
Abstract
Insights into genetic causes of cardiomyopathies have tremendously contributed to the understanding of the molecular basis and pathophysiology of hypertrophic, dilated, arrhythmogenic, restrictive and left ventricular noncompaction cardiomyopathy. More than thousand mutations in approximately 100 genes encoding proteins involved in many different subcellular systems have been identified indicating the diversity of pathways contributing to pathological cardiac remodeling. Moreover, the classical view based on morphology and physiology has been shifted toward genetic and molecular patterns defining the etiology of cardiomyopathies. Today, novel high-throughput genetic technologies provide an opportunity to diagnose individuals based on their genetic findings, sometimes before clinical signs of the disease occur. However, the challenge remains that rapid research developments and the complexity of genetic information are getting introduced into the clinical practice, which requires dedicated guidance in genetic counselling and interpretation of genetic test results for the management of families with inherited cardiomyopathies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cardiomyopathy
- Heart failure
- Sudden death
- Disease genes
- Molecular genetics
- Genetic diagnosis
- Genetic counselling
- Next-generation sequencing
2.1 Introduction
Over the last three decades, genetic research has significantly contributed to the etiology of cardiomyopathies. In fact, most primary cardiomyopathies are influenced by genetic factors predisposing to the development of heart failure, one of the most common causes of death in industrialized countries. Cardiomyopathies (CMPs) are commonly grouped by their morphological signs leading to subtypes called hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), left ventricular noncompaction (LVNC), arrhythmogenic cardiomyopathy (ACM), and restrictive cardiomyopathy (RCM) (Fig. 2.1) [1].

Examples of cardiac magnetic resonance imaging (MRI) pictures of cardiomyopathy subtypes. (a) Short-axis two-chamber view of hypertrophic cardiomyopathy (HCM). Note the thick septal wall of the left ventricle (LV) (white arrow). (b) Long-axis four-chamber view of dilated cardiomyopathy (DCM). Note the enlarged left ventricle. (c) Long-axis four-chamber view of left ventricular noncompaction (LVNC). Note the noncompacted, trabeculated layer of the LV (white arrow). (d) Long-axis four-chamber view of arrhythmogenic cardiomyopathy (ACM). Note the enlarged right ventricle (RV). (e) Long-axis four-chamber view of restrictive cardiomyopathy (RCM). Note the massively enlarged left atrium (LA) (white arrow)
Since, in 1990, the sarcomere gene beta-myosin heavy chain (MYH7) was identified as the first disease gene for HCM, it turned out that the sarcomere plays a major role in the genetic etiology of HCM but also of other CMPs [2, 3]. HCM can primarily be defined as a disease of the sarcomere, but the genetic basis for CMPs has evolved to be diverse. Genetic causes of ACM have been unraveled with disease genes involved in cell-cell contacts called desmosomes [4, 5]. But, in fact, today there are about hundred known disease genes associated with different subtypes of CMPs, which are expressed in various subcellular systems responsible for transcription, cardiac development, energy utilization, electrolyte imbalances, and others. The diversity is making it difficult to define a final common pathway leading from disturbed gene function to the disease phenotype.
Despite the genetic complexity, with the application of high-throughput genetic technology, we are able to analyze large amounts of data in a cost-effective and timely manner which makes it possible to use this technology not only for research but also for comprehensive cardiomyopathy diagnostics [6,7,8]. However, there are challenges that clinicians and their team of health-care providers face in translating research results and comprehensive data sets into the clinic in order to improve patient management. A major challenge is still the classification of variants into pathogenic or disease-causing versus benign. Often variants are defined as “uncertain,” which makes the application for clinical purposes even more difficult [9]. Additionally, when a pathogenic variant has been identified, there can be substantial variation in penetrance, age of onset, and clinical expression of the phenotype, also within the same family. Here, modifying factors such as lifestyle, other genetic and epigenetic factors, pregnancy, and many still unknown entities are playing a role—an ongoing field of research. Moreover, different mutations in the same genes can cause variable clinical entities and even lead to the opposite phenotype (dilated vs. hypertrophic cardiomyopathy). Given the complexity, today the question arises how much a particular genetic diagnosis may help guiding diagnosis and clinical management? There are potential benefits such as differentiating a genetic etiology from other causes of CMPs or allowing the identification of at-risk individuals early, in a preclinical stage. But more importantly, the understanding of the molecular pathways in a translational setting will help to further understand these diseases and dissect more complex disease pathways toward targeted therapies.
This chapter summarizes current knowledge about clinical and molecular genetics of the five main subtypes of inherited cardiomyopathies in a bench-to-bedside approach.
2.2 Hypertrophic Cardiomyopathy (HCM)
Definition
Hypertrophic cardiomyopathy (HCM) is a primary myocardial disorder affecting clinically about 1 in 500 individuals [10]. The pathological sign is an unexplained left ventricular hypertrophy which is not caused by abnormal loading conditions (Fig. 2.1a). Classically, HCM is characterized by asymmetric septal hypertrophy; however there can be any pattern of left ventricular hypertrophy associated with the disease [11,12,13].
Clinical Manifestations
Clinically, the disease is highly variable in presentation and includes diastolic dysfunction, left ventricular outflow tract obstruction (in about 25%), ischemia, and atrial fibrillation. In about 5% of cases, end-stage HCM shows progression to systolic impairments. Many patients are asymptomatic and are diagnosed incidentally, and others may manifest shortness of breath, chest pain, palpitations, or syncope [11]. Disease-related mortality is most often attributable to sudden cardiac death, heart failure, and embolic stroke. However, early data about high mortality rates from tertiary centers from the 1970s and early 1980s, with annual death rates of 2–4% in adults and 4.2–5.9% in children, have been revised. Disease-related mortality in adults is today about 0.5% similar to that of the general population. Decreased mortality is an achievement of more effective risk stratification and the use of the implantable cardioverter-defibrillator for primary prevention of sudden death [14,15,16].
Inheritance
HCM is mainly inherited as an autosomal dominant trait with incomplete penetrance and variable expression of the clinical phenotype. Some cases are explained by de novo mutations, and other apparently sporadic cases can arise due to autosomal recessive, compound heterozygous, or digenic mode of inheritance [17,18,19]. Furthermore, genetic modifiers and environmental and epigenetic factors are likely to influence the phenotype. Consequently, even identical mutations may show distinct degrees and morphology of hypertrophy, pattern of fibrosis, age of onset, and risk for arrhythmias and sudden death [20, 21]. On the other hand, population-based studies looking for variants in HCM-associated sarcomere genes indicated that changes in cardiac morphology and function may also occur without causing overt or typical HCM [22].
Disease Genes
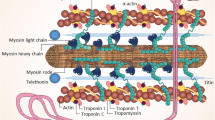
HCM was the first primary cardiomyopathy for which a genetic cause was identified back in 1990. Ever since over thousand mutations in numerous genes have been described. The most common eight genes encoding sarcomere proteins of the thick and thin filaments are beta-myosin heavy chain (MYH7) [2], alpha-tropomyosin (TPM1), cardiac troponin T (TNNT2) [23, 24], cardiac myosin-binding protein C (MYBPC3) [25], myosin regulatory light chain (MYL2), myosin essential light chain (MYL3) [26], cardiac troponin I (TNNI3) [27], and cardiac α-actin (ACTC1) [28]. Two major genes, MYH7 and MYBPC3, account together for up to 75% of all identified mutations, whereas other genes including TNNT2, TNNI3, TPM1, MYL2, MYL3, and ACTC1 each account for a small proportion of cases (1–5%) [29, 30]. Furthermore, rare variants have been described in additional genes encoding proteins of the sarcomere apparatus (MYH6, TNNC1, TTN) and the adjacent Z-disc (ACTN2, CSRP3, TCAP, VCL, NEXN, LDB3, MYOZ2, FLNC, MYPN, ANKRD1) or are involved in calcium homeostasis pathways (PLN, CASQ2, JPH2, CALR3). Some of these genes are less evident for direct pathogenicity and may function as modifiers (Table 2.1, Fig. 2.2a) [31,32,33].

Genes associated with different subtypes of cardiomyopathies according to the year of discovery. Colors indicate the subcellular location and/or functional association. (a) Hypertrophic cardiomyopathy. (b) Dilated cardiomyopathy. (c) Left ventricular noncompaction. (d) Arrhythmogenic cardiomyopathy. (e) Restrictive cardiomyopathy
Genotype-Phenotype Correlation
The penetrance of expressing the phenotype when carrying a mutation appears to be highly variable and can be delayed or incomplete. Often left ventricular hypertrophy (LVH) does not develop until the third decade of life or even after. In addition, penetrance may also differ according to gender with males showing earlier clinical signs than females [21, 34]. Early genotype-phenotype studies described some genes (or particular mutations) with a higher degree of sudden cardiac death (TNNT2, MYH7 p.Arg403Gln) and other genes (MYBPC3) with milder LVH and later age of onset [20, 24]. More recently, reports from the UK suggest a higher risk of sudden death in TNNT2 vs. MYBPC3 families (0.93% vs. 0.46% per year). Overall, currently available phenotypic data could not show clear correlations for specific genes and most of the mutations. Therefore, genotype data have only minor influence on current risk stratification [21, 35, 36].
Despite inherited as an autosomal dominant disease, complex phenotypes with patients carrying more than one mutation or variant exist, and they usually show more severe or earlier phenotypic expression. Such complexity has been reported in 5–7% of cases by Sanger sequencing and is even higher using next-generation sequencing (8–9%). Here, different scenarios have been seen including compound heterozygosity (different heterozygous mutation in each allele of the same gene), digenic cases (heterozygous mutations in two different genes), and also rare homozygosity [37, 38].
Lastly, mutations in classical HCM genes can result in divergent clinical features, also mimicking other forms of cardiomyopathy, in particular LVNC or RCM. Sometimes in the same family, different clinical manifestations may occur [39].
Genetic Counselling and Testing
As indicated above, the majority of HCM cases is inherited as an autosomal dominant trait with a 50% risk for first-degree relatives. Current guidelines of the European Society of Cardiology (ESC) recommend genetic counselling for all patients with HCM, unless an acquired cause is demonstrated. Counselling should be performed by trained health-care professionals working within multidisciplinary teams to help patients understand and manage the psychological, social, professional, ethical, and legal implications of genetic disease [11].
If genetic testing is considered, genetic counselling should be performed to fully inform patients about the benefits and limitations of genetic testing and consequences for them and their families. In patients fulfilling the diagnostic criteria for HCM, genetic testing identifies a pathogenic mutation in up to 65% of cases. Although, nowadays, there is no direct benefit in most of the clinically affected cases having a genetic diagnosis in regard to treatment options and a dedicated prognosis, it is more important for pre-symptomatic testing in the family and reproductive advice. Generally speaking, predictive genetic testing can be performed in asymptomatic relatives of a patient with HCM when a definitive disease-causing mutation has been determined to initiate cascade family screening.
However, in situations where predictive genetic testing is either not available or not considered, first-degree relatives should be offered clinical screening with an ECG and echocardiogram starting at an age of 10 years [40]. Individuals who have nondiagnostic clinical features that are consistent with early disease should be seen initially at intervals of 6–12 months and potentially less frequent depending on progression. As indicated above, age-dependent penetrance requires repeat screening even in individuals with no clinical features, unless a predictive genetic test has been performed and was “negative” for a proven disease-causing mutation [3, 11, 41].
Today, new high-throughput sequencing technologies are more and more used for genetic testing in diseases such as HCM by analyzing the whole exome with the potential identification of a large number of variants of unknown clinical significance (VUS). In contrast to other cardiomyopathies, broader genetic testing has shown only a modest increase in the diagnostic yield in HCM (45–72%) while producing more uncertainty. No matter what sequencing technology is used, for clinical practice, genetic testing should include the eight most commonly implicated sarcomere genes and may also consider genes of potential phenocopies of sarcomeric HCM, in particular when additional or atypical phenotypic signs are present [42, 43].
