplock S, Hopwood JJ, Fuller M (2008) Lipid composition of microdomains is altered in a cell model of Gaucher disease. J Lipid Res 49:1725–1734. https://doi.org/10.1194/jlr.M800092-JLR200
Hemler ME (1998) Integrin associated proteins. Curr Opin Cell Biol 10:578–585. https://doi.org/10.1016/S0955-0674(98)80032-X
Hernández-Corbacho MJ, Jenkins RW, Clarke CJ et al (2011) Accumulation of long-chain glycosphingolipids during aging is prevented by caloric restriction. PLoS One 6. https://doi.org/10.1371/journal.pone.0020411
Herrero MT, Perez-Otaño I, Oset C et al (1993) GM-1 ganglioside promotes the recovery of surviving midbrain dopaminergic neurons in MPTP-treated monkeys. Neuroscience 56:965–972. https://doi.org/10.1016/0306-4522(93)90142-3
Hikita T, Tadano-Aritomi K, Iida-Tanaka N et al (2002) Cationic glycosphingolipids in neuronal tissues and their possible biological significance. Neurochem Res 27:575–581. https://doi.org/10.1023/A:1020259630034
Holleran WM, Takagi Y, Uchida Y (2006) Epidermal sphingolipids: metabolism, function, and roles in skin disorders. FEBS Lett 580:5456–5466
Horres CR, Hannun YA (2012) The roles of neutral sphingomyelinases in neurological pathologies. Neurochem Res 37:1137–1149
Iwabuchi K, Nagaoka I (2002) Lactosylceramide-enriched glycosphingolipid signaling domain mediates superoxide generation from human neutrophils. Blood 100
Iwabuchi K, Handa K, Hakomori SI (1998) Separation of “glycosphingolipid signaling domain” from caveolin- containing membrane fraction in mouse melanoma B16 cells and its role in cell adhesion coupled with signaling. J Biol Chem 273:33766–33773. https://doi.org/10.1074/jbc.273.50.33766
Iwabuchi K, Prinetti A, Sonnino S et al (2008) Involvement of very long fatty acid-containing lactosylceramide in lactosylceramide-mediated superoxide generation and migration in neutrophils. Glycoconj J 25:357–374. https://doi.org/10.1007/s10719-007-9084-6
Jakóbkiewicz-Banecka J, Gabig-Cimińska M, Banecka-Majkutewicz Z et al (2014) Factors and processes modulating phenotypes in neuronopathic lysosomal storage diseases. Metab Brain Dis 29:1–8
Jones ZB, Ren Y (2016) Sphingolipids in spinal cord injury. Int J Physiol Pathophysiol Pharmacol 8:52–69
Kabayama K, Sato T, Saito K et al (2007) Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci U S A 104:13678–13683. https://doi.org/10.1073/pnas.0703650104
Kanoh H, Nitta T, Go S et al (2020) Homeostatic and pathogenic roles of GM 3 ganglioside molecular species in TLR 4 signaling in obesity. EMBO J 39. https://doi.org/10.15252/embj.2019101732
Kawakami Y, Kawakami K, Steelant WFA et al (2002) Tetraspanin CD9 is a “proteolipid,” and its interaction with α3 integrin in microdomain is promoted by GM3 ganglioside, leading to inhibition of laminin-5-dependent cell motility. J Biol Chem 277:34349–34358. https://doi.org/10.1074/jbc.M200771200
Kawamura S, Ohyama C, Watanabe R et al (2001) Glycolipid composition in bladder tumor: a crucial role of gm3 ganglioside in tumor invasion. Int J Cancer 94:343–347. https://doi.org/10.1002/ijc.1482
Kawashima N, Yoon SJ, Itoh K, Nakayama KI (2009) Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J Biol Chem 284:6147–6155. https://doi.org/10.1074/jbc.M808171200
Kim SM, Jung JU, Ryu JS et al (2008) Effects of gangliosides on the differentiation of human mesenchymal stem cells into osteoblasts by modulating epidermal growth factor receptors. Biochem Biophys Res Commun 371:866–871. https://doi.org/10.1016/j.bbrc.2008.04.162
Kischkel FC, Hellbardt S, Behrmann I et al (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a Death-Inducing Signaling Complex (DISC) with the receptor. EMBO J 14:5579–5588
Kishimoto Y, Hiraiwa M, O’Brien JS (1992) Saposins: structure, function, distribution, and molecular genetics. J Lipid Res 33:1255–1267
