
Abstract
The neutral glycosphingolipid lactosylceramide (LacCer) forms lipid rafts (membrane microdomains) coupled with the Src family kinase Lyn on the plasma membranes of human neutrophils; ligand binding to LacCer activates Lyn, resulting in neutrophil functions, such as superoxide generation and migration (Iwabuchi and Nagaoka, Lactosylceramide-enriched glycosphingolipid signaling domain mediates superoxide generation from human neutrophils, Blood 100, 1454–1464, 2002 and Sato et al. Induction of human neutrophil chemotaxis by Candida albicans-derived beta-1,6-long glycoside side-chain-branched beta glycan, J. Leukoc. Biol. 84, 204–211, 2006). Neutrophilic differentiated HL-60 cells (D-HL-60 cells) express almost the same amount of LacCer as neutrophils. However, D-HL-60 cells do not have Lyn-associated LacCer-enriched lipid rafts and lack LacCer-mediated superoxide-generating and migrating abilities. Here, we examined the roles of LacCer molecular species of different fatty acid compositions in these processes. Liquid chromatography-mass spectrometry analyses revealed that the very long fatty acid C24:0 and C24:1 chains were the main components of LacCer (31.6% on the total fatty acid content) in the detergent-resistant membrane fraction (DRM) from neutrophil plasma membranes. In contrast, plasma membrane DRM of D-HL-60 cells included over 70% C16:0-LacCer, but only 13.6% C24-LacCer species. D-HL-60 cells loaded with C24:0 or C24:1-LacCer acquired LacCer-mediated migrating and superoxide-generating abilities, and allowed Lyn coimmunoprecipitation by anti-LacCer antibody. Lyn knockdown by siRNA completely abolished the effect of C24:1-LacCer loading on LacCer-mediated migration of D-HL-60 cells. Immunoelectron microscopy revealed that LacCer clusters were closely associated with Lyn molecules in neutrophils and C24:1-LacCer-loaded D-HL-60 cells, but not in D-HL-60 cells or C16:0-LacCer-loaded cells. Taken together, these observations suggest that LacCer species with very long fatty acids are specifically necessary for Lyn-coupled LacCer-enriched lipid raft-mediated neutrophil superoxide generation and migration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Glycosphingolipids (GSLs) are membrane components consisting of hydrophobic ceramide and hydrophilic sugar moieties [1]. More than 400 species of GSL have been identified to date based on their sugar chain structures [2]. Furthermore, the ceramide structures of each GSL are also highly variable [3]. GSL metabolism and composition are specifically altered during cell proliferation and differentiation in various cell types [2, 4], indicating that expression patterns of GSLs may reflect the functions of those cells. For example, lactosylceramide (LacCer, CDw17; Galβ4Glcβ1Cer) is not expressed on the surface of human metamyelocytes, but is expressed on the plasma membrane of neutrophils with differentiation [5]. Essential functions of GSLs, defining cellular phenotype, are based on their clustering with signal transducers to form lipid rafts (membrane microdomains) [6–9]. Lipid rafts are subdomains of the plasma membrane highly enriched in GSLs, sphingomyelin, glycosylphosphatidylinositol (GPI)-anchored proteins and cholesterol [6, 10]. Several types of membrane-anchored protein partition into lipid rafts [2, 11]. Although lipid rafts have been implicated in a number of important membrane events [2, 8, 12, 13], the molecular mechanisms of GSL-mediated cell functions are still unclear. One of the main issues regards the molecular connection of GSLs or GPI-anchored proteins, inserted into the external leaflet of the membrane bilayer, with signal transduction molecules located at the cytoplasmic side.
LacCer is a neutral GSL, and is associated with a number of key cellular processes. It has been demonstrated that LacCer activates NADPH oxidase to modulate intercellular adhesion molecule-1 expression on human umbilical vein endothelial cells [14], and to induce the proliferation of human aortic smooth muscle cells [15]. Therefore, it is possible that LacCer activates NADPH oxidase, and thereby affects the functions of superoxide-producing cells. LacCer is also a receptor activator of NF-κB ligand and is essential for osteoclastogenesis mediated by macrophage colony stimulation factor [16]. Recently, LacCer has been shown to recruit PCKα/ε and phospholipase A2 to stimulate PECAM-1 expression in human monocytes and adhesion to endothelial cells [17] and to regulate β1-integrin clustering and endocytosis on cell surfaces [18]. LacCer has been shown to bind specifically to several types of pathogenic microorganism, including Escherichia coli, Bordetella pertussis, Bacillus dysenteriae, Propionibacterium freudenreichii, and Candida albicans [19–25], suggesting that LacCer plays roles in the interactions between these microorganisms and host cells.
Neutrophils play important roles in innate immunity. On infection with pathogenic microorganisms, circulating neutrophils immediately migrate toward and phagocytose and kill the microorganisms with microbicidal molecules and by superoxide generation [26]. LacCer accounts for about 70% of the GSL in human neutrophils. These immune cells migrate toward C. Albicans-derived β-glucan (CSBG) [23], and generate superoxide anions via LacCer [12]. Therefore, LacCer is thought to be involved in neutrophil microbicidal functions. On neutrophil plasma membranes, LacCer forms lipid rafts with the Src family kinase Lyn; these rafts are also called LacCer-enriched glycosignaling domains [12]. Dimethyl sulfoxide (DMSO)-treated neutrophilic differentiated human promyelocytic leukemia HL-60 cells (D-HL-60 cells) possess chemotactic and superoxide-generating activities induced by formyl peptide fMLP. Interestingly, D-HL-60 cells do not show superoxide-generating or chemotactic activity induced by anti-LacCer antibodies, although these cells express almost the same amount of LacCer on the plasma membrane as neutrophils. LacCer is localized mainly in the detergent-resistant membrane fractions (DRMs) [12, 27, 28], which are membrane fragments isolated biochemically at low temperature from cellular membranes using non-ionic detergents, such as Triton X-100; there is thought to be a close relationship between rafts and DRMs, and isolation of DRMs is one of the most widely used biochemical methods for studying lipid rafts [6]. Importantly, the Src family kinase Lyn was immunoprecipitated by anti-LacCer antibody from DRMs of neutrophils but not D-HL-60 cells. Furthermore, Lyn was activated by crosslinking of LacCer with anti-LacCer antibody in neutrophils but not D-HL-60 cells. These observations suggested that some essential molecule(s) linking LacCer with Lyn may be lacking in the LacCer-enriched lipid rafts of D-HL-60 cells.
In the present study, we found that C24:0- and C24:1-fatty acid-containing LacCers are the major species of plasma membrane DRM of neutrophils and are minor species of those of D-HL-60 cells. Although exogenously loaded C24:0-, C24:1-, C22:0-, and C16:0-LacCer were incorporated into plasma membrane DRM of D-HL-60 cells, only C24:0- and C24:1-LacCer-loaded D-HL-60 cells showed LacCer-mediated neutrophil superoxide generation and migration. In addition, Lyn activation by LacCer crosslinking and Lyn-coimmunoprecipitation with anti-LacCer antibody were observed only in the C24:0- and C24:1-LacCer-loaded D-HL-60 cells. These observations suggest that C24 fatty acid chains of LacCer play an important role in the formation of LacCer-enriched lipid rafts coupled with Lyn as functional lipid domains responsible for chemotaxis and superoxide generation.
2 Materials and methods
2.1 Antibodies and reagents
Mouse anti-LacCer monoclonal IgMs T5A7 [12] and Huly-m13 (Ancell, Bayport, MN) were used. Mouse anti-Lyn monoclonal IgG and anti-Lyn polyclonal rabbit IgG used here were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-β-actin IgG was from Sigma-Aldrich (St. Louis, MO). Normal mouse IgM and rabbit IgG were obtained from Zymed (San Francisco, CA). Bovine and porcine LacCers (Galβ1-4Glc-ceramides) were from Matreya (Pleasant Gap, PA). Candida albicans-derived-βglucan (CSBG) and Sparassis crispa-derived β-glucan (SCG), isolated as described previously [23], were kindly provided by Drs. Adachi and Ohno (Tokyo University of Pharmacy and Life Science) and Dr. Tamura (Seikagaku Corporation), respectively. Dimethylsulfoxide (DMSO) and N-formylmethionine-leucine-phenylalanine (fMLP) were purchased from Sigma-Aldrich Japan (Tokyo, Japan).
2.2 Synthesis of GSLs and [1-3H]sphingosine
C16:0-(palmitic acid), C22:0-(behenic acid), C24:0-(lignoceric acid), and C24:1 (nervonic acid; C24:1ω9)-LacCer were synthesized from the corresponding lyso compounds. LacCer was prepared by acid hydrolysis of GM1, which was isolated from bovine brain [29]. C16:0- and C22:0-compounds were prepared using commercial C16:0 and C22:0 anhydrides (Sigma-Aldrich). C24:0- and C24:1-compounds were prepared using pentafluorophenol-activated C24:0 and C24:1 fatty acids. LC-MS and NMR analysis indicated that the synthesized compounds used in this study were pure [30, 31]. [1-3H]sphingosine was prepared by specific chemical oxidation of the primary hydroxyl group of sphingosine followed by reduction with sodium boro-[3H]-hydride [32].