Other Genetic Etiologies
There are phenocopies of HCM which can mimic sarcomeric HCM on the basis of cardiac imaging (Table 2.2). In adults, the main non-sarcomeric etiologies are metabolic storage disease such as Danon disease (LAMP2), LVH and Wolff-Parkinson-White syndrome (PRKAG2), Anderson-Fabry disease (GLA), familial TTR amyloidosis (TTR), and some mitochondrial cardiomyopathies. Others are more prevalent in children such as Pompe disease (GAA), Noonan syndrome and Leopard syndrome (PTPN11), or Friedreich ataxia (FXN). To distinguish these diseases from sarcomeric HCM is quite important as an early diagnosis ensures an appropriate management in particular in metabolic diseases [44, 45].
Functional Consequences
Although the genetic basis for HCM is well established, the biochemical and biophysical mechanisms how sarcomere mutations lead to disease remain only partially understood. The majority of mutations described are missense mutations that result in mutant dominant-negative acting “poison” peptides, which are incorporated into the sarcomere and potentially have an adverse effect on sarcomere function [46]. The exception for the “poison” protein thesis is mutations in MYBPC3, which often result in truncated proteins. Here, mutant proteins are often cleared by cell surveillance mechanisms, which leads to a reduced amount of full-length protein resulting in haploinsufficiency and consequently an inadequate amount of functional proteins [47, 48].
Mechanistically, there is substantial evidence that myofilament mutations increase calcium sensitivity and Ca2+ affinity and thereby actin-dependent ATPase activity [49, 50]. Enhanced Ca2+ sensitivity and subsequent defects in calcium homeostasis such as intracellular calcium cycling, sarcoplasmic reticulum [SR] Ca2+ reuptake, and CaMKII-mediated phosphorylation of proteins, including phospholamban, are likely to contribute to the pathogenesis of HCM [51]. A consequence of increased sarcomeric Ca2+ sensitivity is also an enhanced cross-bridge turnover and higher actin-activated ATPase activity. Subsequently, this generates higher energy need to produce a given tension. In accordance with this explanation, several studies have shown mitochondrial abnormalities and an impaired myocardial energy metabolism. Altered Ca2+ handling and energy deficiency appear to be major pathways leading to pathological changes such as hypertrophy and myofibrillar disarray as well as functional features such as impaired relaxation [52, 53].
More recently, understanding of fundamental mechanisms arises hope of more specific, substrate-modulating therapy ranging from metabolic modulators to small molecule effectors and gene therapy rescuing HCM at the mechanistic and biochemical level. Targets are energy utilization and oxidative stress, increased calcium sensitivity, sarcomere function (hypercontractility), and the action potential of the myocytes. However, the challenge exists that different mutations even at the same locus can lead to opposite functional consequences. For example, different mutations in MYH7 exert opposing effects on myofilament calcium sensitivity and contractility [54]. In silico models, such as motility assays using purified recombinant sarcomeric protein constructs containing dedicated mutations, are a way to investigate individual mutations [53]. Mutations in the myosin head region domain lead to hypercontractility and impaired relaxation by assessing the chemo-mechanical cycle in ex vivo preparations of mutated sarcomeric proteins. A small molecule, MYK-461, that decreases sarcomeric power due to inhibition of the myosin adenosine triphosphatase (ATPase) counteracts the effects of the mutation. In mice with hypercontractile mutations in myosin heavy chain, early chronic administration of MYK-461 prevented the development of LVH, myocyte disarray, and fibrosis and normalized expression of profibrotic and mitochondrial genes involved in energy utilization [55].
2.3 Dilated Cardiomyopathy (DCM)
Definition
Dilated cardiomyopathy (DCM) is characterized by left or biventricular dilatation and systolic dysfunction that are not explained by abnormal loading conditions or coronary artery disease (Fig. 2.1b). The disease is a common cause of heart failure leading to heart transplantation and sudden death [1]. Causes of DCM can be genetic, estimated as high as 35–50% [56, 57]; however even non-genetic etiologies may predisposed by genetic factors and interact with extrinsic and environmental factors. Lately, there are suggestions for an updated definition of DCM recognizing a broader clinical spectrum of the disease, in particular in the setting of familial disease and before overt clinical manifestations are present. This includes criteria defining preclinical phases of the disease that may occur in mutation carriers without clinical expression, cases with isolated ventricular dilation, arrhythmic cardiomyopathy, or hypokinetic non-dilated cardiomyopathy [58].
Prevalence data based on echocardiography estimated unexplained DCM at 1:2500; more recently epidemiological and next-generation sequencing data suggest a much higher prevalence, possibly as high as 1:250. The disease incidence is 7 per 100,000, and males are more frequently affected than females (3:1) [59, 60].
Clinical Manifestations
DCM can present as a clinical syndrome of systolic heart failure with reduced ejection fraction (HFrEF) but also with arrhythmias that may lead to sudden death or thromboembolic events. Management of HFrEF should be performed according to current heart failure guidelines; however, despite optimal therapy, the prognosis remains poor for patients showing heart failure symptoms with a 5-year mortality of up to 20%. Cardiac transplantation or other advanced therapies should be considered with progressive DCM, advanced heart failure, or otherwise refractory disease [61,62,63].
Another spectrum of the disease represents unidentified cases without clinical symptoms in early phases of the disease. Genetic approaches offer the possibility to identify individuals at risk in the setting of familial disease. Although not proven by placebo-controlled trials, early medical management may be beneficial, even in symptom-free mutation carriers with only minor cardiac abnormalities [58, 64, 65].
Inheritance
DCM can be inherited as an autosomal dominant, recessive, or X-linked trait; however chromosomal abnormalities or even matrilineal inheritance may also be present.
Most cases of isolated DCM follow autosomal dominant inheritance with age-dependent penetrance and variable clinical expression, which can be delayed until the fifth or sixth decade. This indicates a role for potential modifying genetic, epigenetic, and environmental factors [66, 67]. However, these factors also contribute to the fact that familial DCM is often not recognized, as other more common etiologies are attributed. When compared to other cardiomyopathies, the genetic etiology of DCM is even more heterogenous, which became more obvious since the introduction of next-generation sequencing [68]. To date, more than 50 disease-related genes have been reported, although relatively few are supported by robust segregation analyses or experimental data (Table 2.3; Fig. 2.2b).
Genes Involved in DCM
Pathogenic variants have been reported for a heterogenous group of genes encoding proteins with different functions involved in sarcomere integrity and force transmission, cytoskeletal architecture, cell-cell contacts, nuclear organization, transcription, and ion channel activity. Although extensive genetic heterogeneity is present, titin (TTN) is the most common disease gene accounting for 20–25% of the genetic causes [69,70,71]. The second most prevalent gene is lamin A/C (LMNA) with about 6%, followed by beta-myosin heavy chain (MYH7) [72,73,74]. Several other genes account for 1–2% of familial cases, while many others are less frequent or even reported once. Alterations, such as copy number variations (CNVs), have been reported in association with DCM but also with a low frequency. The most important genes and their role in subcellular systems will be discussed in more detail.
Titin and the Sarcomere
Mutations in the titin gene (TTN) are the main cause of DCM accounting for about 20–25% in familial cases and even up to 18% of sporadic DCM cases. In particular, mutations that truncate the protein have been found to cause disease, although such variants have been also found in 1–2% of the control population [69,70,71]. Titin, the biggest protein in nature, provides the external scaffold interacting with the thin and thick filaments. It is important for sarcomere assembly and provides passive force and elasticity to maintain diastolic and systolic function, respectively. TTN is composed of different domains according to the sarcomere structure (Z-disc, I-band, A-band, M-band). In addition, TTN undergoes extensive alternative splicing and therefore produces many isoforms. Current data suggest that truncating variants in exons that are incorporated into all expressed isoforms, and are part of the final transcript in the heart, are more likely to cause disease (90%), whereas exons, e.g., of the I-band, which are not included in the mature transcript, may not be disease-causing and have been found more frequently in the control population [75].
Mutations in other proteins of the sarcomere are also associated with autosomal dominant DCM such as ACTC1, MYBPC3, MYH6, MYH7, TNNC1, TNNI3, TNNT2, and TPM1, with MYH7 as the most common one [73]. The frequency of sarcomere gene mutations to be associated with DCM is much lower compared to HCM and LVNC. Mechanistically, and in contrast to HCM, mutations are suggested to decrease Ca2+ sensitivity causing a hypocontractile state that leads to systolic dysfunction. For example, mice carrying a HCM-associated mutation (delGlu160) in TNNT2 showed increased Ca2+ sensitivity, whereas a DCM-associated mutation (delLys210) in the same gene showed the opposite, decreased Ca2+ sensitivity [53].
Nuclear Envelope Proteins
DCM-associated mutations are also involving proteins of the nuclear envelope such as lamin A/C (LMNA) located in the nuclear lamina and emerin (EMD), a protein of the inner nuclear membrane. Dominant LMNA mutations account for 6–10% of genetic causes and are frequently associated with arrhythmias and conduction system disturbances [72]. In addition, limb-girdle myopathies are a variable feature. Lamins are involved in nuclear structure support, DNA repair, cell signaling pathway mediation, and chromatin organization. There are two major hypotheses how mutations in LMNA may lead to cardiac dysfunction: (1) disruption and uncoupling of structural proteins at the nuclear lamina that lead to mechanical instability and to an increased susceptibility to mechanical stress and (2) disrupted chromatin organization that impacts directly on gene transcription [76, 77].
EMD is a LEM-domain protein located in the nuclear lamina and has a role in assembly of the nuclear lamina and structural organization of the nuclear envelope. EMD mutations are X-linked and often associated with a skeletal muscle phenotype of Emery-Dreifuss muscular dystrophy [78].
Gene Expression
Genes regulating transcription are also important in the pathogenesis of DCM. Rare mutations have been described in TBX20, NKX2-5, GATA4, GATA6, FOXD4, PRDM16, EYA4, GATAD1, and RBM20. Other associated phenotypes such as congenital heart defects, hearing loss, or more complex syndromic features are often observed (Table 2.3). Variants in most of those genes lead to decreased transcriptional activity influencing a set of genes important for cardiac development and structural remodeling [79,80,81,82,83].
A very interesting gene is RBM20, which encodes a spliceosome protein that regulates pre-mRNA splicing for many genes, including TTN. Interestingly, a mutational hotspot causing highly penetrant DCM alters an arginine-serine-rich region of RBM20, which influences binding with other splicing factors and changes transcript processing [84, 85].
Calcium Handling and Ion Imbalances
Mutations in phospholamban (PLN) are predicted to alter Ca2+ homeostasis. The protein regulates Ca2+ uptake by the SR Ca2+ ATPase (SERCA2a) and inhibits Ca2+ cycling when dephosphorylated [86]. In its phosphorylated state by protein kinase A (PKA), muscle relaxation is enhanced, and beta-agonist activates PKA and enhances relaxation. Mutations in PLN blunt beta-adrenergic activation of PKA and control of Ca2+ cycling leading to decreased contractility [87].
Another important gene is the voltage-gated, type V alpha-subunit of the cardiac sodium channel (SCN5A) which is involved in the cardiac action potential. The channel is responsible for rapid depolarization of the myocardium and the maintenance of impulse conduction. Mutations lead to a variety of arrhythmic disease and also DCM by modifying electrical excitability of the channel leading to disturbances of currents and different ions which are also involved in contraction [88].
The ABCC9 protein is predicted to form ATP-sensitive potassium channels in cardiac and other muscles. Variants in this gene have been also associated with DCM [89].
Z-Disc-Associated Proteins
The sarcomeric Z-disc defines the lateral borders of the sarcomere and consists of actin filaments coming from adjacent sarcomeres which are cross-linked by α-actinin molecules. The Z-disc serves as a nodal point with multiple functions such as intracellular signaling, mechanosensation and mechanotransduction. The Z-disc also links to the T-tubular system and the sarcoplasmic reticulum; moreover, several E3 ubiquitin ligases localized to the Z-disc link this structure to protein turnover and autophagy [32]. To date, mutations in multiple Z-disc proteins or Z-disc-related proteins have been reported to be associated with DCM including ACTN2, ANKRD1, BAG3, CRYAB, CSRP3, LDB3, MYPN, NEBL, NEXN, TCAP, FLH2, FLNC, and DES; however the evidence for clear pathogenicity is variable (Table 2.3) [90]. A typical sarcomeric Z-disc protein is α-actinin 2 (ACTN2), which is important for actin filament localization. The cardiac LIM protein CSRP3 is a protein with multiple interaction partners and is involved in various signal transduction cascades, including mechanosensation and mechanotransduction, calcium metabolism, and myofibrillogenesis. Mutations in CSRP3 can either cause DCM or HCM [91, 92].