Kitatani K, Idkowiak-Baldys J, Hannun YA (2008) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 20:1010–1018
Kniep B, Skubitz KM (1998) Subcellular localization of glycosphingolipids in human neutrophils. J Leukoc Biol 63:83–88. https://doi.org/10.1002/jlb.63.1.83
Kojima N, Shiota M, Sadahira Y et al (1992) Cell adhesion in a dynamic flow system as compared to static system. Glycosphingolipid-glycosphingolipid interaction in the dynamic system predominates over lectin- or integrin-based mechanisms in adhesion of B16 melanoma cells to non-activated endothelial cells. J Biol Chem 267:17264–17270
Kolter T, Sandhoff K (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol 21:81–103. https://doi.org/10.1146/annurev.cellbio.21.122303.120013
Kolter T, Sandhoff K (2006) Sphingolipid metabolism diseases. Biochim Biophys Acta Biomembr 1758:2057–2079
Kracun I, Rosner H, Drnovsek V et al (1991) Human brain gangliosides in development, aging and disease. Int J Dev Biol 35:289–295. https://doi.org/10.1387/ijdb.1814411
Kracun I, Kalanj S, Talan-Hranilovic J, Cosovic C (1992) Cortical distribution of gangliosides in Alzheimer’s disease. Neurochem Int 20:433–438. https://doi.org/10.1016/0197-0186(92)90058-Y
Kreutz F, Frozza RL, Breier AC et al (2011) Amyloid-β induced toxicity involves ganglioside expression and is sensitive to GM1 neuroprotective action. Neurochem Int 59:648–655. https://doi.org/10.1016/j.neuint.2011.06.007
Ladisch S, Kitada S, Hays EF (1987) Gangliosides shed by tumor cells enhance tumor formation in mice. J Clin Invest 79:1879–1882. https://doi.org/10.1172/JCI113031
Ladisch S, Li R, Olson E (1994) Ceramide structure predicts tumor ganglioside immunosuppressive activity. Proc Natl Acad Sci U S A 91:1974–1978. https://doi.org/10.1073/pnas.91.5.1974
Ladisch S, Chang F, Li R et al (1997) Detection of medulloblastoma and astrocytoma-associated, ganglioside G(D3) in cerebrospinal fluid. Cancer Lett 120:71–78. https://doi.org/10.1016/S0304-3835(97)00297-8
Langeveld M, Ghauharali KJM, Sauerwein HP et al (2008) Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. J Clin Endocrinol Metab 93:845–851. https://doi.org/10.1210/jc.2007-1702
Ledeen R, Wu G (2018a) Gangliosides of the nervous system. In: Methods in molecular biology. Humana Press Inc, New York, pp 19–55
Ledeen RW, Wu G (2018b) Gangliosides, α-Synuclein, and Parkinson’s disease. In: Progress in molecular biology and translational science. Elsevier, Oxford, pp 435–454
Ledesma MD, Martin MG, Dotti CG (2012) Lipid changes in the aged brain: effect on synaptic function and neuronal survival. Prog Lipid Res 51:23–35
Lee KH, Feig C, Tchikov V et al (2006) The role of receptor internalization in CD95 signaling. EMBO J 25:1009–1023. https://doi.org/10.1038/sj.emboj.7601016
Li M (2018) Enzyme replacement therapy: a review and its role in treating lysosomal storage diseases. Pediatr Ann 47:e191–e197. https://doi.org/10.3928/19382359-20180424-01
Lill CM, Roehr JT, McQueen MB et al (2009) The PDGene database. Alzheimer Research Forum. Available at: http://www.pdgene.org2
Lu C, Ferzly M, Takagi J, Springer TA (2001) Epitope mapping of antibodies to the C-terminal region of the integrin β 2 subunit reveals regions that become exposed upon receptor activation. J Immunol 166:5629–5637. https://doi.org/10.4049/jimmunol.166.9.5629
Maceyka M, Harikumar KB, Milstien S, Spiegel S (2012) Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 22:50–60
Magistretti PJ, Geisler FH, Schneider JS et al (2019) Gangliosides: treatment avenues in neurodegenerative disease. Front Neurol 10
Maglione V, Marchi P, Di Pardo A et al (2010) Impaired ganglioside metabolism in Huntington’s disease and neuroprotective role of GM1. J Neurosci 30:4072–4080. https://doi.org/10.1523/JNEUROSCI.6348-09.2010
Marquina G, Waki H, Fernandez LE et al (1996) Gangliosides expressed in human breast cancer. Cancer Res 56:5165–5171