2.3 Cell culture and loading with LacCers
Human neutrophils were isolated from heparinized peripheral blood of normal volunteers by PolymorphprepTM (Nycomed Pharma AS, Oslo, Norway) centrifugation, and suspended at 2 × 107 cells/ml in DMEM/F12 medium. The purity of human neutrophils was >95% as estimated by Wright–Giemsa staining.
HL-60 cells were maintained in RPMI1640 medium supplemented with 10% FBS. To induce differentiation into neutrophilic lineage cells, HL-60 cells were cultured with 1.3% DMSO for 7 days [12]. In the case of the metabolic labeling of sphingolipids, DMSO-treated HL-60 cells (D-HL-60, 2 × 107 cells/ml) on day 5 were incubated in RPMI-1640 + 10% FBS for 2 h in the presence of 0.8 nM [1-3H]sphingosine. After washing, cells were further cultured for 48 h in the presence of 1.3% DMSO.
For loading of D-HL-60 cells with LacCers, aliquots of 10 ml of D-HL-60 cells (2 × 107cells/ml) in Dulbecco’s phosphate buffered saline (PBS) were mixed with 10 μl of 0.01–10 mg/ml GSLs in DMSO (final concentration: 0.01–10 μg/ml) for 30 min at 20°C with gentle shaking (30 strokes/min). After incubation, cells were washed twice with 10 ml of PBS + 0.1% BSA to remove non-incorporated or attached LacCers, and then used immediately for analysis.
2.4 Transfection with siRNA
After 5 days of treatment with DMSO, D-HL-60 cells (4 × 106 cells) were transfected with short interfering RNA (siRNA) for human Lyn (siGenome SMARTpool reagent; Dharmacon, Lafayette, CO) and non-targeting siRNA (control siRNA) at a final concentration of 250 nM using an Amaxa Nucleofector with a Nucleofector Kit V in accordance with the manufacturer’s protocol with slight modifications (Amaxa Biosystems, Cologne, Germany). After transfection with siRNA, cells were cultured for 72 h in RPMI1640 medium supplemented with 10% FBS. The cells were then collected and suspended in PBS, and then incubated with C24:1-LacCer for 30 min at 20°C. siRNA-transfected C24:1-LacCer-loaded D-HL-60 cells were subjected to migration assay.
2.5 Molecular species analysis of lipids in DRMs
To isolate plasma membrane and granular DRM, cells (2 × 107 cells/ml) were disrupted by the N2-cavitation method [12], and the plasma membrane and granule fractions were isolated from the cavitates by the differential centrifugation method [33]. Briefly, cells were suspended in relaxation buffer (100 mM KCl, 3mM NaCl, 3.5 mM MgCl2, 1mM ATP-2Na, 10 mM PIPES, pH 7.3) at 2 × 107cells/ml, and disrupted by N2 gas cavitation for 20 min at 350 psi on ice. The cavitated fraction was dropped into EGTA solution (final concentration: 1.25 mM, pH 7.4). The cell homogenates were centrifuged at 500×g for 10 min at 4°C, and the supernatants thus obtained were centrifuged at 8,500×g for 20 min at 4°C to isolate granules (granule fraction). The supernatants thus obtained were centrifuged at 200,000×g for 60 min at 4°C to isolate plasma membranes (plasma membrane fraction). The isolated fractions were resuspended in 1 ml of TNE-T buffer (1% Triton X-100, 10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM DFP, 1/20 v/v Complete) and allowed to stand on ice for 20 min, and then homogenized by 17 strokes in a Dounce homogenizer. The homogenates were mixed with an equal volume of 85% sucrose (w/v) containing 10 mM Tris buffer (pH 7.5), 150 mM NaCl, 5 mM EDTA (Tris–NaCl-EDTA buffer). The resulting diluent was placed at the bottom of a discontinuous sucrose concentration gradient (30–5%) in the same buffer, and then centrifuged for 17 h at 200,000×g at 4°C. After centrifugation, each 1-ml fraction from the top was collected. The LacCer-enriched DRM fraction was isolated from DRM of D-HL-60 cells using a combination of anti-LacCer antibody Huly-m13 and rat anti-mouse IgM-bound Dynabeads (Invitrogen Japan, Tokyo, Japan) as described [12].
Lipid components were extracted with chloroform/methanol (2/1 v/v) solution, and then analyzed by single or two-dimensional high-performance thin-layer chromatography (HPTLC) [12]. The migrated lipids were visualized by primulin spray. In some experiments, to remove glycerophospholipids, lipid extracts were treated with 0.1 M sodium hydroxide in methanol at 40°C for 2 h before HPTLC analysis. Molecular species of LacCer extracted from HPTLC plate were identified by reverse-phase liquid chromatography/electrospray ion trap mass spectrometry (LC-ESI-MSn) analysis as described previously [3]. The LacCer contents of each LacCer band were quantified by densitometric scanning of migrated bands on HPTLC plates using a VersaDoc™ Model 3000 Imaging System (Bio-Rad Japan, Tokyo Japan). Compositions of the recovered LacCer added to the plasma membrane DRM fractions were measured by HPTLC analysis as described above. The abundance of LacCer species in each sample was calculated from the peak area ratios on the LC chart on LC-ESI-MS analysis.
Molecular species of sphingomyelin (SM) were confirmed by LC-ESI-MS/MS analysis. LC separation was performed using Capcellpak C8 UG120, 1.0 mm ID, 150 mm reverse phase column (Shiseido Fine Chemicals, Tokyo, Japan). The mobile phases were as follows: solvent A was 1 mM ammonium acetate in methanol:water (76:24 v/v) and solvent B was 1 mM ammonium acetate in methanol:water (96:4 v/v). The elution program was isocratic elution with 80% B for 5 min, increase to 95% B, and then hold for 30 min. The flow rate was 50 μl/min. The HPLC was connected to a mass spectrometer (TSQ Quantum Ultra AM; ThermoFisher Scientific, San Jose, CA). The MS conditions were as follows: ion transfer tube temperature, 350°C; spray voltage, 4.5 kV; sheath gas pressure, 20 U. The collision gas used for MS/MS experiments was argon at 0.8 mTorr and the collision energy was set to 15 eV plus 0.05 eV per m/z.
Molecular species of phosphatidylcholine (PC) were confirmed by LC-ESI-MS/MS analysis with both positive and negative mode using an LCQ DECA XP ion trap mass spectrometer (ThermoFisher Scientific). Other conditions were the same as described for LacCer. Fatty acyl compositions of PCs were confirmed from MSn spectra.
2.6 Superoxide generation assay
Superoxide generation from D-HL-60 cells was assayed by superoxide dismutase-inhibitable cytochrome c reduction, as described [12]. Aliquots of 100 μl of the loaded or non-loaded D-HL-60 cells (2.5 × 105 cells/100 μl) in phenol red-free RPMI-1640 medium containing 1 mg/ml BSA were incubated with or without 50 μg/ml superoxide dismutase at 37°C for 30 min in 96-well plates, which were pre-coated with anti-LacCer IgM T5A7 or normal IgM. In some experiments, the assay was carried out in the presence of 10 μM PP1. To measure the effect of synthesized LacCer on superoxide generation from cells, non-loaded cells were incubated with several concentrations of synthesized LacCer bovine serum albumin-coated wells at 37°C for 30 min. As positive and negative controls for superoxide generation, non-loaded cells were incubated with 10−7 M fMLP (fMLP) or 0.1% DMSO (vehicle) in bovine serum albumin-coated wells at 37°C for 30 min. The absorbance at 550 nm of the supernatants was measured with a Beckman DU640 spectrophotometer (Beckman Instruments, Fullerton, CA), and the value of cytochrome c reduction was calculated using the formula: E 550 = 2.1 × 104 M−1cm−1 [34].
2.7 Migration assay
Cell migration was assayed using a modified Boyden chamber with cellulose nitrate filters with a pore size of 3 μm (Sartorius, Göttingen, Germany), as described [23]. Briefly, several concentrations of β-glucans and antibodies in RPMI-1640 medium supplemented with 1 mg/ml bovine serum albumin (BSA) were placed into the lower compartment of the chamber. Aliquots of 300 μl of cell suspensions (2.5 × 106 cells/ml) in RPMI-1640 medium containing 1 mg/ml BSA were placed into the upper compartment, and then incubated for 30 min at 37°C in a CO2 incubator. After incubation, the filters were fixed with neutral buffered formalin for 20 min, and then stained with Mayer’s hematoxylin. The distance (μm) from the top of the filter to the furthest two cells in the same focal plane was measured microscopically with a 40× objective in 20 fields across the filter, and the results were expressed as the migration index (relative neutrophil migration: average distance of tested group/average distance of corresponding vehicle control). In each assay, fMLP at a concentration of 50 nM was used as a positive reference chemoattractant.