A protein, which is present at the sarcomeric Z-disc but also as an intermediate filament, is desmin (DES). It provides the link to the desmosomes and to other compartments such as the nucleus [93].
An interesting protein is the co-chaperone BAG3 (Bcl2-associated athanogene 3), which acts as a multifunctional adaptor protein responsible for cell survival, apoptosis, migration, proliferation, macro-autophagy, and proteasomal processes. Mutations causing DCM may influence BAG3-interacting proteins such as HSP70 responsible for protein folding and quality control of newly synthetized proteins [94].
Overall, the Z-disc-associated proteins are diverse and are becoming a “hotspot” for cardiomyopathy-causing mutations; however the spectrum of functional disturbances is various and depending on their specific location and behavior.
Desmosomes
Desmosomes are located at the intercalated discs and part of a junctional structure called “area composita.” Mutations in desmosomal proteins can cause DCM-like phenotypes but are more frequently involved in ACM (see chapter ACM). There is a substantial overlap, in particular in phenotypic features of more left-sided ACM phenotypes and DCM with pronounced arrhythmic features, as seen mainly with mutations in desmoplakin (DSP), but also in other desmosomal proteins [95, 96].
DCM Associated with Other Pathologies
As indicated in Table 2.3, DCM is sometimes associated with other non-cardiac phenotypic features, in particular myopathies. In addition, DCM can also occur as part of a syndrome that often affects children.
Interestingly, there are frequent associations of DCM with either myofibrillar myopathy (MFM), Emery-Dreifuss muscular dystrophy (EDMD), or limb-girdle muscular dystrophy (LGMD). Genetic variations in the same genes can cause isolated DCM and associations with those types of skeletal muscle diseases. Often, but not always, isolated DCM is inherited as an autosomal dominant disease, whereas associated myopathies are X-linked or autosomal recessive.
MFMs are chronic neuromuscular disorders. The morphologic changes in skeletal muscle and sometimes in cardiac muscle result from disintegration of the sarcomeric Z-disc and the myofibrils, followed by abnormal ectopic accumulation of multiple proteins (myofibrillar cytoplasmic inclusions). Mutations in genes encoding desmin (DES), alpha-B-crystallin (CRYAB), dystrophin (DMD), filamin C (FLNC), LIM domain-binding protein 3 (LDB3), and BAG3 [97] are causing MFM and/or DCM.
The two other common myopathies are limb-girdle muscular dystrophy (LGMD) and Emery-Dreifuss muscular dystrophy (EDMD), which are mainly inherited as recessive disorders. EDMD is characterized by myopathic changes in certain skeletal muscles and early contractures at the neck, elbows, and Achilles tendons, as well as cardiac conduction defects. Disease genes for EDMD are emerin (EMD), lamin A/C (LMNA), and nesprin-1 (SYNE1) [98]. Autosomal recessive LGMDs are a heterogenous group of disorders affecting primarily skeletal muscle in various forms; in some, DCM is part of the clinical spectrum such as in LGMD2F, LGMD2E, LGMD2D, and LGMD2G, whereas dominant mutations in LMNA can cause LGMD1B [99].
DCM is an important part of the disease spectrum in Duchenne and Becker muscular dystrophies caused by mainly truncating mutations in the dystrophin gene (DMD). In Duchenne or Becker with a later onset, cardiac features are present in about 95% of cases by the last years of life. Also, female carriers of these X-linked diseases develop DCM in about 15% of cases [100].
Lastly, DCM is also part of inherited syndromes. Some of them are listed in Table 2.3. They are usually associated with a congenital onset, inherited as a recessive or X-linked trait, and affect proteins that play a role in the heart and other tissues.
Genetic Testing and Counselling
The increasing understanding of the genetic basis of DCM and the availability of comprehensive and cost-effective next-generation sequencing technologies have highlighted the importance of considering genetic testing in all patients with DCM, not just those with an obvious family history or a particular phenotype. Today, cardiomyopathy multi-gene panels or even whole-exome sequencing (WES) is available to perform genetic testing. In clinical settings, as opposed to research testing, the focus should be on known genes associated with DCM [101]. The major challenge occurs in interpreting genetic test results to be used in a clinical setting.
Moreover, not only the interpretation of genetic test results but also proper communication to the patients and their families require a clinician specifically trained in medical genetics and cardiology, who is embedded in a team of experts including genetic counsellors, psychologists, and specialized cardiologists. Expert knowledge of advances in testing, emerging data on newly identified variants and clinical correlations, and an understanding of the complex allelic heterogeneity and complicated scenarios such as multiple variants that may influence the disease are particularly important in DCM.
In the scenario that genetic testing results in a pathogenic/likely pathogenic mutation, the mutation can be considered to be tested in relatives at risk for cascade screening in order to facilitate prompt diagnosis, surveillance, and in selected cases preventative treatment. Furthermore, relatives who do not carry the mutation may be discharged from clinical surveillance which has long-range consequences for families and requires a correct review and classification of genetic variants identified by genetic testing.
Strict variant classification according to the ACMG criteria [9] can facilitate a highly accurate diagnostic yield in DCM, with a pathogenic/likely pathogenic variant detection rate of 35.2%, with 47.6% in familial DCM and 25.6% in sporadic cases [102]. However, even with these restrictions, many variants of uncertain significance (VUSs) are identified mainly in genes with weak evidence of being associated with DCM. For example, the interpretation of TTN variants is a big challenge, and pathogenicity depends on additional information and resources, as TTN truncating variants are apparently also present in healthy individuals. There are additional online tools available indicating details in the exon composition of the major TTN transcripts; information whether an exon is constitutively expressed, and other structural features for each exon; as well as the distribution of TTN variants in large published studies of cohorts of DCM patients and controls [71, 75].
Overall, genetic testing is challenging and takes an expert panel to draw the right conclusions. Likewise, there is no doubt about the importance of genetic counselling and clinical surveillance of first-degree relatives of individuals with DCM.
Genotype-Phenotype Correlation
DCM is characterized by marked genetic heterogeneity and variable disease penetrance, which make direct applications to clinical management difficult. Additionally, clinical variability in the phenotype development with the influence of other contributing factors impedes the prediction for a certain genotype [8]. Only few genes such as TTN and LMNA as well as some founder mutations in PLN or MYBPC3 are more common for comprehensive investigations to allow genotype-phenotype studies.
Mutations in LMNA are highly suggestive for progressive conduction disease and ventricular arrhythmias with an increased risk for sudden death. Risk assessment also involves a gender-specific risk with a higher mortality in men compared to women. LMNA-associated DCM demonstrates an age-related penetrance with onset in the third and fourth decades, so that by the seventh decade, penetrance is considered greater than 90–95%. Management implications are to assess for prophylactic implantable cardioverter-defibrillator placement to treat malignant ventricular arrhythmias for patients receiving a pacemaker for LMNA-associated conduction system disease, independent of ejection fraction [76, 103,104,105].
More recently, genotype-phenotype studies for TTN truncation variants suggested a milder phenotype compared to non-TTN-related cardiomyopathies, although the comparison was driven by a direct comparison to LMNA cardiomyopathy which has, as indicated above, a severe phenotype [106, 107]. First findings also suggest a gender-specific difference with regard to median age of onset and prognosis which was more preferable for woman compared to man. In terms of age-dependent penetrance, current data suggest a late age of onset, ranging between about 30% and 50% at the age of 50 and between 80% and 100% at the age of 70 [102, 106].
Severe phenotypes may also appear when additive genetic effects appear, for example, in cases carrying compound heterozygous variants [108].
2.4 Left Ventricular Noncompaction (LVNC)
Definition and Classification
Left ventricular noncompaction (LVNC) is a rare disorder characterized by hypertrophic segments that consist of a thin compacted epicardial layer and a thick noncompacted endocardial layer. The noncompacted layer contains numerous prominent trabeculations and deep intertrabecular recesses [109, 110]. LVNC may be an isolated finding in the absence of any coexisting cardiac anomaly (isolated LVNC) or may be associated with other congenital heart anomalies such as complex congenital heart disease (non-isolated LVNC).
LVNC is a relatively new clinicopathologic condition, first described as such by Chin et al. in 1990 [109]. Based on the predominant myocardial involvement and genetic etiology, LVNC was classified by the American Heart Association (AHA) as a distinct primary cardiomyopathy [1]. The European Society of Cardiology (ESC) recognizes LVNC as an unclassified cardiomyopathy [41]. The ESC questions whether LVNC is a distinct cardiomyopathy or merely a congenital or acquired morphological trait shared by many phenotypically distinct cardiomyopathies [111, 112].
Clinical Manifestation
LVNC presents with a unique congenital cardiac morphology, variable clinical features, and a diverse natural history. The heterogeneity of the clinical features includes both asymptomatic and symptomatic patients with progressive deterioration in cardiac function resulting in congestive heart failure, arrhythmias, thromboembolic events, and sudden cardiac death [113,114,115].
There was a prevalence of 0.014% in patients referred to an echocardiography laboratory in a tertiary referral center [115]. Lately, due to improved imaging techniques, increased awareness, and family screening, the rare entity LVNC is recognized with growing frequency [116, 117]. A substantial proportion of individuals is asymptomatic, suggesting that the true prevalence of LVNC may be higher. Greater extent of, and even excessive, LV trabeculation measured in end-diastole in asymptomatic population-representative individuals by cardiac magnetic resonance imaging (MRI) appeared benign and was not associated with deterioration in LV volumes or function during an almost 10-year period [MESA (Multi-Ethnic Study of Atherosclerosis)] [118]. LVNC was identified as the most frequent cardiomyopathy after DCM and HCM in childhood, with an estimated prevalence of 9% [119] in an Australian cohort and 5% in the US Pediatric Cardiomyopathy Registry [120].
The clinical diagnosis is performed with two-dimensional echocardiography and/or cardiac MRI on the basis of the prominent appearance of LV trabeculae and the ratio between the compacted and noncompacted LV wall (Fig. 2.1c). The diagnosis of LVNC in the symptomatic patient is made at any age, ranging from fetus to old age. Heart failure symptoms are the most common reason for hospital admission. Both systolic and diastolic cardiac dysfunctions have been described. Ventricular tachycardia with a significant risk of cardiac sudden death is frequently found (20–40%). The formation of thrombi in the extensive intertrabecular recesses leading to systemic thromboembolic events is another, although less common, presentation of patients with LVNC. The natural history of LVNC is largely unresolved. As in other cardiomyopathies, index cases represent the most severe spectrum of the disease [114]. LVNC has a high mortality rate and is strongly associated with arrhythmias in children. Preceding cardiac dysfunction or ventricular arrhythmias are associated with increased mortality. Children with normal cardiac dimensions and normal function are at low risk for sudden death [121].
Morphology and Pathogenesis
The noncompacted endocardial layer of the myocardium comprises numerous, excessively prominent ventricular trabeculations with deep intertrabecular recesses. The recesses extend deeply into the trabecular meshwork and end at the thin compacted outer layer. In contrast to “persisting myocardial sinusoids” which were first observed in patients with left or right ventricular outflow tract obstruction, recesses in LVNC have no connection with the coronary circulation and are covered by endocardium throughout the ventricular cavity. The most widely used method for the morphologic diagnosis of LVNC is two-dimensional echocardiography and was established by Jenni et al. [110]. The diagnosis of LVNC is made irrespective of the presence of left ventricular systolic dysfunction or dilatation (DCM) due to hypokinetic segments in the altered LV myocardium. Given the common localization of the noncompacted areas in the apex and the common localization of septal hypertrophy in HCM, the two diagnoses of HCM and LVNC can coexist. LV dilation/dysfunction and LV hypertrophy can therefore be present or absent and do not influence LVNC diagnosis. There may be biventricular noncompaction, but criteria for the diagnosis of noncompacted myocardium involving the right ventricle have not been established. Imaging criteria for the assessment of LVNC are still evolving. A significant proportion of an asymptomatic population free from CVD satisfy all currently used cardiac magnetic resonance imaging diagnostic criteria for LVNC, suggesting that those criteria have poor specificity for LVNC [112].