Martinez Z, Zhu M, Han S, Fink AL (2007) GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry 46:1868–1877. https://doi.org/10.1021/bi061749a
Mayor S, Pagano RE (2007) Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 8:603–612
Mazzulli JR, Xu YH, Sun Y et al (2011) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. https://doi.org/10.1016/j.cell.2011.06.001
McNeill A, Magalhaes J, Shen C et al (2014) Ambroxol improves lysosomal biochemistry in Glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137:1481–1495
Mencarelli C, Martinez-Martinez P (2013) Ceramide function in the brain: when a slight tilt is enough. Cell Mol Life Sci 70:181–203
Meredith GE, Rademacher DJ (2011) MPTP mouse models of Parkinson’s disease: an update. J Parkinsons Dis 1:19–33
Merrill AH (2011) Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev 111:6387–6422
Mielke MM, Lyketsos CG (2010) Alterations of the sphingolipid pathway in Alzheimer’s disease: new biomarkers and treatment targets? NeuroMolecular Med 12:331–340
Miljan EA, Bremer EG (2002) Regulation of growth factor receptors by gangliosides. Sci STKE 2002:re15
Miljan EA, Meuillet EJ, Mania-Farnell B et al (2002) Interaction of the extracellular domain of the epidermal growth factor receptor with gangliosides. J Biol Chem 277:10108–10113. https://doi.org/10.1074/jbc.M111669200
Mitsuzuka K, Handa K, Satoh M et al (2005) A specific microdomain (“glycosynapse 3”) controls phenotypic conversion and reversion of bladder cancer cells through GM3-mediated interaction of α3β1 integrin with CD9. J Biol Chem 280:35545–35553. https://doi.org/10.1074/jbc.M505630200
Miura Y, Kainuma M, Jiang H et al (2004) Reversion of the Jun-induced oncogenic phenotype by enhanced synthesis of sialosyllactosylceramide (GM3 ganglioside). Proc Natl Acad Sci U S A 101:16204–16209. https://doi.org/10.1073/pnas.0407297101
Mo L, Ren Q, Duchemin AM et al (2005) GM1 and ERK signaling in the aged brain. Brain Res 1054:125–134. https://doi.org/10.1016/j.brainres.2005.06.068
Mutoh T, Tokuda A, Miyadai T et al (1995) Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci U S A 92:5087–5091. https://doi.org/10.1073/pnas.92.11.5087
Mutoh T, Hamano T, Yano S et al (2002) Stable transfection of GM1 synthase gene into GM1-deficient NG108-15 cells, CR-72 cells, rescues the responsiveness of Trk-neurotrophin receptor to its ligand, NGF. Neurochem Res 27:801–806. https://doi.org/10.1023/A:1020209008169
Nakayama H, Yoshizaki F, Prinetti A et al (2008) Lyn-coupled LacCer-enriched lipid rafts are required for CD11b/CD18-mediated neutrophil phagocytosis of nonopsonized microorganisms. J Leukoc Biol 83:728–741. https://doi.org/10.1189/jlb.0707478
Nakayama H, Ogawa H, Takamori K, Iwabuchi K (2013) GSL-enriched membrane microdomains in innate immune responses. Arch Immunol Ther Exp 61:217–228
Neudorfer O, Giladi N, Elstein D et al (1996) Occurrence of Parkinson’s syndrome in Type I Gaucher disease. QJM 89:691–694
Ngamukote S, Yanagisawa M, Ariga T et al (2007) Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J Neurochem 103:2327–2341. https://doi.org/10.1111/j.1471-4159.2007.04910.x
Nicotra A, Parvez SH (2002) Apoptotic molecules and MPTP-induced cell death. Neurotoxicol Teratol 24:599–605
Nojiri H, Stroud M, Hakomori S (1991) A specific type of ganglioside as a modulator of insulin-dependent cell growth and insulin receptor tyrosine kinase activity. Possible Association of Ganglioside-Induced Inhibition of insulin receptor function and monocytic differentiation induction in HL-60 cells. J Biol Chem 266:4531–4537
Nojiri H, Yamana H, Shirouzu G et al (2002) Glycotherapy for cancer: remodeling of ganglioside pattern as an effective approach for cancer therapy. Cancer Detect Prev 26:114–120. https://doi.org/10.1016/S0361-090X(02)00033-8