Migration of siRNA-transfected cells was measured by a modification of the method of Boyden using a 48-well microchemotaxis chamber with a polyvinylpyrrolidone-free polycarbonate membrane (pore size: 3 mm; Neuroprobe, Cabin John, MD) in accordance with the manufacturer’s instructions [35]. Chemoattractants or DMEM/F12 medium (22 μl) were added to the lower compartment. The cells were placed in the upper compartment of the chamber (50 μl of a suspension of 106 cells/ml) and allowed to migrate for 1 h at 37°C in a 5% CO2 atmosphere. At the end of the incubation period, the top of the chamber was removed, and the upper side of the membrane was wiped carefully with the rubber scraper provided by the manufacturer. The polycarbonate membrane was fixed in methanol, colored with a Diff-Quick staining kit, mounted on a glass slide, and examined with a light microscope under 400× magnification. The numbers of cells in ten random fields were counted, and the results were expressed as the migration index (relative neutrophil migration: number of cells from tested group/number of cells from corresponding vehicle control). Treatment with fMLP (50 nM) was used as a positive control.
2.8 Assay for Lyn activity in anti-LacCer antibody-treated D-HL-60 cells
Anti-LacCer antibody-induced Lyn activities were assayed essentially as described but with slight modifications [12]. After loading D-HL-60 cells with 0.5 μg/ml C16:0-, C22:0-, C24:0-, C24:1-LacCer, or 0.1% DMSO (vehicle), 5 ml of 2.5 × 106 cells/ml loaded cells in RPMI-1640 medium were incubated in anti-LacCer antibody T5A7-coated 10-cm dishes without or with 10 μM PP1 at 37°C for 5 min. The reaction was terminated by placing the dishes on ice. Then, the cells were lysed in lysis buffer [50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM NaF, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 1/20 v/v Complete™ and 1% Triton X-100], and sonicated for 10 s with 10% due using an ultrasonic disruptor UD-201 (Tominaga Works Ltd., Tokyo, Japan). After centrifugation at 440×g for 5 min, the supernatant was recovered, and pre-cleared by incubation with 50 μl of protein G-Sepharose. Then, the supernatant was incubated with mouse monoclonal anti-Lyn IgG H2 (1 μg IgG/ ml) overnight at 4°C, followed by incubation with 50 μl of protein G-Sepharose beads for 2 h. The immunoprecipitated beads were washed 3 times with lysis buffer B and twice with kinase buffer (30 mM HEPES, pH 7.5, 10 mM MgCl2, 2 mM MnCl2 1 mM CaCl2). The immunoprecipitates were incubated at 37°C for 5 min with 10 μM ATP and 5 μCi of [γ-32P] ATP (3,000 Ci/mmol; NENTM Life Science Products, Boston, MA) in kinase buffer. Samples were placed on ice, centrifuged at 440×g for 5 min, and the Sepharose beads were then boiled with SDS-Sample buffer containing 5% 2-ME. The samples were subjected to 7.5% SDS-PAGE, and blotted onto PVDF membranes. Autoradiography was carried out by exposing the electroblotted membranes to Fuji X-ray film at −80°C with intensifying screens (Dupont Lightning Plus; Dupont, Wilmington, DE). After autoradiography, the blotted membranes were probed with rabbit anti-Lyn IgG, and Lyn was detected using SuperSignal™ reagent.
2.9 Immunoelectron microscopic observation
Immunogold labeling was performed as described with some modifications [36]. Briefly, D-HL-60 cells were incubated with 0.5 μg/ml C16:0- or C24:1-LacCer in DMSO or 0.1% DMSO as a vehicle control for 30 min at 20°C, and then washed with PBS containing 0.1% BSA. The loaded cells and neutrophils were fixed with periodate-lysine paraformaldehyde (PLP) fixative (2% paraformaldehyde 0.1 M lysine in 50 mM sodium phosphate buffer, pH 7.4) for 1 h on ice, infiltrated with 40% polyvinylpyrrolidone (Sigma-Aldrich)/2.3M sucrose/phosphate buffer, embedded, and frozen in liquid nitrogen. Ultrathin cryosections were cut with an Ultracut UCT microtome equipped with the FC-4E cryoattachment (Leica, Wetzlar, Germany) at −110°C and transferred onto nickel grids (150 mesh), blocked, and then incubated with appropriate primary Abs at 4°C overnight. They were then incubated with 5 nm gold-conjugated Goat F(ab′)2 anti-Mouse IgM and 10 nm gold-conjugated Goat F(ab′)2 anti-rabbit IgG (British BioCell, Cardiff, UK) for 2 h. They were re-fixed with 2.5% glutaraldehyde/PB, contrasted with 2% uranyl acetate solution for 30 min, and absorption-stained with 2% polyvinyl alcohol (Sigma-Aldrich) containing 0.2% methylcellulose (Nakalai Tesque, Kyoto, Japan) and 0.2% uranyl acetate for 30 min. The samples were observed with a JEOL 1230 electron microscope (JEOL, Tokyo, Japan). The length of an immunoglobulin molecule has been estimated to be about 10 nm for IgG and 20–30 nm for IgM [37–40]. Thus, we defined a cluster as two 5-nm gold beads at a distance of less than 30 nm. If the distance between 5- and 10-nm gold beads was less than 50 nm, this was taken to indicate that LacCer was associated with Lyn.
2.10 Immunostaining
To examine the effects of anti-LacCer mAb T5A7 on cluster formation of LacCer on plasma membranes, cells were incubated in DMEM/F12 + 0.1% BSA with Alexa 488- or 594-conjugated anti-LacCer antibody T5A7 on ice or 37°C for 5 min, followed by a further incubation for 25 min on ice. In some experiments, cells were incubated at 37°C for 5 min in the presence of 10 μM PP1, and then incubated for a further 25 min on ice. After three washes with ice-cold PBS, cells were fixed with 2% formaldehyde, and then examined with a Zeiss LSM 510 confocal microscope equipped with a Plan-Apochromat ×100 oil DIC objective. The three-dimensional reconstructing images were made using IMARIS software (Bitplane, Zurich, Switzerland).
Expression of LacCer on D-HL-60 cells was measured by flow cytometry using FACSCalibur (BD Biosciences, San Jose, CA) as described [12].
2.11 Statistical analysis
Data are expressed as the means±SD, and were analyzed for significant differences by one-way ANOVA and post hoc test using GraphPad PRISM (GraphPad Software Inc. San Diego, CA). Differences were considered statistically significant at P < 0.05.
3 Results
3.1 Lipid compositions and molecular species of LacCer in plasma membrane DRM of neutrophils and D-HL-60 cells
To compare the lipid compositions of neutrophils and D-HL-60 cells, we performed HPTLC analysis of plasma membrane DRMs from these cells. LacCer of DRM isolated from neutrophil plasma membrane and D-HL-60 cell granules migrated as two bands on HPTLC (Fig. 1a) due to heterogeneity of the ceramide moiety [41]. However, the LacCer upper band of the plasma membrane fraction from D-HL-60 cells was almost undetectable (Fig. 1a and b). Consistent with the data reported by other groups [42, 43], the amount of LacCer in the plasma membrane fraction of neutrophils was 3.7 ± 0.17 (μg/108 cells, mean±SD of three experiments). The amount of LacCer in the D-HL-60 cell plasma membrane was 3.02 ± 0.21 μg /108 cells. Under our experimental conditions, most of the LacCer of neutrophils and D-HL-60 cells was recovered in the DRM fraction [12]. Densitometric analysis revealed that the upper band of LacCer in the plasma membrane fraction accounted for 55.9 ± 8.6% (mean±SD of three experiments) and 9.5 ± 0.9% of total LacCer density in neutrophils and D-HL-60 cells, respectively. LC-ESI-MSn analysis revealed that the upper band of LacCer of neutrophils and D-HL-60 cells was mainly composed of C24:0- and C24:1-LacCer, while the lower band contained LacCer species with shorter fatty acids (Fig. 1c). Based on the peak area ratio of LacCer species on LC-MS/MS analysis, the main plasma membrane LacCer species of neutrophils were C16:0- (22.6% of total, mean of three independent experiments), C24:1- (19.6%), C24:0- (12.0%), and C22:0-LacCer (8.0%), while those of D-HL-60 were C16:0- (70.1% of total), C24:1- (8.2%), and C24:0-LacCer (5.4%) (Fig. 1c). Thus, the most abundant species in D-HL-60 cells is the C16-species, while that in neutrophils is the C24-species. HL-60 cells seem to mainly produce C16:0-LacCer instead of C22 and longer fatty acid-containing LacCers.