There is evidence that LVNC can originate during embryonic development or be acquired later in life. The de novo LV trabeculations in a significant proportion (>25%) of pregnant women suggest that these may occur as a consequence of increased LV loading conditions or other physiological adaptation mechanisms related to pregnancy. Moreover, the general assumption that LVNC is caused by incomplete myocardial compaction during embryogenesis has been questioned [122, 123]. During fetal development, compaction of the ventricular myocardium normally progresses from epicardium to endocardium, from the base to the apex, and from the septum to the lateral wall. An arrest in this process of myocardial morphogenesis would explain the predominant localization of noncompacted myocardium in LVNC. If not reflecting compaction of pre-existing trabeculations, it is certainly possible that noncompaction relates to defects of proliferation of the compacted myocardium. Mutation of PRDM16 causes LVNC in patients, and PRDM16 zebrafish mutants show impaired cardiomyocyte proliferation [79]. Genome editing of PRDM16 leads to proliferation defects in iPSC-CMs, and abnormal TGF-β signaling was suggested as pathological mechanism [124]. Mouse embryos lacking Notch1, systemically or in the endocardium, show defective trabeculation. Molecular analysis of these mutants reveals that expression of endocardial and myocardial trabecular differentiation markers is impaired and ventricular cardiomyocyte proliferation is inhibited. These findings imply that endocardial Notch1 signaling is required for proliferation and differentiation of trabecular myocardium [125].
Disease Genes and Inheritance
Among many sporadic cases, familial recurrence has already been observed in the first reports of isolated LVNC. In familial cases, autosomal dominant inheritance is more common than X-linked inheritance [126]. LVNC has been associated with mutations in almost 20 different genes (Table 2.4 and Fig. 2.2c). Defects in sarcomere genes are the most prevalent genetic cause occurring in 30% of adult patients with isolated LVNC. MYH7 is the most frequent LVNC-associated gene in adult patients with isolated LVNC [127]. Mutations in the sarcomere genes encoding thick (MYH7) [128,129,130,131], intermediate/thick filament associated (MYBPC3) [127, 129, 132], and thin filaments (TNNT2, TPM1, ACTC1) [127, 129, 130, 133] have been described. TTN, encoding for the giant protein Titin that serves as a molecular spring of the sarcomere, has recently been added to the list of sarcomere genes in LVNC [134].
However, mutations in other genes are only rare causes of LVNC in single families. TAZ was the first gene shown to be associated with isolated LVNC by genetic linkage analysis in a family with X-linked inheritance [135]. TAZ encodes for taffazin, a protein involved in the biosynthesis of cardiolipin, an essential component of the inner mitochondrial membrane. A small proportion of familial autosomal dominant LVNC can be explained by mutations in genes encoding proteins of the Z line of the sarcomere, LDB3/ZASP [129, 136] and ACTN2/α-actinin 2 [137]. Two cardiac ion channels HCN4/hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 [138, 139] and SCN5A/cardiac sodium channel alpha-subunit gene [140] lead to LVNC with sinus node dysfunction and arrhythmias, respectively. A component of the nuclear lamina, lamin A/C (LMNA) [141] causes LVNC with cardiac conduction disease. Mutations in the gene MIB1 segregated with autosomal dominant LVNC in two families, and a conditional loss-of-function allele in a mouse model, also led to LVNC [142]. The hypertrabeculation and noncompaction seen in the MIb1 mouse was mimicked in a mouse with inactivation of Jagged1 in the myocardium or Notch1 in the endocardium, suggesting that the Notch1 signaling pathway was involved.
Genetic Syndromes and/or Other Disorders
LVNC is present in a number of neuromuscular disorders, metabolic and mitochondrial disease, congenital malformations, and chromosomal syndromes. Some of these disorders may share pathogenetic mechanisms with LVNC. Alternatively, LVNC might be secondary to other cardiac malformations or even vice versa.
LVNC can occur as part of a syndrome in combination with dysmorphic features and other congenital malformations. Chromosomal deletions have been found on chromosomes 1p36, 1q43, and 5q35 [79]. Left ventricular noncompaction is known to be a part of various syndromes, including the Barth, Noonan, Roifman, Melnick-Needles, Nail-Patella, Toriello-Carey, and other uncommon syndromes [143]. TAZ mutation typically results in Barth syndrome, which is characterized by cardiomyopathy (frequently LVNC), skeletal myopathy, cyclic neutropenia, and 3-methylglutaconic aciduria (a marker of mitochondrial dysfunction) [144]. DSP, encoding for desmoplakin, a gene known for the first recessive human mutation that causes a generalized striate keratoderma particularly affecting the palmoplantar epidermis, woolly hair, and dilated left ventricular cardiomyopathy (Carvajal syndrome), also leads to LVNC with acantholytic palmoplantar keratoderma with autosomal recessive inheritance [145].
The co-occurrence of congenital heart defects (CHD) and noncompaction is seen in children and in adults [144, 146]. DTNA, encoding for α-dystrobrevin, a cytoskeletal component responsible for force transduction, is mutated in patients with hypoplastic left heart syndrome and LVNC [144]. Septal defects and Ebstein anomaly are the most prevalent congenital heart defects in LVNC. Mutations in the cardiac transcription factor NKX2-5 were identified in children with LVNC and atrial septal defects [147]. Noncompaction associated with Ebstein anomaly, a rare type of CHD, has been shown to result from mutations in MYH7 [131]. The association of sarcomere gene defects, cardiomyopathy, and structural CHD still requires further investigation. Very much like HCM and DCM, LVNC has been linked to neuromuscular disorders such as dystrophinopathies and with mitochondrial disease. In a number of muscular dystrophies, “left ventricular hypertrabeculation” was identified [148]. In a study of 113 pediatric patients with mitochondrial disease, LVNC was identified in 13% [149].
Genetic Counselling and Testing
Since extensive family studies showed that the majority of affected relatives are asymptomatic, cardiologic evaluation should include all first-degree relatives irrespective of medical history. Other cardiomyopathies may co-occur within families, like HCM and DCM, so cardiac screening should aim at identifying all cardiomyopathies. Cardiac screening of relatives may show minor abnormalities not fulfilling LVNC criteria. Genetic testing, preferably with a targeted cardiomyopathy gene panel, may lead to the identification of a molecular defect in over 40% of isolated LVNC patients [129].
Molecular studies of LVNC have thus far shown that there are few recurrent mutations. Therefore, it is difficult to establish genotype-phenotype correlations. Additionally, intrafamilial phenotypic variability complicates predictions based on an identified mutation. Compound heterozygous or homozygous truncating sarcomere gene mutations appear to result in a more severe phenotype with childhood onset [132]. Whereas TAZ mutations have been reported in pediatric patients often diagnosed with LVNC during infancy, heterozygous mutations in autosomal dominant LVNC were mainly identified in adult patients.
Probst et al. [127] reported a cohort of 63 LVNC probands, previously studied by Klaassen et al. [130], in which 8 sarcomere genes were analyzed and heterozygous mutations found in 18 (29%) of the probands: 8 mutations were in the MYH7 gene, 5 in MYBPC3, 2 in ACTC1, 2 in TPM1, and 1 in TNNT2. There were no significant differences between mutation-positive and mutation-negative probands in terms of average age, myocardial function, or presence of heart failure or tachyarrhythmias at initial presentation or at follow-up. Probst et al. [127] noted that although 8 of the 15 distinct mutations were novel in this cohort, they were likely not specific to LVNC, because the other 7 mutations had previously been described in patients with other forms of cardiomyopathy, including hypertrophic and dilated forms. In conclusion, molecular analyses of LVNC support the concept of a shared molecular etiology of the different cardiomyopathic phenotypes.
For further reading: Left Ventricular Noncompaction [143] in Clinical Cardiogenetics, Springer International Publishing, H.F. Baars et al. (eds), 2016.
2.5 Arrhythmogenic Cardiomyopathy (ACM)
Definition
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a primary cardiomyopathy characterized by progressive fibro-fatty replacement of the right and left ventricular myocardium (Fig. 2.1d) [150]. In the early eighteenth century, the clinical phenotype of ARVC was first reported by Giovanni Maria Lancisi in a four-generation family, where patients presented with right ventricular dilation or aneurysms and suffered from sudden cardiac death, both typical signs of the disease [151]. During the 1980s, clinical features were systematically described by Marcus and colleagues [152]. Particularly, the central involvement of the right ventricle was described. However, today there is growing evidence that the left ventricle is also affected leading to the term “arrhythmogenic cardiomyopathy” (ACM), which will be also used in this chapter. The estimated disease prevalence ranges from 1:2000 to 1:5000 considering ACM as a rare disease [153].
Clinical Manifestation
Clinical features of the disease are highly variable ranging from unaffected mutation carriers to patients suffering from sudden cardiac death or requiring heart transplantation. Classical symptoms include palpitations, cardiac syncope, and aborted cardiac arrest due to ventricular arrhythmias. Heart failure is rare and may develop in later stages. ACM is a progressive disease which typically manifests in the second to fourth life decade. However, about 20% of cases develop the disease after the age of 50 [154]. Penetrance is age and gender dependent, and clinical manifestations and progression of the disease are highly variable. Men are clinically more often affected than women. The influence of sex hormones and higher physical activity of men may contribute to the disease expression. The clinical diagnosis is challenging, because of the lack of a specific diagnostic test. Hence, the diagnosis is based on fulfilling a set of major and minor criteria proposed by an international task force initially in 1994 [155]. This set of Task Force Criteria (TFC) was highly specific but lacked sensitivity for early forms of ACM and has been revised in 2010 to incorporate new findings and diagnostic modalities [150]. The TFC include evaluation of findings from six different diagnostic categories including cardiac imaging, assessment of electrical alterations, and family history. Management is individualized and focused on prevention of sudden death through use of antiarrhythmic medication and implantable cardioverter-defibrillators and rarely heart transplantation.
Inheritance
An autosomal dominant trait is the most common inheritance pattern for non-syndromic forms of the disease. Today about 40–60% of the genetic causes are known, and plakophilin 2 (PKP2) is the most prevalent disease gene [4]. However, there have been reports of autosomal recessive patterns of almost all desmosomal disease genes; with and without additional syndromic features. For example, mutations in genes like plakoglobin (JUP) and desmoplakin (DSP) lead to ACM, palmoplantar keratoderma, and abnormalities of the hair structure. Carriers with homozygous mutations in desmocollin 2 (DSC2) show an isolated cardiac phenotype (Table 2.5). As seen in other cardiomyopathies, incomplete penetrance and variable clinical expression are also common; even in the same family, the severity of phenotypes demonstrates a wide range. In some cases, different family members develop DCM or ACM [156]. Besides cases with one mutation, there are several reports about compound heterozygous mutations or two or more variants in different genes [157]. Of note, there are also rare cases of de novo mutations [158]. Besides the main genetic drivers, also modifiers such as environmental (e.g., athletic activity) and epigenetic factors might influence the phenotype.
Disease Genes
At the beginning of the 2000s, it was recognized that about 50% of ACM patients carry mutations in five different genes encoding desmosomal proteins: JUP [5], DSP [159], PKP2 [4], DSC2 [160, 161], and DSG2 [162]. Desmosomes are cell-cell junctions, which have important functions for the nano-mechanical coupling of cells [96]. In organs exposed to high mechanical stress like the skin or the heart, cells are connected by these multi-protein complexes. Desmosomes are linked to the intermediate filament system, which is mainly formed in the heart by desmin (DES). Besides cardiac desmosomes, also adhering junctions are important for mechanical coupling of cardiomyocytes. Adhering junctions are linked to actin filaments [163]. Interestingly, ACM-associated mutations were also identified in CTNNA3 and CDH2, which encode two structural proteins of adhering junctions [164, 165]. Therefore, it is suggested that ACM is mainly a disease of the cardiac intercalated disc, as both desmosomes and adhering junctions are located there. In 40–60% of ACM patients, one or more mutations in genes encoding proteins involved in cell adhesion could be identified (Table 2.5; Fig. 2.2d) [158, 166]. In addition to genes, encoding structural proteins involved in cell adhesion of cardiomyocytes, there are some reports about mutations in other genes like RYR2 [167], TGFB3 [168], TMEM43 [169], SCN5A [170], TTN (M [171]), PLN [156], LMNA [172], LDB3 [173], and KCNQ1 [174]. However, mutations in those genes are rare and account for less than 10% (Fig. 2.2d; Table 2.5).