Novgorodov SA, Gudz TI (2009) Ceramide and mitochondria in ischemia/reperfusion. J Cardiovasc Pharmacol 53:198–208. https://doi.org/10.1097/FJC.0b013e31819b52d5
Ohsawa T (1989) Changes of mouse brain gangliosides during aging from young adult until senescence. Mech Ageing Dev 50:169–177. https://doi.org/10.1016/0047-6374(89)90012-2
Ohyama C, Orikasa S, Kawamura S et al (1996) Galactosylgloboside expression in seminoma. Inverse correlation with metastatic potential. Cancer 76. https://doi.org/10.1002/1097-0142(19950915)76:6<1043::AID-CNCR2820760619>3.0.CO;2-A
Ong WY, Herr DR, Farooqui T et al (2015) Role of sphingomyelinases in neurological disorders. Expert Opin Ther Targets 19:1725–1742
Ono M, Handa K, Withers DA, Hakomori S (1999) Motility inhibition and apoptosis are induced by metastasis-suppressing gene product CD82 and its analogue CD9, with concurrent glycosylation. Cancer Res 59:2335–2339
Ono M, Handa K, Sonnino S et al (2001) GM3 ganglioside inhibits CD9-facilitated haptotactic cell motility: Coexpression of GM3 and CD9 is essential in the downregulation of tumor cell motility and malignancy. Biochemistry 40:6414–6421. https://doi.org/10.1021/bi0101998
Ortiz A, Germain DP, Desnick RJ et al (2018) Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 123:416–427
Parenti G, Andria G, Ballabio A (2015) Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med 66:471–486. https://doi.org/10.1146/annurev-med-122313-085916
Perera RM, Zoncu R (2016) The lysosome as a regulatory hub. Annu Rev Cell Dev Biol 32:223–253. https://doi.org/10.1146/annurev-cellbio-111315-125125
Perry RJ, Ridgway ND (2005) Molecular mechanisms and regulation of ceramide transport. Biochim Biophys Acta Mol Cell Biol Lipids 1734:220–234
Piccinini M, Scandroglio F, Prioni S et al (2010) Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol Neurobiol 41:314–340. https://doi.org/10.1007/s12035-009-8096-6
Pitto M, Mutoh T, Kuriyama M et al (1998) Influence of endogenous GM1 ganglioside on TrkB activity, in cultured neurons. FEBS Lett 439:93–96. https://doi.org/10.1016/S0014-5793(98)01344-1
Platt F, Boland B, van der Spoel AC (2012) The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 199:723–734
Polo A, Kirschner G, Guidotti A, Costa E (1994) Brain content of glycosphingolipids after oral administration of monosialogangliosides GM1 and LIGA20 to rats. Mol Chem Neuropathol 21:41–53. https://doi.org/10.1007/BF03160083
Prinetti A, Iwabuchi K, Hakomori SI (1999) Glycosphingolipid-enriched signaling domain in mouse neuroblastoma Neuro2a cells. Mechanism of ganglioside-dependent neuritogenesis. J Biol Chem 274:20916–20924. https://doi.org/10.1074/jbc.274.30.20916
Prinetti A, Chigorno V, Tettamanti G, Sonnino S (2000) Sphingolipid-enriched membrane domains from rat cerebellar granule cells differentiated in culture: a compositional study. J Biol Chem 275:11658–11665. https://doi.org/10.1074/jbc.275.16.11658
Prinetti A, Loberto N, Chigorno V, Sonnino S (2009) Glycosphingolipid behaviour in complex membranes. Biochim Biophys Acta Biomembr 1788:184–193
Rabin SJ, Mocchetti I (1995) GM1 ganglioside activates the high-affinity nerve growth factor receptor trkA. J Neurochem 65:347–354. https://doi.org/10.1046/j.1471-4159.1995.65010347.x
Reczek D, Schwake M, Schröder J et al (2007) LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidase. Cell 131:770–783. https://doi.org/10.1016/j.cell.2007.10.018
Revel-Vilk S, Szer J, Mehta A, Zimran A (2018) How we manage Gaucher disease in the era of choices. Br J Haematol 182:467–480
Riboldi GM, Di Fonzo AB (2019) GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cell 8:364. https://doi.org/10.3390/cells8040364
Riboni L, Bassi R, Prinetti A, Tettamanti G (1996) Salvage of catabolic products in ganglioside metabolism: a study on rat cerebellar granule cells in culture. FEBS Lett 391:336–340. https://doi.org/10.1016/0014-5793(96)00772-7