Distribution of molecular species of LacCer derived from the plasma membrane and granular DRMs of neutrophils and D-HL-60 cells. a HPTLC analysis of plasma membrane and granular DRMs of D-HL-60 cells and neutrophils. Neutrophils and D-HL-60 cells were disrupted by nitrogen cavitation, and the plasma membrane and granular DRMs were isolated from the cavitates. The lipids, which were extracted with chloroform/methanol 2:1 (v/v) from the plasma membrane and granular fractions of D-HL-60 cells (2 × 108 cell equivalents), and the plasma membrane fractions of neutrophils (3 × 108 cell equivalents), were separated by high-performance thin-layer chromatography with chloroform/methanol/water 65:25:4 (v/v/v). The migrated lipids were visualized by primulin spray. b LacCer derived from the plasma membrane and granular DRMs of neutrophils and D-HL-60 cells. The lipids, which were extracted with chloroform/methanol 2:1 (v/v) from the plasma membrane and granular fractions of neutrophils and D-HL-60 cells (3 × 108 cell equivalents), were treated with 0.1 N NaOH in methanol, and then separated by high-performance thin-layer chromatography with chloroform/methanol/water 65:25:4 (v/v/v). The migrated lipids were visualized by primulin spray. c The abundance of LacCer species derived from plasma membrane DRM of neutrophils (open bars) and D-HL-60 cells (closed bars). LacCer bands isolated by HPTLC shown in (a) were recovered by scraping, LacCer was extracted with chloroform/methanol (2:1 v/v), followed by LC-ESI-MSn. The abundance of LacCer species was calculated from the peak area ratios on the LC chart of LC-ESI-MS analysis. Each bar shows the percentage (mean of three independent experiments) of each LacCer molecular species in total LacCer. Upper band, abundance of LacCer species in the upper band as a percentage of the total LacCer species; Lower band, abundance of LacCer species in the lower band as a percentage of total LacCer species; Total, the amount of each LacCer species as a percentage of the total LacCer species. a, Mixture of LacCer(d18:2/C16:0) and LacCer(d18:1/C16:1). Others, the sum of LacCer species, which were less than 1% of total LacCer intensity
The molecular species of sphingomyelin and phosphatidylcholine in the plasma membrane DRMs were not significantly different between neutrophils and D-HL-60 cells (Fig. 2).

Distribution of molecular species of sphingomyelin and phosphatidylcholine derived from plasma membrane DRM of neutrophils and D-HL-60 cells. a Plasma membrane and granular DRM of neutrophils and D-HL-60 cells were isolated using the same method described in Fig. 1a. SM was recovered by scraping gels of the HPTLC plates. The isolated SM was recovered by scraping gels and extracted with chloroform/methanol 2:1 (v/v) solution, followed by analysis of molecular species of SM by LC-ESI-MSn. * m/z 705.6, which was not identified by MS/MS analysis, was predicted as d18:0/C16:0 SM from its molecular mass and the retention time on HPLC. These panels show the representative data set of three independent experiments. b Lipid components of DRM were extracted with chloroform/methanol 2:1 (v/v) solution and then separated by HPTLC with chloroform/methanol/water 65:25:4 (v/v/v). The isolated PC was recovered by scraping gels and extracted with chloroform/methanol 2:1 (v/v) solution, followed by analysis of molecular species of PC by LC-ESI-MSn. These panels show the representative data set of three independent experiments
3.2 Effects of LacCer loading on D-HL-60 cell functions
To understand the possible role of the LacCer species structure, specifically that of C24-species, in the association of Lyn with LacCer-enriched lipid rafts, D-HL-60 cells were loaded with LacCer mixtures from different natural sources or synthetic LacCer species with homogeneous ceramide moieties. As mixtures, we used LacCer derived from bovine milk, which contained 77% C22- or shorter fatty acid chains, and porcine blood cell-derived LacCer, which contained 84% C24- and longer fatty acid chains (Fig. 3a). As shown in Fig. 3b, loading of porcine LacCer onto D-HL-60 cells elicited anti-LacCer antibody T5A7-induced superoxide generation in a dose-dependent manner with a bell-shaped dose response curve, whereas bovine LacCer was ineffective in the induction of superoxide generation by T5A7. These observations suggest that LacCers containing C24 and longer fatty acid chains may be involved in LacCer-meditated superoxide generation from neutrophils.

Effects of natural LacCer loading on superoxide generation of D-HL-60 cells. a The molecular species of porcine blood cell- and bovine milk-derived LacCer. Porcine blood cell (closed bars) and bovine milk (open bars)-derived LacCer were subjected to LC-ESI-MSn analysis as described in Materials and methods. The amounts of each LacCer molecule are shown as the percentages of total LacCer intensity. a, m/z: 988 was indicated to be LacCer (d18:1/C25:0 and d19:1/C24:0). b m/z: 1004 was not identified by MSn analysis, but it was thought to be LacCer (d18:1/Ch25:0) from its molecular mass and the retention time on HPLC. b. D-HL-60 cells were incubated with porcine blood (closed circle) or bovine milk (open circle)-derived LacCer at several concentrations at 20°C for 30 min. fMLP, 10−7 M fMLP; Resting, 0.1% DMSO. Data show the means±SD of three independent experiments. **P < 0.01, ***P < 0.001
Lipid rafts are characterized by liquid-ordered membrane regions, where lipid components are more tightly packed than in the surrounding lipid bilayer [4, 44]. As Lyn is myristoylated and palmitoylated at two N-terminal amino acids [45], LacCer containing C24 and longer fatty acid chains may be associated with Lyn molecules via the fatty acid chains in the neutrophil plasma membrane. To better evaluate the effects of fatty acid length, we loaded neutrophils with synthetic LacCer species containing C16:0, C22:0, C24:0, or C24:1 acyl chains (Fig. 4a), and examined the effects due to incorporation of the added species into the membrane. As shown in Fig. 4b, LacCer-loading per se did not significantly enhance superoxide generation by D-HL-60 cells. In contrast, the anti-LacCer antibody T5A7 significantly induced superoxide generation from C24:0- and C24:1-LacCer-loaded D-HL-60 cells with a bell-shaped dose-response curve (Fig. 4c). However, loading with C16:0- or C22:0-LacCer did not induce LacCer-mediated superoxide generation.

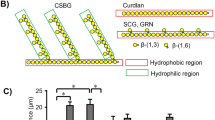
Effects of synthesized LacCer loading on neutrophil functions. a HPTLC analysis of the synthesized LacCer. Synthesized LacCers were analyzed by HPTLC with primulin spray. b Effects of synthesized LacCer on superoxide generation from D-HL-60 cells. Various amounts of C16:0- (closed square), C22:0- (open square), C24:0- (closed triangle), and C24:1- (open triangle) LacCer were incubated with D-HL-60 cells at 37°C for 30 min. fMLP, 10−7 M fMLP; Resting, 0.1% DMSO. Data show the means±SD of 4 independent experiments. c Effects of anti-LacCer antibody on superoxide generation from synthesized LacCer-loaded D-HL-60 cells. Various amounts of C16:0- (closed square), C22:0- (open square), C24:0- (closed triangle), and C24:1- (open triangle) LacCer were loaded onto D-HL-60 cells at 20°C for 30 min. Then the superoxide generating assay was performed as described in the Materials and methods section. fMLP, 10−7 M fMLP; Resting, 0.1% DMSO. Data show the means±SD of four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. d Effects of LacCer loading on β-glucan and anti-LacCer antibody-induced migration of D-HL-60 cells. After loading of D-HL-60 cells, β-glucans or antibodies were assessed for chemotactic activities against the loaded cells by the Boyden chamber method. Resting, 0.1% DMSO. CSBG, 10 μg/ml Candida albicans-derived β-glucan. SCG, 10 μg/ml Sparassis crispa-derived β-glucan. Anti-LacCer IgM, 1 μg/ml T5A7. fMLP, 50 nM fMLP. Each bar shows the mean±SD of five independent experiments. ***P < 0.001
To evaluate the possible role of LacCer species in modulating signaling processes, we examined the effects of LacCer loading on neutrophil migration. Chemotactic migration toward β-glucan is another LacCer-mediated neutrophil function [23]. CSBG, a β-1,3-d-glucan with long 1,6-glucosyl side chain, binds specifically to LacCer, and induces chemotaxis of human neutrophils but not D-HL-60 cells. In contrast, Sparassis crispa-derived β-glucan (SCG), a 1,6-monoglucosyl-branched 1,3- β-d-glucan, cannot induce neutrophil or D-HL-60 cell chemotaxis. As shown in Fig. 5c, C24:0- and C24:1-LacCer-loaded D-HL-60 cells migrated toward not only T5A7 but also CSBG, whereas these loaded cells did not migrate toward SCG or normal IgM. In the case of D-HL-60 cells loaded with C16:0- and C22:0-LacCer, neither T5A7 nor CSBG induced migration. Thus, loading of D-HL-60 cells with C24 fatty acid-containing LacCer is essential for CSBG-induced chemotactic migration.