Cell-Cell Adhesion
McKoy et al. identified the first homozygous 2 bp deletion mutation in JUP in families from the Greek island Naxos [5]. Patients developed ACM in combination with keratosis and woolly hair. The typical triad of features is known in the literature as “Naxos disease” [175]. JUP encodes plakoglobin, a member of the armadillo protein family, which is a structural cytoplasmic component of the desmosome. Nonsense-mediated mRNA decay (NMD) is linked to the pre-mRNA splicing process, and the exon-junction complexes are necessary for the degradation of mRNA carrying a premature termination codon (PTC) [176]. Zhang et al. generated two Jup knock-in mouse models with the same deletion mutation. In one of them, the authors deleted additionally the introns to block NMD. Remarkably, the knock-in mice without the introns were completely healthy [177]. Those findings suggest that potentially “loss of function” mediated by NMD may explain the mechanism due to JUP mutations.
Similar to Naxos disease, Carvajal syndrome is characterized by striate keratoderma, woolly hair, and left ventricular arrhythmic cardiomyopathy. It is caused by mutations in DSP [178]. Rampazzo et al. described the first DSP mutation in dominant ACM [159]. Desmoplakin is a cytolinker protein and connects the cardiac desmosomes with the desmin intermediate filaments. The C-terminal tail domain is responsible for this protein-protein interaction. Desmoplakin forms dimers by the formation of a coiled coil of the Rod domains [179]. Autosomal recessive as well as autosomal dominant DSP mutations were reported in 5–10% of ACM patients (Table 2.5).
In 2004, Gerull et al. identified PKP2 mutations in a large cohort of ACM patients [4]. PKP2 is the most common gene for autosomal dominant ACM, and it accounts for about 20–40% of the known mutations. Most mutations are small deletions, insertions, nonsense, or splice site changes leading to the truncation of the protein. PKP2 encodes plakophilin 2, which is also a member of the Armadillo protein family. Plakophilin 2 binds to the desmosomal cadherins and mediates the molecular interaction with the cytolinker protein desmoplakin [180]. Plakophilin 2 is necessary for the vesicle transport of the desmosomal cadherin desmocollin 2 to the plasma membrane [181]. It is suggested that haploinsufficiency could be the main genetic mode of action. Presumably, NMD and protein degradation pathways are involved in PKP2 haploinsufficiency [182].
Different groups described also mutations in the genes DSC2 [160, 161] and DSG2 [162] encoding for desmosomal cadherins. The extracellular domains of both cadherins mediate the protein-protein interactions in a Ca2+-dependent way by homophilic and presumably also by heterophilic trans-interaction [183]. Besides the description of heterozygous missense and nonsense variants in DSC2 with, so far, lack of clear pathogenicity, there have also been homozygous truncation mutations reported causing a severe biventricular form of ACM without skin and hair features [184, 185]. Interestingly, Wong et al. investigated the clinical phenotype of a founder population with 28 heterozygous and 11 homozygous DSC2-p.Q554X mutation carriers. Almost all of the heterozygous carriers were healthy or presented only with mild clinical symptoms, whereas all homozygous mutation carriers developed ACM [186].
Besides the desmosomal genes, ACM-related mutations were also identified in desmin (DES) [158, 187]. Desmin is the major component of the intermediate filaments in cardiomyocytes, which provides the connection to different cellular organelles like the desmosomes and the costameres. Interestingly, ACM-associated DES mutations disturb the intermediate filament assembly [188].
Recently, mutations in αT-Catenin (CTNNA3) and in N-cadherin (CDH2) have been described [164, 165, 189]. Both proteins are structural components of the adhering junctions. Adhering junctions are cell-cell junctions which are connected to F-actin filaments. However, further studies are required to investigate the frequency and the pathogenic impact of variants/mutations in those genes.
Other Genetic Contributors
Besides mutations affecting cell-cell adhesion proteins, there are also rare mutations reported in genes involved in Ca2+ handling like the RYR2 [167] or PLN [156]. RYR2 encodes the cardiac ryanodine receptor 2 which is mediating the Ca2+ release from the sarcoplasmic reticulum [190], whereas phospholamban (PLN) is a regulator of the sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA), which transports Ca2+ into the sarcoplasmic reticulum [191]. Rare variants were also identified in the 5′- and 3′-untranslated regions of TGFB3, a cytokine from the transforming growth factor family [168]. TGF-β cytokines are involved in fibrotic remodeling process by upregulation of expression of extracellular matrix proteins like different collagens [192]. Merner et al. identified a missense mutation in TMEM43 in a founder population from Newfoundland [169]. Remarkably, this specific mutation leads to nearly complete penetrance [169] and was also identified in ACM patients from different other countries [193]. TMEM43 encodes the nuclear envelope protein luma [194]. The exact biochemical functions and the molecular structure of luma are widely unknown. Additionally, mutations were also identified in LMNA [172], which encodes the nuclear intermediate filament lamin A/C involved in the structural stabilization of the nuclei. Other rare variants were identified in SCN5A [170], encoding the α-subunit of the voltage-gated sodium channel 5, and in KCNQ1 [174], encoding the voltage-gated potassium channel Q1. Mutations in both genes cause inherited arrhythmias like Brugada syndrome or long and short QT syndrome, indicating a genetic overlap of ACM with channelopathies.
Genetic Counselling and Testing
Clinical genetic testing in ACM is in particular challenging and requires to be performed in dedicated cardio-genetic centers, with pre- and post-counselling possibilities. As indicated above, the inheritance pattern of ACM is more complex than previously thought, with frequent requirement for more than one “hit” for fully penetrant disease. More frequent than in other CMPs, patients carry homozygous or compound heterozygous variants in the same gene or digenic/oligogenic variants in a cluster of desmosomal genes. The broad use of large gene panels or whole-exome sequencing often results in a high number of variants of unknown clinical significance (VUSs), which should be carefully classified according to guidelines as suggested by the ACMG [9]. Additionally, genetic testing should be performed with the consideration that even a positive genetic test result assigned as a pathogenic or likely pathogenic mutation may not be the only genetic contributor to the phenotype in the patient and/or family.
However, genetic counselling should be done in all families with a confirmed case of ACM, and first-degree relatives should be screened according to the TFC by ECG, echocardiography, Holter ECG, and signal-averaged ECG starting at the age of 10 years, and this should be regularly repeated [40]. In case there is a reliable genetic test result (pathogenic or likely pathogenic mutation) available, this may be used for cascade family screening, although cardiologic evaluation may still continue at a lower level as a negative genetic result may not exclude any genetic predisposition. Lifestyle advice and reproductive counselling on the risk of transmission to offspring should be also provided. Restriction from competitive sports activity and strenuous physical exercise is strongly recommended to prevent disease onset and progression.
Despite some progress in the understanding of the functional impact of certain mutations, there is still a gap of knowledge of all contributing factors resulting in the clinical picture of ACM. Hopefully, the increasing genetic knowledge will also lead to more profound biochemical and cellular understanding of the pathomechanisms toward the development of more efficient molecular therapies.
2.6 Restrictive Cardiomyopathy (RCM)
Definition
Restrictive cardiomyopathy (RCM) is characterized by an abnormal left ventricular filling pattern, diastolic dysfunction but normal wall thickness, and usually preserved systolic function. The restrictive filling pattern is caused by an increased muscle stiffness leading to atrial enlargement and an increase of ventricular end-diastolic blood pressure (Fig. 2.1e). RCM can be classified as a rare primary cardiomyopathy, often affecting children but also adults. However, there are also secondary causes known, such as storage or infiltrative disorders [41, 195].
Clinical Manifestation and Inheritance
Patients typically develop heart failure symptoms such as dyspnea and fatigue [196]. Preserved LV ejection fraction, normal or mildly increased left and right ventricular wall thicknesses, restrictive diastolic filling patterns and severe atrial enlargement are common echocardiographic findings. The general prognosis is poor, patients often requiring heart transplantation. Clinically, the differentiation between RCM and constrictive pericarditis imposed by external pericardial constraint can be challenging. Secondary RCM can be part of systemic diseases, e.g., a mineralization disorder called pseudoxanthoma elasticum (PXE), caused by ABCC6 mutations [197] or of cardiac amyloidosis associated with TTR mutations [198]. Additionally, metabolic diseases such as Pompe, Fabry, and Danon disease as well as Friedreich ataxia can be associated with LVH (Table 2.2) but can also present as RCM. Primary RCM is mainly a familial disease and inherited as an autosomal dominant trait (Table 2.6).
Disease Genes
RCM is a rare disease and the proportion of genetic etiology remains unknown. However, Kostareva et al. investigated 24 non-related index patients using broad next-generation sequencing panels and identified in about 54% of them a pathogenic or likely pathogenic mutation [199]. Overall, mutations in 13 different genes have been reported (Fig. 2.2e, Table 2.6). Most of those genes encode for sarcomeric or cytoskeletal proteins. Of note, there is also a genetic overlap with other primary cardiomyopathies, in particular with HCM.
Sarcomeric Proteins
Sarcomeres are the essential contracting units of striated muscle cells consisting of thick and thin filaments, which are connected by Z-bands. First mutations associated with familial RCM were identified in the TNNI3 gene, encoding cardiac troponin I (cTnI) [200]. cTnI inhibits the ATPase function of the actin-myosin complex during the contraction cycle [201]. Mutations in cardiac troponin C (TNNC1, cTnC) and troponin T (TNNT2, cTnT), encoding the two other cardiac components of the troponin complex, were also identified in RCM patients [202, 203]. cTnC is the Ca2+-binding and cTnT is the tropomyosin-binding subunit of the troponin complex, mediating the molecular interaction of thin and thick filaments during the contraction cycles [204]. Other sarcomeric disease genes are ACTC1 and MYH7 [205, 206]. Recently, Wu et al. identified a family with three affected patients carrying a nonsense mutation in MYBPC3, which encodes the myosin-binding protein C and binds to the thick filaments [207]. Peled and colleagues identified a de novo missense mutation (p.Y7621C) in titin (TTN) causing RCM [208]. Furthermore, other RCM-associated mutations were found in genes encoding myopalladin (MYPN) and α-actinin 2 (ACTN2) [199, 209]. α-Actinin 2 is the major structural component of the Z-bands, and myopalladin is a binding partner. Myopalladin is involved in the regulation and maintenance of sarcomeres close to the Z-disc [210].
Cytoskeletal Proteins
The second group of RCM-associated disease genes includes different cytoskeletal structural proteins like desmin (DES) [211], filamin C (FLNC) [212], and αB-Crystallin (CRYAB) [213]. DES mutations cause besides RCM a wide spectrum of different other cardiac and skeletal myopathies including myofibrillar myopathy (MFM). Desmin is the major component of the cardiac intermediate filaments and connects different cell organelles like desmosomes, Z-bands, costameres, and presumably also the nuclei with the cytoskeleton [214]. αB-Crystallin is a member of the small heat-shock protein family and binds directly to the intermediate filaments suggesting a stabilizing function, and filamin C is an actin cross-linking protein, which is involved in the integrin-mediated cell-extracellular matrix adhesion [215]. Interestingly, mutations in those three genes cause also myofibrillar myopathy (MFM), characterized by toxic protein aggregation within the skeletal myocytes [216]. There is some evidence that an abnormal protein aggregation and accumulation in the myocardial tissue might contribute to the increased stiffness of the ventricular walls in RCM.