Ringerike T, Blystad FD, Levy FO et al (2002) Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J Cell Sci 115:1331–1340
Rocha EM, Smith GA, Park E et al (2015) Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol 2:433–438. https://doi.org/10.1002/acn3.177
Roepstorff K, Thomsen P, Sandvig K, Van Deurs B (2002) Sequestration of epidermal growth factor receptors in non-caveolar lipid rafts inhibits ligand binding. J Biol Chem 277:18954–18960. https://doi.org/10.1074/jbc.M201422200
Rogers J, Harding C, Mayo A et al (1996) Stratum corneum lipids: the effect of ageing and the seasons. Arch Dermatol Res 288:765–770. https://doi.org/10.1007/BF02505294
Rothblat DS, Schneider JS (1998) Effects of GM1 ganglioside treatment on dopamine innervation of the striatum of MPTP-treated mice. In: Annals of the New York Academy of Sciences. Blackwell Publishing Inc, pp 274–277
Roux A, Muller L, Jackson SN et al (2016) Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. J Neurosci Methods 272:19–32. https://doi.org/10.1016/j.jneumeth.2016.02.004
Russo D, Parashuraman S, D’Angelo G (2016) Glycosphingolipid–protein interaction in signal transduction. Int J Mol Sci 17:1732
Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10:623–635
Sajja VSSS, Jablonska A, Haughey N et al (2018) Sphingolipids and microRNA changes in blood following blast traumatic brain injury: an exploratory study. J Neurotrauma 35:353–361. https://doi.org/10.1089/neu.2017.5009
Samarani M, Loberto N, Soldà G et al (2018) A lysosome–plasma membrane–sphingolipid axis linking lysosomal storage to cell growth arrest. FASEB J 32:5685–5702. https://doi.org/10.1096/fj.201701512RR
Sandhoff K (2016) Neuronal sphingolipidoses: membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie 130:146–151
Sasaki N, Itakura Y, Toyoda M (2015) Ganglioside GM1 contributes to the state of insulin resistance in senescent human arterial endothelial cells. J Biol Chem 290:25475–25486. https://doi.org/10.1074/jbc.M115.684274
Satoh M, Handa K, Saito S et al (1996) Disialosyl galactosylgloboside as an adhesion molecule expressed on renal cell carcinoma and its relationship to metastatic potential. Cancer Res 56:1932–1938
Satoh M, Nejad FM, Ohtani H et al (2000) Association of renal cell carcinoma antigen, disialylgalactosylgloboside, with c-Src and Rho A in clustered domains at the surface membrane. Int J Oncol 16:529–536. https://doi.org/10.3892/ijo.16.3.529
Satoh M, Ito A, Nojiri H et al (2001) Enhanced GM3 expression, associated with decreased invasiveness, is induced by Brefeldin A in bladder cancer cells. Int J Oncol 19:723–731
Saulino MF, Schengrund C-L (1994) Differential accumulation of gangliosides by the brains of MPTP-lesioned mice. J Neurosci Res 37:384–391. https://doi.org/10.1002/jnr.490370310
Scandroglio F, Venkata JK, Loberto N et al (2008) Lipid content of brain, brain membrane lipid domains, and neurons from acid sphingomyelinase deficient mice. J Neurochem 107:329–338. https://doi.org/10.1111/j.1471-4159.2008.05591.x
Schengrund CL (2015) Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem Sci 40:397–406
Schengrund C-L, Prouty C (1988) Oligosaccharide portion of GM1 enhances process formation by S20Y neuroblastoma cells. J Neurochem 51:277–282. https://doi.org/10.1111/j.1471-4159.1988.tb04867.x
Schnaar RL (2016) Gangliosides of the vertebrate nervous system. J Mol Biol 428:3325–3336
Schneider JS (2018) Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PLoS One 13. https://doi.org/10.1371/journal.pone.0199189
Schneider JS, DiStefano L (1994) Oral administration of semisynthetic sphingolipids promotes recovery of striatal dopamine concentrations in a murine model of parkinsonism. Neurology 44:748–750. https://doi.org/10.1212/wnl.44.4.748
Schneider JS, Pope A, Simpson K et al (1992) Recovery from experimental parkinsonism in primates with GM1 ganglioside treatment. Science 256:843–846. https://doi.org/10.1126/science.1350379