Lipid composition analysis of plasma membrane DRM fractions of synthetic LacCer-loaded D-HL-60 cells. a LacCer in DRM fractions separated by sucrose density gradient centrifugation. DRMs were isolated from the plasma membranes of LacCer-loaded cells, and lipids in each DRM fraction were analyzed with primulin spray. b Two-dimensional HPTLC analysis of LacCer-enriched DRM. D-HL-60 cells were metabolically labeled with [1-3H]-sphingosine. Plasma membrane DRMs were immunoprecipitated with anti-LacCer antibody Huly-m13. Lipids were extracted from the immunoprecipitates and analyzed by two-dimensional HPTLC. Plates were sprayed with primulin to visualize all lipids, while radioactive sphingolipids were visualized by autoradiography (autoradiogram). PE phosphatidylethanolamine, PC phosphatidylcholine, SM sphingomyelin, Chol cholesterol. Arrows indicate the migrated LacCer
Loading of homogeneous species allowed us to obtain detailed biochemical information on the new lipid raft organization. We analyzed the LacCer content and pattern in the plasma membrane DRM from C16:0-, C22:0-, C24:0- and C24:1-LacCers loaded D-HL-60 cells. The LacCer content in the plasma membrane DRM after loading cells with these LacCer species at 0.54 μM were 3.07 ± 0.36, 3.30 ± 0.26, 3.40 ± 0.29 and 3.39 ± 0.27 μg/108 cells, respectively. These values are slightly, but not significantly, higher than that in the DRM from unloaded cells. In addition, we did not find LacCer in the detergent-solubilized membrane belonging to the heavy fractions separated by sucrose gradient ultracentrifugation (data not shown). This confirms previous observations on the displacement of endogenous LacCer by the exogenously added LacCer species and taken up by the cells [46, 47]. The cells were washed extensively after loading to remove all the LacCer loosely bound to the cell surface but not inserted into the membrane. To confirm that the added LacCer species became components of the plasma membranes, we performed detailed analysis of the LacCer species pattern by HPTLC. The chromatographic behavior of LacCer species is highly dependent on the length of fatty acid, as shown above (see Fig. 1). Figure 5a clearly shows that the C24:0- and C24:1-LacCer species became the main species in the plasma membrane DRM from loaded cells, and replaced the original C16:0-LacCer, the main LacCer species in D-HL-60 cells. According to the intensity of the densitometric spots in unloaded and loaded cells, we calculated that the C24-LacCer species covered over 55% of the total LacCer species in loaded cells.
To evaluate whether loading with LacCer affects the lipid composition of LacCer-enriched DRM in D-HL-60 cells, we analyzed the lipid composition of immunoprecipitates obtained from DRM using the anti-LacCer antibody Huly-m13. Two-dimensional HPTLC analysis showed that the immunoprecipitates from the DRM of loaded D-HL-60 cells were composed mainly of sphingomyelin (SM), cholesterol, phosphatidylcholine (PC), phosphatidylethanolamine (PE), and a very minor amount of LacCer, and that the lipid patterns were essentially the same as neutrophils [12] (Fig. 5b). Experiments carried out on cells metabolically labeled with tritiated sphingosine before the loading process, confirmed that LacCer loading did not affect the sphingolipid composition of the LacCer-enriched DRM of D-HL-60 cells.
3.3 Association of LacCer with Lyn
LacCer-dependent Lyn activation is necessary for the biological functions mediated via LacCer-enriched lipid rafts [12, 23]. The Src family kinase inhibitor PP1 completely inhibited anti-LacCer antibody-induced superoxide generation from D-HL-60 cells after loading with C24:0-, C24:1-, and porcine LacCer (Fig. 6a). To further substantiate the involvement of Lyn in LacCer-mediated signal transduction, we evaluated the activation of Lyn after treatment of D-HL-60 cells with the anti-LacCer antibody T5A7; D-HL-60 cells were incubated with T5A7 for 5 min, and autophosphorylation of Lyn was then measured. T5A7 treatment increased the phosphorylation of Lyn in D-HL-60 cells loaded with C24:0- and C24:1-LacCer, but not C16:0- or C22:0-LacCer (Fig. 6b). Furthermore, T5A7-induced Lyn phosphorylation of C24:1-LacCer-loaded D-HL-60 cells was inhibited by treatment with PP1. These results suggest that the presence of C24-LacCer is necessary for LacCer-mediated activation of Lyn in D-HL-60 cells. To determine whether the Lyn molecule associates with LacCer in the LacCer-loaded D-HL-60 cells, the DRM isolated from the plasma membrane fraction of D-HL-60 cells loaded with different LacCer species were immunoprecipitated with the anti-LacCer antibody Huly-m13. Notably, Lyn was detected in immunoprecipitates with anti-LacCer antibody recovered from D-HL-60 DRM loaded with C24:0-, C24:1-, and porcine LacCer, but not C16:0-, C22:0-, or bovine milk LacCer (Fig. 6c). These results suggest that C24 fatty acid chains are crucial for association of the Lyn molecule with LacCer, which is responsible for LacCer-mediated outside-in signal transduction, and this association is indispensable for LacCer-enriched lipid raft-mediated neutrophil superoxide generation and migration [12, 23]. Thus, to determine whether signaling through Lyn is critical for the LacCer-mediated functions of C24:1-LacCer-loaded D-HL-60 cells, we performed Lyn gene knockdown with short interfering RNA (siRNA) in D-HL-60 cells. Lyn siRNA, but not control siRNA, reduced the level of Lyn gene expression by about 62% (Fig. 6d). Under these conditions, C24:1-LacCer loading significantly enhanced the migration of D-HL-60 cells toward CSBG and anti-LacCer mAb T5A7 in control siRNA-transfected D-HL-60 cells (Fig. 6d). In contrast, loading with C24:1-LacCer did not enhance migration of Lyn siRNA-transfected D-HL-60 cells. These observations clearly indicated that Lyn is crucial for C24-LacCer-dependent D-HL-60 cell migration. It has been demonstrated that Src family kinases are not involved in fMLP-induced neutrophil migration [48]. Consistent with these results, C24:1-LacCer-loaded Lyn knockdown D-HL-60 cells migrated toward fMLP (Fig. 6e).

Association of LacCer with Lyn in C24-LacCer loaded D-HL-60 cells. a Effects of PP1 on anti-LacCer antibody-induced superoxide generation from LacCer-loaded D-HL-60 cells. D-HL-60 cells loaded with different LacCer species were incubated without (open bars) or with normal IgM (closed bars), anti-LacCer antibody T5A7 (hatched bars), or T5A7 in the presence of 10 μM PP1 (shaded bars). Data show the means±SD of three independent experiments. **P < 0.01, ***P < 0.001. b Induction of Lyn activation by anti-LacCer antibody. After loading of D-HL-60 cells with different LacCer species, cells were incubated on anti-LacCer antibody T5A7-coated dishes at 37°C for 5 min. After incubation, phosphorylation of Lyn was analyzed by immunoprecipitation with mouse anti-Lyn IgG, followed by SDS-PAGE and blotting onto PVDF membranes. After autoradiography (p-Lyn), the blotted membranes were probed with anti-Lyn rabbit IgG (Lyn). C24:1/PP1, C24:1-LacCer-loaded cells were incubated in the presence of 10 μM PP1. c Co-immunoprecipitation of Lyn with anti-LacCer antibody in C24:0- and C24:1-LacCer-loaded but not C16:0- or C22:0-loaded D-HL-60 cells. After loading of D-HL-60 cells with different LacCer species or 0.1% DMSO (vehicle), plasma membrane DRM were isolated, and immunoprecipitated with anti-LacCer IgM Huly-m13. The immunoprecipitates were also analyzed by SDS-PAGE/immunoblotting using rabbit anti-Lyn IgG. d, e Effects of Lyn siRNA on LacCer-mediated chemotaxis of C24:1-LacCer-loaded D-HL-60 cells. After 5 days of treatment with DMSO, HL-60 cells (4 × 106 cells) were transfected with Lyn siRNA or non-targeting siRNA (control siRNA), and cultured for a further 72 h. Total cell lysates were subjected to SDS-PAGE/immunoblotting with anti-Lyn IgG for evaluation of knockdown efficiency (d). The transfected D-HL-60 cells were loaded with 0.5 μg/ml C24:1-LacCer for 30 min at 20°C, and then migration assay was performed (e). Resting, 0.1% DMSO. CSBG, 10 μg/ml Candida albicans-derived β-glucan. SCG, 10 μg/ml Sparassis crispa-derived β-glucan. Anti-LacCer IgM, 1 μg/ml T5A7. fMLP, 50 nM fMLP. Data are presented as means±SD of three independent experiments. ***P < 0.001
3.4 Distribution of LacCer and Lyn in LacCer-loaded D-HL-60 cells
Biochemical analyses suggested that Lyn is closely associated with LacCer-enriched lipid rafts in neutrophils and C24-LacCer-loaded D-HL-60 cells but not non-loaded, C16:0-, or C22:0-LacCer-loaded D-HL-60 cells (Fig. 6). To further analyze the effects of LacCer loading on the distribution of LacCer and Lyn on the plasma membrane, we performed immunoelectron microscopy. Cellular GSLs are easily solved in alcohols, such as ethanol. Therefore, to observe the association of LacCer with Lyn on the plasma membrane in situ, immunoelectron microscopy with ultrathin cryosections, which were prepared without organic solvents used by routine electron microscopy, was performed [36]. Neutrophils and D-HL-60 cells expressed LacCer (detected by 5-nm gold particles) as clusters on the plasma membrane (Fig. 7a–d). These clusters in neutrophils and D-HL-60 cells were 43.2 ± 12.0 nm (mean±SD of 480 clusters) and 36.4 ± 8.6 nm (185 clusters) in diameter, respectively (Table 1). In neutrophils, 24% of LacCer clusters on the plasma membrane were associated with Lyn (detected by 10-nm gold particles) (Fig. 7b, Table 1). In contrast, it was difficult to find Lyn-associated LacCer clusters in D-HL-60 cells (Fig. 7c). Loading of C16:0- and C24:1-LacCer increased the sizes of LacCer clusters on D-HL-60 cells (Table 1). Importantly, 27% of LacCer clusters were associated with Lyn in C24:1-LacCer-loaded D-HL-60 cells, whereas we found no Lyn-associated LacCer clusters in C16:0-LacCer loaded D-HL-60 cells (Fig. 7e–h, Table 1). The distances between LacCer and Lyn (5- and 10-nm gold particles) in Lyn-associated LacCer clusters of neutrophils and C-24:1-LacCer-loaded D-HL-60 cells were 22.2 ± 11.7 nm (mean±SD of 117 clusters) and 14.7 ± 7.1 nm (49 clusters), suggesting that Lyn is located adjacent to LacCer in these LacCer clusters on both neutrophils and C24:1-LacCer-loaded D-HL-60 cells.