Genetic Counselling and Testing
Because RCM is less common than other primary cardiomyopathies, the knowledge about prevalence and genetic associations are currently limited or even missing. Most reports about disease genes are based on single families or patients. As indicated above, there is substantial overlap in the genetic etiology, but also clinical presentation with other cardiomyopathies. Moreover, some genes are also involved in skeletal muscle disease, in particular MFM. Formal genetic counselling should be performed in all familial cases, and repeated clinical screenings of first-degree relatives are recommended using ECG, Holter ECG, and echocardiogram [40]. Secondary forms of RCM and constrictive pericarditis should be considered as a differential diagnosis as some metabolic and systemic diseases may lead to instituting specific therapy. As indicated for other cardiomyopathies, genetic testing should be performed for clinical purposes using NGS panels focusing on known genes. Broader approaches such as whole-exome analysis should be considered for research studies. However, in single patients, the classification of novel variants will be challenging and often leading to VUS. Over time more genetic and phenotypic data on bigger cohorts, genotype-phenotype studies, and further functional validation are required to provide more certain recommendations.
2.7 Translational Perspectives
Classical genetic approaches such as linkage analysis and positional cloning in combination with newer technologies such as next-generation sequencing have provided dramatic progress in unraveling the genetic causes of cardiomyopathies but have also demonstrated the complexity of human disease. What we previously thought are monogenic diseases are now much more complex diseases where the primary mutation appears to be a “more or less” important contributor. Despite making some progress in the understanding of disease mechanisms and the gain of knowledge about molecular changes of specific genes and mutations, there are still a lot of unknowns in the pathogenesis of cardiomyopathies—to understand how a mutated gene leads to clinical expression. Prospectively, this inspiring field of research provides remarkable opportunities to dissect what are the genetic and non-genetic contributors in the early and late disease process, how does different disease expression develop, and how to disrupt disease progression or even prevent full clinical expression.
From the clinical and translational perspective, many more questions should be answered to translate research findings to the benefit of patient care. We still have a “genetic gap”—overall about 50% of the genetic causes of CMPs remain unknown. How do we handle the increasing number of variants of unknown significance? Is there a way to translate research information about specific mutations and how they alter protein function directly to the benefit of patients to improve health care, rather than identifying just family members at risk?
Last but not least, someday there is the hope that genetic cardiomyopathies are not only treated but may also be cured. Given the current advances in genome editing technologies and other personalized genetic and molecular approaches, also outlined in other chapters, this day may be not so far away.
References
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–16. https://doi.org/10.1161/CIRCULATIONAHA.106.174287.
Geisterfer-Lowrance AAT, Kass S, Tanigawa G, Vosberg H-P, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell. 1990;62(5):999–1006. https://doi.org/10.1016/0092-8674(90)90274-I.
Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105(4):397–408. https://doi.org/10.1093/cvr/cvv025.
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36(11):1162–4. https://doi.org/10.1038/ng1461.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet (London, England). 2000;355(9221):2119–24.
Genomes Project, C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. https://doi.org/10.1038/nature09534.
Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, et al. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7(6):751–9. https://doi.org/10.1161/CIRCGENETICS.113.000578.
Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106(2):127–39. https://doi.org/10.1007/s00392-016-1033-6.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23. https://doi.org/10.1038/gim.2015.30.
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92(4):785–9. https://doi.org/10.1161/01.cir.92.4.785.
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–79. https://doi.org/10.1093/eurheartj/ehu284.
Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58(25):e212–60. https://doi.org/10.1016/j.jacc.2011.06.011.
Nagueh SF, Mahmarian JJ. Noninvasive cardiac imaging in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2006;48(12):2410–22. https://doi.org/10.1016/j.jacc.2006.07.065.
Maron BJ, Rowin EJ, Casey SA, Garberich RF, Maron MS. What do patients with hypertrophic cardiomyopathy die from? Am J Cardiol. 2016a;117(3):434–5. https://doi.org/10.1016/j.amjcard.2015.11.013.
Maron BJ, Rowin EJ, Casey SA, Lesser JR, Garberich RF, McGriff DM, Maron MS. Hypertrophic cardiomyopathy in children, adolescents, and young adults associated with low cardiovascular mortality with contemporary management strategies. Circulation. 2016b;133(1):62–73. https://doi.org/10.1161/circulationaha.115.017633.
Maron BJ, Rowin EJ, Casey SA, Link MS, Lesser JR, Chan RH, et al. Hypertrophic cardiomyopathy in adulthood associated with low cardiovascular mortality with contemporary management strategies. J Am Coll Cardiol. 2015;65(18):1915–28. https://doi.org/10.1016/j.jacc.2015.02.061.
Charron P, Carrier L, Dubourg O, Tesson F, Desnos M, Richard P, et al. Penetrance of familial hypertrophic cardiomyopathy. Genet Couns. 1997;8(2):107–14.
Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42(10):e59. https://doi.org/10.1136/jmg.2005.033886.
Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287(10):1308–20.
Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, et al. Mutations in the gene for cardiac myosin-binding protein c and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338(18):1248–57. https://doi.org/10.1056/nejm199804303381802.
Page SP, Kounas S, Syrris P, Christiansen M, Frank-Hansen R, Andersen PS, et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet. 2012;5(2):156–66. https://doi.org/10.1161/circgenetics.111.960831.
Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5(4):391–9. https://doi.org/10.1161/CIRCGENETICS.112.962928.
Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg H-P, et al. α-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77(5):701–12. https://doi.org/10.1016/0092-8674(94)90054-X.
Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11(4):434–7.
Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11(4):438–40.
Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13(1):63–9.
Kimura A, Harada H, Park J-E, Nishi H, Satoh M, Takahashi M, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16(4):379–82.
Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, et al. α-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103(10):R39–43. https://doi.org/10.1172/JCI6460.
Andersen PS, Havndrup O, Hougs L, Sørensen KM, Jensen M, Larsen LA, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30(3):363–70. https://doi.org/10.1002/humu.20862.
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy. Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107(17):2227–32. https://doi.org/10.1161/01.cir.0000066323.15244.54.
Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol. 2016;68(25):2871–86. https://doi.org/10.1016/j.jacc.2016.08.079.
Knoll R, Buyandelger B, Lab M. The sarcomeric Z-disc and Z-discopathies. J Biomed Biotechnol. 2011;2011:569628. https://doi.org/10.1155/2011/569628.
Landstrom AP, Ackerman MJ. Beyond the cardiac myofilament: hypertrophic cardiomyopathy-associated mutations in genes that encode calcium-handling proteins. Curr Mol Med. 2012;12(5):507–18.
Millat G, Bouvagnet P, Chevalier P, Dauphin C, Simon Jouk P, Da Costa A, et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet. 2010;53(5):261–7. https://doi.org/10.1016/j.ejmg.2010.07.007.
Anan R, Greve G, Thierfelder L, Watkins H, McKenna WJ, Solomon S, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest. 1994;93(1):280–5. https://doi.org/10.1172/JCI116957.
Pasquale F, Syrris P, Kaski JP, Mogensen J, McKenna WJ, Elliott P. Long-term outcomes in hypertrophic cardiomyopathy caused by mutations in the cardiac troponin T gene. Circ Cardiovasc Genet. 2012;5(1):10–7. https://doi.org/10.1161/circgenetics.111.959973.
Alpert NR, Mohiddin SA, Tripodi D, Jacobson-Hatzell J, Vaughn-Whitley K, Brosseau C, et al. Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy-chain mutations. Am J Physiol Heart Circ Physiol. 2005;288(3):H1097–102. https://doi.org/10.1152/ajpheart.00650.2004.
Fourey D, Care M, Siminovitch KA, Weissler-Snir A, Hindieh W, Chan RH, et al. Prevalence and clinical implication of double mutations in hypertrophic cardiomyopathy: revisiting the gene-dose effect. Circ Cardiovasc Genet. 2017;10(2):e001685. https://doi.org/10.1161/circgenetics.116.001685.
Olivotto I, d’Amati G, Basso C, Van Rossum A, Patten M, Emdin M, et al. Defining phenotypes and disease progression in sarcomeric cardiomyopathies: contemporary role of clinical investigations. Cardiovasc Res. 2015;105(4):409–23. https://doi.org/10.1093/cvr/cvv024.
Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31(22):2715–26. https://doi.org/10.1093/eurheartj/ehq271.
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270–6. https://doi.org/10.1093/eurheartj/ehm342.
Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, et al. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. 2013;21(9):918–28. https://doi.org/10.1038/ejhg.2012.283.
Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. 2013;50(4):228–39. https://doi.org/10.1136/jmedgenet-2012-101270.
Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115(6):773–81. https://doi.org/10.1161/circulationaha.106.621185.
Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno-Blanes J, Helio T, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(19):1448–58. https://doi.org/10.1093/eurheartj/ehs397.
Sata M, Ikebe M. Functional analysis of the mutations in the human cardiac beta-myosin that are responsible for familial hypertrophic cardiomyopathy. Implication for the clinical outcome. J Clin Invest. 1996;98(12):2866–73. https://doi.org/10.1172/jci119115.
Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108(6):751–64. https://doi.org/10.1161/circresaha.110.231670.
van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy. Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119(11):1473–83. https://doi.org/10.1161/circulationaha.108.838672.
Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101(12):1266–73. https://doi.org/10.1161/circresaha.107.156380.
Robinson P, Mirza M, Knott A, Abdulrazzak H, Willott R, Marston S, et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem. 2002;277(43):40710–6. https://doi.org/10.1074/jbc.M203446200.
Guinto PJ, Haim TE, Dowell-Martino CC, Sibinga N, Tardiff JC. Temporal and mutation-specific alterations in Ca2+ homeostasis differentially determine the progression of cTnT-related cardiomyopathies in murine models. Am J Physiol Heart Circ Physiol. 2009;297(2):H614–26. https://doi.org/10.1152/ajpheart.01143.2008.
Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res. 2011;109(1):86–96. https://doi.org/10.1161/circresaha.111.242974.
Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106(6):1236–49. https://doi.org/10.1016/j.bpj.2014.02.011.
Kirschner SE, Becker E, Antognozzi M, Kubis HP, Francino A, Navarro-Lopez F, et al. Hypertrophic cardiomyopathy-related beta-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am J Physiol Heart Circ Physiol. 2005;288(3):H1242–51. https://doi.org/10.1152/ajpheart.00686.2004.
Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–21. https://doi.org/10.1126/science.aad3456.
Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31(1):186–94.
Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol. 2011;108(8):1171–6. https://doi.org/10.1016/j.amjcard.2011.06.022.
Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850–8. https://doi.org/10.1093/eurheartj/ehv727.
Codd MB, Sugrue DD, Gersh BJ, Melton LJ. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation. 1989;80(3):564–72. https://doi.org/10.1161/01.cir.80.3.564.
Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49(1):46–53. https://doi.org/10.1038/ng.3719.. http://www.nature.com/ng/journal/v49/n1/abs/ng.3719.html#supplementary-information
Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013;309(9):896–908. https://doi.org/10.1001/jama.2013.1363.
Kober L, Thune JJ, Nielsen JC, Haarbo J, Videbaek L, Korup E, et al. Defibrillator implantation in patients with nonischemic systolic heart failure. N Engl J Med. 2016;375(13):1221–30. https://doi.org/10.1056/NEJMoa1608029.
McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14(8):803–69. https://doi.org/10.1093/eurjhf/hfs105.
Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am Heart J. 2007;154(3):596–602. https://doi.org/10.1016/j.ahj.2007.05.014.
Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143(2):108–15.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47. https://doi.org/10.1038/nrcardio.2013.105.
Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MR. Genetic causes of dilated cardiomyopathy. Prog Pediatr Cardiol. 2014;37(1–2):13–8. https://doi.org/10.1016/j.ppedcard.2014.10.003.
Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123–35. https://doi.org/10.1093/eurheartj/ehu301.
Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201–4. https://doi.org/10.1038/ng815.
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28. https://doi.org/10.1056/NEJMoa1110186.
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7(270):270ra276. https://doi.org/10.1126/scitranslmed.3010134.
Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341(23):1715–24. https://doi.org/10.1056/nejm199912023412302.
Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343(23):1688–96. https://doi.org/10.1056/nejm200012073432304.
McNally EM, Puckelwartz MJ. Genetic variation in cardiomyopathy and cardiovascular disorders. Circ J. 2015;79(7):1409–15. https://doi.org/10.1253/circj.CJ-15-0536.