Schneider JS, Sendek S, Daskalakis C, Cambi F (2010) GM1 ganglioside in Parkinson’s disease: results of a five year open study. J Neurol Sci 292:45–51. https://doi.org/10.1016/j.jns.2010.02.009
Schneider JS, Gollomp SM, Sendek S et al (2013) A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J Neurol Sci 324:140–148. https://doi.org/10.1016/j.jns.2012.10.024
Schneider JS, Cambi F, Gollomp SM et al (2015a) GM1 ganglioside in Parkinson’s disease: pilot study of effects on dopamine transporter binding. J Neurol Sci 356:118–123. https://doi.org/10.1016/j.jns.2015.06.028
Schneider JS, Seyfried TN, Choi HS, Kidd SK (2015b) Intraventricular sialidase administration enhances GM1 ganglioside expression and is partially neuroprotective in a mouse model of Parkinson’s disease. PLoS One 10. https://doi.org/10.1371/journal.pone.0143351
Schneider JS, Aras R, Williams CK et al (2019) GM1 ganglioside modifies α-Synuclein toxicity and is neuroprotective in a rat α-Synuclein model of Parkinson’s disease. Sci Rep 9. https://doi.org/10.1038/s41598-019-42847-x
Schuchman EH (2007) The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis 30:654–663
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1. https://doi.org/10.1101/cshperspect.a006189
Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14:283–296
Sidransky E (2005) Gaucher disease and parkinsonism. Mol Genet Metab 84:302–304
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11:986–998. https://doi.org/10.1016/S1474-4422(12)70190-4
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39
Sokolova TV, Zakharova IO, Furaev VV et al (2007) Neuroprotective effect of ganglioside GM1 on the cytotoxic action of hydrogen peroxide and amyloid β-peptide in PC12 cells. Neurochem Res 32:1302–1313. https://doi.org/10.1007/s11064-007-9304-2
Sokolova T, Rychkova MP, Avrova N (2014) Protective effect of GM1 ganglioside against toxic action of glutamate on cerebellar granule cells. J Evol Biochem Physiol 50:399–401
Sonderfeld S, Conzelma E, Schwarzmann G et al (1985) Incorporation and metabolism of ganglioside GM2 in skin fibroblasts from normal and GM2 gangliosidosis subjects. Eur J Biochem 149:247–255. https://doi.org/10.1111/j.1432-1033.1985.tb08919.x
Sonnino S, Prinetti A (2013) Membrane domains and the “lipid raft” concept. Curr Med Chem 20:4–21
Sonnino S, Ghidoni R, Marchesini S, Tettamanti G (1979) Cytosolic gangliosides: occurrence in calf brain as ganglioside-protein complexes. J Neurochem 33:117–121. https://doi.org/10.1111/j.1471-4159.1979.tb11713.x
Sonnino S, Prinetti A, Mauri L et al (2006) Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem Rev 106:2111–2125
Sonnino S, Chiricozzi E, Ciampa MG et al (2017) Serum antibodies to glycans in peripheral neuropathies. Mol Neurobiol 54:1564–1567
Spassieva S, Bieberich E (2016) Lysosphingolipids and sphingolipidoses: Psychosine in Krabbe’s disease. J Neurosci Res 94:974–981
Spychalska J, Smoleńska-Sym G, Zdebska E et al (2003) Quantitative analysis of LacCer/CDw17 in human myelogenous leukaemic cells. Cell Mol Biol Lett 8:911–917
Steelant WF, Kawakami Y, Ito A et al (2002) Monosialyl-Gb5 organized with cSrc and FAK in GEM of human breast carcinoma MCF-7 cells defines their invasive properties. FEBS Lett 531:93–98. https://doi.org/10.1016/S0014-5793(02)03484-1
Stern MB, Lang A, Poewe W (2012) Toward a redefinition of Parkinson’s disease. Mov Disord 27:54–60
Stoffel W (1970) Studies on the biosynthesis and degradation of sphingosine bases. Chem Phys Lipids 5:139–158. https://doi.org/10.1016/0009-3084(70)90014-9
Sugiura Y, Shimma S, Konishi Y et al (2008) Imaging mass spectrometry technology and application on ganglioside study; visualization of age-dependent accumulation of C20-ganglioside molecular species in the mouse hippocampus. PLoS One 3. https://doi.org/10.1371/journal.pone.0003232