Electron micrograph showing the results of double-immunolabeling cytochemistry using anti-LacCer and anti-Lyn antibodies. Neutrophils (a, b), 0.1% DMSO-treated (Vehicle; c, d), C16:0- (e, f), or C24:1-LacCer-loaded (g, h) D-HL-60 cells were fixed, and ultra-thin sections of these cells were incubated with anti-LacCer IgM Huly-m13 along with rabbit polyclonal IgG to Lyn. As a secondary antibody, we used 5-nm gold-conjugated anti-mouse IgM and 10-nm gold-conjugated anti-rabbit IgG (Fab′)2. Hatched circles indicate LacCer clusters on the plasma membrane
The lipid raft-preferring molecules, such as GSLs, become clustered upon stimulation, resulting in induction of larger and stabilized lipid rafts, probably by coalescence of small lipid rafts or cholesterol-GSL complexes in the receptor clusters [49]. Confocal microscopy showed that LacCer was observed as clusters on the plasma membranes of neutrophils on ice (Fig. 8a). Upon incubation for 5 min at 37°C with anti-LacCer antibody T5A7, LacCer formed large clusters on the plasma membrane, and this cluster formation was inhibited by treatment with PP1. These results suggest that crosslinking of Lyn-associated LacCer-enriched lipid rafts mediates Lyn-dependent outside-in signaling to form large clusters of LacCer-enriched lipid rafts. LacCer also formed clusters on the plasma membrane of D-HL-60 cells on ice (Fig. 8a, vehicle). However, treatment with T5A7 at 37°C did not affect LacCer cluster size on the plasma membranes of these cells. After loading with C24:0- and C24:1-LacCer, LacCer formed large clusters on the plasma membrane upon incubation for 5 min at 37°C with T5A7, as observed in the case of neutrophils. PP1 inhibited the formation of large clusters in C24:1-LacCer loaded D-HL-60 cells by antibody treatment at 37°C. In contrast, anti-LacCer antibody treatment did not induce the formation of large LacCer clusters in D-HL-60 cells loaded with C16:0- or C22:0-LacCer. Flow cytometric analysis showed that LacCer expression was unaffected by incubation temperature (Fig. 8b), indicating that the amounts of LacCer on the plasma membranes of D-HL-60 cells and neutrophils were not significantly altered by treatment with anti-LacCer antibody. Anti-LacCer antibodies have been shown to activate neutrophils through LacCer-associated Lyn in neutrophils [12, 23]. In this context, PP1 inhibited the formation of large LacCer clusters on the plasma membrane by incubation at 37°C not only in neutrophils, but also in D-HL-60 cells loaded with C-24:1-LacCer, suggesting that the formation of large LacCer clusters induced by crosslinking with anti-LacCer antibody in cells containing C24-LacCer can be induced dependent on Lyn activation.

Expression of LacCer on plasma membranes of D-HL-60 cells. a Three-dimensional reconstruction images of LacCer on the plasma membranes of LacCer-loaded D-HL-60 cells. D-HL-60 cells were incubated with 0.5 μg/ml C16:0-, C22:0-, C24:0-, and C24:1-LacCers or 0.1% DMSO as a vehicle control (vehicle) for 30 min at 20°C. The loaded cells were incubated with Alexa-594-conjugated anti-LacCer IgM T5A7 on ice or 37°C for 5 min, followed by incubation for a further 25 min on ice. 37°C/PP1, incubation with Alexa-594-conjugated T5A7 at 37°C for 5 min in the presence of 10 μM PP1. bar, 5 μm. b Expression of LacCer on LacCer-loaded D-HL-60 cells. LacCers or 0.1% DMSO-treated D-HL-60 cells and neutrophils as indicated in (a) were incubated with Alexa-488-conjugated T5A7 on ice (black lines) or 37°C (red lines) for 5 min, followed by incubation for a further 25 min on ice. The dotted line in the vehicle panel shows normal IgM staining on ice
4 Discussion
The results of the present study indicated that LacCer species containing C24:0- and C24:1-fatty acids can reconstruct LacCer-enriched lipid rafts coupled with Lyn and restore LacCer-mediated neutrophil superoxide generation and migration in D-HL-60 cells. Plasma membrane LacCer of neutrophils was composed mainly of C16:0-, C24:1-, C24:0-, and C22:0-LacCer, while that of D-HL-60 cells consisted mainly of C16:0-LacCer and had low contents of C24-fatty acid chains (Fig. 1). Although all LacCer species were incorporated into the plasma membrane DRM of D-HL-60 cells (Fig. 5), C24:0-, and C24:1-LacCer, but not C16:0- or C22:0-LacCer-loaded cells showed LacCer-dependent functions (Figs. 4 and 6). Without stimulation of cells, Lyn was co-immunoprecipitated with anti-LacCer antibody from the plasma membrane DRM of neutrophils that naturally have high C24:0- and C24:1-LacCer contents, and of C24:0- and C24:1-LacCer-loaded D-HL-60 cells that originally contain only small amounts of C24:0- and C24:1-LacCer (Fig. 6). In contrast, Lyn was not co-immunoprecipitated with anti-LacCer antibody in C16- or C22-LacCer-loaded D-HL-60 cells. Furthermore, immunoelectron microscopic observations indicated that loading with C24:1-LacCer, but not C16:0-LacCer, resulted in association of LacCer clusters with Lyn (Fig. 7). These observations suggest that the presence of Lyn-coupled LacCer-enriched lipid rafts is indispensable for LacCer-mediated neutrophil superoxide generation and migration, and that the presence of a 24-carbon fatty acid chain in the LacCer molecule is necessary for the functional connection with Lyn in LacCer-enriched lipid rafts. This association occurred under basal conditions in neutrophils, which naturally contain C24-LacCer, and in D-HL-60 cells after loading with C24-LacCer. Loading with LacCer containing fatty acid chains with shorter chain lengths, such as C16 and C22, did not confer LacCer-mediated biological functions on D-HL-60 cells. In the case of D-HL-60 cells, a higher content of C16:0-LacCer in the plasma membrane may interfere with the connection of LacCer with Lyn in the LacCer-enriched lipid rafts. Contents of cholesterol and other lipids in the anti-LacCer antibody-immunoprecipitated plasma membrane DRM of D-HL-60 cells were not affected by the loading of LacCer species with different fatty acid structures (Fig. 5). The association of LacCer with Lyn in the neutrophil plasma membrane was not disrupted by cholesterol-scavenging reagents, such as methyl-β-cyclodextrin and nystatin [12]. Therefore, it seems that the connection of LacCer with Lyn is not affected by cholesterol and other lipid components of LacCer-enriched lipid rafts.