Akinrinade O, Alastalo TP, Koskenvuo JW. Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet. 2016;90(1):49–54. https://doi.org/10.1111/cge.12741.
Brayson D, Shanahan CM. Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus. 2017;8(1):17–33. https://doi.org/10.1080/19491034.2016.1260798.
Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113(3):357–69. https://doi.org/10.1172/JCI19448.
Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323–7. https://doi.org/10.1038/ng1294-323.
Arndt AK, Schafer S, Drenckhahn JD, Sabeh MK, Plovie ER, Caliebe A, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet. 2013;93(1):67–77. https://doi.org/10.1016/j.ajhg.2013.05.015.
Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, et al. Mutations in cardiac T-box factor gene <em>TBX20</em> are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81(2):280–91. https://doi.org/10.1086/519530.
Williams T, Hundertmark M, Nordbeck P, Voll S, Arias-Loza PA, Oppelt D, et al. Eya4 induces hypertrophy via regulation of p27kip1. Circ Cardiovasc Genet. 2015;8(6):752–64. https://doi.org/10.1161/circgenetics.115.001134.
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li RG, et al. GATA6 loss-of-function mutations contribute to familial dilated cardiomyopathy. Int J Mol Med. 2014;34(5):1315–22. https://doi.org/10.3892/ijmm.2014.1896.
Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu YJ, et al. A novel NKX2-5 loss-of-function mutation predisposes to familial dilated cardiomyopathy and arrhythmias. Int J Mol Med. 2015;35(2):478–86. https://doi.org/10.3892/ijmm.2014.2029.
Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18(5):766–73. https://doi.org/10.1038/nm.2693.
Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest. 2014;124(8):3419–30. https://doi.org/10.1172/jci74523.
Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299(5611):1410–3. https://doi.org/10.1126/science.1081578.
Abrol N, de Tombe PP, Robia SL. Acute inotropic and lusitropic effects of cardiomyopathic R9C mutation of phospholamban. J Biol Chem. 2015;290(11):7130–40. https://doi.org/10.1074/jbc.M114.630319.
McNair WP, Sinagra G, Taylor MRG, Di Lenarda A, Ferguson DA, Salcedo EE, et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011;57(21):2160–8. https://doi.org/10.1016/j.jacc.2010.09.084.
Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36(4):382–7.
Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–6. https://doi.org/10.1111/j.1752-8062.2008.00017.x.
Geier C, Gehmlich K, Ehler E, Hassfeld S, Perrot A, Hayess K, et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet. 2008;17(18):2753–65. https://doi.org/10.1093/hmg/ddn160.
Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG, et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab. 2003;80(1-2):207–15.
Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342(11):770–80. https://doi.org/10.1056/nejm200003163421104.
Arimura T, Ishikawa T, Nunoda S, Kawai S, Kimura A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat. 2011;32(12):1481–91. https://doi.org/10.1002/humu.21603.
Garcia-Pavia P, Syrris P, Salas C, Evans A, Mirelis JG, Cobo-Marcos M, et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart. 2011;97(21):1744–52. https://doi.org/10.1136/hrt.2011.227967.
Patel DM, Green KJ. Desmosomes in the heart: a review of clinical and mechanistic analyses. Cell Commun Adhes. 2014;21(3):109–28. https://doi.org/10.3109/15419061.2014.906533.
Behin A, Salort-Campana E, Wahbi K, Richard P, Carlier RY, Carlier P, et al. Myofibrillar myopathies: state of the art, present and future challenges. Rev Neurol (Paris). 2015;171(10):715–29. https://doi.org/10.1016/j.neurol.2015.06.002.
Sommerville RB, Vincenti MG, Winborn K, Casey A, Stitziel NO, Connolly AM, Mann DL. Diagnosis and management of adult hereditary cardio-neuromuscular disorders: a model for the multidisciplinary care of complex genetic disorders. Trends Cardiovasc Med. 2017;27(1):51–8. https://doi.org/10.1016/j.tcm.2016.06.005.
Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313(10):2121–33. https://doi.org/10.1016/j.yexcr.2007.03.028.
Kamdar F, Garry DJ. Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol. 2016;67(21):2533–46. https://doi.org/10.1016/j.jacc.2016.02.081.
Pua CJ, Bhalshankar J, Miao K, Walsh R, John S, Lim SQ, et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res. 2016;9(1):3–11. https://doi.org/10.1007/s12265-016-9673-5.
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36(34):2327–37. https://doi.org/10.1093/eurheartj/ehv253.
Hershberger RE, Morales A. LMNA-related dilated cardiomyopathy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R). Seattle (WA): University of Washington, Seattle; 1993.
Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52(15):1250–60. https://doi.org/10.1016/j.jacc.2008.06.044.
van Rijsingen IAW, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European Cohort Study. J Am Coll Cardiol. 2012;59(5):493–500. https://doi.org/10.1016/j.jacc.2011.08.078.
Franaszczyk M, Chmielewski P, Truszkowska G, Stawinski P, Michalak E, Rydzanicz M, et al. Titin truncating variants in dilated cardiomyopathy – prevalence and genotype-phenotype correlations. PLoS One. 2017;12(1):e0169007. https://doi.org/10.1371/journal.pone.0169007.
Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41(5):771–80.
Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet. 2013;21(10):1105–11. https://doi.org/10.1038/ejhg.2013.16.
Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82(2):507–13.
Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart. 2001;86(6):666–71.
Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J. 2011;32(12):1446–56. https://doi.org/10.1093/eurheartj/ehq508.
Weir-McCall JR, Yeap PM, Papagiorcopulo C, Fitzgerald K, Gandy SJ, Lambert M, et al. Left ventricular noncompaction: anatomical phenotype or distinct cardiomyopathy? J Am Coll Cardiol. 2016;68(20):2157–65. https://doi.org/10.1016/j.jacc.2016.08.054.
Ichida F, Hamamichi Y, Miyawaki T, Ono Y, Kamiya T, Akagi T, et al. Clinical features of isolated noncompaction of the ventricular myocardium: long-term clinical course, hemodynamic properties, and genetic background. J Am Coll Cardiol. 1999;34(1):233–40.
Murphy RT, Thaman R, Blanes JG, Ward D, Sevdalis E, Papra E, et al. Natural history and familial characteristics of isolated left ventricular non-compaction. Eur Heart J. 2005;26(2):187–92. https://doi.org/10.1093/eurheartj/ehi025.
Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36(2):493–500.
Belanger AR, Miller MA, Donthireddi UR, Najovits AJ, Goldman ME. New classification scheme of left ventricular noncompaction and correlation with ventricular performance. Am J Cardiol. 2008;102(1):92–6. https://doi.org/10.1016/j.amjcard.2008.02.107.
Kohli SK, Pantazis AA, Shah JS, Adeyemi B, Jackson G, McKenna WJ, et al. Diagnosis of left-ventricular non-compaction in patients with left-ventricular systolic dysfunction: time for a reappraisal of diagnostic criteria? Eur Heart J. 2008;29(1):89–95. https://doi.org/10.1093/eurheartj/ehm481.
Zemrak F, Ahlman MA, Captur G, Mohiddin SA, Kawel-Boehm N, Prince MR, et al. The relationship of left ventricular trabeculation to ventricular function and structure over a 9.5-year follow-up: the MESA study. J Am Coll Cardiol. 2014;64(19):1971–80. https://doi.org/10.1016/j.jacc.2014.08.035.
Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348(17):1639–46. https://doi.org/10.1056/NEJMoa021737.
Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the pediatric cardiomyopathy registry. J Card Fail. 2015;21(11):877–84. https://doi.org/10.1016/j.cardfail.2015.06.381.
Brescia ST, Rossano JW, Pignatelli R, Jefferies JL, Price JF, Decker JA, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation. 2013;127(22):2202–8. https://doi.org/10.1161/CIRCULATIONAHA.113.002511.
Anderson RH, Jensen B, Mohun TJ, Petersen SE, Aung N, Zemrak F, et al. Key questions relating to left ventricular noncompaction cardiomyopathy: is the emperor still wearing any clothes? Can J Cardiol. 2017;33(6):747–57. https://doi.org/10.1016/j.cjca.2017.01.017.
Freedom RM, Yoo SJ, Perrin D, Taylor G, Petersen S, Anderson RH. The morphological spectrum of ventricular noncompaction. Cardiol Young. 2005;15(4):345–64. https://doi.org/10.1017/S1047951105000752.
Kodo K, Ong SG, Jahanbani F, Termglinchan V, Hirono K, InanlooRahatloo K, et al. iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat Cell Biol. 2016;18(10):1031–42. https://doi.org/10.1038/ncb3411.
Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12(3):415–29. https://doi.org/10.1016/j.devcel.2006.12.011.
Sasse-Klaassen S, Gerull B, Oechslin E, Jenni R, Thierfelder L. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am J Med Genet A. 2003;119A(2):162–7. https://doi.org/10.1002/ajmg.a.20075.
Probst S, Oechslin E, Schuler P, Greutmann M, Boye P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4(4):367–74. https://doi.org/10.1161/CIRCGENETICS.110.959270.
Budde BS, Binner P, Waldmuller S, Hohne W, Blankenfeldt W, Hassfeld S, et al. Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta-myosin heavy chain gene. PLoS One. 2007;2(12):e1362. https://doi.org/10.1371/journal.pone.0001362.
Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, et al. The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet. 2010;3(3):232–9. https://doi.org/10.1161/CIRCGENETICS.109.903898.
Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117(22):2893–901. https://doi.org/10.1161/CIRCULATIONAHA.107.746164.
Postma AV, van Engelen K, van de Meerakker J, Rahman T, Probst S, Baars MJ, et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ Cardiovasc Genet. 2011;4(1):43–50. https://doi.org/10.1161/CIRCGENETICS.110.957985.
Wessels MW, Herkert JC, Frohn-Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, et al. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet. 2015;23(7):922–8. https://doi.org/10.1038/ejhg.2014.211.
Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007;28(16):1953–61. https://doi.org/10.1093/eurheartj/ehm239.
Hastings R, de Villiers CP, Hooper C, Ormondroyd L, Pagnamenta A, Lise S, et al. Combination of whole genome sequencing, linkage, and functional studies implicates a missense mutation in titin as a cause of autosomal dominant cardiomyopathy with features of left ventricular noncompaction. Circ Cardiovasc Genet. 2016;9(5):426–35. https://doi.org/10.1161/CIRCGENETICS.116.001431.
Bleyl SB, Mumford BR, Thompson V, Carey JC, Pysher TJ, Chin TK, Ward K. Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet. 1997;61(4):868–72. https://doi.org/10.1086/514879.
Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42(11):2014–27.
Bagnall RD, Molloy LK, Kalman JM, Semsarian C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med Genet. 2014;15:99. https://doi.org/10.1186/s12881-014-0099-0.
Milano A, Vermeer AM, Lodder EM, Barc J, Verkerk AO, Postma AV, et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol. 2014;64(8):745–56. https://doi.org/10.1016/j.jacc.2014.05.045.
Schweizer PA, Schroter J, Greiner S, Haas J, Yampolsky P, Mereles D, et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol. 2014;64(8):757–67. https://doi.org/10.1016/j.jacc.2014.06.1155.
Shan L, Makita N, Xing Y, Watanabe S, Futatani T, Ye F, et al. SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol Genet Metab. 2008;93(4):468–74.
Hermida-Prieto M, Monserrat L, Castro-Beiras A, Laredo R, Soler R, Peteiro J, et al. Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. Am J Cardiol. 2004;94(1):50–4. https://doi.org/10.1016/j.amjcard.2004.03.029.
Luxan G, Casanova JC, Martinez-Poveda B, Prados B, D'Amato G, MacGrogan D, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. 2013;19(2):193–201. https://doi.org/10.1038/nm.3046.
Hoedemaekers YM, Klaassen S. Left ventricular noncompaction. In: Baars HF, Doevendans PAFM, Houweling A, Tintelen JP, editors. Clinical cardiogenetics. Berlin: Springer; 2016. p. 113–35. https://doi.org/10.1007/978-3-319-44203-7.
Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001;103(9):1256–63.