Svennerholm L (1994) Ganglioside loss is a primary event in Alzheimer disease type I. Prog Brain Res 101:391–404. https://doi.org/10.1016/S0079-6123(08)61965-2
Svennerholm L, Gottfries C-G (1994) Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (Type I) and demyelination in late-onset form (Type II). J Neurochem 62:1039–1047. https://doi.org/10.1046/j.1471-4159.1994.62031039.x
Svennerholm L, Gottfries CG, Blennow K et al (1990) Parenteral administration of GM1 ganglioside to presenile Alzheimer patients. Acta Neurol Scand 81:48–53. https://doi.org/10.1111/j.1600-0404.1990.tb00930.x
Tagami S, Inokuchi JI, Kabayama K et al (2002) Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem 277:3085–3092. https://doi.org/10.1074/jbc.M103705200
Tayebi N, Walker J, Stubblefield B et al (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 79:104–109. https://doi.org/10.1016/S1096-7192(03)00071-4
Tettamanti G (2004) Ganglioside/glycosphingolipid turnover: new concepts. Glycoconj J 20:301–317
Thudichum JLW (1884) A treatise on the chemical constitution of the brain: based throughout upon original researches. Bailliere, Tindall &, Cox, London.
Todeschini AR, Dos Santos JN, Handa K, Hakomori SI (2007) Ganglioside GM2-tetraspanin CD82 complex inhibits Met and its cross-talk with integrins, providing a basis for control of cell motility through glycosynapse. J Biol Chem 282:8123–8133. https://doi.org/10.1074/jbc.M611407200
Valdes-Gonzalez T, Goto-Inoue N, Hirano W et al (2011) New approach for glyco- and lipidomics – molecular scanning of human brain gangliosides by TLC-Blot and MALDI-QIT-TOF MS. J Neurochem 116:678–683
Valsecchi M, Palestini P, Chigorno V, Sonnino S (1996) Age-related changes of the ganglioside long-chain base composition in rat cerebellum. Neurochem Int 28:183–187. https://doi.org/10.1016/0197-0186(95)00069-0
Varki A, Cummings RD, Aebi M et al (2015) Symbol nomenclature for graphical representations of glycans. Glycobiology 25:1323–1324
Walkley SU (2004) Secondary accumulation of gangliosides in lysosomal storage disorders. Semin Cell Dev Biol 15:433–444. https://doi.org/10.1016/j.semcdb.2004.03.002
Walkley SU, Vanier MT (2009) Secondary lipid accumulation in lysosomal disease. Biochim Biophys Acta, Mol Cell Res 1793:726–736
Wang G, Bieberich E (2018) Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv Biol Regul 70:51–64
Wang XQ, Sun P, Paller AS (2002) Ganglioside induces caveolin-1 redistribution and interaction with the epidermal growth factor receptor. J Biol Chem 277:47028–47034. https://doi.org/10.1074/jbc.M208257200
Wang RY, Bodamer OA, Watson MS, Wilcox WR (2011) Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med 13:457–484. https://doi.org/10.1097/GIM.0b013e318211a7e1
Watanabe R, Ohyama C, Aoki H et al (2002) Ganglioside G(M3) overexpression induces apoptosis and reduces malignant potential in murine bladder cancer. Cancer Res 62:3850–3854
Wennekes T, Van Den Berg RJBHN, Boot RG et al (2009) Glycosphingolipids – nature, function, and pharmacological modulation. Angew Chemie Int Ed 48:8848–8869
Wertz PW (2018) Naturally occurring ω-hydroxyacids. Int J Cosmet Sci 40:31–33
White AB, Givogri MI, Lopez-Rosas A et al (2009) Psychosine accumulates in membrane microdomains in the brain of Krabbe patients, disrupting the raft architecture. J Neurosci 29:6068–6077. https://doi.org/10.1523/JNEUROSCI.5597-08.2009
Wong K, Sidransky E, Verma A et al (2004) Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 82:192–207. https://doi.org/10.1016/j.ymgme.2004.04.011
Wu G, Lu ZH, Xie X, Ledeen RW (2004) Susceptibility of cerebellar granule neurons from GM2/GD2 synthase-null mice to apoptosis induced by glutamate excitotoxicity and elevated KCl: Rescue by GM1 and LIGA20. Glycoconj J 21:305–313. https://doi.org/10.1023/B:GLYC.0000046273.68493.f7