Glycosphingolipids participate in the stabilization of lipid rafts [28] and determination of membrane asymmetry. Asymmetry is a well-established property of the plasma membranes, which depends on the different lipid compositions of the two leaflets of the membrane bilayer. GSLs belong to the external membrane leaflet, while phosphatidylinositol, phosphatidylethanolamine, and phosphatidylserine are mainly distributed in the cytoplasmic leaflet [11, 50]. Sphingolipid-enriched lipid rafts are more tightly packed than the surrounding non-raft phase of the bilayer containing complex lipid species carrying 16–18 carbon acyl chains [44, 51], which, due to their high transition temperature, reduce membrane fluidity and favor cholesterol segregation within the same membrane domains [4]. The assembly of cholesterol could induce vacant pockets in the central part of the membrane, thus allowing interdigitation of longer alkyl chains [49]. Minor interdigitation of the longer alkyl chains usually occurs at the center of the membrane core, and its occurrence increases markedly when the maximum length of the hydrophobic chain exceeds the half thickness of the membrane. The thickness of lipid rafts has been estimated to be about 5 nm [52]. According to previous calculations [53], C24:0- and C24:1-LacCer species have a hydrophobic maximum length of about 3.2 nm, about 44% longer than the half hydrophobic thickness of the membrane. NMR studies of artificial lipid bilayers indicated that C24 fatty acid chains of LacCer interdigitated with fatty acids of the opposing monolayer [54]. Moreover, differential scanning calorimetry and Langmuir film balance experiments indicated that not only C24:0- but also C24:1-LacCer show the condensed and chain-ordered phase behavior [55]. Our data clearly indicate that the C24:0- and C24:1-LacCer but not C16:0- or C22:0-LacCer hydrophobic chains reconstruct the LacCer-mediated Lyn phosphorylation in D-HL-60 cells, which have only trace amounts of C24-LacCers (Fig. 6). Thus, one possibility is that the protrusion into the cytoplasmic membrane leaflet of the hydrophobic chains of LacCer could be so pronounced as to allow direct van der Waals interactions between the acyl chains of Lyn and those of LacCer. Actually, direct connections between gangliosides present in the external leaflet of the plasma membrane and proteins of the cytoplasmic plasma membrane face, such as Src-family protein kinases, caveolin, and tubulin, have been demonstrated in cells by loading with radioactive gangliosides containing a photoactivable group at the end of the acyl chain; radioactive proteins cross-linked with the ganglioside could be isolated from the DRM and characterized [56–58].
Several pathogenic microorganisms, such as C. albicans, bind selectively to LacCer among the GSLs [23, 25], indicating that the oligosaccharide chain of LacCer is also required for LacCer-mediated biological functions. Taken together, it seems that the oligosaccharide chains of GSLs play a role in specific ligand interactions on the cell surface, whereas very long fatty acid chains of GSLs function as key elements for the functional organization of GSL-enriched lipid rafts, which may be formed by lipid interdigitation. Further studies are required to elucidate the molecular machinery responsible for the association between fatty acid chains of LacCer and Lyn in LacCer-enriched lipid rafts.
Abbreviations
- DMSO:
-
Dimethyl sulfoxide
- D-HL-60 cells:
-
DMSO-treated neutrophilic differentiated human promyelocytic leukemia HL-60 cells
- fMLP:
-
Formyl peptide (N-formyl-methionyl-leucyl-phenylalanine)
- CSBG:
-
Candida albicans-derived β-glucan
- SCG:
-
Sparassis crispa-derived β-glucan
- SM:
-
Sphingomyelin
- PC:
-
Phosphatidylcholine
- PE:
-
Phosphatidylethanolamine
References
Degroote, S., Wolthoorn, J., van Meer, G.: The cell biology of glycosphingolipids. Semin. Cell. Dev. Biol. 15, 375–387 (2004)
Hakomori, S.: Structure, organization, and function of glycosphingolipids in membrane. Curr. Opin. Hematol. 10, 16–24 (2003)
Kaga, N., Kazuno, S., Taka, H., Iwabuchi, K., Murayama, K.: Isolation and mass spectrometry characterization of molecular species of lactosylceramides using liquid chromatography-electrospray ion trap mass spectrometry. Anal. Biochem. 337, 316–324 (2005)
Sonnino, S., Prinetti, A., Mauri, L., Chigorno, V., Tettamanti, G.: Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem. Rev. 106, 2111–2125 (2006)
Brackman, D., Lund-Johansen, F., Aarskog, D.: Expression of leukocyte differentiation antigens during the differentiation of HL-60 cells induced by 1,25-dihydroxyvitamin D3: comparison with the maturation of normal monocytic and granulocytic bone marrow cells. J. Leukoc. Biol. 58, 547–555 (1995)
Brown, D.A., London, E.: Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochem. Biophys. Res. Commun. 240, 1–7 (1997)
Iwabuchi, K., Handa, K., Hakomori, S.: Separation of “glycosphingolipid signaling domain” from caveolin-containing membrane fraction in mouse melanoma B16 cells and its role in cell adhesion coupled with signaling. J. Biol. Chem. 273, 33766–33773 (1998)
Iwabuchi, K., Yamamura, S., Prinetti, A., Handa, K., Hakomori, S.: GM3-enriched microdomain involved in cell adhesion and signal transduction through carbohydrate-carbohydrate interaction in mouse melanoma B16 cells. J. Biol. Chem. 273, 9130–9138 (1998)
Yamamura, S., Handa, K., Hakomori, S.: A close association of GM3 with c-Src and Rho in GM3-enriched microdomains at the B16 melanoma cell surface membrane: a preliminary note. Biochem. Biophys. Res. Commun. 236, 218–222 (1997)
Okada, Y., Mugnai, G., Bremer, E.G., Hakomori, S.: Glycosphingolipids in detergent-insoluble substrate attachment matrix (DISAM) prepared from substrate attachment material (SAM). Their possible role in regulating cell adhesion. Exp. Cell Res. 155, 448–456 (1984)
Simons, K., Ikonen, E.: Functional rafts in cell membranes. Nature 387, 569–572 (1997)
Iwabuchi, K., Nagaoka, I.: Lactosylceramide-enriched glycosphingolipid signaling domain mediates superoxide generation from human neutrophils. Blood 100, 1454–1464 (2002)
Mukherjee, S., Maxfield, F.R.: Membrane domains. Annu. Rev. Cell Dev. Biol. 20, 839–866 (2004)
Arai, T., Bhunia, A.K., Chatterjee, S., Bulkley, G.B.: Lactosylceramide stimulates human neutrophils to upregulate Mac-1, adhere to endothelium, and generate reactive oxygen metabolites in vitro. Circ Res 82, 540–547 (1998)
Bhunia, A.K., Han, H., Snowden, A., Chatterjee, S.: Redox-regulated signaling by lactosylceramide in the proliferation of human aortic smooth muscle cells. J. Biol. Chem. 272, 15642–15649 (1997)
Iwamoto, T., Fukumoto, S., Kanaoka, K., Sakai, E., Shibata, M., Fukumoto, E., Inokuchi Ji, J., Takamiya, K., Furukawa, K., Furukawa, K., Kato, Y., Mizuno, A.: Lactosylceramide is essential for the osteoclastogenesis mediated by macrophage-colony-stimulating factor and receptor activator of nuclear factor-kappa B ligand. J. Biol. Chem. 276, 46031–46038 (2001)
Gong, N., Wei, H., Chowdhury, S.H., Chatterjee, S.: Lactosylceramide recruits PKCalpha/epsilon and phospholipase A2 to stimulate PECAM-1 expression in human monocytes and adhesion to endothelial cells. Proc Natl Acad Sci USA 101, 6490–6495 (2004)
Sharma, D.K., Brown, J.C., Cheng, Z., Holicky, E.L., Marks, D.L., Pagano, R.E.: The glycosphingolipid, lactosylceramide, regulates beta1-integrin clustering and endocytosis. Cancer Res. 65, 8233–8241 (2005)
Abul-Milh, M., Paradis, S.E., Dubreuil, J.D., Jacques, M.: Binding of Actinobacillus pleuropneumoniae lipopolysaccharides to glycosphingolipids evaluated by thin-layer chromatography. Infect. Immun. 67, 4983–4987 (1999)
Angstrom, J., Teneberg, S., Milh, M.A., Larsson, T., Leonardsson, I., Olsson, B.M., Halvarsson, M.O., Danielsson, D., Naslund, I., Ljungh, A., Wadstrom, T., Karlsson, K.A.: The laactosylceramide binding specificity of Helicobacter pylori. Glycobiology 8, 297–309 (1998)
Hahn, P.Y., Evans, S.E., Kottom, T.J., Standing, J.E., Pagano, R.E., Limper, A.H.: Pneumocystis carinii cell wall beta-glucan induces release of macrophage inflammatory protein-2 from alveolar epithelial cells via a lactosylceramide-mediated mechanism. J. Biol. Chem. 278, 2043–2050 (2003)