Williams T, Machann W, Kuhler L, Hamm H, Muller-Hocker J, Zimmer M, et al. Novel desmoplakin mutation: juvenile biventricular cardiomyopathy with left ventricular non-compaction and acantholytic palmoplantar keratoderma. Clin Res Cardiol. 2011;100(12):1087–93. https://doi.org/10.1007/s00392-011-0345-9.
Stahli BE, Gebhard C, Biaggi P, Klaassen S, Valsangiacomo Buechel E, Attenhofer Jost CH, et al. Left ventricular non-compaction: prevalence in congenital heart disease. Int J Cardiol. 2013;167(6):2477–81. https://doi.org/10.1016/j.ijcard.2012.05.095.
Ouyang P, Saarel E, Bai Y, Luo C, Lv Q, Xu Y, et al. A de novo mutation in NKX2.5 associated with atrial septal defects, ventricular noncompaction, syncope and sudden death. Clin Chim Acta. 2011;412(1-2):170–5. https://doi.org/10.1016/j.cca.2010.09.035.
Finsterer J, Stollberger C, Towbin JA. Left ventricular noncompaction cardiomyopathy: cardiac, neuromuscular, and genetic factors. Nat Rev Cardiol. 2017;14(4):224–37. https://doi.org/10.1038/nrcardio.2016.207.
Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925–31. https://doi.org/10.1542/peds.2004-0718.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121(13):1533–41. https://doi.org/10.1161/CIRCULATIONAHA.108.840827.
Lancisi GM. De motu cordis et aneurysmatibus opus postumum. 1740.
Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384–98.
Romero J, Mejia-Lopez E, Manrique C, Lucariello R. Arrhythmogenic right ventricular cardiomyopathy (ARVC/D): a systematic literature review. Clin Med Insights Cardiol. 2013;7:97–114. https://doi.org/10.4137/CMC.S10940.
Bhonsale A, Te Riele A, Sawant AC, Groeneweg JA, James CA, Murray B, et al. Cardiac phenotype and long-term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm. 2017;14(6):883–91. https://doi.org/10.1016/j.hrthm.2017.02.013.
McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215–8.
van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14(11):1199–207. https://doi.org/10.1093/eurjhf/hfs119.
Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55(6):587–97. https://doi.org/10.1016/j.jacc.2009.11.020.
Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19(23):4595–607. https://doi.org/10.1093/hmg/ddq387.
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71(5):1200–6. https://doi.org/10.1086/344208.
Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79(6):1081–8. https://doi.org/10.1086/509044.
Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79(5):978–84. https://doi.org/10.1086/509122.
Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113(9):1171–9. https://doi.org/10.1161/CIRCULATIONAHA.105.583674.
Vermij SH, Abriel H, van Veen TA. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc Res. 2017;113(3):259–75. https://doi.org/10.1093/cvr/cvw259.
Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10(2):e001605. https://doi.org/10.1161/CIRCGENETICS.116.001605.
van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34(3):201–10. https://doi.org/10.1093/eurheartj/ehs373.
Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12(6):861–8. https://doi.org/10.1093/europace/euq104.
Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10(3):189–94.
Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65(2):366–73. https://doi.org/10.1016/j.cardiores.2004.10.005.
Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–21. https://doi.org/10.1016/j.ajhg.2008.01.010.
Erkapic D, Neumann T, Schmitt J, Sperzel J, Berkowitsch A, Kuniss M, et al. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace. 2008;10(7):884–7. https://doi.org/10.1093/europace/eun065.
Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124(8):876–85. https://doi.org/10.1161/CIRCULATIONAHA.110.005405.
Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33(9):1128–36. https://doi.org/10.1093/eurheartj/ehr451.
Lopez-Ayala JM, Ortiz-Genga M, Gomez-Milanes I, Lopez-Cuenca D, Ruiz-Espejo F, Sanchez-Munoz JJ, et al. A mutation in the Z-line Cypher/ZASP protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clin Genet. 2015;88(2):172–6. https://doi.org/10.1111/cge.12458.
Xiong Q, Cao Q, Zhou Q, Xie J, Shen Y, Wan R, et al. Arrhythmogenic cardiomyopathy in a patient with a rare loss-of-function KCNQ1 mutation. J Am Heart Assoc. 2015;4(1):e001526. https://doi.org/10.1161/JAHA.114.001526.
Protonotarios N, Tsatsopoulou A. Naxos disease: cardiocutaneous syndrome due to cell adhesion defect. Orphanet J Rare Dis. 2006;1:4. https://doi.org/10.1186/1750-1172-1-4.
Brogna S, Wen J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol. 2009;16(2):107–13. https://doi.org/10.1038/nsmb.1550.
Zhang Z, Stroud MJ, Zhang J, Fang X, Ouyang K, Kimura K, et al. Normalization of Naxos plakoglobin levels restores cardiac function in mice. J Clin Invest. 2015;125(4):1708–12. https://doi.org/10.1172/JCI80335.
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9(18):2761–6.
Green KJ, Stappenbeck TS, Parry DA, Virata ML. Structure of desmoplakin and its association with intermediate filaments. J Dermatol. 1992;19(11):765–9.
Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J Biol Chem. 2002;277(12):10512–22. https://doi.org/10.1074/jbc.M108765200.
Nekrasova OE, Amargo EV, Smith WO, Chen J, Kreitzer GE, Green KJ. Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J Cell Biol. 2011;195(7):1185–203. https://doi.org/10.1083/jcb.201106057.
Kirchner F, Schuetz A, Boldt LH, Martens K, Dittmar G, Haverkamp W, et al. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ Cardiovasc Genet. 2012;5(4):400–11. https://doi.org/10.1161/CIRCGENETICS.111.961854.
Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, Shapiro L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc Natl Acad Sci USA. 2016;113(26):7160–5. https://doi.org/10.1073/pnas.1606272113.
Gerull B, Kirchner F, Chong JX, Tagoe J, Chandrasekharan K, Strohm O, et al. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet. 2013;6(4):327–36. https://doi.org/10.1161/CIRCGENETICS.113.000097.
Simpson MA, Mansour S, Ahnood D, Kalidas K, Patton MA, McKenna WJ, et al. Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology. 2009;113(1):28–34. https://doi.org/10.1159/000165696.
Wong JA, Duff HJ, Yuen T, Kolman L, Exner DV, Weeks SG, Gerull B. Phenotypic analysis of arrhythmogenic cardiomyopathy in the Hutterite population: role of electrocardiogram in identifying high-risk desmocollin-2 carriers. J Am Heart Assoc. 2014;3(6):e001407. https://doi.org/10.1161/JAHA.114.001407.
van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6(11):1574–83. https://doi.org/10.1016/j.hrthm.2009.07.041.
Brodehl A, Hedde PN, Dieding M, Fatima A, Walhorn V, Gayda S, et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J Biol Chem. 2012;287(19):16047–57. https://doi.org/10.1074/jbc.M111.313841.
Turkowski KL, Tester DJ, Bos JM, Haugaa KH, Ackerman MJ. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit Heart Dis. 2017;12(2):226–35. https://doi.org/10.1111/chd.12462.
Bers DM, Perez-Reyes E. Ca channels in cardiac myocytes: structure and function in Ca influx and intracellular Ca release. Cardiovasc Res. 1999;42(2):339–60.
Kranias EG, Bers DM. Calcium and cardiomyopathies. Subcell Biochem. 2007;45:523–37.
Ma Y, Zou H, Zhu XX, Pang J, Xu Q, Jin QY, et al. Transforming growth factor beta: a potential biomarker and therapeutic target of ventricular remodeling. Oncotarget. 2017;8(32):53780–90. https://doi.org/10.18632/oncotarget.17255.
Milting H, Klauke B, Christensen AH, Musebeck J, Walhorn V, Grannemann S, et al. The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. Eur Heart J. 2015;36(14):872–81. https://doi.org/10.1093/eurheartj/ehu077.
Bengtsson L, Otto H. LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J Cell Sci. 2008;121(Pt 4):536–48. https://doi.org/10.1242/jcs.019281.
Garcia MJ. Constrictive pericarditis versus restrictive cardiomyopathy? J Am Coll Cardiol. 2016;67(17):2061–76. https://doi.org/10.1016/j.jacc.2016.01.076.
Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336(4):267–76. https://doi.org/10.1056/NEJM199701233360407.
Schulz V, Hendig D, Szliska C, Gotting C, Kleesiek K. Novel mutations in the ABCC6 gene of German patients with pseudoxanthoma elasticum. Hum Biol. 2005;77(3):367–84.
Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–300. https://doi.org/10.1161/CIRCULATIONAHA.111.078915.
Kostareva A, Kiselev A, Gudkova A, Frishman G, Ruepp A, Frishman D, et al. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS One. 2016;11(9):e0163362. https://doi.org/10.1371/journal.pone.0163362.
Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest. 2003;111(2):209–16. https://doi.org/10.1172/JCI16336.
Filatov VL, Katrukha AG, Bulargina TV, Gusev NB. Troponin: structure, properties, and mechanism of functioning. Biochemistry (Mosc). 1999;64(9):969–85.
Peddy SB, Vricella LA, Crosson JE, Oswald GL, Cohn RD, Cameron DE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117(5):1830–3. https://doi.org/10.1542/peds.2005-2301.
Ploski R, Rydzanicz M, Ksiazczyk TM, Franaszczyk M, Pollak A, Kosinska J, et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am J Med Genet A. 2016;170(12):3241–8. https://doi.org/10.1002/ajmg.a.37860.
Marques MA, de Oliveira GA. Cardiac troponin and tropomyosin: structural and cellular perspectives to unveil the hypertrophic cardiomyopathy phenotype. Front Physiol. 2016;7:429. https://doi.org/10.3389/fphys.2016.00429.
Karam S, Raboisson MJ, Ducreux C, Chalabreysse L, Millat G, Bozio A, Bouvagnet P. A de novo mutation of the beta cardiac myosin heavy chain gene in an infantile restrictive cardiomyopathy. Congenit Heart Dis. 2008;3(2):138–43. https://doi.org/10.1111/j.1747-0803.2008.00165.x.
Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94(11):1478–84. https://doi.org/10.1136/hrt.2007.134684.
Wu W, Lu CX, Wang YN, Liu F, Chen W, Liu YT, et al. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosin-binding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J Am Heart Assoc. 2015;4(7):e001879. https://doi.org/10.1161/JAHA.115.001879.
Peled Y, Gramlich M, Yoskovitz G, Feinberg MS, Afek A, Polak-Charcon S, et al. Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol. 2014;171(1):24–30. https://doi.org/10.1016/j.ijcard.2013.11.037.
Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21(9):2039–53. https://doi.org/10.1093/hmg/dds022.
Bang ML, Mudry RE, McElhinny AS, Trombitas K, Geach AJ, Yamasaki R, et al. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153(2):413–27.
Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006;8(5):477–83. https://doi.org/10.1016/j.ejheart.2005.11.003.
Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat. 2016;37(3):269–79. https://doi.org/10.1002/humu.22942.
Brodehl A, Gaertner-Rommel A, Klauke B, Grewe SA, Schirmer I, Peterschroder A, et al. The novel alphaB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum Mutat. 2017;38(8):947–52. https://doi.org/10.1002/humu.23248.
Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119(7):1806–13. https://doi.org/10.1172/JCI38027.
Jahed Z, Shams H, Mehrbod M, Mofrad MR. Mechanotransduction pathways linking the extracellular matrix to the nucleus. Int Rev Cell Mol Biol. 2014;310:171–220. https://doi.org/10.1016/B978-0-12-800180-6.00005-0.
Olive M, Kley RA, Goldfarb LG. Myofibrillar myopathies: new developments. Curr Opin Neurol. 2013;26(5):527–35. https://doi.org/10.1097/WCO.0b013e328364d6b1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest
Brenda Gerull declares that she has no conflict of interest. Sabine Klaassen declares that she has no conflict of interest. Andreas Brodehl declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gerull, B., Klaassen, S., Brodehl, A. (2019). The Genetic Landscape of Cardiomyopathies. In: Erdmann, J., Moretti, A. (eds) Genetic Causes of Cardiac Disease. Cardiac and Vascular Biology, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-030-27371-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-27371-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27370-5
Online ISBN: 978-3-030-27371-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)