Wu G, Lu ZH, Wang J et al (2005) Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane-permeant analog of GM1. J Neurosci 25:11014–11022. https://doi.org/10.1523/JNEUROSCI.3635-05.2005
Wu G, Lu ZH, Kulkarni N, Ledeen RW (2012) Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J Neurosci Res 90:1997–2008. https://doi.org/10.1002/jnr.23090
Yamamoto N, Matsubara T, Sato T, Yanagisawa K (2008) Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid β-protein fibrillogenesis. Biochim Biophys Acta Biomembr 1778:2717–2726. https://doi.org/10.1016/j.bbamem.2008.07.028
Yamashita T, Hashiramoto A, Haluzik M et al (2003) Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci U S A 100:3445–3449. https://doi.org/10.1073/pnas.0635898100
Yanagisawa M, Ariga T, Yu RK (2010) Cytotoxic effects of GM1 ganglioside and amyloid β-peptide on mouse embryonic neural stem cells. ASN Neuro 2:49–56. https://doi.org/10.1042/AN20090063
Yang R, Wang Q, Min L et al (2013) Monosialoanglioside improves memory deficits and relieves oxidative stress in the hippocampus of rat model of Alzheimer’s disease. Neurol Sci 34:1447–1451. https://doi.org/10.1007/s10072-012-1263-y
Yates AJ, Rampersaud A (1998) Sphingolipids as receptor modulators: an overview. Ann N Y Acad Sci 845:57–71. https://doi.org/10.1111/j.1749-6632.1998.tb09662.x
Yednak MA, Bremer EG (1994) Preferential binding of the epidermal growth factor receptor to ganglioside GM3 coated plates. Mol Chem Neuropathol 21:369–378. https://doi.org/10.1007/BF02815362
Yo S, Hamamura K, Mishima Y et al (2019) Deficiency of GD3 synthase in mice resulting in the attenuation of bone loss with aging. Int J Mol Sci 20. https://doi.org/10.3390/ijms20112825
Yoon SJ, Nakayama KI, Hikita T et al (2006a) Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc Natl Acad Sci U S A 103:18987–18991. https://doi.org/10.1073/pnas.0609281103
Yoon SJ, Nakayama KI, Takahashi N et al (2006b) Interaction of N-linked glycans, having multivalent GlcNAc termini, with GM3 ganglioside. Glycoconj J 23:639–649. https://doi.org/10.1007/s10719-006-9001-4
Yoshizaki F, Nakayama H, Iwahara C et al (2008) Role of glycosphingolipid-enriched microdomains in innate immunity: microdomain-dependent phagocytic cell functions. Biochim Biophys Acta Gen Subj 1780:383–392
Yuyama K, Sun H, Sakai S et al (2014) Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J Biol Chem 289:24488–24498. https://doi.org/10.1074/jbc.M114.577213
Zesiewicz TA (2019) Parkinson disease. Contin Lifelong Learn Neurol 25:896–918
Zhang X, Kiechle FL (2004) Review: glycosphingolipids in health and disease. Ann Clin Lab Sci 34:3–13
Zhou Q, Hakomori S, Kitamura K, Igarashi Y (1994) GM3 directly inhibits tyrosine phosphorylation and de-N-acetyl-GM3 directly enhances serine phosphorylation of epidermal growth factor receptor, independently of receptor-receptor interaction. J Biol Chem 269:1959–1965
Zhu Y, Yang J, Jiao S, Ji T (2013) Ganglioside-monosialic acid (GM1) prevents oxaliplatin-induced peripheral neurotoxicity in patients with gastrointestinal tumors. World J Surg Oncol 11. https://doi.org/10.1186/1477-7819-11-19
Zunke F, Moise AC, Belur NR, et al (2018) Reversible conformational conversion of α-Synuclein into toxic assemblies by glucosylceramide. Neuron 97:92–107.e10. https://doi.org/10.1016/j.neuron.2017.12.012
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Chiricozzi, E. et al. (2021). Glycosphingolipids. In: Lauc, G., Trbojević-Akmačić, I. (eds) The Role of Glycosylation in Health and Disease. Advances in Experimental Medicine and Biology, vol 1325. Springer, Cham. https://doi.org/10.1007/978-3-030-70115-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-70115-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-70114-7
Online ISBN: 978-3-030-70115-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)