Karlsson, K.A.: Animal glycolipids as attachment sites for microbes. Chem. Phys. Lipids 42, 153–172 (1986)
Sato, T., Iwabuchi, K., Nagaoka, I., Adachi, Y., Ohno, N., Tamura, H., Seyama, K., Fukuchi, Y., Nakayama, H., Yoshizaki, F., Takamori, K., Ogawa, H.: Induction of human neutrophil chemotaxis by Candida albicans-derived beta-1,6-long glycoside side-chain-branched beta-glucan. J. Leukoc. Biol. 80, 204–211 (2006)
Saukkonen, K., Burnette, W.N., Mar, V.L., Masure, H.R., Tuomanen, E.I.: Pertussis toxin has eukaryotic-like carbohydrate recognition domains. Proc Natl Acad Sci USA 89, 118–122 (1992)
Zimmerman, J.W., Lindermuth, J., Fish, P.A., Palace, G.P., Stevenson, T.T., DeMong, D.E.: A novel carbohydrate-glycosphingolipid interaction between a beta-(1–3)-glucan immunomodulator, PGG-glucan, and lactosylceramide of human leukocytes. J. Biol. Chem. 273, 22014–22020 (1998)
Greenberg, S., Grinstein, S.: Phagocytosis and innate immunity. Curr. Opin. Immunol. 14, 136–145 (2002)
Brown, D.A., Rose, J.K.: Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533–544 (1992)
Parkin, E.T., Turner, A.J., Hooper, N.M.: Differential effects of glycosphingolipids on the detergent-insolubility of the glycosylphosphatidylinositol-anchored membrane dipeptidase. Biochem. J. 358, 209–216 (2001)
Palestini, P., Allietta, M., Sonnino, S., Tettamanti, G., Thompson, T.E., Tillack, T.W.: Gel phase preference of ganglioside GM1 at low concentration in two-component, two-phase phosphatidylcholine bilayers depends upon the ceramide moiety. Biochim. Biophys. Acta. 1235, 221–230 (1995)
Acquotti, D., Sonnino, S.: Use of nuclear magnetic resonance spectroscopy in evaluation of ganglioside structure, conformation, and dynamics. Methods Enzymol. 312, 247–272 (2000)
Mauri, L., Casellato, R., Kirschner, G., Sonnino, S.: A procedure for the preparation of GM3 ganglioside from GM1-lactone. Glycoconj. J. 16, 197–203 (1999)
Tokyokuni, T., Nisar, M., Dean, B., Hakomori, S.: A facile and regiospecific titration of sphingosine: synthesis of (2S,3R,4E)-2-amino-4-octadecene-1,3-diol-1-3H. J, Labeled Compd. Radiopharm. 29, 567–574 (1991)
Vaissiere, C., Le Cabec, V., Maridonneau-Parini, I.: NADPH oxidase is functionally assembled in specific granules during activation of human neutrophils. J. Leukoc. Biol. 65, 629–634 (1999)
Someya, A., Nagaoka, I., Iwabuchi, K., Yamashita, T.: Comparison of O2(-)-producing activity of guinea-pig eosinophils and neutrophils in a cell-free system. Comp. Biochem. Physiol. [B] 100, 25–30 (1991)
Brkovic, A., Pelletier, M., Girard, D., Sirois, M.G.: Angiopoietin chemotactic activities on neutrophils are regulated by PI-3K activation. J. Leukoc. Biol. 81, 1093–1101 (2007)
Kurihara, H., Anderson, J.M., Farquhar, M.G.: Increased Tyr phosphorylation of ZO-1 during modification of tight junctions between glomerular foot processes. Am. J. Physiol. 268, F514–524 (1995)
Dimmock, E., Franks, D., Glauert, A.M.: The location of blood group antigen A on cultured rabbit kidney cells as revealed by ferritin-labelled antibody. J. Cell Sci. 10, 525–533 (1972)
Munn, E.A., Bachmann, L., Feinstein, A.: Structure of hydrated immunoglobulins and antigen-antibody complexes. Electron microscopy of spray-freeze-etched specimens. Biochim. Biophys. Acta. 625, 1–9 (1980)
Parkhouse, R.M., Askonas, B.A., Dourmashkin, R.R.: Electron microscopic studies of mouse immunoglobulin M; structure and reconstitution following reduction. Immunology 18, 575–584 (1970)
Plowman, S.J., Muncke, C., Parton, R.G., Hancock, J.F.: H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 102, 15500–15505 (2005)
Macher, B.A., Klock, J.C.: Isolation and chemical characterization of neutral glycosphingolipids of human neutrophils. J. Biol. Chem. 255, 2092–2096 (1980)
Symington, F.W., Murray, W.A., Bearman, S.I., Hakomori, S.: Intracellular localization of lactosylceramide, the major human neutrophil glycosphingolipid. J. Biol. Chem. 262, 11356–11363 (1987)
Kniep, B., Skubitz, K.M.: Subcellular localization of glycosphingolipids in human neutrophils. J. Leukoc. Biol. 63, 83–88 (1998)
Rajendran, L., Simons, K.: Lipid rafts and membrane dynamics. J. Cell Sci. 118, 1099–1102 (2005)
Resh, M.D.: Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1451, 1–16 (1999)
Kobayashi, T., Shinnoh, N., Goto, I., Kuroiwa, Y., Okawauchi, M., Sugihara, G., Tanaka, M.: Galactosylceramide- and lactosylceramide-loading studies in cultured fibroblasts from normal individuals and patients with globoid cell leukodystrophy (Krabbe’s disease) and GM1-gangliosidosis. Biochim. Biophys. Acta. 835, 456–464 (1985)
Martin, S.F., Williams, N., Chatterjee, S.: Lactosylceramide is required in apoptosis induced by N-Smase. Glycoconj. J. 23, 147–157 (2006)
Nijhuis, E., Lammers, J.W., Koenderman, L., Coffer, P.J.: Src kinases regulate PKB activation and modulate cytokine and chemoattractant-controlled neutrophil functioning. J. Leukoc. Biol. 71, 115–124 (2002)
Kusumi, A., Koyama-Honda, I., Suzuki, K.: Molecular dynamics and interactions for creation of stimulation-induced stabilized rafts from small unstable steady-state rafts. Traffic 5, 213–230 (2004)
Zuvic-Butorac, M., Muller, P., Pomorski, T., Libera, J., Herrmann, A., Schara, M.: Lipid domains in the exoplasmic and cytoplasmic leaflet of the human erythrocyte membrane: a spin label approach. Eur. Biophys. J. 28, 302–311 (1999)
Prinetti, A., Chigorno, V., Prioni, S., Loberto, N., Marano, N., Tettamanti, G., Sonnino, S.: Changes in the lipid turnover, composition, and organization, as sphingolipid-enriched membrane domains, in rat cerebellar granule cells developing in vitro. J. Biol. Chem. 276, 21136–21145 (2001)
Allende, D., Vidal, A., McIntosh, T.J.: Jumping to rafts: gatekeeper role of bilayer elasticity. Trends Biochem. Sci. 29, 325–330 (2004)
Tanford, C.: The Hydrophobic Effect: Formation of Micelles and Biological Membranes. Ohn Wiley and Sons Inc, New York (1980)
Grant, C.W., Mehlhorn, I.E., Florio, E., Barber, K.R.: A long chain spin label for glycosphingolipid studies: transbilayer fatty acid interdigitation of lactosyl ceramide. Biochim. Biophys. Acta 902, 169–177 (1987)
Li, X.M., Momsen, M.M., Brockman, H.L., Brown, R.E.: Lactosylceramide: effect of acyl chain structure on phase behavior and molecular packing. Biophys. J. 83, 1535–1546 (2002)
Fra, A.M., Masserini, M., Palestini, P., Sonnino, S., Simons, K.: A photo-reactive derivative of ganglioside GM1 specifically cross-links VIP21-caveolin on the cell surface. FEBS Lett. 375, 11–14 (1995)
Prinetti, A., Marano, N., Prioni, S., Chigorno, V., Mauri, L., Casellato, R., Tettamanti, G., Sonnino, S.: Association of Src-family protein tyrosine kinases with sphingolipids in rat cerebellar granule cells differentiated in culture. Glycoconj. J. 17, 223–232 (2000)
Palestini, P., Pitto, M., Tedeschi, G., Ferraretto, A., Parenti, M., Brunner, J., Masserini, M.: Tubulin anchoring to glycolipid-enriched, detergent-resistant domains of the neuronal plasma membrane. J. Biol. Chem. 275, 9978–9985 (2000)
Acknowledgements
We are grateful to Dr. Sen-itiroh Hakomori (University of Washington) for his encouragement and invaluable comments throughout this study. We thank Dr. Hiroshi Tamura (Seikagaku Corporation) and Drs. Yoshiyuki Adachi and Naohito Ohno (Tokyo University of Pharmacy and Life Science) for providing Candida albicans-derived β-glucan and Sparassis crispa-derived β-glucan, respectively. We also thank Dr. Irwin D. Bernstein at Fred Hutchinson Cancer Research Center, Seattle, WA, USA, for important contributions.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported in part by a grant-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (16017293) to K.I., by COFIN-PRIN 2004 to A.P., and by “High-Tech Research Center” Project for Private Universities: matching fund subsidy.
An erratum to this article can be found at http://dx.doi.org/10.1007/s10719-008-9110-3
Rights and permissions
About this article
Cite this article
Iwabuchi, K., Prinetti, A., Sonnino, S. et al. Involvement of very long fatty acid-containing lactosylceramide in lactosylceramide-mediated superoxide generation and migration in neutrophils. Glycoconj J 25, 357–374 (2008). https://doi.org/10.1007/s10719-007-9084-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-007-9084-6